

Abstract
The transmembrane mucin MUC1 is overexpressed in most ductal carcinomas, and its overexpression is frequently associated with metastatic progression. MUC1 can drive tumor initiation and progression via interactions with many oncogenic partners, including β-catenin, the epidermal growth factor receptor (EGFR) and Src. The decoy peptide PMIP (Protein transduction domain MUC1 Inhibitory Peptide) has been shown to inhibit the tumor promoting activities of MUC1 in breast and lung cancer, including cell growth and invasion, and its usage suppresses metastatic progression in mouse models of breast cancer. To further characterize the reduced metastasis observed upon PMIP treatment, we performed motility assays and observed that PMIP inhibits cell motility of breast cancer cells. To determine the mechanism by which PMIP inhibits motility, we evaluated changes in global gene transcription upon PMIP treatment, and identified a number of genes with altered expression in response to PMIP. Among these genes is the metastatic mediator, c-Met, a transmembrane tyrosine kinase that can promote cell scattering, migration and invasion. To further investigate the role of c-Met in MUC1-dependent metastatic events, we evaluated the effects of MUC1 expression and EGFR activation on breast cancer cell scattering, branching and migration. We found that MUC1 strongly promoted all of these events and this effect was further amplified by EGF treatment. Importantly, the effect of MUC1 and EGF on these phenotypes was dependent upon c-Met activity. Overall, these results indicate that PMIP can block the expression of a key metastatic mediator, further advancing its potential use as a clinical therapeutic.
Keywords: MUC1, Epidermal Growth Factor Receptor, therapy, c-Met, migration, breast cancer
Introduction
MUC1 is a hetero-dimeric O-glycosylated transmembrane mucin highly expressed in ductal carcinomas, including carcinomas of the breast, lung, pancreas, colon and ovary [1–5]. MUC1 expression has been shown to correlate with increased severity of disease and metastatic progression in gastric, prostate, breast and pancreatic cancer in addition to thymic epithelial tumors [6–11] and MUC1 cellular mislocalization has been shown to correlate with disease progression in non-small cell lung cancer [12]. In addition, MUC1 expression is increased specifically in metastatic colorectal cancer [13], highlighting the importance of MUC1 for cancer progression and metastasis.
MUC1 has been implicated as a driver of metastatic progression, via specific activities of both the extracellular domain and its intracellular domain (MUC1-C) [14–16]. The extracellular domain of MUC1 has been shown to increase endothelial cell adhesion and promote intravasation and extravasation through interactions with ICAM-1 and galectin-3 [17, 18]. In addition, localization of MUC1 can alter integrin-dependent binding and cell adhesion [19]. Furthermore, MUC1 can drive invasion and migration via the upregulation of matrix metalloproteinase 13 (MMP13), a protein that aids in cell invasion [20], and through the induction of epithelial to mesenchymal transition in pancreatic cancer cells [21]. These analyses demonstrate a role for MUC1 in promoting cell migration, invasion, and metastasis. In addition, MMTV-MUC1 transgenic models of breast cancer displayed lung metastasis that was dependent upon expression of the cytoplasmic portion of MUC1 [22].
MUC1-C has been shown to interact with a number of oncogenic proteins, promoting either their membrane function or activity in the nucleus [23–28]. One such interaction is via the Epidermal Growth Factor Receptor (EGFR), wherein MUC1 expression promotes the oncogenic properties of EGFR by prolonging the activity of EGFR and promoting its nuclear function [29, 30]. Studies in the WAP-TGFα model of EGFR-dependent breast cancer showed that loss of Muc1 expression suppresses tumor formation by greater than 60% and significantly blocks early hyperplasia (note: mouse Muc1 and human MUC1 are denoted as such). In addition, loss of Muc1 expression completely abrogates metastatic progression in this model [31]. Interactions between MUC1 and EGFR reduce the ligand-dependent degradation of EGFR, and alter EGFR trafficking from the lysosome, resulting in preferential recycling to the plasma membrane [29]. In addition, MUC1 co-expression promotes retro-translocation of EGFR to the nucleus, where interaction with MUC1 promotes the binding of EGFR to the CCND1 promoter [30]. MUC1-C has also been shown to interact with β-catenin and p120-catenin to promote their translocation to the nucleus and their activity as transcriptional cofactors [16, 32]. Overall, MUC1 promotes the intracellular localization and activity of a number of proto-oncogenes, including EGFR, FGFR, PDGFRβ, β-catenin, p120 catenin, src, estrogen receptor, p53, HSP70 and HSP90 [14–16, 29, 30, 32–35].
In previous studies, we have generated a MUC1 ‘decoy’ peptide to block protein-protein interactions between MUC1 and EGFR and MUC1 and β-catenin. This was accomplished by synthesizing a 15-amino acid region of MUC1-C that was previously shown to be required for interactions between these proteins in tandem with a Cell Penetrating Peptide (CPP) [36–39]. The CPP allows adjacent peptide sequences to be taken up into cells across the plasma membrane, where interactions with endogenous intracellular proteins can occur. The MUC1 peptide was termed Protein Transduction Domain – MUC1 Inhibitory Peptide (PMIP), and in vitro studies demonstrated that treatment of breast cancer cells with PMIP resulted in an inhibition of interaction between MUC1 and EGFR as well as an inhibition of the colocalization between MUC1 and β-catenin [39]. PMIP significantly inhibited the growth and invasion of breast cancer cell lines in vivo and the initiation and progression of mammary gland tumors in the MMTV-pyMT transgenic model. In animals treated with PMIP, analysis of remaining mammary glands and tumors revealed a reduction in MUC1 expression after treatment. In addition, PMIP significantly suppressed the ability of primary breast tumors to form secondary metastasis in a MDA-MB-231 orthotopic model of breast cancer [39]. Subsequent to this work, Klinge et al., reported PMIP treatment of lung cancer cells resulted in a decrease in proliferation, decreased Estrogen Receptor α (ERα)-dependent gene transcription, and altered subcellular localization of MUC1, ERα and ERβ [40].
In the current study, we investigated the mechanism by which MUC1 promotes metastatic progression and whether PMIP could inhibit this phenotype. Analysis of MUC1-induced migration in Matrigel revealed an induction of both migration and cell scattering. Using microarray technology, we identified c-Met mRNA as being significantly downregulated by PMIP. Further characterization of c-Met regulation demonstrated a role for c-Met in driving MUC1 and EGFR-dependent migration and scattering.
Methods
Microarray
BT20 breast cancer cells were treated for one hour with 50μM PMIP, 50μM control peptide, or peptide vehicle (PBS) and RNA was collected after 24 hours. Six CodeLink Human Whole Genome Bioarrays were hybridized, and data analysis was performed by the University of Arizona Genomics Facility Core. The False Discovery Rate method of statistical significance was employed to interpret the data [41], in addition to the GeneSpring program by Agilent.
RT-PCR
RNA was extracted from cells using the RNeasy Mini Kit (Qiagen). The Superscript III First-Strand Synthesis System for RT-PCR was used to generate cDNA (Invitrogen). Polymerase chain reaction was performed using Crimson Taq DNA Polymerase (New England Biolabs) and the following gene-specific primers: MET: sense 5′-ACTCCCCCTGAAAACCAAAGCC-3′, antisense 5′-GGCTTACACTTCGGGCACTTAC-3′; ACTB: sense 5′-AAGAGAGGCATCCTCACCCT-3′, antisense 5′-TACATGGCTGGGGTGTTGAA-3′ [42]. Cycling conditions were: MET: denaturing at 95°C for 30 s, annealing at 62°C for 30 s, and extension at 68°C for 30 s; ACTB: denaturing at 95°C for 30 s, annealing at 58°C for 30 s, and extension at 68°C for 30 s. The PCR products were separated on a 1% agarose gel. Gels were imaged and densitometry performed using CareStream Molecular Imaging Software (v 5.0.4.46).
qRT-PCR
RNA was extracted from cells using the RNeasy Mini Kit (Qiagen) and cDNA was generated through the use of Transcriptor First Strand cDNA Synthesis Kit (Roche). qPCR was performed using the following primers synthesized by Operon: MET: 5′-AAATGTGCATGAAGCAGGAA-3′ and 5′-TCTCTGAATTAGAGCGATGTTGA-3′; ALAS1: 5′-GCCTCTGCAGTCCTCAGC-3′ and 5′-AACAACACTCTCCATGTTCAGG-3′. Cycling conditions were: initial denaturing at 95°C for 2min, 55 cycles of: denaturing at 95°C for 10 sec, annealing at 62–52°C for 10 sec (temperature was reduced 0.5°C per cycle), and extension at 72°C for 5 sec, final extension at 72°C for 2 min, and a final cooling to 37°C for 2 min. Products were detected using Roche Universal Probe Library gene-specific probes. PCR was performed on a Lightcycler 2.0 (Roche).
Cell culture
MDA-MB-231 cells were acquired from the American Type Culture Collection (ATCC) and grown in RPMI 1640 (Cellgro) with 10% FBS (PAA Laboratories) and 1% Pen/Strep (Cellgro). Cells were selected and grown with 1 mg/mL G418 (Omega Scientific, GN-04). BT20 cells were acquired from ATCC and grown in EMEM (ATCC) with 10% FBS and 1% Pen/Strep. All cells were grown in 5% CO2.
Plasmids and transfection reagents
MDA-MB-231 cells were transfected with pCDNA3 plasmid containing the full length MUC1 cDNA, which was a kind gift from MA Hollingsworth (University of Nebraska Medical Center). Lipofectamine 2000 (Invitrogen) was used to transfect constructs into cells according to the manufacturer’s specifications.
Reagents and antibodies
Human recombinant EGF was from Fisher. SU11274 was from EMD Chemicals. Human recombinant HGF was from R&D Systems. PMIP was produced by GenScript (sequence: NH2-YARAAARQARAPYEKVSAGNGGSSLS-COOH) and reconstituted in PBS at a concentration of 1mmol/L. MUC-1 antibody was obtained from Thermo Scientific (AB-5), and β-actin antibody was obtained from Sigma (AC-15).
Western blot
Cells were lysed in cell lysis buffer (20 mM Tris, pH 7.5, 150 mM NaCl, 1% NP40, 5 mM EDTA, pH 8.0, and 10 mM sodium fluoride, 2 mM sodium orthovanadate, 50 μM ammonium molybdate, and Complete (Roche) protease inhibitors). Cell lysates were vortexed briefly, centrifuged, and the supernatant stored at −80°C. Protein concentrations were determined by BCA assay (Pierce). Proteins were separated by SDS-PAGE and transferred onto PVDF membrane (Millipore). Membrane was blocked in 3% nonfat milk in PBS (0.1% Tween20 (Fisher Scientific)) or 3% BSA in TBS (0.1% Tween20 (Fisher Scientific)) and immunoblotted. The membrane was then treated with Super Signal West Pico Chemiluminscent Substrate (Pierce), visualized on Full Speed Blue film (American Digital Imaging & Medical Products, Inc.), and developed with a Konica SRX-101A. Blot images were manipulated as follows: images were converted to black and white and brightness was adjusted using Adobe Photoshop Elements Version 9.0.
Scattering assay
MDA-MB-231 cells were seeded onto Matrigel at a density of 6.0 × 103 cells per 8-well chamber and incubated in RPMI with 10%FBS, 1% Pen/Strep, and 1 μg/mL G418. Cells were treated with 10 ng/mL EGF, 20 ng/mL HGF, both ligands, or were left untreated. Media and treatment were changed every other day. To count single scattered cells, cells less than 50 pixels in diameter were considered a single cell. We performed a two-tailed student t-test in order to define statistical significance. Images were manipulated as follows: all images were converted to black and white, shadows were lightened, highlights were darkened, and brightness and contrast were adjusted using Adobe Photoshop Elements Version 9.0.
Scratch assay
MDA-MB-231 cells and BT20 cells were seeded and allowed to grow to complete confluence. A scratch was introduced and media was replaced with complete media containing either 10 ng/mL EGF, 50 μM PMIP, 5 μM SU11274 or no treatment. Cells were observed at 6 hours (MDA-MB-231) or 24 hours (BT20) after scratching. Image J software was used to analyze the area of the scratch, and we performed a two-tailed student t-test to determine statistical significance. Images were manipulated as follows: all images were converted to black and white, shadows were lightened, highlights were darkened, and brightness and contrast were adjusted using Adobe Photoshop Elements Version 9.0.
Results
PMIP inhibits cell motility
MUC1 promotes EGFR-dependent transformation in the WAP-TGFα model of breast cancer, inhibits ligand-dependent degradation of EGFR and promotes EGFR-dependent cyclin D1 expression [29–31]. The cell penetrating peptide to the MUC1-cytoplasmic domain, PMIP, blocks MUC1 and EGFR interactions and inhibits tumor progression in both the MMTV-pyMT transgenic model of breast cancer and the scid-MDA-MB-231 orthotopic model of breast cancer [39]. In addition to blocking tumor progression, PMIP treatment results in a reduction of metastasis in the scid-MDA-MB-231 mouse model [39]. In order to determine whether inhibition of metastasis is due to decreased cell motility upon PMIP treatment, we performed an in vitro wound healing assay. For this assay, parental BT20 breast cancer cells or MDA-MB-231 breast cancer cells transfected with either a CMV-MUC1 overexpression construct (hereafter referred to as MDA-MB-231-MUC1) or the CMV empty vector (MDA-MB-231-control) were used (Figure 1N). Cells were seeded on Collagen IV-coated tissue culture plastic, allowed to grow to complete confluence and then a scratch was introduced into the monolayer. Complete media alone (Figure 1G and H), complete media plus 10 ng/mL EGF (Figure 1A–D and I–J), or complete media plus 10 ng/mL EGF and 50 μM PMIP (Figure 1E–F and K–L) was added to cells. Activation of phospho-tyrosine by EGF was observed in EGF-treated groups, although PMIP treatment reduced total pEGFR/EGFR levels (Figure 1P) as was previously shown [39]. Cells were observed at either 6 hours (MDA-MB-231) or 24 hours (BT20) after treatment to assess motility into the scratch. Six hours after treatment, it was found that MDA-MB-231-MUC1 cells filled the scratch 23% more than MDA-MB-231-control cells (p=0.002), and PMIP treatment reduced this behavior back to baseline (p=0.0004) (Figure 1M). 24 hours after treatment of BT20 cells, PMIP treatment reduced EGF-dependent migration by 28% (p=0.017) (Figure 1O). This indicates that MUC1 is a driver of cell motility, and that PMIP blocks this activity. These data indicate that PMIP functions to block EGF-dependent migration.
Figure 1.

PMIP treatment inhibits in vitro wound healing. MDA-MB-231-control or MDA-MB-231-MUC1 cells (A–F), or BT20 cells (G–L) were grown to complete confluence on Collagen IV-coated tissue culture plates and a scratch was introduced. Cells were untreated (G and H), treated with 10 ng/mL EGF alone (A–D and I–J) or EGF and 50 μM PMIP (E, F, K, and L). Area filled was determined by demarcating cell front as demonstrated by dotted line and measuring the area of the center space. Area filled at end of experiment is quantified in M (for MDA-MB-231) and O (for BT20). MUC1 expression in MDA-MB-231-MUC1 and MDA-MB-231-control cells is shown in N. Phospho-tyrosine expression in response to EGF (Santa Cruz PY99), EGFR (Santa Cruz 1005-G), and MUC1 (NeoMarkers AB-5) expression are shown in P. Scale bar indicates 100 μm. *p<0.05, ** p< 0.01, *** p< 0.001.
PMIP treatment results in downregulation of c-Met transcript
To analyze a potential mechanism by which PMIP reduces metastasis, we performed a Codelink Whole Human Genome Bioarray in BT20 breast cancer cells. Cells were treated with PMIP, cell penetrating peptide (PTD-4) or vehicle (PBS) for 24 hours, RNA was collected and hybridized to the array (Figure 2A). We observed differential regulation greater than 3-fold in ninety-seven genes affecting a variety of processes with a majority of the downregulated genes involved in cell cycle, cell proliferation, and signal transduction (Figure 2A and B, and Supplemental Figure 1). Among the downregulated genes was MET, which was downregulated 3-fold with PMIP treatment. To verify that MET expression was regulated by PMIP treatment, we performed semi-quantitative RT-PCR on BT20 cells treated with 10 μM or 20 μM PMIP, compared MET expression to untreated cells, and found a dose-dependent reduction of MET expression in response to PMIP treatment (Figure 2C). Additionally, we performed qRT-PCR on BT20 cells treated with 50 μM PMIP, the same concentration that was used in the microarray study. We compared MET expression in these cells to cells treated with vehicle (PBS) and found a significant reduction in MET expression upon PMIP treatment (Figure 2D). Next, the effect of PMIP treatment on MET expression was evaluated by RT-PCR in MCF10A cells, a non-transformed breast epithelial cell line (Figure 2E). While MET is expressed in MCF10A, no significant reduction was found upon PMIP treatment, indicating that the effect of PMIP may be tumor-specific. These data indicate that one potential mechanism by which PMIP blocks metastasis is through the down-regulation of MET.
Figure 2.

PMIP treatment changes gene transcription. BT20 breast cancer cells (A–D) or MCF10A immortalized breast epithelial cells (E) were treated with 50 μM PMIP, control peptide (TAT), or vehicle (PBS) and analyzed for differences in gene expression. (A) Induction of gene expression (red) and repression of gene expression (green) are shown for the ninety-seven expression changes detected. The accession number for c-Met is highlighted in yellow. (B) Changes in gene expression grouped by biological process. (C) RNA was extracted from untreated BT20 cells or cells treated with PMIP and analyzed for relative MET expression by RT-PCR. MET expression is normalized to ACTB expression. (D) RNA was extracted from BT20 cells treated with PMIP or vehicle (PBS) and analyzed for relative MET expression by qRT-PCR. MET expression is normalized to ALAS1 expression. (E) RNA was extracted from MCF10A cells treated with PMIP or vehicle (PBS) and analyzed for relative MET expression by RT-PCR. * p<0.05, ** p<0.01, *** p<0.001, NS=no significance.
Inhibition of c-Met activity decreases cell motility
PMIP is hypothesized to act by blocking MUC1-dependent functions in the cell. To determine whether MUC1-dependent migration relied on c-Met activity, we performed an in vitro wound healing assay on Collagen Type IV and Fibronectin extracellular matrix substrates with the c-Met kinase inhibitor, SU11274. MDA-MB-231-control and MDA-MB-231-MUC1 cells were seeded onto Collagen Type IV or Fibronectin and scratched as described in Figure 1. To determine the effect of c-Met activity on MUC1-dependent migration, cells were either treated with DMSO vehicle and 10 ng/mL EGF (Figure 3A, B, C and D) or supplemented with 5 μM SU11274 (Figure 3E and F). As seen before, MDA-MB-231-MUC1 cells exhibited greater wound healing capacity than MDA-MB-231-control cells (Figure 3D vs 3B). Addition of SU11274 significantly decreased scratch filling in MUC1-overexpressing cells (Figure 3F vs 3D), demonstrating that MUC1-driven cell motility requires activation of c-Met. On Collagen Type IV, MUC1 increased migration by 19% (p=0.002), which was repressed by c-Met inhibitor back to baseline levels (p=0.0002). On Fibronectin, MUC1 increased migration by 36% (p=0.032), which again, was repressed by c-Met inhibitor to control levels (p=0.033) (Figure 3G and 3H). In addition to evaluating migration in MDA-MB-231 cells, migration was also assessed in BT20 cells which were treated with either complete media (Figure 3I and J), complete media plus 10 ng/mL EGF (Figure 3K and L), or complete media plus 10 ng/mL EGF and 5 μM c-Met inhibitor (Figure 3M and N). Analysis of c-Met activity revealed an induction of phospho-Met in response to EGF compared to control (Figure 3P middle lane vs left lane), while this induction is blocked by the c-Met kinase inhibitor (Figure 3P right lane vs middle lane). Note that treatment with either EGF or the c-Met kinase inhibitor does not alter total MUC1 levels. When evaluated in BT20 cells (Figure 3I–N), EGF-stimulated migration (on both Collagen IV and Fibronectin) was again blocked by Met inhibitor. As seen in MDA-MB-231 cells, the c-Met kinase inhibitor blocks wound healing in BT20 cells. On Collagen IV, the c-Met kinase inhibitor reduced migration of BT20 cells by 28% (p=0.013) (data not shown), and on Fibronectin, migration was reduced by 27% (p=0.0001) (Figure 3O).
Figure 3.

c-Met kinase inhibitor (SU11274) inhibits MUC1-dependent in vitro wound healing. MDA-MB-231-control and MDA-MB-231-MUC1 cells (A–F) or BT20 breast cancer cells (I–N) were plated and scratched as described in Figure 1 on both Collagen IV and Fibronectin substrates, after which cells were treated with either DMSO vehicle (A and B), complete media (I and J), 10 ng/mL EGF (A–D and K–L), or 10 ng/mL EGF plus 5 μM SU11274 (E–F and M–N), a selective c-Met kinase inhibitor. Images shown are on Collagen IV substrate. Area filled was determined as in Figure 1. Quantification of area filled is shown in G and H (MDA-MB-231) and O (BT20). (P) Protein lysates were created from BT20 cells grown on Fibronectin overnight in either complete media (left lane), complete media plus 10 ng/mL EGF (middle lane), or complete media plus 10 ng/mL EGF and 5 μM SU11274 (right lane). 40 μg of lysate per treatment were separated by SDS-PAGE and probed by immunoblot to evaluate c-Met phosphorylation (Cell Signaling 3D7), total c-Met (Santa Cruz C-28), and MUC1 expression (NeoMarkers AB-5). Scale bar represents 100 μm. * p<0.05, ** p<0.01, *** p<0.001.
MUC1 expression and EGF activation promote cell scattering and the formation of invasive branching
One of the hallmarks of c-Met activity is its ability to induce cell scattering. To evaluate the effects of MUC1 and EGF on cell scattering, cells were plated in Matrigel and evaluated for single-cell scattering away from primary colonies. For this assay, MDA-MB-231 cells were seeded in Matrigel and grown for thirteen days in the presence of 10% serum. During this time, cells formed colonies and dispersed from the periphery of the colonies through the surrounding matrix (Figure 4, arrowheads). To determine the effect of MUC1 expression on scattering in this assay, MDA-MB-231-MUC1 cells or MDA-MB-231-control cells were seeded in Matrigel and single cells were counted in the matrix surrounding the cell colonies. We observed that MDA-MB-231-MUC1 exhibited greater cell scattering than MDA-MB-231-control cells (Figure 4B and D vs A and C, arrowheads, and quantified in Figure 4E). We also noticed that colonies of MDA-MB-231-MUC1 cells occasionally had branches invading into the surrounding Matrigel matrix, whereas we did not see these branches in MDA-MB-231-control cells. (Figure 4B and D vs A and C, arrows). These results show that MUC1 promotes cell scattering in breast cancer cells grown on Matrigel.
Figure 4.

MUC1 expression and EGF treatment promote cell scattering and matrix invasion. MDA-MB-231-control (A,C) or MDA-MB-231-MUC1 (B,D) were seeded on Matrigel in complete media (A,B) or complete media with 10ng/mL EGF (C,D). Arrowheads indicate single cells, and arrows indicate invasive protrusions. Images were taken after 8 days, and (E) single cells around the cell colonies were counted after 13 days for eight images per treatment in three independent experiments. Shown is quantification from one experiment, though similar results were found for all three. Scale bar represents 50 μm. * p<0.05, ** p<0.01, ***p<0.001
To evaluate the role of EGFR activity on MUC1-dependent cell scattering, we treated MDA-MB-231 cells with 10ng/ml human recombinant EGF to stimulate EGFR and evaluated scattering as described above. We found that EGF-treated MDA-MB-231-MUC1 cells exhibited more cell scattering than MDA-MB-231-MUC1 cells grown in the absence of EGF (Figure 4C and D vs A and B, arrowheads, and quantified in Figure 4E). We also observed that EGF-treated MDA-MB-231-control cell colonies possessed invasive branching similar to those seen in MDA-MB-231-MUC1 cells without EGF treatment, and the invasive phenotype of the cell colonies was most apparent in cells both overexpressing MUC1 and treated with EGF (Figure 4C and D vs A and B, arrows). These results indicate that EGFR activation promotes scattering similar to that seen with MUC1 expression in three-dimensional culture.
c-Met activation drives invasive branching selectively in MUC1-overexpressing cells
Our observation that MUC1 and EGF induced cell branching in addition to cell scattering prompted us to evaluate the branching phenotype. We treated MDA-MB-231-MUC1 and MDA-MB-231-control cells in Matrigel with no exogenous ligand (Figure 5A and E), 10 ng/mL EGF (Figure 5B and F), 20 ng/mL HGF, the Hepatocyte Growth Factor and the ligand for c-Met (Figure 5C and G), or both 20 ng/mL HGF and 10 ng/mL EGF (Figure 5D and H). MDA-MB-231-MUC1 cells were found to form branches in the presence of EGF or HGF (Figure 5F and G, arrows). While MUC1 expression was required for the branching phenotype induced by either ligand alone, both ligands together could synergize and form branches in the absence of MUC1 (Figure 5D, arrows). These data indicate that MUC1 and EGFR together promote cell scattering and branching, a phenotype that is enhanced by the c-Met ligand, HGF.
Figure 5.

HGF stimulation drives invasive branching. MDA-MB-231-control (A–D) and MDA-MB-231-MUC1 (E–H) cells were seeded in Matrigel with 10% serum alone (A and E), 10% serum and 10 ng/mL EGF (B and F), 10% serum and 20 ng/mL HGF (C and G) or 10% serum and both 20 ng/mL HGF and 10 ng/mL EGF (D and H). Arrows indicate cell branching. Cells were imaged after five days of growth. Scale bar represents 50 μm.
MUC1 and EGF-dependent cell scattering is due to activated c-Met
To explore whether the MUC1 and EGF-driven scattering effects observed in three-dimensional culture were due to c-Met activity, we treated MDA-MB-231-MUC1 cells with 10ng/ml EGF and a selective c-Met kinase inhibitor, SU11274, or DMSO vehicle and cells were imaged after eight days of treatment. In MDA-MB-231-MUC1 cells treated with EGF, scattering (Figure 6B, arrowheads), and branching (Figure 6B, arrows) were observed as shown in Figures 4 and 5. Alternatively, the c-Met kinase inhibitor completely blocked the MUC1 and EGF- induced phenotype (Figure 6C). Importantly, SU11274 does not affect proliferation in MDA-MB-231-MUC1 or MDA-MB-231-control cells (Supplemental Figure 2). These results indicated that MUC1 and EGFR-dependent cell scattering is dependent upon c-Met kinase activity.
Figure 6.
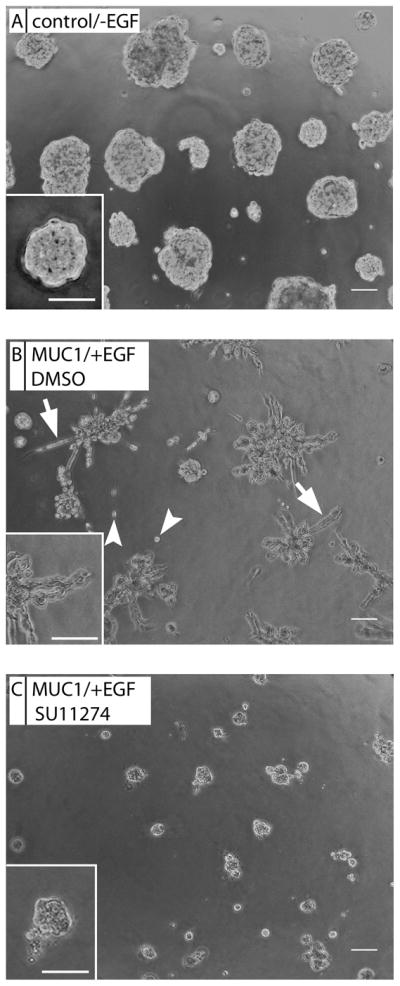
c-Met kinase inhibitor (SU11274) inhibits MUC1 and EGF-driven cell scattering and invasive phenotype. MDA-MB-231-control cells (A) or a MDA-MB-231-MUC1 cells (B) were seeded in Matrigel. MDA-MB-231-MUC1 cells were treated with 10ng/mL EGF and DMSO (B) or 5 μM SU11274, a selective c-Met kinase inhibitor (C). Arrowheads indicate single cells, and arrows indicate branching. Images were taken after 8 days. Scale bar represents 50 μm.
Discussion
In the current study, we have identified c-Met as an important mediator of MUC1-dependent migration, scattering and branching of breast cancer cells. Microarray analysis of genes with altered expression in response to PMIP treatment identified a number of potential metastatic mediators, including c-Met. In BT20 breast cancer cells RT-PCR verified that PMIP downregulates c-Met expression in a dose-dependent fashion. Further evaluation of c-Met determined that MUC1 expression resulted in an increase in cell migration on Type IV collagen, and this increase in migration can be inhibited by the selective c-Met inhibitor, SU11274. Furthermore, when placed in Matrigel, cell scattering and branching is increased dramatically upon MUC1 overexpression and EGF treatment, and this phenotype is also blocked by treatment with the c-Met kinase inhibitor.
Previously, MUC1 was shown to promote cell motility and invasion through upregulation of MMP13 and through the induction of epithelial to mesenchymal transition (EMT) in pancreatic cell lines [20, 21]. The breast cancer cells we utilized in this study, MDA-MB-231 have a mesenchymal phenotype which is not dependent on MUC1. The second cell line utilized, BT20, have an epithelial phenotype, and this was not altered by either MUC1 downregulation or PMIP treatment (data not shown). It is possible that the EMT phenotype previously identified is specific to pancreatic cell lines. In the microarray analysis we did not find that MMP13 transcription was altered following PMIP treatment (data not shown). Alternatively, we did find LAMA5 was downregulated by PMIP treatment (Supplementary Fig. 1), and LAMA5 has been shown to be involved in cell migration [43]. Whether LAMA5 is a direct or indirect target of MUC1 will be the subject of future studies. Overall, we initially identified ninety-seven genes significantly upregulated/downregulated at the level of the microarray, and further analyzed six targets by qRT-PCR. Of these six, only MET and LAMA5 were confirmed by qRT-PCR in 3 or more replicates (data not shown).
Our data indicate that MUC1 expression can drive c-Met-dependent scattering and branching. These data are in opposition to a previous study in which MUC1 was shown to interact with and be phosphorylated by Met [34]. In that study, the authors observed that MUC1 expression reduced MMP1 and decreased cell invasion in pancreatic cells [44]. Whether this is a tissue-type specific response, or whether c-Met negatively regulates MUC1 is unclear. Of note, all of our c-Met-dependent migration required EGF, which may be an important difference between our experiments and those in the previous study. In the current study, we have shown that MUC1 promotes the expression of c-Met while EGF promotes the activation of c-Met. Together, MUC1 expression and EGFR activation would result in the activated expression of c-Met, driving metastatic progression. We previously demonstrated that PMIP treatment increases EGF-dependent degradation of EGFR [39]. This data, in combination with the current study, may indicate that PMIP blocks metastatic progression by blocking both c-Met expression and its activation via EGFR.
Overall, the body of literature indicates that both MUC1 and c-Met promote metastatic phenotypes [45–48]. A recent study by Matsubara et al. that demonstrate that MUC1, EGFR and c-Met are coordinately upregulated in aggressive lung cancer patient samples [49]. This human data correlates with both our current study demonstrating that MUC1 expression can promote c-Met activity and with our previous study demonstrating that MUC1 promotes EGFR protein stabilization[29]. We are currently investigating the mechanism by which MUC1 and/or EGFR promote increased MET expression.
Using metastatic breast cancer cells, we demonstrated that MUC1 promotes cell motility using an in vitro wound healing assay. This behavior is reversed upon treatment with PMIP, illustrating that MUC1 promotion of metastatic behavior can be targeted therapeutically. This is the first demonstration of the therapeutic targeting of MUC1 activity as it relates specifically to metastasis. Recently, Raina et al demonstrated another decoy peptide, GO-201, that is designed to block MUC1 multimerization and nuclear translocation. Results from that study indicated a loss of tumor growth, but no effects on metastatic potential were reported [50]. Interestingly, [51] recently found that PMIP blocks smoke-induced EMT in airway epithelial cells, another mechanism by which PMIP may be capable of blocking metastasis.
Previous work regarding the role of MUC1 in promoting metastatic progression indicated that the extracellular domain of MUC1 was required for its function in this process [52]. None of our studies separated the effects of the extracellular domain of MUC1 from MUC1-C per se, in that we either overexpressed the intact protein or knocked down the entire protein via siRNA. While PMIP is designed to block protein-protein interactions between MUC1 and EGFR and/or MUC1 and beta-catenin, we cannot rule out the possibility that PMIP also affects the localization of the MUC1 or EGFR as a whole and therefore alters the interactions of MUC1 with proteins at its extracellular domain.
Downregulation of c-Met by PMIP demonstrated a reduction in an important oncogene and metastasis promoter [53]. In addition, c-Met is known to play a role in resistance of EGFR-positive cancers to EGFR-directed therapies, such as specific tyrosine kinase inhibitors and monoclonal antibodies. Bean et al assessed c-Met expression in EGFR-expressing tumors that are resistant to EGFR-targeted therapies and compared them to EGFR-expressing tumors that had never been exposed to therapies. In their study, they found that 21% of the tumors with acquired resistance overexpressed c-Met while only 3% of the unexposed tumors overexpressed c-Met [54]. This study implicated a c-Met dependent mechanism for acquiring resistance to EGFR-targeted therapies. Conversely, it has also been seen that EGFR activation can contribute to resistance to c-Met-directed therapies. Bachleitner-Hofmann et al examined the effect of a c-Met kinase inhibitor on c-Met-overexpressing gastric cancer cell lines. They found that inhibition of c-Met prevented receptor tyrosine kinase crosstalk; specifically, EGFR and HER3 downstream signaling was reduced upon c-Met kinase inhibition. However, they found that stimulating cells with EGF effectively circumvented c-Met kinase inhibition by activating downstream signaling pathways, such as MAPK and PI3K, describing one mechanism by which c-Met kinase inhibition can be thwarted[55]. Therefore, it may prove that targeting the activity of both of these receptors may prevent this resistance from occurring.
Supplementary Material
Acknowledgments
Financial support: NIH P30CA023074 (JAS), NIH R01CA102113 (JAS), Biochemistry and Molecular and Cellular Biology Training Grant T32GM008659 (TMH), Cancer Biology Training Grant T32CA009213 (BGB)
We would like to thank Matthew Hart and Hsin-Yuan Su for critical reading of this manuscript, and we are grateful to M.A. Hollingsworth for the MUC1 expression vector. CodeLink microarrays were provided by a grant from the University of Arizona Genomics Facility Core. This work was supported by the University of Arizona Biochemistry and Molecular Biology Training Grant (T32GM008659), Cancer Biology Training Grant (T32CA009213), the Arizona Cancer Center (NIH P30CA023074), and the National Institutes of Health (NIH R01CA102113).
Footnotes
Conflict of Interest: JAS is also the chief scientific officer of Arizona Cancer Therapeutics
References
- 1.Baldus SE, et al. Expression of MUC1, MUC2 and oligosaccharide epitopes in breast cancer: Prognostic significance of a sialylated MUC1 epitope. International Journal of Oncology. 2005;27(5):1289–1297. [PubMed] [Google Scholar]
- 2.Feng H, et al. Expression of MUC1 and MUC2 mucin gene products in human ovarian carcinomas. Japanese Journal of Clinical Oncology. 2002;32(12):525–529. doi: 10.1093/jjco/hyf111. [DOI] [PubMed] [Google Scholar]
- 3.Guddo F, et al. Depolarised expression of episialin (EMA, MUC1) in lung adenocarcinoma is associated with tumor progression. Anticancer Research. 1998;18(3B):1915–1920. [PubMed] [Google Scholar]
- 4.Xu HL, et al. Expression of KL-6/MUC1 in pancreatic ductal carcinoma and its potential relationship with beta-catenin in tumor progression. Life Sciences. 2011;88(23–24):1063–1069. doi: 10.1016/j.lfs.2011.03.021. [DOI] [PubMed] [Google Scholar]
- 5.Ajioka Y, Allison LJ, Jass JR. Significance of MUC1 and MUC2 mucin expression in colorectal cancer. Journal of Clinical Pathology. 1996;49(7):560–564. doi: 10.1136/jcp.49.7.560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kaira K, et al. MUC1 expression in thymic epithelial tumors: MUC1 may be useful marker as differential diagnosis between type B3 thymoma and thymic carcinoma. Virchows Archiv. 2011;458(5):615–620. doi: 10.1007/s00428-011-1041-x. [DOI] [PubMed] [Google Scholar]
- 7.Zhang HK, et al. Expression of mucins and E-cadherin in gastric carcinoma and their clinical significance. World Journal of Gastroenterology. 2004;10(20):3044–3047. doi: 10.3748/wjg.v10.i20.3044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hinoda Y, et al. Increased expression of MUC1 in advanced pancreatic cancer. Journal of Gastroenterology. 2003;38(12):1162–1166. doi: 10.1007/s00535-003-1224-6. [DOI] [PubMed] [Google Scholar]
- 9.Arai T, et al. Expression of sialylated MUC1 in prostate cancer: Relationship to clinical stage and prognosis. International Journal of Urology. 2005;12(7):654–661. doi: 10.1111/j.1442-2042.2005.01112.x. [DOI] [PubMed] [Google Scholar]
- 10.Lacunza E, et al. MUC1 oncogene amplification correlates with protein overexpression in invasive breast carcinoma cells. Cancer Genetics and Cytogenetics. 2010;201(2):102–110. doi: 10.1016/j.cancergencyto.2010.05.015. [DOI] [PubMed] [Google Scholar]
- 11.Wang L, et al. Expression of MUC1 in primary and metastatic human epithelial ovarian cancer and its therapeutic significance. Gynecologic Oncology. 2007;105(3):695–702. doi: 10.1016/j.ygyno.2007.02.004. [DOI] [PubMed] [Google Scholar]
- 12.Nagai S, et al. A novel classification of MUC1 expression is correlated with tumor differentiation and postoperative prognosis in non-small cell lung cancer. Journal of thoracic oncology: official publication of the International Association for the Study of Lung Cancer. 2006;1(1):46–51. [PubMed] [Google Scholar]
- 13.Matsuda K, et al. Clinical significance of MUC1 and MUC2 mucin and p53 protein expression in colorectal carcinoma. Japanese Journal of Clinical Oncology. 2000;30(2):89–94. doi: 10.1093/jjco/hyd023. [DOI] [PubMed] [Google Scholar]
- 14.Raina D, et al. MUC1 oncoprotein suppresses activation of the ARF-MDM2-p53 pathway. Cancer Biology & Therapy. 2008;7(12):1959–1967. doi: 10.4161/cbt.7.12.6956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wei XL, Xu H, Kufe D. MUC1 oncoprotein stabilizes and activates estrogen receptor alpha. Molecular cell. 2006;21(2):295–305. doi: 10.1016/j.molcel.2005.11.030. [DOI] [PubMed] [Google Scholar]
- 16.Li YQ, et al. The epidermal growth factor receptor regulates interaction of the human DF3/MUC1 carcinoma antigen with c-Src and beta-catenin. Journal of Biological Chemistry. 2001;276(38):35239–35242. doi: 10.1074/jbc.C100359200. [DOI] [PubMed] [Google Scholar]
- 17.Zhao QC, et al. Circulating Galectin-3 Promotes Metastasis by Modifying MUC1 Localization on Cancer Cell Surface. Cancer Research. 2009;69(17):6799–6806. doi: 10.1158/0008-5472.CAN-09-1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rahn JJ, et al. MUC1 mediates transendothelial migration in vitro by ligating endothelial cell ICAM-1. Clinical & Experimental Metastasis. 2005;22(6):475–483. doi: 10.1007/s10585-005-3098-x. [DOI] [PubMed] [Google Scholar]
- 19.Wesseling J, et al. Episialin (MUC1) overexpression inhibits integrin-mediated cell adhesion to extracellular matrix components. The Journal of cell biology. 1995;129(1):255–65. doi: 10.1083/jcb.129.1.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ye Q, et al. MUC1 induces metastasis in esophagealsquamous cell carcinoma by upregulating matrix metalloproteinase 13. Laboratory Investigation. 2011;91(5):778–787. doi: 10.1038/labinvest.2011.12. [DOI] [PubMed] [Google Scholar]
- 21.Roy LD, et al. MUC1 enhances invasiveness of pancreatic cancer cells by inducing epithelial to mesenchymal transition. Oncogene. 2011;30(12):1449–59. doi: 10.1038/onc.2010.526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schroeder JA, et al. MUC1 overexpression results in mammary gland tumorigenesis and prolonged alveolar differentiation. Oncogene. 2004;23(34):5739–5747. doi: 10.1038/sj.onc.1207713. [DOI] [PubMed] [Google Scholar]
- 23.Rajabi H, et al. MUC1-C Oncoprotein Induces TCF7L2 Transcription Factor Activation and Promotes Cyclin D1 Expression in Human Breast Cancer Cells. Journal of Biological Chemistry. 2012;287(13):10703–10713. doi: 10.1074/jbc.M111.323311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ahmad R, et al. MUC1-C Oncoprotein Functions as a Direct Activator of the Nuclear Factor-kappa B p65 Transcription Factor. Cancer Research. 2009;69(17):7013–7021. doi: 10.1158/0008-5472.CAN-09-0523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ahmad R, et al. MUC1-C Oncoprotein Promotes STAT3 Activation in an Autoinductive Regulatory Loop. Science Signaling. 2011;4(160) doi: 10.1126/scisignal.2001426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schroeder JA, et al. Transgenic MUC1 interacts with epidermal growth factor receptor and correlates with mitogen-activated protein kinase activation ill the mouse mammary gland. Journal of Biological Chemistry. 2001;276(16):13057–13064. doi: 10.1074/jbc.M011248200. [DOI] [PubMed] [Google Scholar]
- 27.Li YQ, et al. The c-Src tyrosine kinase regulates signaling of the human DF3/MUC1 carcinoma-associated antigen with GSK3 beta and beta-catenin. Journal of Biological Chemistry. 2001;276(9):6061–6064. doi: 10.1074/jbc.C000754200. [DOI] [PubMed] [Google Scholar]
- 28.Yamamoto M, et al. Interaction of the DF3/MUC1 breast carcinoma-associated antigen and beta-catenin in cell adhesion. Journal of Biological Chemistry. 1997;272(19):12492–12494. doi: 10.1074/jbc.272.19.12492. [DOI] [PubMed] [Google Scholar]
- 29.Pochampalli MR, el Bejjani RM, Schroeder JA. MUC1 is a novel regulator of erbB1 receptor trafficking. Oncogene. 2007;26(12):1693–1701. doi: 10.1038/sj.onc.1209976. [DOI] [PubMed] [Google Scholar]
- 30.Bitler BG, Goverdhan A, Schroeder JA. MUC1 regulates nuclear localization and function of the epidermal growth factor receptor. Journal of Cell Science. 2010;123(10):1716–1723. doi: 10.1242/jcs.062661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pochampalli MR, Bitler BG, Schroeder JA. Transforming growth factor alpha-dependent cancer progression is modulated by Muc1. Cancer Research. 2007;67(14):6591–6598. doi: 10.1158/0008-5472.CAN-06-4518. [DOI] [PubMed] [Google Scholar]
- 32.Li YQ, Kufe D. The human DF3/MUC1 carcinoma-associated antigen signals nuclear localization of the catenin p120(ctn) Biochemical and Biophysical Research Communications. 2001;281(2):440–443. doi: 10.1006/bbrc.2001.4383. [DOI] [PubMed] [Google Scholar]
- 33.Ren J, et al. MUC1 oncoprotein functions in activation of fibroblast growth factor receptor signaling. Molecular Cancer Research. 2006;4(11):873–883. doi: 10.1158/1541-7786.MCR-06-0204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Singh PK, et al. Platelet-derived growth factor receptor beta-mediated phosphorylation of MUC1 enhances invasiveness in pancreatic adenocarcinoma cells. Cancer Research. 2007;67(11):5201–5210. doi: 10.1158/0008-5472.CAN-06-4647. [DOI] [PubMed] [Google Scholar]
- 35.Ren J, et al. MUC1 oncoprotein is targeted to mitochondria by heregulin-induced activation of c-Src and the molecular chaperone HSP90. Oncogene. 2006;25(1):20–31. doi: 10.1038/sj.onc.1209012. [DOI] [PubMed] [Google Scholar]
- 36.Dietz GPH, Bahr M. Delivery of bioactive molecules into the cell: The Trojan horse approach. Molecular and Cellular Neuroscience. 2004;27(2):85–131. doi: 10.1016/j.mcn.2004.03.005. [DOI] [PubMed] [Google Scholar]
- 37.Schwarze SR, et al. In vivo protein transduction: Delivery of a biologically active protein into the mouse. Science. 1999;285(5433):1569–1572. doi: 10.1126/science.285.5433.1569. [DOI] [PubMed] [Google Scholar]
- 38.Ho A, et al. Synthetic protein transduction domains: Enhanced transduction potential in vitro and in vivo. Cancer Research. 2001;61(2):474–477. [PubMed] [Google Scholar]
- 39.Bitler BG, et al. Intracellular MUC1 Peptides Inhibit Cancer Progression. Clinical Cancer Research. 2009;15(1):100–109. doi: 10.1158/1078-0432.CCR-08-1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Klinge CM, et al. Targeting the Intracellular MUC1 C-terminal Domain Inhibits Proliferation and Estrogen Receptor Transcriptional Activity in Lung Adenocarcinoma Cells. Molecular Cancer Therapeutics. 2011;10(11):2062–2071. doi: 10.1158/1535-7163.MCT-11-0381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(16):9440–9445. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Imaizumi Y, et al. Expression of the c-Met proto-oncogene and its possible involvement in liver invasion in adult T-cell leukemia. Clinical cancer research: an official journal of the American Association for Cancer Research. 2003;9(1):181–7. [PubMed] [Google Scholar]
- 43.Kawataki T, et al. Laminin isoforms and their integrin receptors in glioma cell migration and invasiveness: Evidence for a role of alpha5-laminin(s) and alpha3beta1 integrin. Experimental Cell Research. 2007;313(18):3819–31. doi: 10.1016/j.yexcr.2007.07.038. [DOI] [PubMed] [Google Scholar]
- 44.Singh PK, et al. Phosphorylation of MUC1 by Met modulates interaction with p53 and MMP1 expression. The Journal of biological chemistry. 2008;283(40):26985–95. doi: 10.1074/jbc.M805036200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yang E, Hu XF, Xing PX. Advances of MUC1 as a target for breast cancer immunotherapy. Histology and histopathology. 2007;22(8):905–22. doi: 10.14670/HH-22.905. [DOI] [PubMed] [Google Scholar]
- 46.Gherardi E, et al. Targeting MET in cancer: rationale and progress. Nature reviews Cancer. 2012;12(2):89–103. doi: 10.1038/nrc3205. [DOI] [PubMed] [Google Scholar]
- 47.Hu XF, et al. MUC1 cytoplasmic tail: a potential therapeutic target for ovarian carcinoma. Expert review of anticancer therapy. 2006;6(8):1261–71. doi: 10.1586/14737140.6.8.1261. [DOI] [PubMed] [Google Scholar]
- 48.Yuan Z, et al. Down-regulation of MUC1 in cancer cells inhibits cell migration by promoting E-cadherin/catenin complex formation. Biochemical and Biophysical Research Communications. 2007;362(3):740–6. doi: 10.1016/j.bbrc.2007.08.074. [DOI] [PubMed] [Google Scholar]
- 49.Matsubara D, et al. Co-activation of epidermal growth factor receptor and c-MET defines a distinct subset of lung adenocarcinomas. The American journal of pathology. 2010;177(5):2191–204. doi: 10.2353/ajpath.2010.100217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Raina D, et al. Direct targeting of the mucin 1 oncoprotein blocks survival and tumorigenicity of human breast carcinoma cells. Cancer Research. 2009;69(12):5133–41. doi: 10.1158/0008-5472.CAN-09-0854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang L, et al. Cigarette Smoke Disrupts the Integrity of Airway Adherens Junctions through the Aberrant Interaction of P120-catenin with the Cytoplasmic Tail of MUC1. The Journal of pathology. 2012 doi: 10.1002/path.4070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Satoh S, et al. Enhancement of metastatic properties of pancreatic cancer cells by MUC1 gene encoding an anti-adhesion molecule. International journal of cancer. Journal international du cancer. 2000;88(4):507–18. doi: 10.1002/1097-0215(20001115)88:4<507::aid-ijc1>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 53.Birchmeier C, et al. Met, metastasis, motility and more. Nature Reviews Molecular Cell Biology. 2003;4(12):915–925. doi: 10.1038/nrm1261. [DOI] [PubMed] [Google Scholar]
- 54.Bean J, et al. MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(52):20932–20937. doi: 10.1073/pnas.0710370104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bachleitner-Hofmann T, et al. HER kinase activation confers resistance to MET tyrosine kinase inhibition in MET oncogene-addicted gastric cancer cells. Molecular Cancer Therapeutics. 2008;7(11):3499–3508. doi: 10.1158/1535-7163.MCT-08-0374. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.