

Abstract
Multipotential stromal cells, or mesenchymal stem cells, (MSC) have ben proposed as aids in regenerating bone and adipose tissues, as these cells form osteoblasts and adipocytes. A major obstacle to this use of MSC is the initial loss of cells post-implantation. This cell death in part, is due to ubiquitous non-specific inflammatory cytokines such as FasL generated in the implant site. Our group previously found that soluble epidermal growth factor (sEGF) promotes MSC expansion. Further, tethering EGF onto a two-dimensional surface (tEGF) altered MSC responses, by restricting epidermal growth factor receptor (EGFR) to the cell surface, causing sustained activation of EGFR, and promoting survival from FasL-induced death. sEGF by causing internalization of EGFR does not support MSC survival. However, for tEGF to be useful in bone regeneration, it needs to allow for MSC differentiation into osteoblasts while also protecting emerging osteoblasts from apoptosis. tEGF did not block induced differentiation of MSCs into osteoblasts, or adipocytes, a common default MSC-differentiation pathway. MSC-derived pre-osteoblasts showed increased Fas levels and became more susceptible to FasL induced death, which tEGF prevented. Differentiating adipocytes underwent a reduction in Fas expression and became resistant to FasL-induced death, with tEGF having no further survival effect. tEGF protected undifferentiated MSC from combined insults of FasL, serum deprivation and physiologic hypoxia. Additionally, tEGF was dominant in the face of sEGF to protect MSC from FasL-induced death. Our results suggest that MSCs and differentiating osteoblasts need protective signals to survive in the inflammatory wound milieu and that tEGF can serve this function.
Keywords: Epidermal growth factor, Mesenchymal stem cells, Multipotential Stromal Cells, Osteoblasts, Adipocytes
1. Introduction
Adult human MSC have the potential in vitro to form a variety of cell types including osteoblasts, chondrocytes and adipocytes [1-2], but clinical success in regenerating connective tissues from these cells has been relatively elusive. Although the use of freshly aspirated marrow during bone grafting has shown improvement, many studies have shown that transplanted cells likely do not contribute to bone growth. In MSC based regenerative experiments in animal models, most of the positive effects seen post delivery of MSC occur due to trophic effects secondary to MSC release of growth factors, and not due to MSC differentiation. The MSC-derived growth factors support development of extracellular matrix and new blood vessels in cells intrinsic to the wounded tissues; but most evidence suggests that differentiated cells in the regenerated tissue do not arise from differentiation of transplanted MSC [3-5]. This is in part because most of the implanted MSC are lost within 48 hours and less than 1% of implanted cells are detectable after 7 days [6-9]. The loss in cell numbers can be due to several adverse factors in the ischemic wound environment including ROS [10], hypoxia [11], nutrient deprivation [12] and presence of first-phase inflammatory cytokines like FasL and TRAIL to which MSC are most susceptible [13].
We previously reported that sEGF could be used to increase MSC numbers while not affecting differentiation of these cells in the absence or presence of external differentiation cues [14]. However, when challenged with pro-apoptotic cytokines, sEGF only provided protection to MSC for a short time frame most likely due to rapid internalization of EGFR [19] and on prolonged exposure actually enhanced death in MSC, likely due to activation of mitogenic stimulus [13]. Surface-restricted EGFR with persistent activation restricted to the peri-plasma membrane space is known to signal in a qualitatively distinct manner for motility and survival over mitogenesis in other cell types [13, 15]. To accomplish this mode of survival in MSC, EGF was tethered using a polyethylene oxide based polymer onto a two-dimensional glass surface on which MSCs were grown. This restriction of EGFR to the MSC membrane caused prolonged downstream EGFR signaling and increased survival in the presence of death signals like FasL [13,16]. Thus, this approach became a candidate for promoting survival of implanted MSCs in the wound milieu.
Survival of implanted MSC in an undifferentiated state is only part of the story; the MSCs must subsequently differentiate into the desired tissues, and do so within this inflammatory field. Thus, in this study we investigated whether MSC in the process of differentiating towards a specific lineage were similar to or divergent from undifferentiated MSC in terms of susceptibility to inflammatory signals, since receptors and protein composition in cells undergo major changes during the progress of differentiation. We looked specifically at differentiating osteoblasts and adipocytes with a long-term perspective of cranio-facial reconstruction and healing of critical sized defects in long bones. FasL was used as a representative cytokine since undifferentiated MSCs die the most when its receptor Fas is activated, either directly or by transactivation [13,17,18,19]. We found not only that differentiating osteoblasts need external survival cues, but also that the tEGF signal from the engineered surfaces persists for 30 days and protects both pre-osteoblasts and osteoblasts in the presence of FasL. Further we challenged MSC with low oxygen, serum deprivation and FasL, a situation likely to be extant in the wound, and found that tEGF provided ad survival advantage to the cells under these multiple unfavorable conditions.
2. Materials and Methods
2.1 Reagents
α-MEM without ribonucleotides or deoxyribonucleotides was obtained from Gibco (Carlsbad, CA), DMEM 1g/l glucose was from Cellgro (Mediatech, Washington, DC), fetal bovine serum (FBS) was from Atlanta Biologicals (Norcross, GA) and adipogenic differentiation media was from Lonza (Walkersville, MD). Human recombinant epidermal growth factor [354052] was from BD Biosciences (Franklin Lakes, NJ), murine epidermal growth factor was from Peprotech [#315-09] (Rocky Hill, NJ), and human soluble recombinant Super-FasL [ALX-522-020-3005] was from Enzo Lifesciences (Plymouth Meeting, PA). RNeasy Minikit and QuantiTect Reverse Transcription kit was form Qiagen (Valencia, CA). Brilliant SYBR Green qPCR Master Mix was from Stratagene (Santa Clara, CA). Poly ADP ribose polymerase (PARP) [9542] and Fas [4233] antibodies were form Cell Signaling (Boston, MA), fatty acid binding protein 4 (FABP4) [SAB2104636] and GAPDH [G5945] antibodies were from Sigma Aldrich (St. Louis, MO), EGFR antibody [sc-365829] was from Santa Cruz Biotechnology (Santa Cruz, CA), and Caspase inhibitor z-VAD-FMK [G7231] was from Promega (Madison, WI).
2.2 Cell Culture
Two human bone marrow derived MSC cell preparations were used for the study. The first is an immortalized human multipotent stromal cell line (imhMSC) [20], a kind gift from Dr. Junya Toguchida, Kyoto University, Japan. The cells were derived from a single clone and immortalized by expression of hTERT. Immortalized hMSC were used for standardization, consistency and to work with a pure population that can be differentiated into multiple lineages from a common progenitor. These cells were cultured in proliferation media comprising DMEM 1g/l glucose with 10% FBS, 1mM Sodium pyruvate, 1μM non-essential amino acids and 100units/ml penicillin-streptomycin. The second is a primary human bone marrow multipotential stromal cell isolate (prhMSC) provided by Dr. Darwin Prockop (Texas A&M). Proliferation media for these cells was composed of α-MEM without ribonucleotides or deoxyribonucleotides with 16.5% FBS, and 2mM L-glutamine. Immortalized hMSC between 6 and 15 passages, and prhMSC within passage 4 were used for experiments. These passages were tested as positive for MSC being undifferentiated by qPCR of osteogenic and adipogenic markers as well as von Kossa staining for osteogenesis and Oil Red O staining for adipogenesis. Immortalized hMSC were found to undergo differentiation into osteoblasts and adipocytes at a rate similar to primary human MSC.
Proliferation media maintains the two MSC cell preparations in the undifferentiated state and the cells are refers to as Day 0. When cells at Day 0 established complete cell-cell contact on the culture surface, media was changed to either osteogenic differentiation media comprising proliferation media plus 50μM ascorbic acid, 100μM glycerol-2-phosphate and 100nM dexamethasone, or adipogenic differentiation media from Lonza consisting of differentiation inducers indomethacin, isobutyl methyl xanthine and insulin. imhMSC were grown on all three surfaces to standardize experiments: mock (treated but without EGF added for tethered), tEGF and tissue culture plastic. Experiments were repeated with prhMSC, first on tissue culture plastic and select experiments on mock and tEGF surfaces, as mentioned in the text. For culture of cells at 4% and 1% oxygen, a Biospherix (Lacona, NY) workstation and incubators were used so that the cells remained in constant oxygen concentrations during passaging and experimentation.
2.3 Mock and tEGF surface preparation
To create tEGF surfaces, EGF was attached to surfaces composed of a silanized glass coverslip spin-coated with poly methyl methacrylate-graft-polyethylene oxide (PMMA-g-PEO) polymer in two different weight percentages (33% and 22%) as previously described [13,21]. The 33% PEO component of the polymer covalently binds EGF through an activated 4-nitrophenyl chloroformate group at its free apical end, while the 22% PEO component increases adhesiveness if MSC to the polymer coated glass surface by allowing for adsorption of proteins, The higher weight polymer was mixed with the lower weight polymer in the ratio of 60:40 to achieve a concentration of 5000-7000EGF per μm2 of the glass coverslip while allowing for cell attachment and spread. This concentration of EGF allows for saturation of EGFR found in low numbers in MSC: as low as 7,300 in imhMSC [14].
Mock surfaces consist of all components of the tEGF surface, minus the EGF itself to simulate a similar environment to MSC. They are made up of PMMA-g-PEO polymer spin-coated onto silanized glass coverslips with no activated 4-nitrophenylcholoroformate groups or attached murine EGF. Murine EGF is used due to the presence of only one tertiary amine at the N-terminus that helps link EGF based on amine chemistry. Biologically murine and human EGF are indistinguishable, with murine EGF shown to induce downstream EGFR pathways similar to human EGF in human MSC [13]. Mock or tEGF surfaces were placed in wells of a 12-well plate, coated with 3μg/ml of Collagen I in PBS for 2 hours, followed by blocking with 1% BSA in PBS and UV sterilization for 30 minutes before MSC were seeded.
2.4 Immunoblots
To check for responses of MSC to FasL, cells were grown on tissue culture plastic. After treatment of cells with cytokines at defined time-points, both floating cells in the media and cells left on the culture plate were combined and lysed in SDS lysis buffer (0.1M Tris-HCl, 4% SDS, 0.2% Bromophenol Blue and 5% β-mercaptopethanol). Cell lysates were separated by electrophoresis on polyacrylamide gels in SDS-Laemmli buffers, and transferred electrophoretically to polyvinylidine difluoride-derivatized nylon. After the blots were blocked with 5% milk in PBS, target proteins were probed with primary antibodies at 1:1000 for PARP or 1: 10000 for GAPDH. Immunoblots for PARP were not performed on MSC grown on either mock or tEGF surfaces since the cells left on the surface after treatment with death cytokines gave insufficient protein. FLICA was used to analyze MSC death on surfaces. For determination of EGFR and Fas levels on differentiating MSC, imhMSC and prhMSC grown on tissue culture plastic or mock surfaces were lysed and separated by electrophoresis as mentioned above and blots probed with 1: 500 EGFR antibody or 1:2000 Fas antibody in 1% milk in PBS, followed by horseradish peroxidase-conjugated secondary antibodies, and the bound peroxidase was visualized by enhanced chemiluminescence (WBKLS0500, Millipore, Billerica, MA).
2.5 Fluorochrome inhibitor of caspase assay (FLICA)
FLICA consists of cell permeable and non-cytotoxic inhibitors of caspase-3 bound to sulforhodamine, which emits red fluorescence when bound to active caspase-3. MSCs were grown on tissue culture plastic, mock or tEGF surfaces and treated with cytokines. 30 minutes prior to the lapse of time-treatment with cytokines, FLICA reagent (#APT 503, Millipore, Billerica, MA) and Hoescht 33342 dye was added to cells in media and incubated for 30 minutes at 37°C. Media was aspirated, cells washed with wash buffer and live cells imaged for sulforhodamine fluorescence. Red fluorescence indaicative of active caspase3 was measured by Image J analysis software (National Institutes of Health). To account for cell loss, intensity of FLICA was normalized to the number of cells remaining on the chamber slide. To account for differences in the optical properties of the surfaces over time (matrix and cell accumulation), the intensity was also normalized to background for the control diluent treatment at that time. The bar graphs account for these differences and thus may appear slightly discrepant with the visualization of photomicrographs.
2.6 Real time quantitative PCR Analysis
Total RNA form MSC was isolated using guanidine isothiocyanate denaturation followed by silica gel binding (RNeasy kit, Qiagen, Valencia, CA). 1μg of RNA was reverse transcribed into first strand c-DNA using QuantiTect Reverse Transcription kit (Qiagen). Real time PCR was performed on a MX3000P instrument (Startagene) using 1μl of cDNA and 12.5μl of Brilliant Green qPCR Master Mix in a total volume of 25μl with 1nM of forward and reverse primers: GAPDH Forward Primer: 5′-GAGTCAACGGATTTGGTCGT-3′; GAPDH Reverse Primer: 5′-TTCATTTTGGAGGGATCTCG-3′; Fas Forward Primer: 5′-TGAAGGACATGGCTTAGAAGTG-3′; Fas Reverse Primer: 5′-GGTGCAAGGGTCACAGTGTT-3′; EGFR Forward Primer 5′-AGGACCAAGCAACATGGTCA-3′; EGFR Reverse Primer 5′-CCTTGCAGCTGTTTTCACCT-3′; Collagen I Forward Primer 5′-CAATGCTGCCCTTTCTGCTCCTTT-3′; Collagen I Reverse Primer 5′-CACTTGGGTGTTTGAGCATTGCCT-3′; PPARG2 Forward Primer 5′-CACAAGAACAGATCCAGTGGTTGC-3′; PPARG2 Reverse Primer 5′-CAATTGCCATGAGGGAGTTGGAAG-3′; Osteocalcin Forward Primer 5′-GTTTATTTGGGAGCAGCTGGGATG-3′; Osteocalcin Reverse Primer 5′-GTTTATTTGGGAGCAGCTGGGATG-3′; Runx2 Forward Primer 5′-CCTCGGAGAGGTACCAGATG-3′; Runx2 Reverse Primer 5′- TTCCCGAGGTCCATCTACTG-3′ The GAPDH primers were the normalization controls, at an annealing temperatures of 55°C and 45 cycles. All reactions were performed in triplicates and the fold change in transcript levels was calculated based on ΔΔCt method with GAPDH as reference. Product size was confirmed using agarose gel electrophoresis.
2.7 Differentiation Assays
Oil Red O Assay was used to stain adipocytes. Cells were washed in PBS, fixed in 4% formaldehyde for 20 minutes, placed in 60% iso-propanol for 5 minutes, followed by addition of 3mg/ml Oil Red O in 3:2 isopropanol: water for 10 minutes. Cells were washed and photographed under bright field.
For silver (von Kossa) satin of hydroxyapatite, cells were washed in PBS, fixed in 4% formaldehyde for 20 minutes and placed in a 1% silver nitrate in water under UV light for 10 minutes. Cells were washed in sodium thiosulphate and photographed under transmitted light.
2.8 Statistical Analysis
Cell apoptosis was measured using paired t test. p<0.05 was stated as statistically significant.
3. Results
3.1 MSC susceptibility to FasL-induced cell death continues as cells differentiate into osteoblasts
Our long-term aim is to use MSC to reconstruct critical sized defects in bone that do not heal by combining a source of MSC with a scaffold. For regeneration of bone to occur, both proliferating MSC and differentiating pre-osteoblasts derived from MSC must survive exposure to inflammatory cytokines present at the wound site. To test if differentiating osteoblasts died in the presence of FasL, we first established culture medium conditions that drove cells toward the osteogenic phenotype as determined by accumulation of hydroxyapatite (assessed via von Kossa staining) and expression of osteogenic markers (as assessed by real-time qPCR for Runx2 and Osteocalcin) after 30 days (Figure 1A-B and Supplemental Figure 1A). Immortalized hMSC on tissue culture plastic (data not shown), imhMSC on mock surfaces or prhMSC on tissue culture plastic differentiating into osteoblasts, displayed hydroxyapatite deposition after 15 and 30 days in osteogenic differentiation media (Figure 1A). Real-time PCR showed increases in the early osteogenic marker Runx2 and late osteogenic marker Osteocalcin in imhMSC on mock surfaces on Day 6 and Day 21 (Figure 1B) as well as prhMSC on tissue culture plastic on Days 15 and 30 of osteogenic differentiation (Supplemental Figure 1A).
Figure 1. MSC forming osteoblasts continue to be susceptible to inflammatory cytokines like FasL.
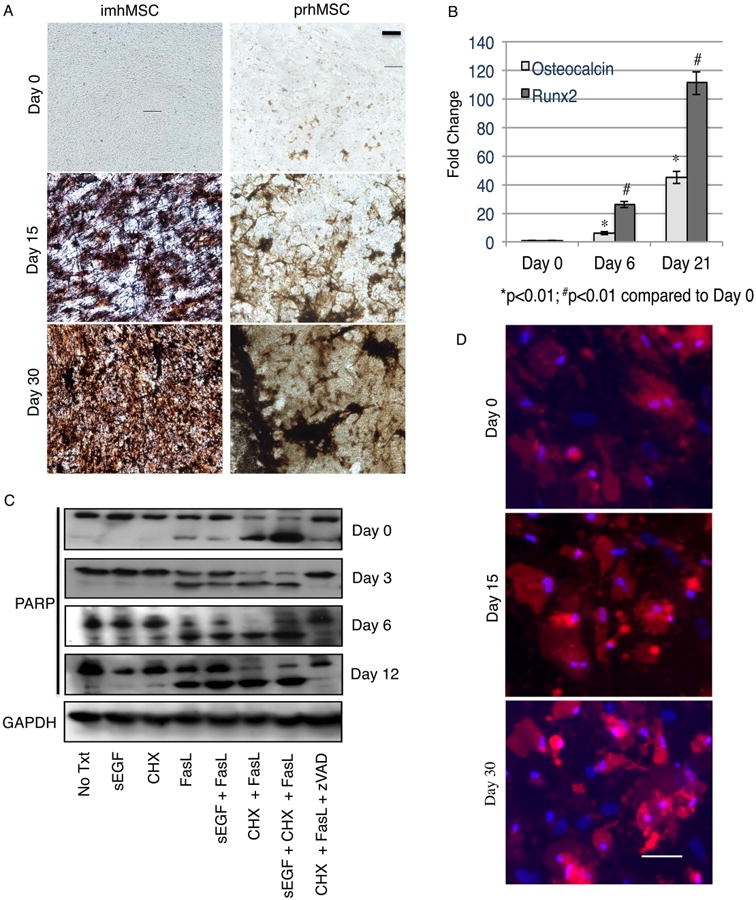
Immortalized hMSC on mock surface (Magnification 20×, Scale bar 10μm) and prhMSC on tissue culture plastic (Magnification 20×, Scale bar 10μm) depositing hydroxyapatite by von Kossa (A). Change in expression of osteogenic markers by qPCR during osteogenic differentiation of imhMSC on mock surfaces (B). Immunoblot for full length and cleaved PARP in differentiating imhMSC grown on tissue culture plastic and treated with cytokines for 8 hours (C). Caspase 3 positive cells (red) and Hoescht 33342 (blue) stained prhMSC during progress of osteogenic differentiation (Magnification 10×, Scale bar 50m). Graphs of mean ± s.e.m and representative photomicrographs of cells, of two independent experiments.
After defining the osteogenic differentiation protocol, we subjected differentiating imhMSC on tissue culture plastic to FasL cytokine treatment to test for susceptibility to cell death. Differentiating imhMSC on tissue culture plastic were treated with FasL on Day 0, Day 3, Day 6 and Day 12 for 8 hours and apoptosis assessed by the amount of cleaved PARP. After 12 days the high levels of matrix and/or fat interfered with immnoblotting and thus survival was determined only using FLICA. The 8 hour time period was chosen based on optimum detection of cells undergoing caspase 3 activation and apoptosis (Supplemental Figure 2A). On Day 0, while imhMSC were still undifferentiated, treatment with FasL alone caused PARP cleavage. Cell death was enhanced when cells were treated with FasL and cycloheximide (CHX), a protein synthesis inhibitor. Cycloheximide was added with FasL to increase cell stress and to mimic a starved, non-perfused wound environment. sEGF was unable to protect undifferentiated imhMSC from FasL induced cell death as previously observed [13]. The pan-caspase inhibitor zVAD-FMK significantly reduced degradation of PARP in the presence of CHX and FasL, confirming MSCs undergo apoptotic cell death in the presence of FasL. Controls with only CHX or sEGF did not cause imhMSCs to undergo PARP degradation and apoptosis.
Immortalized hMSCs differentiating into osteoblasts continued to undergo apoptosis in the presence of only FasL or CHX and FasL. sEGF did not protect differentiating MSC from FasL or CHX and FasL (Figure 1C). Immortalized hMSC are a true multipotential cell line since they were immortalized from a single clone and all cells of the population show similarities in differentiation and susceptibility to death cytokines. prhMSC on the other hand, although isolated to contain mainly cells with stem cell like properties, are a heterogenous stromal cell population; hence, a heterogeneous response to differentiation cues or death signals is expected in this population. To assess possible heterogeneity in response to FasL in this cell population, we used an in situ FLICA protocol after 12 hr of prhMSC treatment with FasL, at Day 0, as well as at Day 15 and Day 30 (Figure 1D). We saw that undifferentiated prhMSC, as well as differentiating pre-osteoblasts and differentiated osteoblasts are susceptible to death with FasL. The 12 hour time-point was chosen for further study of prhMSC death since this time-point showed caspase 3 activation, while still allowing for cell attachment to the culture surface (Supplemental Figure 2B). We were able to conclude that differentiating osteoblasts are susceptible to FasL-induced cell death similar to undifferentiated MSC.
3.2 tEGF protects differentiating osteoblasts from FasL induced cell death
Although our group had shown previously that tEGF protected undifferentiated MSC against FasL induced cell death by restricting EGFR to the cell membrane and prolonging downstream EGFR signaling [13], it was unknown whether the tEGF signal persisted to longer times scales required to protect differentiating MSC-derived osteoblasts from FasL-mediated death. Immortalized hMSC were either grown on mock or tEGF surfaces and were treated with FasL and/or other factors for 8 hours. Tethered EGF protected differentiating osteoblasts at Day 15 and differentiated osteoblasts on Day 30 in the presence of CHX and FasL (Figure 2A). Soluble EGF was unable to protect cells on mock surfaces from CHX and FasL induced apoptosis on Days 15 and Day 30 similar to what was seen on Day 0.
Figure 2. tEGF protects differentiating osteogenic cells from death signals.
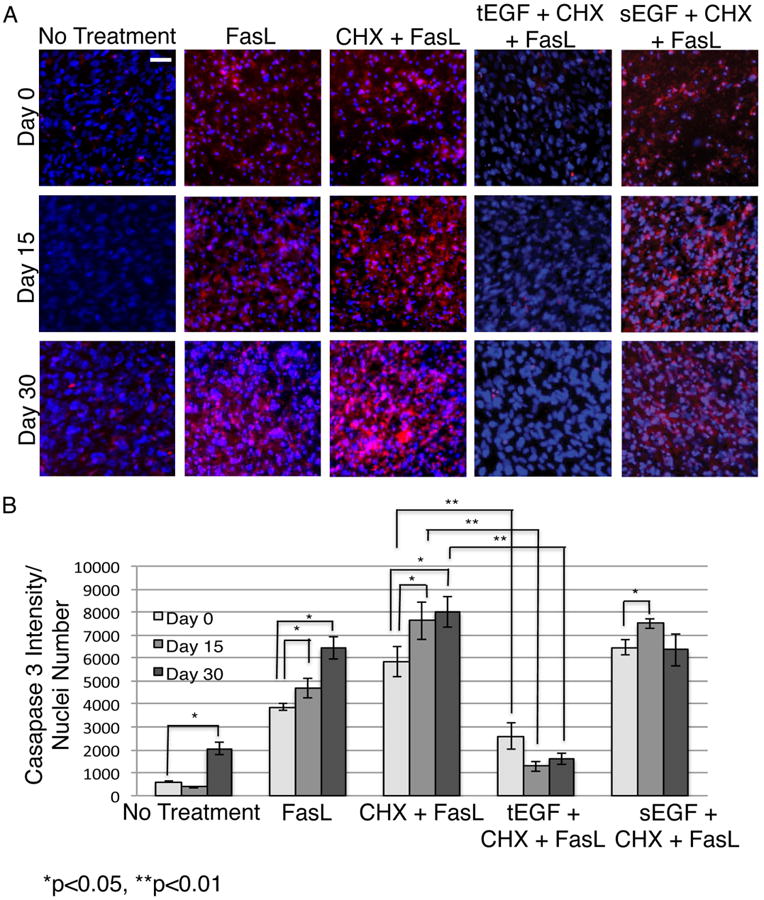
Fluorochrome inhibitor of caspase assay (FLICA) stained imhMSC (Magnification 10×, Scale bar 50μm) grown on mock/tEGF surfaces under osteogenic conditions, after 8 hours of treatment with cytokines on Day 0/ Day 15/ Day 30 (A). Quantification of FLICA intensity normalized to cell numbers and optical background (B). Shown are representative photomicrographs of cells and graphs of mean ± s.e.m of two independent experiments.
Caspase3 intensity measured in relation to total cell numbers showed continued cell death with FasL alone or CHX and FasL as differentiation into osteoblasts progressed. While sEGF was unable to protect against this death, tEGF significantly reduced FasL-induced apoptosis (Figure 2B). Tethered EGF therefore appears to be an appealing candidate for improving survival in both undifferentiated MSC and in differentiating osteoblasts.
3.3 MSC susceptibility to FasL induced cell death decreases as cells differentiate into adipocytes
Although stimulation of EGFR with tEGF increased survival of differentiating osteoblasts and did not impair their differentiation, MSC can differentiate down multiple lineages. One of the main concerns for clinical use of MSC is the activation of one of the default pathways of adipocyte formation, hence, we investigated whether differentiating adipocytes also undergo enhanced cell death during the progress of differentiation in the presence of FasL in a manner similar to differentiating osteoblasts. We first established protocols for adipocytic differentiation. Confluent undifferentiated imhMSC and prhMSC placed in adipocyte differentiating media displayed a collection of tiny oil droplets seen by Oil Red O stain on Day 12, which coalesced to form larger droplets in adipocytes on Day 20, indicating white fat cell formation (Figure 3A). This progression of oil droplet deposition was seen in imhMSC or prhMSC cells differentiating on tissue culture plastic or mock surfaces (Figure 3A). Real time PCR of adipogenesis markers peroxisome proliferator activated receptor γ (PPAR γ) and alkaline lipid binding protein (ALBP) showed increased expression with the advancement of adipogenic differentiation in both imhMSC grown on mock surfaces (Figure 3C) or prhMSC grown on tissue culture plastic (Supplemental Figure 1B). Adipogenesis was further confirmed in imhMSC grown on mock surfaces by immunoblot of fatty acid binding protein 4 (FABP4) that increased with progress of differentiation into adipocytes. The average nuclear size of differentiated adipocytes was significantly smaller than that of undifferentiated imhMSC indicating accumulation of fat droplets (Supplemental Figure 1C).
Figure 3. MSC forming adipose cells become more resistant to inflammatory cytokines like FasL.
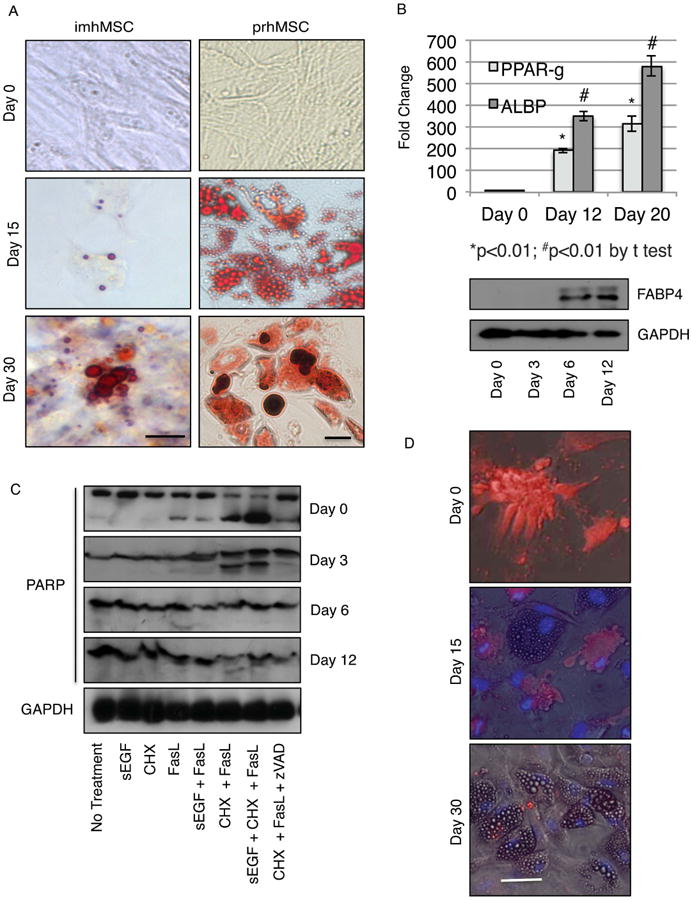
Immortalized human MSC (imhMSC) on mock surface (Magnification 40×, Scale bar 10μm) and isolated primary human MSC (prhMSC) on tissue culture plastic (Magnification 20×, Scale bar 10μm) differentiating into adipose cells by Oil Red O (A). Change in expression of adipogenic markers by qPCR and immunoblot during adipogenic differentiation of imhMSC on mock surfaces (B). Immunoblot for full length and cleaved PARP in differentiating imhMSC grown on tissue culture plastic and treated with cytokines for 8 hours (C). Caspase 3 positive cells (red) and Hoescht 33342 (blue) stained overlaid with phase contrast images of prhMSC during progress of adipogenic differentiation (Magnification 10×, 50μm). Graphs of mean ± s.e.m and representative photomicrographs of cells, of two independent experiments.
With adipocytic differentiation protocols established, we next subjected differentiating imhMSC on tissue culture plastic to FasL cytokine treatment to test for susceptibility of the differentiating adipocytes to cell death. Cells on Days 0, 3, 6 and 12 of differentiation regimen were exposed to FasL for 8 hours and cell death determined by an immunoblot for the caspase-3 substrate PARP. As the imhMSCs underwent differentiation into adipocytes, on Day 3, Day 6 and Day 12 there was lesser-cleaved PARP in the presence of both FasL alone or CHX and FasL. Addition of sEGF to FasL or CHX and FasL did not alter the response of pre-adipocytes to FasL. The negative control of zVAD-FMK along with CHX and FasL showed no apoptosis. Treatment with only sEGF or CHX did not cause death of differentiating imhMSC. These results indicate that MSC differentiating into adipocytes become less susceptible to apoptotic death by FasL.
With prhMSC, we hypothesized that not all cells of the prhMSC population might react to differentiation cues or death signals in the same manner. The cells within the prhMSC population differentiated at slightly dysynchronous rates and extents. When we tested cell death of differentiating prhMSC under FasL treatment by the in situ FLICA assay, we observed that undifferentiated cells died after 12 hours of treatment; on Day 15 only cells without oil droplets underwent cell death, and on Day 30, when most cells had oil droplets, there was very little cell death, occurring only in undifferentiated stromal cells (Figure 3D). The 12-hour incubation period for treatment with FasL was chosen based on a time response of prhMSC to FasL (Supplemental Figure 2B). To ensure that the resistance of adipocytes to FasL was not simply a short time window effect, we extended treatment of differentiating adipocytes on Days 15 and 30 with FasL from 12 hours to 24 hours and looked for apoptosis by FLICA. Even at 24 hours of treatment with FasL, cells with oil droplets did not undergo apoptosis (Supplemental Figure 3). This confirmed that adipose cells were resistant to inflammatory cytokines like FasL.
3.4 tEGF protects MSC, while differentiating adipocytes become resistant to FasL
Since differentiating adipocytes are resistant to FasL we wanted to see if tEGF altered this acquired resistance to cell death in the presence of cytokines. Immortalized hMSC were differentiated on mock or tEGF surfaces and subjected to treatment with FasL for 8 hours. Caspase3 activity was analyzed as a measure of cell apoptosis by FLICA assay. Cells on Day 0 showed caspase3 activation with CHX and FasL which sEGF was unable to prevent. imhMSC grown on tEGF surfaces and treated with CHX and FasL on the other hand were protected from caspase3 activation. Differentiating adipocytes on Day 15 and Day 20 showed lesser caspase3 activation and apoptosis on surfaces similar to outcomes on tissue culture plastic surfaces. Cells on Days 15 and 20 grown on tEGF surfaces did not show a change in cell death in the presence of CHX and FasL (Figure 4A). Intensity of caspase3 fluorescence was normalized to cell numbers to account for the loss of cells nearing late apoptosis and these values across the various treatments represented in Figure 4B. We were able to conclude that MSC differentiating into adipocytes become resistant to death signals as they begin accumulating fat deposits and tEGF signaling from the biomaterial surfaces does not have any effect on this resistance to cell death.
Figure 4. tEGF does not alter resistance of adipose cells to death signals.
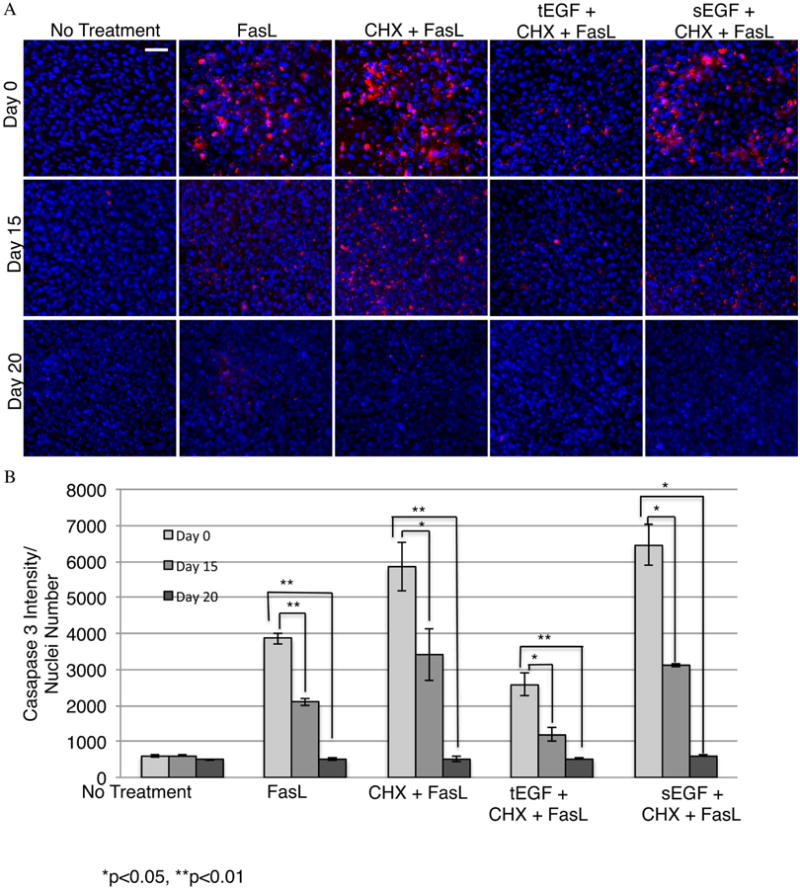
Fluorochrome inhibitor of caspase assay (FLICA) stained imhMSC (Magnification 10×, Scale bar 50μm) grown on mock/tEGF surfaces under adipogenic conditions, after 8 hours of treatment with cytokines on Day 0/ Day 15/ Day 20 (A). Quantification of FLICA intensity normalized to cell numbers and optical background (B). Shown are representative photomicrographs of cells and graphs are of mean ± s.e.m of two independent experiments.
3.5 Changes in receptor expression with differentiation correlates with differences in susceptibility to death
We initially looked at death in differentiating MSC based on the hypothesis that differentiating cells would react to cytokines and death factors differently from MSC. This is because during the progress of differentiation, MSC will undergo changes in various death factor receptors as well as pro- and anti-apoptotic proteins. Since we used FasL as a representative death cytokine, we looked at expression of its receptor Fas as MSC differentiated into fat and bone in vitro. Fas levels in imhMSC differentiating into adipocytes decreased on Days 0, 6 and 12 by immunoblot (Figure 5A) and real time qPCR (Figure 5B) as well as in adipocytes derived from prhMSC (Supplemental Figure 1D). Conversely Fas levels in differentiating osteoblasts derived from imhMSC increased as seen by immunoblot (Figure 5C). Real time PCR showed increases in Fas in differentiating osteoblasts derived from imhMSC (Figure 5D) as well as prhMSC (Supplemental Figure 1E). These results suggest that changes in death receptor levels alter the susceptibility of differentiating MSC and make adipocytes less susceptible and osteoblasts more vulnerable to the death cytokine FasL.
Figure 5. Changes in protein receptor levels during differentiation contribute to altered reactions to death signals.
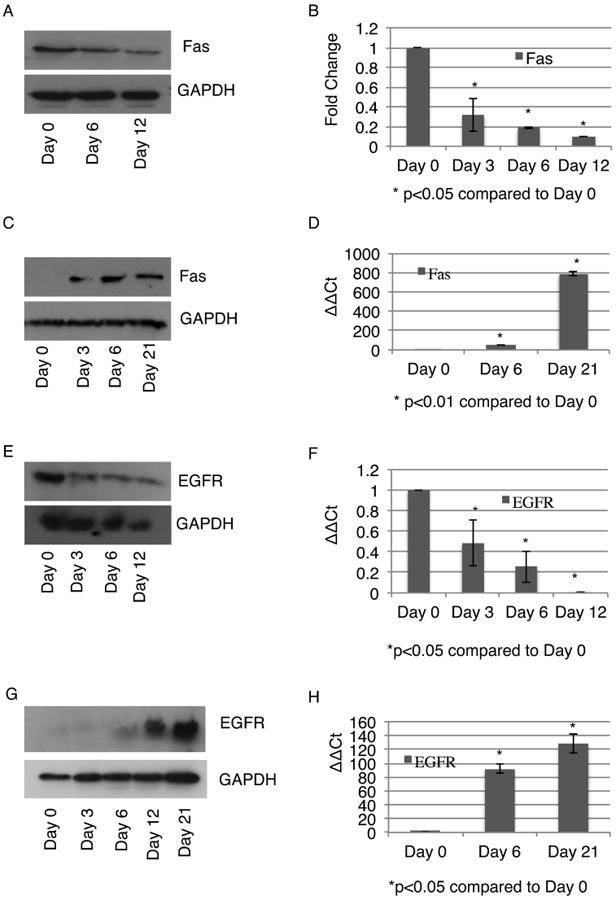
Change in levels of Fas in differentiating adipocytes grown on tissue culture plastic by immunoblot (A) and real time qPCR (B). Change in levels of Fas in differentiating osteoblasts grown on tissue culture plastic by immunoblot (C) and real time qPCR (D). Change in levels of EGFR in differentiating adipocytes grown on tissue culture plastic by immunoblot (E) and real time qPCR (F). Change in levels of EGFR in differentiating osteoblasts grown on tissue culture plastic by immunoblot (E) and real time qPCR (F). Representative immunoblots and graphs are of mean ± s.e.m from two independent experiments.
Since tEGF offered protection to not only MSC but also differentiating osteoblasts in the presence of FasL, we looked at changes in EGFR levels with the progress of MSC differentiation. imhMSC differentiating into adipocytes lost EGFR expression as seen by immunoblot (Figure 5E) and real time PCR (Figure 5F). Similar loss in EGFR expression was seen in prhMSC differentiating into adipocytes (Supplemental Figure 1D). The absence of EGFR expression might explain why treatment with tEGF does not affect either adipocyte death or survival in the presence of FasL. imhMSC differentiating into osteoblasts on the other hand show increasing expression of EGFR with advancement of differentiation by immunoblot (Figure 5G) as well as real time PCR (Figure 5H), as seen in differentiating prhMSC (Supplemental Figure 1E). Increased EGFR levels allowed for continued protection offered by tEGF both to pre-osteoblasts and differentiated osteoblasts in the presence of FasL.
3.6 tEGF does not block induced osteogenic or adipogenic differentiation, and osteogenic differentiation is faster at 4% oxygen
The previous experiments were all performed at ambient oxygen concentrations of 21%. However oxygen levels in the bone are comparatively lower at 4-7%. We needed to confirm osteogenesis and adipogenesis of MSC on tEGF surfaces in the presence of differentiation inducers, under ambient oxygen levels as well as physiologic normal levels of 4% oxygen. Immortalized hMSC were grown on tEGF surfaces either in proliferation media alone or osteogenic media for 15 or 30 days, and mineralization assessed by von Kossa staining. MSC underwent osteogenic differentiation on tEGF surfaces in the presence of differentiation media at both 4% and 21% oxygen, however osteogenesis was achieved faster at 4% oxygen as observed by hydroxyapatite deposition on Day 15 (Figure 6A). tEGF by itself in the absence of differentiation inducers did not cause osteogenic differentiation of imhMSC.
Figure 6. tEGF does not inhibit osteogenesis or adipogenesis in the presence of differentiation media; Faster osteogenesis occurs at 4% oxygen.
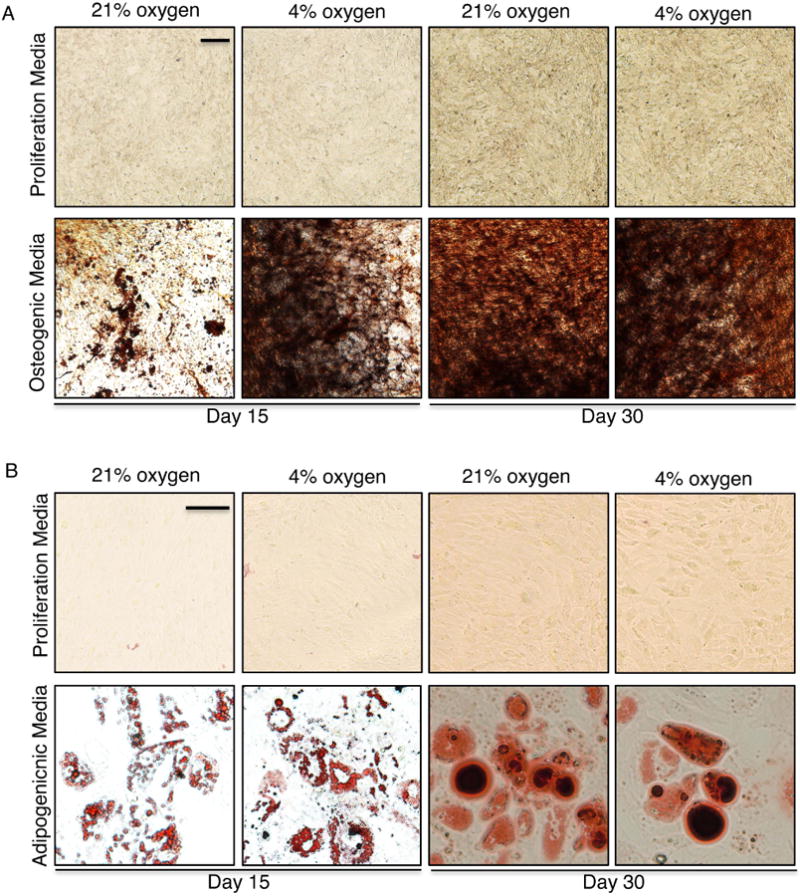
Primary hMSC (Magnification 20×, Scale bar 50μm) on tEGF surface in the presence of proliferation or osteogenic media at 21% or 4% oxygen, stained for von Kossa (A). Immortalized hMSC (Magnification 40×, Scale bar 10μm) on tEGF surface in the presence of proliferation or adipogenic media at 21% or 4% oxygen, stained for Oil Red O (B)
Similarly tEGF did not inhibit adipogenic differentiation in the presence of adipogenic differentiation inducers and did not cause adipogenic differentiation in the absence of differentiation inducers as seen by Oil Red O staining (Figure 6B). There was no difference in the rate of fat droplet deposition at 21% oxygen or 4% oxygen.
3.7 tEGF protects MSC from combined threats of hypoxia, serum deprivation and FasL
Although the Fas pathway has been shown by several groups to be the leading cause of MSC death via cytokine action (and representative of other apoptosis-inducing death cytokines), there are several other challenges in the ischemic wound bed, the two major ones being low oxygen and nutrient deprivation. To stimulate such conditions in vitro, imhMSC were treated with FasL in the absence or presence of serum under 21%, 4% and 1% oxygen. While 21% oxygen reflects ambient lab conditions, 4% oxygen simulates physiologic normoxia in the bone and 1% oxygen is representative of physiologic hypoxia. We found that 8 hours after treatment with FasL in the presence or absence of serum, caspase3 activation at 4% oxygen was lesser than caspase3 activation at 21% oxygen (Figure 7A). imhMSC at 1% oxygen though were the most susceptible to FasL induced cell death both in the presence or absence of serum. tEGF was able to protect undifferentiated imhMSC from FasL and serum deprivation induced apoptosis at all three concentrations of oxygen.
Figure 7. Tethered EGF is protective of MSC under FasL treatment, serum deprivation, low oxygen, and in the presence of sEGF.
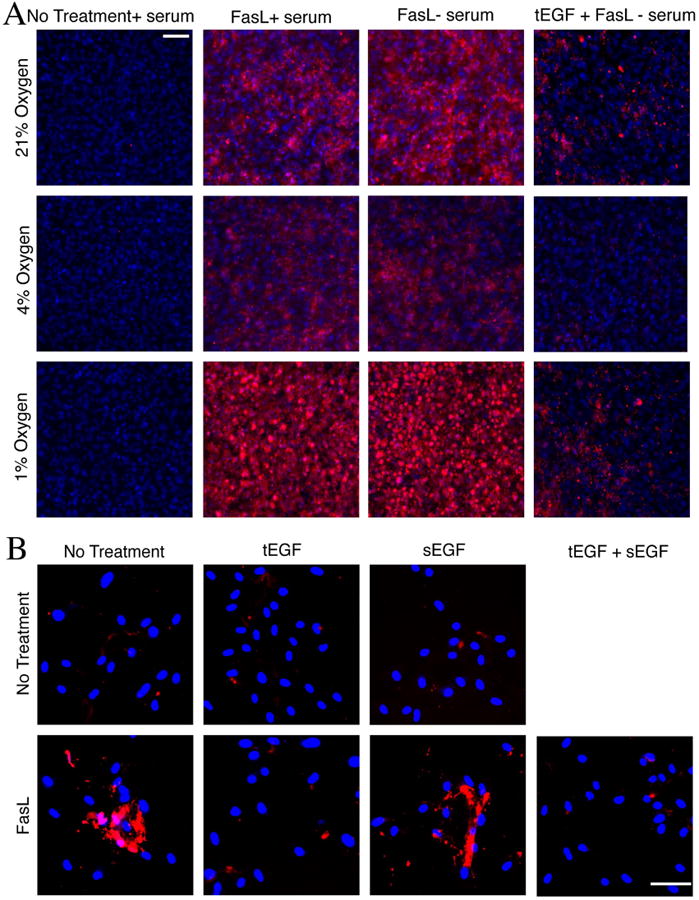
(A) Undifferentiated MSCs are more susceptible to FasL and serum deprivation mediated cell death at 1% oxygen, which tEGF is able to ablate. Fluorescent caspase inhibitor (FLICA) stained undifferentiated imhMSC (Magnification 10×, Scale bar 10μm) grown on tissue culture plastic/tEGF surfaces after 24 hours with or without serum and/ or 8 hours of treatment with FasL. (B) Presence of sEGF does not inhibit the protection rendered by tEGF to FasL treated prhMSCs (Magnification 10×, Scale bar 50μm)
One confounding variable in situ is that ligands for the EGFR are present at high levels in wound environments. Thus we tested whether the tEGF might be displaced and thus not functional in the presence of soluble ligands. This was not expected as our group has shown previously that labeled sEGF when added to prhMSCs grown on tEGF surfaces shows very little sEGF internalization, compared to internalization of sEGF added to prhMSC grown on mock surfaces or tissue culture plastic [16]. This is because both these factors wound bind EGFR with the same affinity, but tEGF being spatially restricted would have a much higher avidity. Also sEGF added to prhMSC grown on tEGF does not cause a new peak in Erk signaling, indicating that sEGF does not compete and replace tEGF from EGFR on prhMSC [16]. We wanted to confirm however that the presence of sEGF did not hinder the protective effects offered by tEGF to MSC in the presence of FasL. Unless treatments of tEGF were required, prhMSC were grown on mock surfaces. FasL and sEGF were added to prhMSC grown on tEGF surface for 12 hours, followed by a FLICA analysis for apoptosis. We found that sEGF added to prhMSC grown on tEGF surfaces did not limit the protection conferred by tEGF in the presence of FasL (Figure 7B). While addition of FasL by itself caused apoptosis, and presence of tEGF protected from FasL apoptosis as seen earlier, presence of sEGF or tEGF by themselves did not cause prhMSC apoptosis. Since sEGF is a ligand known to bind EGFR with the strongest affinity, we were able to conclude that the protective effects of tEGF must still be persistent in a wound bed in the presence of various other EGFR ligands.
4. Discussion
Successful deployment of MSC in regeneration of tissues such as bone requires incorporation, expansion and finally differentiation of the implanted MSC into osteoblasts, followed by deposition of bone matrix and mineral (hydroxyapatite). However, in vivo trials in humans have achieved limited success [22], perhaps due to death of transplanted MSC as observed in animal studies [7].
Our group has proposed using EGF, a growth factor known to promote proliferation, survival and motility in several cell types [23-25] to aid in the course of MSC bone formation [26]. EGF increases MSC proliferation while not affecting differentiation in the absence of differentiation cues [14]. This is advantageous since it would allow for sufficient ex vivo expansion of MSCs that are generally found in scarce numbers of one in a million bone marrow cells. It would also eliminate the use of serum for expansion ex vivo, eliminating variability and immunogenic reactions on implantation [26]. However, when we probed MSC survival, we found that MSC were most susceptible to FasL and TRAIL induced cell death [13] and thus constitute a threat to MSC survival. Factors like TNF-α [18], INF-γ [18] and TRAIL [13] have been shown to kill MSC both in vitro and in vivo, however the most potent death initiator in MSC is reportedly the Fas-FasL pathway [13,17,18,19], with FasL expressed by both natural killer cells [49] and T-lymphocytes [19]. These death cytokines in combination with hypoxia and nutrient deprivation and reactive oxygen species at the ischemic wound site could be severely detrimental to MSCs. EGF delivered in its physiological soluble form was unable to prevent MSC from undergoing death in the presence of these pro-death cytokines. We reasoned that survival signaling via sEGF might not be occurring since EGF would be driving proliferation that sensitizes cells to death signals, and over the longer term would lead to downregulation of EGFR levels limiting the efficacy of survivals signals.
Of interest, differential localization of activated EGFR in the cell can alter the strength of downstream signals to cause varied effects [28-30]. EGFR activated Erk when localized to the cell membrane triggers motility preferentially compared to EGFR activated Erk in the cytoplasm which activates cellular proliferation [31]. Further, the Erk and AKT signaling elicited from membrane restricted EGFR are more tonic and consistent with survival over proliferative signaling [32].
We therefore changed the mode of delivery of EGF based on an earlier designed model system that maintains EGF in both its active conformation as well as a mobile state [33]. The model also increases ligand availability by preventing ligand depletion, a system known to enhance downstream effects such as mitogenesis [34]. EGF was delivered on a PEO tether attached to the underlying polymer substrate in a manner that created local clusters of EGF presented to cells in the context of an adhesive surface. tEGF localized EGFR on MSC to the cell surface and promoted survival of undifferentiated MSC in the presence of FasL which sEGF was unable to accomplish [13].
Addition of sEGF to MSC during the progress of osteogenic differentiation promotes bone formation [35]. However cells grown on tEGF in the presence of osteoinductive factors further fastens this differentiation process seen by increased alkaline phosphatase activity at Day 7 and greater mineralization at Day 21 [16]. This increased osteogenesis is attributed to increased ratios of phosphorylated EGFR to total EGFR. Tethered EGF caused restriction of phosphorylated EGFR to the cell membrane, where it actively signals, preventing it from being internalized and simultaneously prevented production of new EGFR [16].
The major question that remained in evaluation of our designed biomaterial was whether tEGF would help protect differentiating MSC in the presence of inflammatory cytokines. This is significant since differentiating MSC would not respond the same way to death factors as undifferentiated MSC, which tEGF is known to protect. Others and we have shown that undifferentiated MSC are susceptible to Fas induced cell death one of the major death pathways activated during first phase of inflammation [13,17,18]. We have also shown that FasL not only increased MSC death by activating caspases, but also promotes production of intracellular ROS in MSC, causing cell death [19].
We addressed the death of MSC differentiating into osteoblasts since our ultimate goal is repairing critical sized bone wounds. We found that MSC differentiating into osteoblasts increased expression of Fas, the receptor for FasL and continued to die significantly like undifferentiated MSC, emphasizing the need for continued protection of MSC differentiating into bone cells. Use of tEGF surfaces as a substratum for growth of MSC differentiating into bone, showed continued protection to both pre-osteoblasts and osteoblasts derived from MSC similar to its protective effect in undifferentiated MSC. Continued protection offered by tEGF correlated with presence of EGFR expression in differentiating osteoblasts. Of note, the decrease in death by day 30 hold promise that fully differentiated osteocytes will not be readily killed by inflammatory cytokines after the supporting materials dissipates in the body. Thus, the tEGF appears to protect through the critical period of engraftment and osteogenic differentiation when the cells are most susceptible to the wound environment.
A major concern during repair is the accumulation of fat droplets in MSC and formation of adipose cells, one of the main default pathways, which may hinder healthy bone development. When MSC were induced to form adipose cells and tested for death in the presence of FasL, we found MSC became more resistant to FasL induced death. This corresponded with decrease in Fas levels. tEGF did not affect this reduction in cell death since levels of its receptor EGFR decreased with the progress of adipogenic differentiation. The main issue that likely contributes to adipocyte survival is the fact that adipocytes, more specifically white adipocytes prevalent in adult tissue are generally resistant to death cytokines and serum deprivation, with expression of the anti-apoptotic protein Bcl2 increasing as pre-adipocytes differentiate into adipocytes [36].
One of the best ways to prevent adipocyte formation is to use an MSC population that specifically enriches for the osteoblast lineage. An example of such a population is the Mx1+ stromal population [37]. Alternatively specific integrins like α-4 and α-5 promoting MSC homing and differentiation into osteoblasts can be activated to augment bone growth from MSC [38]. Alternatively adipocyte formation can be directly addressed. MSCs overexpressing growth factors like PDGF-B have been shown to strongly inhibit differentiation into adipocytes and can easily be included in a designed scaffold along with tEGF to promote healthy bone formation [39]. So also antagonists to proteins specifically expressed in differentiating adipocytes, and promoting survival of differentiating adipocytes like PPAR-Y [40] might prove helpful in reducing the number of adipocytes formed. Taken together, an osteo-inductive and anti-adipogenic environment needs to be continually maintained until a vascularized, non-inflammatory environment is attained to obtain appropriate bone differentiation.
Lastly, in preparation for in vivo studies, we determined whether the tEGF survival signal was sufficiently robust to withstand the multiple insults of death cytokines, nutrient deprivation and hypoxia, as well as competitive effects of sEGF; as EGFR ligands are present in wounds beds. We found that not only did the hMSC differentiate under these conditions but also that tEGF was successful in staving off apoptosis. Obviously, much work including optimization of presentation, cell number and preconditioning for in vivo experimentation are required before moving such studies to the preclinical stages.
The literature provides guidance for steps forward. For instance, there currently are diverse reports on the importance of number of MSC implanted and the outcome of bone formation based on prior exposure to osteogenic differentiation agents. While some report exposure to osteogenic differentiation inducers cause better bone formation [41,42], others indicate that exposure to such inducers does not change the outcome of osteogenesis [43]. It is also suggested that bone formation increases with increasing numbers of MSCs, but the relationship is sigmoidal, with a threshold number of MSCs required for good bone formation [44]. Few modes of delivering MSC have shown to be promising for increasing survival, these include hydrogels that quench reactive oxygen species [45-46], scaffolds containing pre-determined mixtures of extracellular matrix (ECM) proteins [47], or anisotrophic scaffolds that mimic ECM and contain entrapped growth factors [48]. In short, our findings herein set the stage for further work combining these considerations with a cyto-protective substrate to reach the goal of cell to tissue replacement therapy.
5. Conclusions
Herein we describe a two-dimensional biomaterial that uses tethered EGF to restrict signaling of the EGFR receptor to the surface of MSC, altering the balance of signaling of EGFR to make MSC more resistant to inflammatory death signals (and other wound bed challenges), both in undifferentiated MSC and differentiating MSC. This manipulation does not alter the differentiation potential or rate of the MSC. This biomaterial serves as a foundation for the development of a 3D bioengineered tEGF scaffold that will serve both as a carrier and a signaling device for MSC to aid in the regeneration of non-healing bone wounds.
Supplementary Material
Supplemental Figure 1. MSC display changes in markers for differentiation as well as cell surface receptors during progress of differentiation. Change in levels of osteogenic markers during progress of prhMSC osteogenic differentiation on tissue culture plastic (A). Change in level of adipogenic markers by qPCR during adipogenic differentiation of prhMSC on tissue culture plastic (B). Change in nuclear size during progress of adipogenic differentiation of imhMSC on tissue culture plastic (C). Change in the expression of EGFR and Fas in prhMSC during adipogenic (D) and osteogenic differentiation (E) on tissue culture plastic.
Supplemental Figure 2. Time course of death of MSC in the presence of FasL. Fluorochrome inhibitor of caspase assay (FLICA) stained imhMSC (A) and prhMSC (B) after treatment with FasL for defined time-points. Shown are representative photomicrographs of cells of two independent experiments.
Supplemental Figure 3. Time course of death of differentiating adipocytes in the presence of FasL. Fluorochrome inhibitor of caspase assay (FLICA) and Hoescht 33342 images of prhMSC in adipogenic media on Day 15 (A) and Day 30, overlaid with phase contrast image to identify fat droplets (B) treated with FasL for 0, 8 and 24 hours. Shown are representative photomicrographs of cells of two independent experiments.
Acknowledgments
Contract Grant Sponsor: National Institute of General Medical Sciences, and National Institute of Dental and Craniofacial Research (GM069668, DE019523).
M.R.: conception and design, performed research, data analysis and interpretation, wrote manuscript; H.B.: Data analysis, image processing; L.S.: Prepared tEGF surfaces; L.G.: conception and design, data interpretation, financial support; A.W.: conception and design, data interpretation, edited manuscript, financial support
References
- 1.Pittenger MF, Mackay AM, Beck SC, et al. Multilineage potential of adult human mesenchymal stem cells. Science. 1999;284(5411):143–147. doi: 10.1126/science.284.5411.143. [DOI] [PubMed] [Google Scholar]
- 2.Jones E, Yang X. Mesenchymal stem cells and bone regeneration: current status. Injury. 2011;42(6):562–568. doi: 10.1016/j.injury.2011.03.030. [DOI] [PubMed] [Google Scholar]
- 3.Caplan AI, Dennis JE. Mesenchymal stem cells as trophic mediators. J Cell Biochem. 2006;98(5):1076–1084. doi: 10.1002/jcb.20886. [DOI] [PubMed] [Google Scholar]
- 4.Caplan AI. Why are MSCs therapeutic? New data: new insight. J Pathol. 2009;217(2):318–324. doi: 10.1002/path.2469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Uccelli A, Prockop DJ. Why should mesenchymal stem cells (MSCs) cure autoimmune diseases? Curr Opin Immunol. 2010;22(6):768–774. doi: 10.1016/j.coi.2010.10.012. [DOI] [PubMed] [Google Scholar]
- 6.van der Bogt KE, Schrepfer S, Yu J, et al. Comparison of transplantation of adipose tissue- and bone marrow-derived mesenchymal stem cells in the infarcted heart. Transplantation. 2009;87(5):642–652. doi: 10.1097/TP.0b013e31819609d9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rodrigues M, Griffith LG, Wells A. Growth factor regulation of proliferation and survival of multipotential stromal cells. Stem Cell Res Ther. 2010;1(4):32. doi: 10.1186/scrt32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Semont A, Mouiseddine M, et al. Mesenchymal stem cells improve small intestinal integrity through regulation of endogenous epithelial cell homeostasis. Cell Death Differ. 2010;17(6):952–961. doi: 10.1038/cdd.2009.187. [DOI] [PubMed] [Google Scholar]
- 9.Zimmermann CE, Gierloff M, et al. Survival of Transplanted Rat Bone Marrow-Derived Osteogenic Stem Cells In Vivo. Tissue Eng Part A. 2011;17(7-8):1147–56. doi: 10.1089/ten.TEA.2009.0577. [DOI] [PubMed] [Google Scholar]
- 10.Wei H, Li Z, Hu S, et al. Apoptosis of mesenchymal stem cells induced by hydrogen peroxide concerns both endoplasmic reticulum stress and mitochondrial death pathway through regulation of caspases, p38 and JNK. J Cell Biochem. 2010;111(4):967–978. doi: 10.1002/jcb.22785. [DOI] [PubMed] [Google Scholar]
- 11.Zhu W, Chen J, Cong X, et al. Hypoxia and serum deprivation-induced apoptosis in mesenchymal stem cells. Stem Cells. 2006;24(2):416–425. doi: 10.1634/stemcells.2005-0121. [DOI] [PubMed] [Google Scholar]
- 12.Kim WK, Meliton V, Bourquard Ne. Hedgehog signaling and osteogenic differentiation in multipotent bone marrow stromal cells are inhibited by oxidative stress. J Cell Biochem. 2010;111(5):1199–1209. doi: 10.1002/jcb.22846. [DOI] [PubMed] [Google Scholar]
- 13.Fan VH, Tamama K, Au A, et al. Tethered epidermal growth factor provides a survival advantage to mesenchymal stem cells. Stem Cells. 2007;25(5):1241–1251. doi: 10.1634/stemcells.2006-0320. [DOI] [PubMed] [Google Scholar]
- 14.Tamama K, Fan VH, Griffith LG, et al. Epidermal growth factor as a candidate for ex vivo expansion of bone marrow-derived mesenchymal stem cells. Stem Cells. 2006;24(3):686–695. doi: 10.1634/stemcells.2005-0176. [DOI] [PubMed] [Google Scholar]
- 15.Iyer AK, Tran KT, Borysenko CW, et al. Tenascin cytotactin epidermal growth factor-like repeat binds epidermal growth factor receptor with low affinity. J Cell Physiol. 2007;211(3):748–758. doi: 10.1002/jcp.20986. [DOI] [PubMed] [Google Scholar]
- 16.Platt MO, Roman AJ, Wells A, et al. Sustained epidermal growth factor receptor levels and activation by tethered ligand binding enhances osteogenic differentiation of multi-potent marrow stromal cells. J Cell Physiol. 2009;221(2):306–317. doi: 10.1002/jcp.21854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gotherstrom C, Lundqvist A, Duprez IR, et al. Fetal and adult multipotent mesenchymal stromal cells are killed by different pathways. Cytotherapy. 2011;13(3):269–278. doi: 10.3109/14653249.2010.523077. [DOI] [PubMed] [Google Scholar]
- 18.Liu Y, Wang L, Kikuiri T, et al. Mesenchymal stem cell-based tissue regeneration is governed by recipient T lymphocytes via IFN-gamma and TNF-alpha. Nat Med. 2011;17(12):1594–1601. doi: 10.1038/nm.2542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rodrigues M, Turner O, Stolz D, et al. Production of reactive oxygen species by multipoint stromal cells/ mesenchymal stem cells upon exposure to FasL. Cell Transplant. 2012 doi: 10.3727/096368912X639035. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Okamoto T, Aoyama T, Nakayama T, et al. Clonal heterogeneity in differentiation potential of immortalized human mesenchymal stem cells. Biochem Biophys Res Commun. 2002;295(2):354–361. doi: 10.1016/s0006-291x(02)00661-7. [DOI] [PubMed] [Google Scholar]
- 21.Wu S, Wells A, Griffith LG, et al. Controlling multipotent stromal cell migration by integrating “course-graining” materials and “fine-tuning” small molecules via decision tree signal-response modeling. Biomaterials. 2011;32(30):7524–7531. doi: 10.1016/j.biomaterials.2011.06.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Patterson TE, Kumagai K, Griffith L, et al. Cellular strategies for enhancement of fracture repair. J Bone Joint Surg Am. 2008;90(Suppl 1):111–119. doi: 10.2106/JBJS.G.01572. [DOI] [PubMed] [Google Scholar]
- 23.Iwabu A, Smith K, Allen FD, et al. Epidermal growth factor induces fibroblast contractility and motility via a protein kinase C delta-dependent pathway. J Biol Chem. 2004;279(15):14551–14560. doi: 10.1074/jbc.M311981200. [DOI] [PubMed] [Google Scholar]
- 24.Leloup L, Shao H, Bae YH. m-Calpain activation is regulated by its membrane localization and by its binding to phosphatidylinositol 4,5-bisphosphate. J Biol Chem. 2010;285(43):33549–33566. doi: 10.1074/jbc.M110.123604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shao H, Wu C, Wells A. Phosphorylation of alpha-actinin 4 upon epidermal growth factor exposure regulates its interaction with actin. J Biol Chem. 2010;285(4):2591–2600. doi: 10.1074/jbc.M109.035790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Marcantonio NA, Boehm CA, Rozic RJ, et al. The influence of tethered epidermal growth factor on connective tissue progenitor colony formation. Biomaterials. 2009;30(27):4629–4638. doi: 10.1016/j.biomaterials.2009.05.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tamama K, Kawasaki H, Wells A. Epidermal growth factor (EGF) treatment on multipotential stromal cells (MSCs). Possible enhancement of therapeutic potential of MSC. J Biomed Biotechnol. 2010:795385. doi: 10.1155/2010/795385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wells A, Welsh JB, Lazar CS, et al. Ligand-induced transformation by a noninternalizing epidermal growth factor receptor. Science. 1990;247(4945):962–964. doi: 10.1126/science.2305263. [DOI] [PubMed] [Google Scholar]
- 29.Haugh JM, Huang AC, Wiley HS, et al. Internalized epidermal growth factor receptors participate in the activation of p21(ras) in fibroblasts. J Biol Chem. 1999;274(48):34350–34360. doi: 10.1074/jbc.274.48.34350. [DOI] [PubMed] [Google Scholar]
- 30.Wells A. EGF receptor. Int J Biochem Cell Biol. 1999;31(6):637–643. doi: 10.1016/s1357-2725(99)00015-1. [DOI] [PubMed] [Google Scholar]
- 31.Chen P, Xie H, Wells, A Mitogenic signaling from the egf receptor is attenuated by a phospholipase C-gamma/protein kinase C feedback mechanism. Mol Biol Cell. 1996;7(6):871–881. doi: 10.1091/mbc.7.6.871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Platt MO, Wilder CL, Wells A, et al. Multipathway kinase signatures of multipotent stromal cells are predictive for osteogenic differentiation: tissue-specific stem cells. Stem Cells. 2009;27(11):2804–2814. doi: 10.1002/stem.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kuhl PR, Griffith-Cima LG. Tethered epidermal growth factor as a paradigm for growth factor-induced stimulation from the solid phase. Nat Med. 1996;2(9):1022–1027. doi: 10.1038/nm0996-1022. [DOI] [PubMed] [Google Scholar]
- 34.Reddy CC, Wells A, Lauffenburger DA. Receptor-mediated effects on ligand availability influence relative mitogenic potencies of epidermal growth factor and transforming growth factor alpha. J Cell Physiol. 1996;166(3):512–522. doi: 10.1002/(SICI)1097-4652(199603)166:3<512::AID-JCP6>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 35.Kratchmarova I, Blagoev B, Haack-Sorensen M, et al. Mechanism of divergent growth factor effects in mesenchymal stem cell differentiation. Science. 2005;308(5727):1472–1477. doi: 10.1126/science.1107627. [DOI] [PubMed] [Google Scholar]
- 36.Nisoli E, Cardile A, Bulbarelli A, et al. White adipocytes are less prone to apoptotic stimuli than brown adipocytes in rodent. Cell Death Differ. 2006;13:2154–2156. doi: 10.1038/sj.cdd.4401956. [DOI] [PubMed] [Google Scholar]
- 37.Park D, Spencer JA, Koh BI, et al. Endogenous bone marrow MSCs are dynamic, fate restricted participants in bone maintenance and regeneration. Cell Stem Cell. 2012;10(3):259–272. doi: 10.1016/j.stem.2012.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Guan M, Yao W, Liu R, et al. Directing mesenchymal stem cells to bone to augment formation and increase bone mass. Nat Med. 2012;18(3):456–62. doi: 10.1038/nm.2665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fierro FA, Kalomoiris S, Sondergaard CS, et al. Effects on proliferation and differentiation of multipotent bone marrow stromal cells engineered to express growth factors for combined cell and gene therapy. Stem Cells. 2011;(11):1727–37. doi: 10.1002/stem.720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Takeshi I, Reiko T, Marchand S, et al. Peroxisome proliferator-activated receptor γ is required in mature white and brown adipocytes for their survival in the mouse. PNAS. 2004;13:4534–4547. doi: 10.1073/pnas.0400356101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Song IH, Caplan AI, Dennis JE. Dexamethasone inhibition of confluence-induced apoptosis in human mesenchymal stem cells. J Orthop Res. 2009;27(2):216–221. doi: 10.1002/jor.20726. [DOI] [PubMed] [Google Scholar]
- 42.Song IH, Caplan AI, Dennis JE. In vitro dexamethasone pretreatment enhances bone formation of human mesenchymal stem cells in vivo. J Orthop Res. 2009;27(7):916–921. doi: 10.1002/jor.20838. [DOI] [PubMed] [Google Scholar]
- 43.Kuznetsov SA, Mankani MH, Robey PG. In vivo formation of bone and haematopoietic territories by transplanted human bone marrow stromal cells generated in medium with and without osteogenic supplements. J Tissue Eng Regen Med. 2011 doi: 10.1002/term.515. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mankani MH, Kuznetsov SA, Robey PG. Formation of hematopoietic territories and bone by transplanted human bone marrow stromal cells requires a critical cell density. Exp Hematol. 2007;35(6):995–1004. doi: 10.1016/j.exphem.2007.01.051. [DOI] [PubMed] [Google Scholar]
- 45.Wong VW, Rustad KC, Glorzbach JP, et al. Pullulan hydrogels improve mesenchymal stem cell delivery into high-oxidative-stress wounds. Macromol Biosci. 2011;11(11):1458–66. doi: 10.1002/mabi.201100180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rustad KC, Wong VW, Sorkin M, et al. Enhancement of mesenchymal stem cell angiogenic capacity and stemness by a biomimetic hydrogel scaffold. Biomaterials. 2012;33(1):80–90. doi: 10.1016/j.biomaterials.2011.09.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hamrahi VF, Goverman J, Jung W, et al. In vivo molecular imaging of murine embryonic stem cells delivered to a burn wound surface via Integra® scaffolding. J Burn Care Res. 2012 doi: 10.1097/BCR.0b013e3182331d1c. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang F, Li Z, Tamama K, et al. Fabrication and characterization of prosurvival growth factor releasing, anisotropic scaffolds for enhanced mesenchymal stem cell survival/growth and orientation. Biomacromolecules. 2009;10(9):2609–2618. doi: 10.1021/bm900541u. [DOI] [PubMed] [Google Scholar]
- 49.Yamaza T, Miura Y, Bi Y, et al. Pharmacologic stem cells based intervention as a new approach to osteoporosis treatment in rodents. PLoS One. 2008;3(7):e2615. doi: 10.1371/journal.pone.0002615. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. MSC display changes in markers for differentiation as well as cell surface receptors during progress of differentiation. Change in levels of osteogenic markers during progress of prhMSC osteogenic differentiation on tissue culture plastic (A). Change in level of adipogenic markers by qPCR during adipogenic differentiation of prhMSC on tissue culture plastic (B). Change in nuclear size during progress of adipogenic differentiation of imhMSC on tissue culture plastic (C). Change in the expression of EGFR and Fas in prhMSC during adipogenic (D) and osteogenic differentiation (E) on tissue culture plastic.
Supplemental Figure 2. Time course of death of MSC in the presence of FasL. Fluorochrome inhibitor of caspase assay (FLICA) stained imhMSC (A) and prhMSC (B) after treatment with FasL for defined time-points. Shown are representative photomicrographs of cells of two independent experiments.
Supplemental Figure 3. Time course of death of differentiating adipocytes in the presence of FasL. Fluorochrome inhibitor of caspase assay (FLICA) and Hoescht 33342 images of prhMSC in adipogenic media on Day 15 (A) and Day 30, overlaid with phase contrast image to identify fat droplets (B) treated with FasL for 0, 8 and 24 hours. Shown are representative photomicrographs of cells of two independent experiments.