

Abstract
Sphingosine 1-phosphate (S1P) is a lysophospholipid signaling molecule that regulates important biological functions, including lymphocyte trafficking and vascular development, by activating G protein-coupled receptors for S1P, namely S1P1 through S1P5. Here we map the S1P3 binding pocket with a novel allosteric agonist (CYM-5541), an orthosteric agonist (S1P), and a novel bitopic antagonist (SPM-242). With a combination of site-directed mutagenesis, ligand competition assay, and molecular modeling, we concluded that S1P and CYM-5541 occupy different chemical spaces in the ligand binding pocket of S1P3. CYM-5541 allowed us to identify an allosteric site where Phe263 is a key gate-keeper residue for its affinity and efficacy. This ligand lacks a polar moiety and the novel allosteric hydrophobic pocket permits S1P3 selectivity of CYM-5541 within the highly similar S1P receptor family. On the other hand, a novel S1P3-selective antagonist, SPM-242, in the S1P3 pocket occupies the ligand binding spaces of both S1P and CYM-5541, showing its bitopic mode of binding. Therefore, our coordinated approach with biochemical data and molecular modeling, based on our recently published S1P1 crystal structure data in a highly conserved set of related receptors with a shared ligand, provides a strong basis for the successful optimization of orthosteric, allosteric, and bitopic modulators of S1P3.
INTRODUCTION
Sphingosine 1-phosphate (S1P) is a lysophospholipid signaling molecule that regulates important biological functions, including lymphocyte trafficking, endothelial development/integrity, heart rate, and vascular tone/maturation (1–6). S1P is synthesized intracellularly by phosphorylation of sphingosine, and secreted to plasma and interstitial fluids through a spinster channel (7); extracellular signals are then transduced by five related G protein-coupled receptors (GPCR) for S1P; S1P1 through S1P5. These group A (rhodopsin-like) GPCRs form part of the EDG (endothelial differentiation gene) cluster on the GPCR phylogenetic tree together with lysophosphatidic acid receptors and the cannabinoid receptors.
S1P receptors are involved in many biological processes and many disease progresses from autoimmunity (3, 8) to atherosclerosis and cancer (9, 10). Despite significant homologies in their sequence and structure, S1P1-5 have varied tissue distribution, different arrays of downstream G protein coupling, various biochemical and physiological functions, and different receptor fates (4, 11). In order to understand the role of individual receptor subtypes, the search for subtype-selective agonists and antagonists has been one of the focuses of our S1P research. However, the number of well-characterized ligands that interact highly selectively with S1P receptor subtypes is very limited. Five S1P receptor subtypes share high sequence homology within the orthosteric site; thus targeting the orthosteric site would not yield high selectivity (12). The development of receptor subtype-selective ligands of an allosteric nature would help elucidate the function of each receptor subtype. Furthermore, this approach would aid in the design of more specific drugs targeted for the selective receptor subtype with minimized side effects through the other subtypes.
S1P3 receptor couples promiscuously to Gi, Gq, and G12/13 proteins (13–16) and is medically significant. Distributed on cardiac myocytes and cardiac fibroblasts in vivo, it plays a critical role in cardiovascular physiology (14, 16), especially pace-making activity. In the adaptive immune system, S1P3 is highly expressed on marginal zone B cells (17), and deletion of S1P3 leads to disorganization of the marginal zone of spleen (13) with alterations in B cell responses (18–21). The receptor is also significantly expressed on dendritic cells in vivo. In settings of overwhelming dysregulation of innate immunity, such as lipopolysaccharide (LPS) challenge or cecal-ligation-puncture sepsis in rodents, poor outcome is S1P3-dependent. For example, protection from LPS has been demonstrated in S1P3-deletant mice (22) or by blocking monoclonal antibodies to the receptor (23).
GPCRs are now viewed as dynamic structures that adopt multiple biologically relevant conformations (12). Orthosteric and allosteric ligands can preferentially stabilize distinct active states at a given GPCR, leading to discrete signaling activities. This has been termed functional selectivity or stimulus bias because the ligand-specific conformation may lead to signal complex-biased or pathway-biased effects (24). The differential signaling is expected to provide novel therapeutics with selectivity in drug action. We report here an S1P3-selective agonist, CYM-5541. Characterization of this molecular probe has yielded interesting results. The probe is a full agonist of the S1P3 but does not contain the polar headgroup moieties that play a critical role in S1P binding. Additionally, we find that CYM-5541 does not compete with the native ligand in competition studies. With a combination of site-directed mutagenesis, ligand competition assay and molecular modeling, we concluded that the ligand binds to a distinct allosteric binding site of the S1P3 receptor.
In order to improve affinity and selectivity, researchers have recently attempted to engineer “bitopic ligands” where orthosteric and allosteric pharmacophores are linked (24). Bitopic ligands will provide improved affinity through additional interactions, and they will also provide improved selectivity by engaging less conserved regions across a family of receptors. Bitopic ligands can also lead to a signal complex-biased pathway by selecting a preferred receptor conformation. We have characterized a bitopic antagonist that is highly selective for S1P3. The bitopism can allow us to gain new insights into the biology of S1P receptors by focusing on the advantages of both orthosteric and allosteric properties.
The first lipid GPCR structure at the atomic level (S1P1) was recently solved by our group (25) and it provided us a fundamental basis for modeling S1P3. We employed the newly solved S1P1 structure and conducted S1P3/ligand molecular modeling and found it to be remarkably consistent with the biochemical data. We now propose that S1P and CYM-5541 occupy differential chemical spaces in the ligand binding pocket of S1P3. The S1P3 ligand binding pocket is defined by two distinct binding regions; one region around transmembrane (TM) helices 1/3 binds to S1P, and a lower hydrophobic region around TM6 binds to CYM-5541.
The novel allosteric binding site may account for the S1P3 selectivity of CYM-5541 and may open up the possibility of drug design and lead optimization with subtype selectivity for members of other GPCR families as well. As shown in this report, the combination of the chemistry, cell biology, and structural biology provides a platform for mapping pathways in physiology and pathology that can lead to the discovery of therapeutic targets.
RESULTS AND DISCUSSION
CYM-5541 is a full agonist
In order to achieve subtype selectivity, we and others (26–28) have hypothesized that finding allosteric pockets within the receptor can lead to potent and selective chemical probes. There are alternate binding mode agonists (CYM-5442) for S1P1 but no true allosteric agonist (25, 29). There are potential interaction sites around the S1P3 orthosteric binding pocket that make this approach feasible.
From the initial screening, a series of dicyclohexyl amides were found to be selective for S1P3 and inactive on S1P1 (30). Further optimization led to the discovery of CYM-5541 (also designated as chemical probe ML249 from the NIH Molecular Libraries Initiative), a novel S1P3-selective agonist (Fig. 1A). In order to examine the binding mode of the agonist to S1P3 in stable cell lines, we established Jump-In cell lines with either WT or mutant S1P3. Jump-In integration is irreversible and only a single copy of the gene of interest is integrated at the same site in every cell.
Figure 1.

Structures of S1P3-selective agonist and antagonist. (A) CYM-5541 (N,N-dicyclohexyl(5-cyclopropylisoxazol-3-yl)carboxamide). (B) SPM-242 ((+)-2-amino-4-(2-chloro-4-((3-hydroxyphenyl)thio)phenyl)-2-(hydroxymethyl)butyl dihydrogen phosphate).
S1P has been shown to activate the p42/p44 mitogen-activated protein kinase (MAPK) cascade, leading to phosphorylation of extracellular signal-regulated kinase (7, 31–33). WT S1P3 Jump-In stable CHO cells were stimulated with increasing concentrations of either S1P or CYM-5541 and were examined for their ERK phosphorylation (Fig. 2). CYM-5541 is a full agonist, able to reach the maximum level of ERK phosphorylation that was observed with S1P. CYM-5541 has an EC50 of between 72 and 132 nM and exhibits exquisite selectivity over other S1P receptor subtypes in vitro: S1P1 EC50 > 10 μM, S1P2 EC50 > 50 μM, S1P4 EC50 > 50 μM, and S1P5 EC50 > 25 μM (34). CYM-5541 also shows selectivity over a large panel of protein targets, with no significant activities, in the Ricerca profiling panel of 55 GPCRs, ion channels, and transporters.
Figure 2. Ligand-induced ERK phosphorylation in WT S1P3 Jump-In stable cell lines (Mean ± SEM; n=3), revealing CYM-5541 is a full agonist of S1P3.
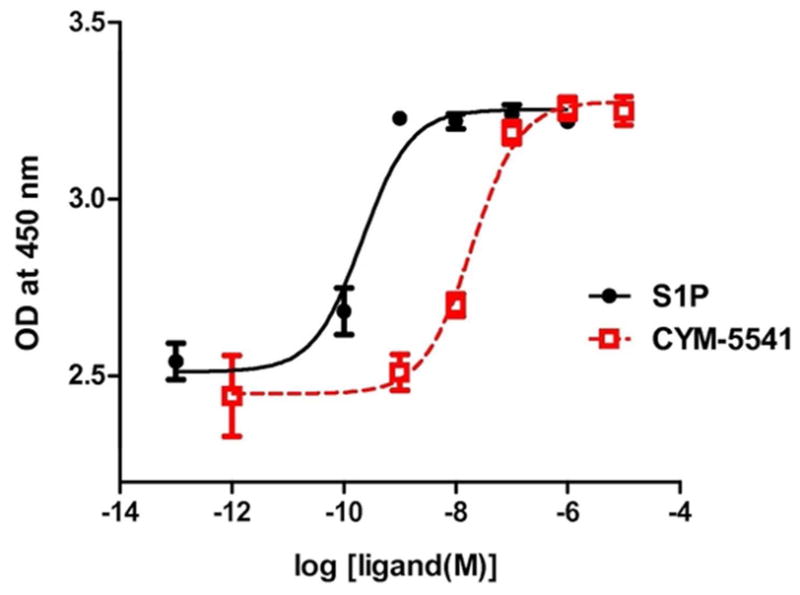
Targetable Jump-In™ TI™ CHO-K cell lines were used to integrate a single copy of S1P3 in a site-specific manner. WT Jump-In stable cell lines were stimulated with increasing concentrations of either S1P or CYM-5541.
CYM-5541 is an allosteric agonist
[33P]S1P binding of WT, W256L, and F263L mutant S1P3 was competed by adding increasing doses of its agonists and an antagonist. F263L is a loss-of-function mutation in S1P3, while L276F provides a gain-of-function in S1P1, helping to define L276 in S1P1 as a gate-keeper residue (30, 35). With increasing concentrations of cold S1P, [33P]S1P binding was competitively reversed in a dose dependent manner in WT, W256L, and F263L, as expected (Fig. 3B). S1P3-selective antagonist SPM-242 (Fig. 1B) also reversed [33P]S1P binding competitively in a dose dependent manner in all cell lines (Fig. 3C). On the other hand, CYM-5541 was unable to compete for [33P]S1P binding in all cell lines (Fig. 3A). This suggests that the S1P and CYM-5541 binding pockets are significantly different.
Figure 3. Radioligand binding and competition.

WT, W256L, and F263L Jump-In stable cell lines were incubated with 0.1 nM [33P]S1P in the presence of increasing concentrations of (A) CYM-5541, (B) S1P, and (C) SPM-242. [33P]S1P binding was competitively reversed with S1P and SPM-242 in all cell lines tested whereas CYM-5541 was unable to compete for [33P]S1P binding (Mean ± SEM; n=3).
Furthermore, unlike the natural agonist S1P, CYM-5541 does not have a headgroup, and therefore did not require the R114 residue for activating S1P3-dependent ERK phosphorylation (Supporting Figure S1). Both WT and M98A mutant S1P3 Jump-In CHO cells revealed agonist dose-dependent ERK phosphorylation by both S1P and CYM-5541. However, R114A mutant was unable to respond to S1P due to lack of the charged R114 that provides a critical charge interaction between the guanidium side-chain of the arginine and the phosphate of the S1P headgroup. On the other hand, CYM-5541-induced ERK phosphorylation was comparable for WT and R114A, indicating that R114 is not a key residue for CYM-5541-binding pocket. These results show that CYM-5541 binding does not overlap with the headgroup interactions of S1P.
F263 is a key gate-keeper residue for the allosteric binding site
Based on the previously published S1P1 crystal structure and homology modeling (25, 30, 36), we made a series of minimal point mutations along the hydrophobic ligand binding pocket to see if any of the mutants result in defective binding or signaling in response to S1P or CYM-5541. Receptor mutagenesis and functional assay revealed that F263 is a key gate-keeper residue for the allosteric binding site. In the ERK phosphorylation assay, the F263L mutation shifted CYM-5541-induced receptor activation but not S1P-induced activation, confirming that CYM-5541 makes a non-overlapping interaction (Fig. 4A). The W256L mutation did not affect either S1P-or CYM-5541-induced receptor activation (Fig. 4B), as opposed to its effect on S1P1 and CYM-5442 (25).
Figure 4. Receptor mutagenesis and ligand-induced ERK phosphorylation.

(A) ERK phosphorylation assay revealed that F263L mutation shifted CYM-5541-induced receptor activation but not S1P-induced activation. (B) W256L mutation did not affect either S1P- or CYM-5541-induced receptor activation (Mean ± SEM; n=3). Data is a representative of four independent experiments.
The ELISA results were confirmed by MAPmates multiplex assay (Millipore) (Supporting Figure S2). Using pERK1/2 (Thr185/Tyr187) antibodies, F263L S1P3 was shown to result in significantly diminished receptor activation by CYM-5541, compared to WT S1P3. These data confirm that F263 in S1P3 plays a critical role in CYM-5541-induced receptor activation. This differential requirement for S1P- and CYM-5541-induced receptor activation further suggests that receptor interactions are distinctly different.
We have further evaluated the allosteric agonist CYM-5541 in receptor assays coupled to Gi and Gq, using ERK phosphorylation, calcium flux, β-arrestin, and NFAT-β-lactamase reporter assays. In contrast to S1P1 receptor where up to 3 logs of bias have been documented in the ERK phosphorylation (31) and the β-arrestin-dependent receptor polyubiquitination assays (37), no such differences were observed when comparing CYM-5541 to S1P on the S1P3 receptor.
S1P3-selective antagonist SPM-242 bridges the S1P and CYM-5541 pockets
SPM-242 is an S1P3-selective antagonist and was synthesized by Kohno et al. (Fig. 1B; See Methods). SPM-242 has been profiled against the full panel of S1P receptors and was selective for S1P3 (Supporting Table S3). In the ERK phosphorylation assay, it significantly right-shifted the concentration response curve of S1P (Fig. 5A) by 152-fold, demonstrating its competitive inhibition. SPM-242 also shifted the concentration response curve of CYM-5541 to a lesser extent (6-fold). This was confirmed in an S1P3- NFAT β–lactamase reporter assay; SPM-242 right-shifted the concentration response curve of CYM-5541 in a dose-dependent manner (Fig. 5B). The EC50 derived from the concentration response curves of CYM5541 in the presence of increasing concentrations of SPM-242 were best fit to the Gaddum/Schild equation yielding a Ki of 0.25 nM (38). The rightward shift of the concentration response curves with full maximal efficacy is indicative of competitive antagonism of CYM-5541 by SPM-242. The fact that SPM-242 was able to compete for CYM-5541 as well as S1P suggests that SPM-242 may span both orthosteric and allosteric binding pockets, thus having a bitopic binding mode (24).
Figure 5. SPM-242, an S1P3-selective antagonist, is a bitopic antagonist.

(A) SPM-242 shifted the dose-response curve of S1P and CYM-5541 (ERK phosphorylation assay). (B) SPM-242 (concentrations ranging from 10−9.5 to 10−7 M) shifted the dose-response curve of CYM-5541 in a dose-dependent manner (NFAT β–lactamase reporter assay) (Mean ± SEM; n=3).
Based on the experimental results, the S1P3 ligand binding pocket seems to be able to accommodate both S1P and CYM-5541. It may be a big pocket with two regions: one region around TM1/3 binds to S1P, and a lower hydrophobic region around TM6 binds to CYM-5541. SPM-242, as a bitopic antagonist, may span both of those regions. The structure of S1P is more flexible in that it can bind to the pocket in the presence of CYM-5541 whereas SPM-242, with a more rigid structure, would have to displace CYM-5541 in order to antagonize S1P3.
Homology modeling of S1P3
A homology model was produced for S1P3 by using the S1P1 structure as obtained from PDB (3V2W, resolution 3.35Å) and the Uniprot sequence of S1PR3_Human (accession Q99500). The alignment score of S1P1–S1P3 was 0.035 and RMSD (C- alpha atoms of the aligned chains) was 0.882. RMSD is explained by the identity of two proteins of 58% and a 5% gap in the structures. The S1P3 receptor model showed that disulfide bonds in the extracellular loops EL2 and EL3 were aligned with the corresponding S1P1 disulfide bonds. Importantly, the S1P binding region of S1P3 was well aligned with S1P1 (Supporting Figure S4) with a RMSD (alpha-C) of 0.325. The receptor structures were optimized by minimization and molecular dynamics. Mutant forms were generated from the optimized WT forms followed by energy minimization. Optimized structures were used further for docking studies.
Key elements unanticipated by previous models include unexpected H-bond interactions and the very tightly ordered N-terminal structure with a conserved disulfide hairpin ordering the alpha-helices.
Docking studies of WT and mutant S1P3 with agonists and an antagonist
Ligand-receptor binding models were generated as described in Methods. The final ligand-receptor complexes were ranked by glide score, emodel, and MM GB/SA energies, based on known interactions. The energetically most favorable and comparable poses are reported to visualize likely binding modes in the S1P1 and S1P3 receptors.
In S1P3, the S1P headgroup interacts with R114 and E115 while the hydrocarbon alkyl tail interacts in the hydrophobic pocket (Fig. 6A). CYM-5541 sits in the hydrophobic pocket close to F263 (Fig. 6B) with no apparent headgroup interaction. Co-docking of S1P and CYM-5541 suggested that the receptor pocket could spatially expand in the lower region of the hydrophobic pocket to accommodate CYM-5541 in addition to S1P (Fig. 6C). A top-down view from the extracellular surface also show the co-docked S1P and CYM-5541 with the non-overlapping allosteric binding site for CYM-5541 (Fig. 6D). Our biochemical data on calcium flux dependent β-lactamase assay strongly support this docking model. When both S1P and CYM-5541 were added to S1P3-CHO cells, the orthosteric and allosteric agonist responses were additive and no synergy was observed in terms of left shift in the efficacy curves (Fig. 6E). The additive effect shown in Fig. 6E clearly suggests that CYM-5541 is a true allosteric agonist.
Figure 6. Visualization of the receptor binding pocket by homology modeling and docking. Three dimensional plot of S1P3 binding to (A) S1P, (B) CYM-5541, and (C and D) both.


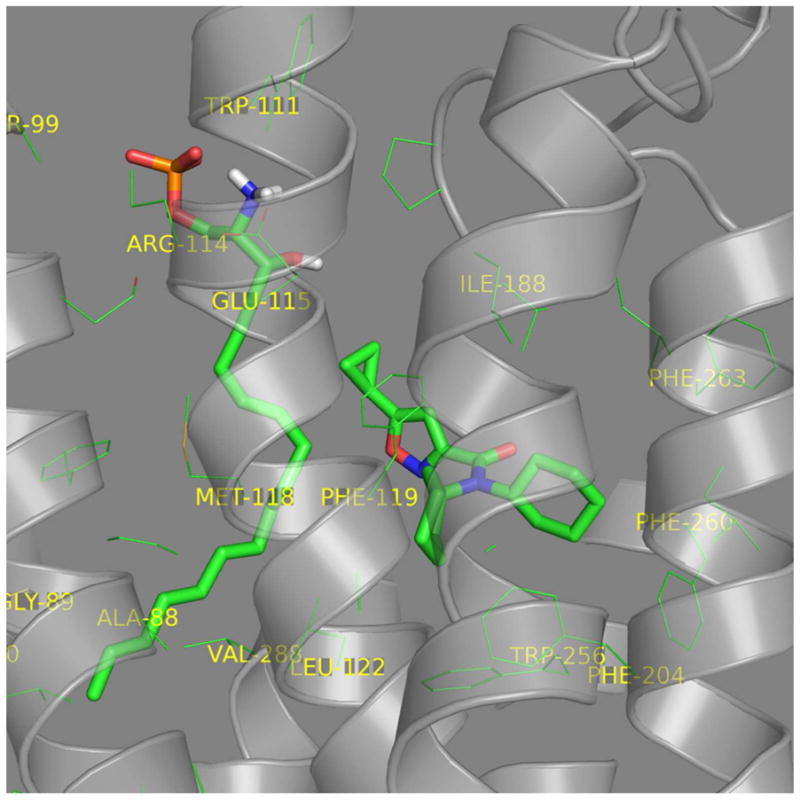
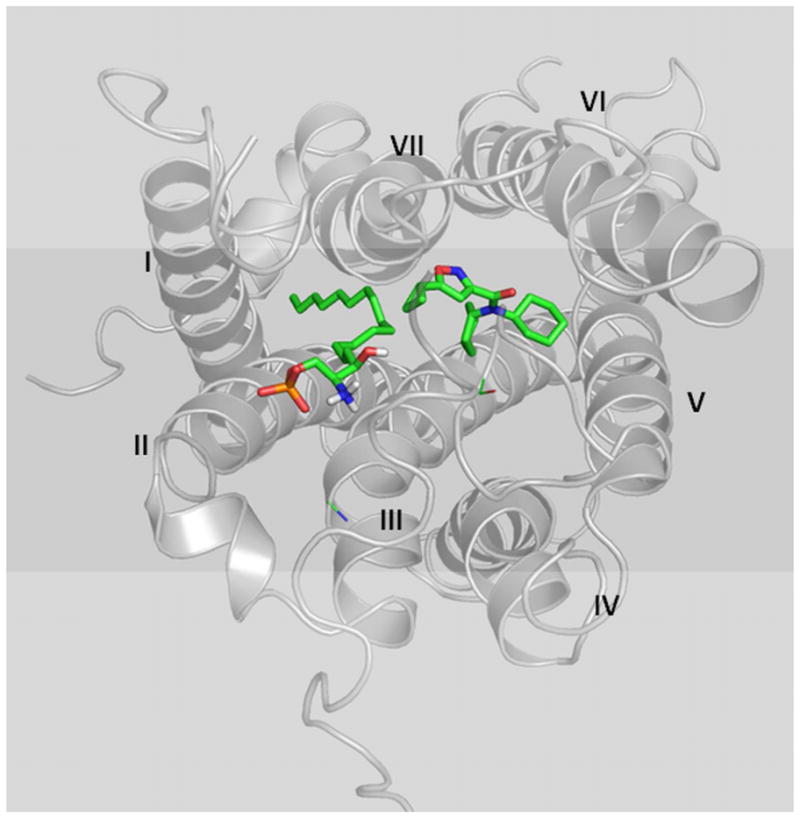

(A) S1P headgroup interacting with R114 and E115. (B) CYM-5541 in the hydrophobic pocket. (C) S1P and CYM-5541 co-docked to S1P3. In the presence of S1P, the pocket opens up in the lower hydrophobic region adjusting CYM-5541 (after 5ns MD optimization). (D) Top view from the extracellular surface with helix orientation identical to the other panels. Detailed modeling and docking procedures are described in Methods section. (E) Calcium response assay upon co-application of S1P and CYM-5541 to S1P3-CHO cells. When both S1P and CYM-5541 were added to S1P3-CHO cells, calcium release response was increased, indicating their additive responses (Mean ± SEM; n=9).
The average distances between the key amino acids and CYM-5541 in the S1P3 WT pocket indicate that it binds in close proximity to F263 compared to W256. Molecular dynamics simulations show that in the presence of S1P, it moves away from F263 closer towards W256 in the hydrophobic region. Average distances obtained over several nanosecond-long simulations of the S1P3-CYM-5541 complex and S1P3 co-docked with CYM-5541 and S1P are shown in Supporting Table S5 (also see Fig. 6C).
Docking studies and relative binding free energy calculations support stronger binding affinity of CYM-5541 to S1P3 compared to S1P1 (Table 1). We report Glide XP scores because hydrophobic interactions dominate the binding of CYM-5541 analogs to the receptor (30). The data is compatible with the mutagenesis data shown in Fig. 4, indicating CYM-5541 ligand in the S1P3 pocket requires F263 but not W256.
Table 1.
CYM-5541 binding energetics to S1P3 WT and mutants.
| Receptor | Glide Score (XP*) | Relative Binding Free Energies*** | ||||
|---|---|---|---|---|---|---|
| WT | W256L/W269L | F263L | WT | W256L/W269L | F263L | |
| S1P3 | −12.15 | −10.20 | −8.13** | −44.02 | −42.99 | −36.89 |
| S1P1 | −9.79 | −9.04 | - | −40.04 | −37.66 | - |
XP score was chosen because hydrophobic interactions are dominant for CYM-5541.
Docking score of the S1P3 WT pose of CYM5541 after minimization: The exact WT pose was invalid (i.e. interferes with Tyr92).
Computed using MM-GBSA (includes ligand and receptor strain).
Computed relative binding affinities of CYM-5541 in the S1P3 W256L mutant receptor (Supporting Figure S6) are only slightly higher compared to the WT receptor (Table 1) suggesting that W256 is not required for binding of CYM-5541 and S1P3 receptor activation. The same ligand pose in the S1P3 F263L mutant receptor shows significantly worse energetics (docking score-based binding affinity and relative binding free energy by MM-GB/SA; Table 1 and Supporting Figure S7). We also observed flipped poses of CYM-5541 in the F263L mutant receptor in many of the frames extracted from the MD simulations for cross-docking studies of the ligand receptor interactions (to account for receptor flexibility). In contrast, the CYM-5541 docking poses in S1P3 WT and also the W256L mutant receptors were stable across the simulation frames. These results further support that the F263L mutation, in contrast to W256L and WT, destabilizes binding of CYM-5541 (Supporting Figure S6). In all of these cases, docking scores for CYM-5541 in the F263L mutant receptor remain significantly above (worse energetics) the WT scores (Supporting Table S8). To conclude, these results support that F263 is required for binding of CYM-5541 to the S1P3 receptor.
When docked into the S1P3 pocket, S1P3 antagonist SPM-242 overlaps with S1P in terms of the headgroup and some hydrophobic interactions while it also picks up some aromatic interactions near F263, interrupting CYM-5541 binding (Fig. 7). Therefore, SPM-242 in the S1P3 pocket occupies the ligand binding spaces of both S1P and CYM-5541, showing its bitopic mode of binding, consistent with our hypothesis based on the signaling data (Fig. 5).
Figure 7. Visualization of the orthosteric, allosteric, and bitopic ligand interactions based upon the biochemistry, mutagenesis, homology modeling, and molecular dynamic simulations. Dual inhibition of SPM-242 is illustrated.

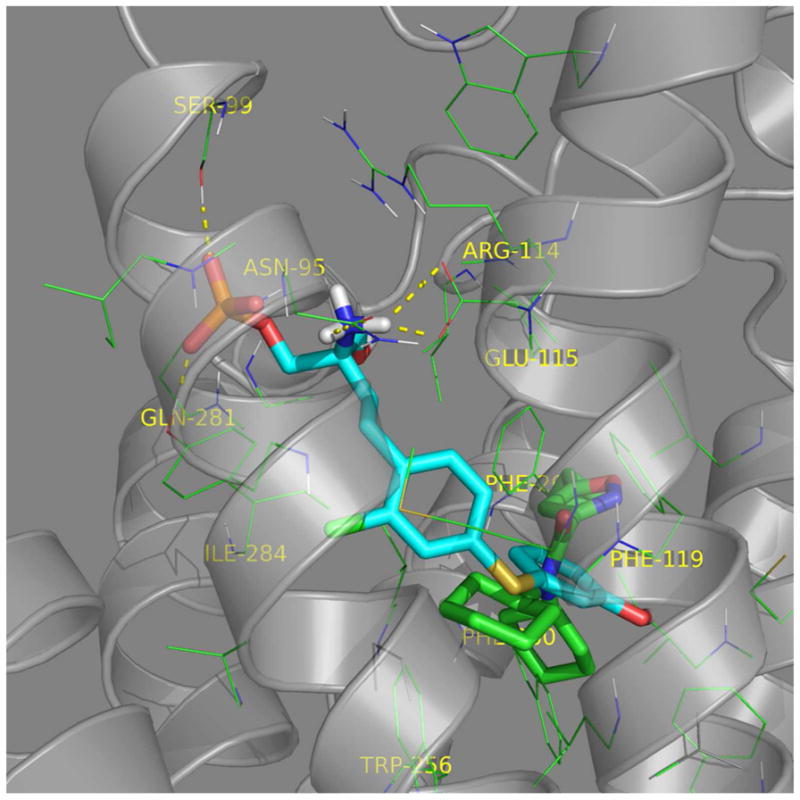
(A) S1P (green) and SPM-242 (cyan) overlap in the S1P3 binding pocket. (B) CYM-5541 (green) and SPM-242 (cyan) overlap in the S1P3 binding pocket.
Conclusions
Our biochemical data as well as molecular modeling strongly suggest that S1P and CYM-5541 occupy different chemical spaces in the ligand binding pocket of S1P3. CYM-5541 allowed us to identify an allosteric site where F263 is a key gate-keeper residue for its affinity and efficacy. The novel allosteric hydrophobic pocket may account for the S1P3 selectivity of CYM-5541. However, despite its great selectivity and stability, CYM-5541 has some limitations. It has low solubility and moderate potency, which limits its DMPK profiling and ultimately its use in animal testing. In addition, SPM-242 has a challenge of its phosphate-ester bond, which is extremely labile in biological systems. Its hydrophobic, amino-phosphate zwitterion also has low solubility, which limits its application for in vivo delivery. Therefore, these chemical probes with therapeutic potential need further optimization as in vivo chemical probes of S1P3 function. Overall, our coordinated approach maximizing crystal structure data in a highly conserved set of related receptors with a shared ligand, together with mutagenesis, provides a potential basis for the successful optimization of orthosteric and allosteric modulators of S1P3.
METHODS
S1P Receptor Agonists and Antagonist
S1P was purchased from BioMol Research Laboratories. The selective S1P3 agonist CYM-5541, N,N-dicyclohexyl(5-cyclopropylisoxazol-3-yl)carboxamide, was synthesized in Dr. Edward Roberts’ lab (The Scripps Research Institute). The compound synthesis procedure and characterization data are provided (Supporting Scheme S9). [33P]S1P was from American Radiolabeled Chemicals, Inc. The selective S1P3 antagonist SPM-242, (+)-2-amino-4-(2-chloro-4-((3-hydroxyphenyl)thio)phenyl)-2-(hydroxymethyl)butyl dihydrogen phosphate, was synthesized by Kohno et al. (Kyorin Pharmaceutical Co.). As shown in the Supporting Scheme S10, 1 (100 mg, 0.19 mmol) was dissolved in CH2Cl2 (2 mL) and Iodotrimethylsilane (105 uL, 0.74 mmol) was slowly added at room temperature under argon atmosphere. After stirring for 5 h at room temperature, the reaction mixture was purified by column chromatography (Silica gel 60 Silanised, H2O : CH3CN = 9:1 to 3:1) to give SPM-242 (63 mg, 0.145 mmol) as a white powder. The compound characterization data are provided (Supporting Scheme S10).
Cell Lines and Plasmids
Plasmid encoding N-terminal, triple HA-tagged S1P3 fusion protein in pcDNA3.1 was purchased from cDNA.org. S1P3 receptor mutants were generated by overlapping PCR mutagenesis, and sequences were verified prior to use.
Jump-In™ TI™ CHO-K parental cells, Gateway cloning vectors (pDONR 221, pJTI R4 DEST, and pJTI R4 Int), and enzymes (BP clonase II and LR clonase) were purchased from Invitrogen Corp. Triple HA-tagged WT and mutant S1P3 were first cloned into entry clones using the BP recombination reaction. They were retargeted into an appropriate pJTI R4 DEST vector to yield a pJTI R4 EXP retargeting expression vector. Jump-In TI CHO-K cells were transfected with both pJTI R4 EXP S1P1 and pJTI R4 Int vector for retargeting. After expanding retargeted cells, retargeted Jump-In TI cells were selected using blasticidin at 10 μg/mL for 4 weeks.
ELISA for p44/42 MAPK Phosphorylation
Ligand-mediated ERK phosphorylation was measured using PathScan Phospho-p44/42 MAPK (Thr202/Tyr204) Sandwich ELISA kit (Cell Signaling Technology). Cells expressing WT or mutant S1P3 were serum-starved for 4 hr. In the antagonist experiments, cells were either pre-incubated with SPM-242 at 1 uM for 15 min before agonist treatment or 1 uM SPM-242 was pre-mixed with agonists. Cells were then stimulated for 5 min (determined to give maximal ERK phosphorylation for all agonists) with increasing concentrations of S1P or CYM-5541 and phosphorylation of p44/42 MAPK was assayed according to manufacturer’s protocol. The dose response curves for agonist-mediated p44/42 MAPK phosphorylation were analyzed and EC50 was determined using Prism (Graphpad Software).
[33P]S1P Radioligand Binding Assay
Jump-In TI CHO-K cells (5 × 105) stably expressing WT or mutant S1P3 were serum-starved for 4 hrs. They were then incubated at 4 °C for 30 min in the binding buffer containing 20 mM Tris-HCl (pH 7.5), 100 mM NaCl, 15 mM NaF, 0.5 mM EDTA, 1 mM Na3VO4, 0.5% fatty acid-free bovine serum albumin, and protease inhibitor mixture (Roche) with 0.1 nM [33P]S1P and increasing concentrations of S1P, SPM-242, or CYM-5541. Cells were washed three times with cold binding buffer. Cell-bound radioactivity was measured by lysing the cells with 0.5% SDS followed by liquid scintillation counting. The raw data was normalized so that the level of [33P]S1P bound to each cell line (WT or mutant) in the absence of competing ligand was referenced as 100% for its own cell line.
Calcium response assay
A CHO cell line stably transfected with human S1P3 receptor and nuclear factor of activated T cell β-lactamase (NFAT-BLA) reporter construct was used. The growth medium consisted of Dulbecco’s Modified Eagle’s Media containing 10% v/v heat inactivated bovine growth serum, 0.1 mM NEAA, 1 mM Sodium Pyruvate, 25 mM HEPES, 2 mg/mL 5 mM L-Glutamine, 0.2 mg/mL Hygromycin B and 1x penicillin-streptomycin. Prior to the start of the assay, cells were suspended to a concentration of 1 × 106/mL in phenol red free Dulbecco’s Modified Eagle’s Media containing 0.5% charcoal/dextran treated fetal bovine serum, 0.1 mM NEAA, 1 mM Sodium Pyruvate, 25 mM HEPES, and 5 mM L-Glutamine. The cells were then dispensed in 384-well plates and incubated overnight at 37°C in 5% CO2. The next day, S1P dilution series with six different concentrations of CYM-5541 (ranging EC0 to EC80) were added. The S1P or CYM-5541 alone was also added to the appropriate control wells. Plates were then incubated at 37°C in 5% CO2 for 4 hours. After the incubation, the GeneBLAzer’s fluorescent substrate mixture containing 200 mM probenicid was added. After 2 hours of incubation at room temperature, plates were read on the EnVision plate reader (PerkinElmer Lifesciences) at an excitation wavelength 405 nm and emission wavelengths of 535 nm & 460 nm.
S1P1/S1P3 Receptor Structure WT and Mutant Models
The initial S1P1 receptor structure was taken from the x-ray co-crystal structure (PDB code 3V2W, resolution 3.35Å) (25). The structure was prepared using the protein preparation workflow in Maestro (39) to assign hydrogens, optimize hydrogen bonds and to perform constraint minimization. The homology model of S1P3 was built using the Uniprot sequence S1PR3_Human (accession Q99500) in Prime (39). This initial S1P3 model was optimized using the same protein preparation workflow above. Both S1P1 and the S1P3 models with the antagonist sphingolipid mimic ML5 ligand (25) were then optimized using a multi-step all-atom minimization and molecular dynamics (MD) simulation implemented in the software package Desmond (DE Shaw Research) (40). Prior to the MD multi-step simulation, a membrane bilayer model (POPC 300K) was added to both the S1P1 and S1P3 models. The system was set up using the OPLS-AA force field, the TIP4P explicit solvent model in an orthorhombic simulation box 10Å distance in all directions and adding counter ions. Simulations were performed at 300K and 1.01325 bar using the NPT ensemble class. All other settings were default. The production simulation time was 12 ns. Simulations were run on an IBM E-server 1350 cluster (36 nodes of 8 Xeon 2.3 GHZ cores and 12 GB of memory). Several later simulation frames were extracted from the S1P1 and S1P3 simulations based on conformational diversity, low (stable) RMSD, and a stable ML5 (ligand) pose with maximum H-bonds. These frames were then used to generate W269L mutant receptor for S1P1 and W256L and F263L mutant receptor structures for S1P3. To avoid clashing side chains, constraint minimization (39) was performed for the WT and mutant S1P1 and S1P3 receptor structures. These structures were then used for further modeling.
Ligand Receptor Binding Models
Using the optimized S1P1 and S1P3 WT and mutant receptor models above, we generated initial binding poses for the ligands of CYM-5441 and S1P as follows. Ligands were prepared using ligprep (39) to generate ionization states (pH=7) and stereoisomers resulting in a single representation for S1P and CYM-5441 and two representations for SPM242. Ligands were initially docked into the receptor structures using the Induced Fit Docking (IFD) (39) protocol with default settings. The IFD protocol includes a constraint receptor minimization step followed by initial flexible Glide docking of the ligand using a softened potential to generate an ensemble of poses. For each pose, the nearby receptor structure is then refined using Prime. Each ligand is then re-docked (using Glide) into its corresponding optimized low-energy receptor structure and ranked by Glide score. For S1P, we required at least two hydrogen bond interactions with two (of the three) polar receptor side chains known to interact with S1P (R120/114, E121/115, R292/K286; S1P1/S1P3). For CYM-5541 and SPM-242, no constrains were used. The best pose with highest IFD score obtained for each ligand was again subjected to MD simulation (3–5 ns production runs) for further optimization of the protein ligand complex. The MD protocol includes a multi-step procedure of minimizations and short MD runs followed by the production MD simulation. The same parameter and settings as described above were used. Poses were stable during the production MD runs. The final frames of these simulations were then used for docking of ligands after constraint minimization (39). Ligands were re-docked using Glide SP and XP with default potential and other settings. The best pose of the ligand was selected based on the Glide scores, known interactions (e.g. head group) and visual inspection. MM-GB/SA implemented in Prime was performed to calculate the relative binding free energies for the studied ligands. Receptor flexibility cutoff was set to 4Å around the ligand. 3D plots were produced using PyMol.
To evaluate the energetics of the best pose of CYM-5541 in S1P3 WT in comparison to the mutants, the ligand was cloned into the mutated receptors and scored in place using Glide XP. In the case of F263L the complex was minimized using MacroModel (39) with default settings prior to docking because the initial docking score was invalid due to an unfavorable Van der Waals interaction with Y92.
Supplementary Material
Acknowledgments
This work was supported by the NIH National Institute of Allergy and Infectious Diseases [Grants U01-AI074564, R01-AI055509], the NIH National Institute of Mental Health [Grant U54-MH084512], Grant GM-094618 (R. Stevens, TSRI), and SFP 1799 funding from Kyorin Pharmaceutical Co., Japan.
Footnotes
Supporting Information Available: This material includes Supporting Figures S1, S2, S4, S6 and S7; Supporting Tables S3, S5 and S8; and Supporting Schemes S9 and S10 describing the detailed syntheses and full NMR characterizations of CYM-5541 and SPM-242 respectively is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES CITED
- 1.Hopson KP, Truelove J, Chun J, Wang Y, Waeber C. S1P activates storeoperated calcium entry via receptor- and non-receptor-mediated pathways in vascular smooth muscle cells. American journal of physiology Cell physiology. 2011;300:C919–926. doi: 10.1152/ajpcell.00350.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Igarashi J, Michel T. Sphingosine-1-phosphate and modulation of vascular tone. Cardiovascular Research. 2009;82:212–220. doi: 10.1093/cvr/cvp064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rosen H, Gonzalez-Cabrera P, Marsolais D, Cahalan S, Don AS, Sanna MG. Modulating tone: the overture of S1P receptor immunotherapeutics. Immunological Reviews. 2008;223:221–235. doi: 10.1111/j.1600-065X.2008.00645.x. [DOI] [PubMed] [Google Scholar]
- 4.Rosen H, Gonzalez-Cabrera PJ, Sanna MG, Brown S. Sphingosine 1-phosphate receptor signaling. Annual review of biochemistry. 2009;78:743–768. doi: 10.1146/annurev.biochem.78.072407.103733. [DOI] [PubMed] [Google Scholar]
- 5.Rosen H, Sanna MG, Cahalan SM, Gonzalez-Cabrera PJ. Tipping the gatekeeper: S1P regulation of endothelial barrier function. Trends Immunol. 2007;28:102–107. doi: 10.1016/j.it.2007.01.007. [DOI] [PubMed] [Google Scholar]
- 6.Schwab SR, Cyster JG. Finding a way out: lymphocyte egress from lymphoid organs. Nat Immunol. 2007;8:1295–1301. doi: 10.1038/ni1545. [DOI] [PubMed] [Google Scholar]
- 7.Osborne N, Brand-Arzamendi K, Ober EA, Jin SW, Verkade H, Holtzman NG, Yelon D, Stainier DY. The spinster homolog, two of hearts, is required for sphingosine 1-phosphate signaling in zebrafish. Current biology : CB. 2008;18:1882–1888. doi: 10.1016/j.cub.2008.10.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Iwasaki A, Medzhitov R. A new shield for a cytokine storm. Cell. 2011;146:861–862. doi: 10.1016/j.cell.2011.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stevenson CE, Takabe K, Nagahashi M, Milstien S, Spiegel S. Targeting sphingosine-1-phosphate in hematologic malignancies. Anti-cancer agents in medicinal chemistry. 2011;11:794–798. doi: 10.2174/187152011797655122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Weigert A, Weichand B, Brune B. S1P Regulation of Macrophage Functions in The Context of Cancer. Anti-cancer agents in medicinal chemistry. 2011 doi: 10.2174/187152011797655096. [DOI] [PubMed] [Google Scholar]
- 11.Marsolais D, Rosen H. Chemical modulators of sphingosine-1-phosphate receptors as barrier-oriented therapeutic molecules. Nat Rev Drug Discov. 2009;8:297–307. doi: 10.1038/nrd2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Canals M, Sexton PM, Christopoulos A. Allostery in GPCRs: ‘MWC’ revisited. Trends Biochem Sci. 2011;36:663–672. doi: 10.1016/j.tibs.2011.08.005. [DOI] [PubMed] [Google Scholar]
- 13.Girkontaite I, Sakk V, Wagner M, Borggrefe T, Tedford K, Chun J, Fischer KD. The sphingosine-1-phosphate (S1P) lysophospholipid receptor S1P3 regulates MAdCAM-1+ endothelial cells in splenic marginal sinus organization. The Journal of experimental medicine. 2004;200:1491–1501. doi: 10.1084/jem.20041483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Means CK, Xiao CY, Li Z, Zhang T, Omens JH, Ishii I, Chun J, Brown JH. Sphingosine 1-phosphate S1P2 and S1P3 receptor-mediated Akt activation protects against in vivo myocardial ischemia-reperfusion injury. American journal of physiology Heart and circulatory physiology. 2007;292:H2944–2951. doi: 10.1152/ajpheart.01331.2006. [DOI] [PubMed] [Google Scholar]
- 15.Sensken SC, Staubert C, Keul P, Levkau B, Schoneberg T, Graler MH. Selective activation of G alpha i mediated signalling of S1P3 by FTY720-phosphate. Cellular signalling. 2008;20:1125–1133. doi: 10.1016/j.cellsig.2008.01.019. [DOI] [PubMed] [Google Scholar]
- 16.Waeber C, Blondeau N, Salomone S. Vascular sphingosine-1-phosphate S1P1 and S1P3 receptors. Drug news & perspectives. 2004;17:365–382. doi: 10.1358/dnp.2004.17.6.829028. [DOI] [PubMed] [Google Scholar]
- 17.Cinamon G, Zachariah MA, Lam OM, Foss FW, Jr, Cyster JG. Follicular shuttling of marginal zone B cells facilitates antigen transport. Nat Immunol. 2008;9:54–62. doi: 10.1038/ni1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cyster JG. Settling the thymus: immigration requirements. The Journal of experimental medicine. 2009;206:731–734. doi: 10.1084/jem.20090458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cyster JG, Schwab SR. Sphingosine-1-phosphate and lymphocyte egress from lymphoid organs. Annual review of immunology. 2012;30:69–94. doi: 10.1146/annurev-immunol-020711-075011. [DOI] [PubMed] [Google Scholar]
- 20.Pereira JP, Kelly LM, Cyster JG. Finding the right niche: B-cell migration in the early phases of T-dependent antibody responses. International immunology. 2010;22:413–419. doi: 10.1093/intimm/dxq047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang X, Cho B, Suzuki K, Xu Y, Green JA, An J, Cyster JG. Follicular dendritic cells help establish follicle identity and promote B cell retention in germinal centers. The Journal of experimental medicine. 2011;208:2497–2510. doi: 10.1084/jem.20111449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Niessen F, Schaffner F, Furlan-Freguia C, Pawlinski R, Bhattacharjee G, Chun J, Derian CK, Andrade-Gordon P, Rosen H, Ruf W. Dendritic cell PAR1-S1P3 signalling couples coagulation and inflammation. Nature. 2008;452:654–658. doi: 10.1038/nature06663. [DOI] [PubMed] [Google Scholar]
- 23.Harris GL, Creason MB, Brulte GB, Herr DR. In vitro and in vivo antagonism of a G protein-coupled receptor (S1P3) with a novel blocking monoclonal antibody. PloS one. 2012;7:e35129. doi: 10.1371/journal.pone.0035129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Valant C, Robert Lane J, Sexton PM, Christopoulos A. The best of both worlds? Bitopic orthosteric/allosteric ligands of g protein-coupled receptors. Annu Rev Pharmacol Toxicol. 2012;52:153–178. doi: 10.1146/annurev-pharmtox-010611-134514. [DOI] [PubMed] [Google Scholar]
- 25.Hanson M, Roth C, Jo E, Griffith M, Scott F, Reinhart G, Desale H, Clemons B, Cahalan SM, Schurer SC, Sanna MG, Han G, Kuhn P, Rosen H, Stevens R. Crystal Structure of a Lipid G-protein Coupled Receptor. Science. 2012;335:851–855. doi: 10.1126/science.1215904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cahalan SM, Gonzalez-Cabrera PJ, Sarkisyan G, Nguyen N, Schaeffer MT, Huang L, Yeager A, Clemons B, Scott F, Rosen H. Actions of a picomolar short-acting S1P agonist in S1P-eGFP knock-in mice. Nature chemical biology. 2011;7:254–256. doi: 10.1038/nchembio.547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Conn PJ, Christopoulos A, Lindsley CW. Allosteric modulators of GPCRs: a novel approach for the treatment of CNS disorders. Nat Rev Drug Discov. 2009;8:41–54. doi: 10.1038/nrd2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Melancon BJ, Hopkins CR, Wood MR, Emmitte KA, Niswender CM, Christopoulos A, Conn PJ, Lindsley CW. Allosteric modulation of seven transmembrane spanning receptors: theory, practice, and opportunities for central nervous system drug discovery. Journal of medicinal chemistry. 2012;55:1445–1464. doi: 10.1021/jm201139r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gonzalez-Cabrera PJ, Jo E, Sanna MG, Brown S, Leaf N, Marsolais D, Schaeffer MT, Chapman J, Cameron M, Guerrero M, Roberts E, Rosen H. Full pharmacological efficacy of a novel S1P1 agonist that does not require S1P-like headgroup interactions. Mol Pharmacol. 2008;74:1308–1318. doi: 10.1124/mol.108.049783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schurer SC, Brown SJ, Gonzalez-Cabrera PJ, Schaeffer MT, Chapman J, Jo E, Chase P, Spicer T, Hodder P, Rosen H. Ligand-binding pocket shape differences between sphingosine 1-phosphate (S1P) receptors S1P1 and S1P3 determine efficiency of chemical probe identification by ultrahigh-throughput screening. ACS Chem Biol. 2008;3:486–498. doi: 10.1021/cb800051m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jo E, Sanna MG, Gonzalez-Cabrera PJ, Thangada S, Tigyi G, Osborne DA, Hla T, Parrill AL, Rosen H. S1P1-selective in vivo-active agonists from high-throughput screening: off-the-shelf chemical probes of receptor interactions, signaling, and fate. Chemistry and Biology. 2005;12:703–715. doi: 10.1016/j.chembiol.2005.04.019. [DOI] [PubMed] [Google Scholar]
- 32.Rakhit S, Conway AM, Tate R, Bower T, Pyne NJ, Pyne S. Sphingosine 1-phosphate stimulation of the p42/p44 mitogen-activated protein kinase pathway in airway smooth muscle. Role of endothelial differentiation gene 1, c-Src tyrosine kinase and phosphoinositide 3-kinase. Biochemical Journal. 1999;338(Pt 3):643–649. [PMC free article] [PubMed] [Google Scholar]
- 33.Sato K, Tomura H, Igarashi Y, Ui M, Okajima F. Possible involvement of cell surface receptors in sphingosine 1-phosphate-induced activation of extracellular signalregulated kinase in C6 glioma cells. Molecular Pharmacology. 1999;55:126–133. doi: 10.1124/mol.55.1.126. [DOI] [PubMed] [Google Scholar]
- 34.NIH. From The Lab To The Clinic: Discovery From Molecular Libraries Program Enters Clinical Trials. 2011. [Google Scholar]
- 35.Deng Q, Clemas JA, Chrebet G, Fischer P, Hale JJ, Li Z, Mills SG, Bergstrom J, Mandala S, Mosley R, Parent SA. Identification of Leu276 of the S1P1 receptor and Phe263 of the S1P3 receptor in interaction with receptor specific agonists by molecular modeling, site-directed mutagenesis, and affinity studies. Mol Pharmacol. 2007;71:724–735. doi: 10.1124/mol.106.029223. [DOI] [PubMed] [Google Scholar]
- 36.Fujiwara Y, Osborne DA, Walker MD, Wang DA, Bautista DA, Liliom K, Van Brocklyn JR, Parrill AL, Tigyi G. Identification of the hydrophobic ligand binding pocket of the S1P1 receptor. Journal of Biological Chemistry. 2007;282:2374–2385. doi: 10.1074/jbc.M609648200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gonzalez-Cabrera PJ, Hla T, Rosen H. Mapping pathways downstream of sphingosine 1-phosphate subtype 1 by differential chemical perturbation and proteomics. The Journal of biological chemistry. 2007;282:7254–7264. doi: 10.1074/jbc.M610581200. [DOI] [PubMed] [Google Scholar]
- 38.Kenakin TP. Pharmacologic Analysis of Drug-Receptor Interaction. Raven Press Books, Ltd; New York: 1987. [Google Scholar]
- 39.MacroModel, version 9.9. Schrodinger, LLC; New York, NY: 2011. [Google Scholar]
- 40.Bowers KJ, Chow E, Xu H, Dror RO, Eastwood MP, Gregersen BA, Klepeis JL, Kolossváry I, Moraes MA, Sacerdoti FD, Salmon JK, Shan Y, Shaw DE. Scalable Algorithms for Molecular Dynamics Simulations on Commodity Clusters. Proceedings of the ACM/IEEE Conference on Supercomputing (SC06); Tampa, Florida, USA. 2006. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.