

Abstract
Synaptic inhibition in the amygdala actively participates in processing emotional information. To improve the understanding of interneurons in amygdala networks it is necessary to characterize the GABAergic cell types, their connectivity and physiological roles. We used a mouse line expressing a green fluorescent protein (GFP) under the neuropeptide Y (NPY) promoter. Paired recordings between presynaptic NPY-GFP-expressing (+) cells and postsynaptic principal neurons (PNs) of the basolateral amygdala (BLA) were performed. The NPY-GFP+ neurons displayed small somata and short dendrites embedded in a cloud of highly arborized axon, suggesting a neurogliaform cell (NGFC) type. We discovered that a NPY-GFP+ cell evoked a GABAA receptor-mediated slow inhibitory postsynaptic current (IPSC) in a PN and an autaptic IPSC. The slow kinetics of these IPSCs was likely caused by the low concentration and spillover of extracellular GABA. We also report that NGFCs of the BLA fired action potentials phase-locked to hippocampal theta oscillations in anaesthetized rats. When this firing was re-played in NPY+-NGFCs in vitro, it evoked a transient depression of the IPSCs. Presynaptic GABAB receptors and functional depletion of synaptic vesicles determined this short-term plasticity. Synaptic contacts made by recorded NGFCs showed close appositions, and rarely identifiable classical synaptic structures. Thus, we report here a novel interneuron type of the amygdala that generates volume transmission of GABA. The peculiar functional mode of NGFCs makes them unique amongst all GABAergic cell types of the amygdala identified so far.
Key points
The amygdala is a key brain structure implicated in processing emotions and is thought to underlie anxiety-related behaviours.
Interneurons of the basolateral amygdala provide constant silencing of principal cells, and are proven to be active cellular elements during emotional processing.
We report here a novel interneuron type of the amygdala, termed neurogliaform cell (NGFC), and study its function by a combination of in vivo and in vitro techniques.
NGFCs provide a peculiar cell-to-cell form of communication via volume transmission of GABA.
In-depth analysis of specific neuron type is likely to improve the understanding of amygdala circuits in health and disease.
Introduction
The basolateral complex of the amygdala (BLA) is one of the most important sites of integration during fear conditioning (Herry et al. 2008). This nucleus is a cortical-like structure consisting of glutamatergic principal neurons (PNs) and local GABAergic inhibitory neurons (Pape & Pare, 2010). The interaction between these two cell classes is thought to determine the computation of emotional memories in the BLA. The role of GABAergic cells is particularly important, as their inhibitory action provides constant silencing of PNs of the BLA (Gaudreau & Pare, 1996). Suppression of inhibition can increase the firing rate and synchronization of PNs during fear (Pare & Collins, 2000). Conversely, pharmacological impairment of GABAergic transmission induces anxiogenic-like effects (Makkar et al. 2010).
In cortical structures, diverse interneuron types regulate the activity of PNs, therefore governing the state of neuronal assemblies (Klausberger & Somogyi, 2008). Specialized types of interneurons release GABA on target-specific subcellular domains of postsynaptic cells with high temporal precision (Klausberger & Somogyi, 2008). Moreover, the inhibitory postsynaptic currents (IPSCs) evoked by different interneuron types and recorded in postsynaptic cells show marked kinetic diversity (Buhl et al. 1994; Nusser et al. 2001). Some phasic IPSCs are very fast (Bartos et al. 2002), but others are 5–10 times slower (rise time >3 ms and decay time >30 ms; Tamas et al. 2003). The slower events have been referred to as GABAA,slow (Capogna & Pearce, 2011). This signal has been detected in many brain areas, including the hippocampus (Pearce, 1993; Banks et al. 1998; Price et al. 2005, 2008; Prenosil et al. 2006; Karayannis et al. 2010; Armstrong et al. 2011; Markwardt et al. 2011), neocortex (Tamas et al. 2003; Olah et al. 2007, 2009; Szabadics et al. 2007; Wozny & Williams, 2011; Suzuki & Bekkers, 2012), striatum (Ibanez-Sandoval et al. 2011; English et al. 2012), thalamus (Huntsman et al. 1999; Schofield & Huguenard, 2007), cerebellum (Crowley et al. 2009) and brainstem (Kuo et al. 2009). The role of GABAA,slow is likely to differ from that of the classical fast inhibition (Capogna & Pearce, 2011). This includes: modulation of NMDA-dependent synaptic plasticity (Kapur et al. 1997); control of glutamate release in the neocortex (Olah et al. 2009); participation in hippocampal theta oscillations (Fuentealba et al. 2010); depolarization of newborn granule cells in the dentate gyrus (Markwardt et al. 2011); gating nicotinic inputs onto spiny projection neurons of striatum (English et al. 2012). A specific type of interneuron, termed the neurogliaform cell (NGFC), is a source of GABAA,slow in the neocortex, hippocampus and striatum (Tamas et al. 2003; Price et al. 2005; Ibanez-Sandoval et al. 2011), in addition to the related Ivy cell type in the hippocampus (Fuentealba et al. 2008).
NGFCs express the neuropeptide Y (NPY; Price et al. 2005; Karagiannis et al. 2009; Ibanez-Sandoval et al. 2011). Some GABAergic cells of the BLA are known to express this peptide (McDonald, 1989) but their identity, connectivity and roles remain entirely unknown. Because of the unique function of GABAA,slow, we asked whether NPY+ cells of the BLA could trigger this signal and studied their connectivity.
We found here that a NPY+ GABAergic cell, with dendritic and axonal patterns typical of the NGFC, is a source of GABAA,slow in the BLA. We also describe the firing patterns of NGFCs in vivo and the mechanisms underlying the slow IPSCs. The data suggest that low concentration and spillover of GABA to extrasynaptic sites are likely to underlie the slow kinetics of the IPSCs.
Methods
Acute slice preparation
All procedures involving animals were performed according to methods approved by the UK Home Office and The Animals (Scientific Procedures) Act 1986. The authors have read, and the experiments comply with, the policies and regulations of The Journal of Physiology (Drummond, 2009). Acute coronal slices were prepared from bacterial artificial chromosome (BAC) transgenic mice that express the humanized Renilla green fluorescent protein (hrGFP) under the control of the mouse NPY promoter (stock 006417; The Jackson Laboratory, Bar Harbor, ME, USA). Animals (postnatal day 16–25) were decapitated under deep isoflurane anaesthesia (4% in O2), and their brains were rapidly removed and placed in ice-cold sucrose-containing artificial cerebrospinal fluid (ACSF) cutting solution containing (in mm): sucrose, 75; NaCl, 87; NaHCO3, 25; KCl, 2.5; NaH2PO4, 1.25; CaCl2, 0.5; MgCl2, 7; glucose, 25; saturated with 95% O2, 5% CO2, at pH 7.3–7.4. Slices (325 μm thickness) including amygdala were cut (Microm HM 650 V; Thermo Fisher Scientific Inc., Germany) and transferred on a nylon mesh where they were maintained in a chamber initially containing sucrose ACSF cutting solution at 37°C for 30 min. During this period the cutting solution was substituted with normal ACSF consisting of (in mm): NaCl, 130; NaHCO3, 24; KCl, 3.5; NaH2PO4, 1.25; CaCl2, 2.5; MgSO4, 1.5; glucose, 10; saturated with 95% O2, 5% CO2, at pH 7.3. Slices were then resting at room temperature (18–22°C).
Electrophysiology and analysis
Acute slices were secured under a nylon mesh, submerged and superfused with ACSF in a chamber mounted on the stage of an upright microscope (Axioskop; Zeiss, Jena, Germany). Slices were visualized with a 40×/0.1 NA or 60×/0.9 NA water-immersion objective (LUMPlanFl; Olympus, Tokyo, Japan) coupled with infrared and differential interference contrast optics linked to a video camera (CCD Camera C7500; Hamamatsu, Hamamatsu City, Japan). A mercury vapour short-arc lamp (100 W, N HBC 103; Zeiss) was connected to the epifluorescence system to visualize the GFP-expressing neurons. Somatic whole-cell patch-clamp recordings (at ∼33°C) were made from visually identified cells using borosilicate glass capillaries (GC120F, 1.2 mm o.d.; Clarke Electromedical Instruments, Reading, UK), pulled on a DMZ puller (Zeitz-instrumente GmbH, Munich, Germany) and filled with a filtered intracellular solution consisting of (in mm): potassium gluconate, 42; KCl, 88; ATP-Mg, 4; GTP-Na2, 0.3; Na2-phosphocreatine, 10; Hepes, 10; and 0.5% w/v biocytin (all from Sigma-Aldrich Co. Ltd, Poole, UK), osmolarity 270–280 mosmol l−1 without biocytin, pH 7.3 adjusted with KOH. The equilibrium potential for Cl− ions (ECl) was −11 mV; the IPSC polarity was inward at the holding potential (Vh) of −65 mV. In a subset of experiments, EGTA (10 mm; Sigma-Aldrich) was added to the internal solution of the presynaptic cell. A caesium-based intracellular solution was used to block K+ channels consisting of (in mm): caesium methansulphonate, 42; CsCl, 88; Hepes, 10; sodium phosphocreatine, 10; Mg-ATP, 4; Na-GTP, 0.3; and 0.5% w/v biocytin (all from Sigma-Aldrich), osmolarity 270–280 mosmol l−1 without biocytin, pH 7.3 adjusted with CsOH. Biocytin was added to allow post hoc visualization of the recorded neurons. Resistance of the patch pipettes was 4–5 MΩ. Recordings were accepted only if the initial seal resistance was greater than 1 GΩ and series resistance did not change by more than 20% throughout the recording period. No correction was made for the junction potential between the pipette and the ACSF. All electrophysiological signals were amplified (10 mV pA−1; EPC9/2 amplifier HEKA Electronik, Lambrecht, Germany, PULSE software), low pass filtered at 2.9 kHz, digitized at 5 or 10 kHz. The amplifier was controlled with a personal computer running the PULSE data acquisition and analysis program (HEKA). Currents/voltages were analysed offline with Pulsefit (HEKA) and IGOR Pro5.05 (WaveMetrics, Inc., Lake Oswego, OR, USA). The input resistance (Rin) was calculated from the slope of a line fitted to the subthreshold range on a plot of the injected current versus the steady-state membrane voltage when a family of hyperpolarizing and depolarizing current injections was applied (range, −30/+120 pA). The apparent membrane time constant (τ) was calculated by fitting a single exponential to the response of the cell to a current injection of −50 pA in current-clamp mode. Membrane capacitance was calculated as τ/Rin. To study the kinetics of action potentials, a depolarizing current step (3 ms, 100–150 pA) was applied. Action potential half-width, peak amplitude and membrane afterhyperpolarization (AHP) were measured from the initial point of the action potential raising phase by a user-defined program in IGOR. The adaptation index was calculated as the ratio between the first and last inter-spike intervals evoked by a 1 s depolarizing current pulse. Presynaptic and postsynaptic neurons were voltage-clamped at Vh=−65 mV. After obtaining whole-cell configuration, we waited 5 min before starting a protocol to ensure diffusion of internal solution into the recorded neurons. Action currents were elicited in a presynaptic cell (3 ms, 100 pA) and the corresponding unitary IPSC (uIPSC) was recorded in the synaptically coupled postsynaptic neuron or in the presynaptic neuron as an autaptic IPSC. The inter-stimuli interval was usually 120 s to ensure stable and non-depressing IPSCs during repeated stimulation, consistent with other NGFC-mediated IPSCs (Tamas et al. 2003; Price et al. 2005). A longer interval of 180 s was used as the interval between 5 Hz or in vivo firing (see below) stimulation. Kinetic analysis and exponential fitting of synaptic responses were performed in Igor Pro. In a subset of recordings, evoked action current partially masked the presence of the rising phase of the autaptic IPSC. In this case, the rise time was estimated from a line fitted to the remaining trace, only if it constituted more than 20% of the total peak amplitude. Spontaneous IPSCs (sIPSCs) were recorded at Vh=−65 mV for 60 s and analysed using Minianalysis software (Synaptosoft, Decatur, GA, USA). The peak amplitude, the 20–80% rise time and decay time were calculated by fitting a single exponential in each trace. The decay of the averaged postsynaptic and autaptic uIPSCs could be also fit with a double exponential, and the weighted decay time constant was calculated using the following formula: τw=τ1 A1+τ2(1 −A1), where τw is the weighted decay time constant, τ1 and τ2 are the time constants of the first and second exponential functions, respectively, and A1 is the proportion of the peak amplitude of the averaged uIPSC that is contributed by τ1.
In vivo firing patterns of NGFCs were recorded in three anaesthetized rats (250–350 g) with the juxtacellular/extracellular method. Briefly, animals were anaesthetized with intraperitoneal injections of urethane (1.30 g (kg body weight)−1) supplemented with ketamine and xylazine, (10–15 and 1–1.5 mg kg−1, respectively) as needed. Neuronal activities in the right BLA and dCA1 (stratum oriens-pyramidale) were recorded with glass pipettes filled with 1.5% Neurobiotin in 0.5 m NaCl (14.5–17 MΩ resistance). Glass electrode signals were referenced against a wire implanted subcutaneously in the neck. The electrocorticogram (ECoG) was recorded via a steel screw in contact with the dura mater above the ipsilateral prefrontal cortex. All data were analysed off-line using Spike2 software, as recently reported (Bienvenu et al. 2012). Preferred theta phase and modulation depth (length of the mean vector) for each cell were determined using Rayleigh's test. Homogeneity of preferred phases across cells was tested using Moore's test (Zar, 1999). Grand average and modulation depths were calculated using Bachelet's method (Zar, 1999).
A series of 11 action potentials recorded in a NGFC during hippocampal theta oscillations in vivo was injected into whole-cell patch-clamped cells as voltage pulses (1 ms, 100 pA) using Matlab (The Mathworks, Inc., Natick, MA, USA) and Pulse (HEKA) software. The stimuli evoked a train of action currents in vitro that exactly matched the sequence of action potentials detected in vivo.
All values are expressed as means ± SEM. Statistical tests and analyses were performed using SPSS 17.0 (SPSS, Chicago, IL, USA). Data are presented as mean ± SEM; non-parametrical two-tailed tests (Mann–Whitney U test, Wilcoxon's single ranks test, Kruskal–Wallis test) were used to assess the significance of differences between groups of data obtained in vitro.
Histological processing
After recordings, the slices were immersed at 4°C for 14–18 h in a fixative solution containing: 4% paraformaldehyde, 15% (v/v) saturated picric acid and 0.1 m phosphate buffer (PB; pH 7.2–7.4). Gelatin-embedded slices were re-sectioned into 60 μm-thick sections. Sections containing biocytin-filled cells were incubated in avidin-biotinylated horseradish peroxidase complex (1:100 dilution; ABC Kit; Vector Laboratories, Burlingame, CA, USA) followed by a peroxidase reaction using diaminobenzidine (DAB; Sigma-Aldrich; 0.05%) as the chromogen and 1% H2O2 as the substrate. Sections were then mounted on gelatin-coated slides, air-dried, immersed in xylene-based mounting medium (Entellan; Merck, Damstadt, Germany) and cover-slipped. Neurons were drawn using a drawing tube attached to a light microscope (63× oil objective 1.4 NA).
Regarding the experiments in vivo, neurons were filled with neurobiotin using the juxtacellular labelling method (Pinault, 1996). After neurobiotin was allowed to be transported in the neurons (0.5–6.5 h after cell juxtacellular labelling), rats (n= 3) were perfused with fixative (4% paraformaldehyde, 15% v/v saturated picric acid and 0.1% glutaraldehyde in 0.1 m PB at pH ∼7.4). Brain processing and immunohistochemical analysis were performed as recently reported (Bienvenu et al. 2012). Antibodies used in this study and indications of their specificities are listed in Supplementary Table 1.
In addition, adult NPY-GFP+ mice (n= 5) were perfused with phosphate-buffered saline (PBS) followed by fixative (4% paraformaldehyde, 15% v/v saturated picric acid and 0.05% glutaraldehyde in 0.1 m PB at pH ∼7.2) to test NPY and GFP co-localization.
Electron microscopy (EM)
After recordings, slices were immersed at 4°C for 14–18 h in a fixative solution containing: 4% paraformaldehyde, 0.05 glutaraldehyde, 15% (v/v) saturated picric acid and 0.1 m PB (pH 7.2–7.4). Slices were incubated in 1% H2O2 to block endogenous peroxidase activity, embedded in gelatin, and re-sectioned at 60 μm thickness. After cryoprotection in 0.1 m PB with 10% and 20% sucrose (at 4°C for 1 h), the sections were freeze-thawed in liquid nitrogen. Sections were incubated in avidin-biotinylated horseradish peroxidase complex (1:100 dilution) followed by a peroxidase reaction using DAB (Sigma-Aldrich; 0.05%) as the chromogen and 1% H2O2 as the substrate. Sections were post-fixed in 1% osmium tetroxide (Oxkem, Reading, UK) in 0.1 m PB for ∼45 min. The sections were dehydrated through a graded series of ethanol dilutions (50%, 70%, 90%, 95%, 100%; 2 × 10 min for all dilutions). To increase contrast in the EM, 1% uranyl acetate was included in the 70% ethanol solution (∼45 min). Next, sections were washed (2 × 20 min) in propylene oxide and infiltrated with resin for ∼8 h (Durcupan; Fluka Chemical, St. Louis, MO, USA). Sections were mounted in resin on glass microscope slides and polymerized at 60°C for 24 h. The somata, axon and dendrites of labelled neurons were examined under a light microscope at high magnification. Areas of interest were cut out from the sections and re-embedded in Durcupan blocks for sectioning. Consecutive ultrathin (70 nm) sections were collected on Pioloform-coated single-slot copper grids. The ultrathin sections were then contrasted with lead citrate for 1–2 min and examined using a Philips CM-100 EM (Philips, Eindhoven, The Netherlands).
Chemicals and drugs
All drugs were applied to the slice via bath solution. Salts used for the patch pipette and ACSF were obtained either from VWR International (Lutterworth, UK) or Sigma-Aldrich. Kynurenic acid (3 mm; Sigma-Aldrich) was added routinely to ACSF. Drugs obtained from Tocris Bioscience (Bristol, UK) were added at the following concentrations: SR95531 (also termed gabazine, 5 μm); (1,2,5,6-tetrahydropyridin-4-yl)methylphosphinic acid (TPMPA, 75 μm); dextran (1.25 mm); CGP35348 (50 μm); Cl218872 (1 μm); L655708 (25 nm); EGTA (10 mm, intracellularly applied); SKF89976A (25 μm). RO4938581 (500 nm) was donated by Hoffmann-LaRoche (Basel, Switzerland).
Results
NGFCs are a source of slow phasic inhibition in the BLA
The great majority of hippocampal NGFCs express NPY (Price et al. 2005). Therefore, we recorded NPY-GFP+ cells, filled them with biocytin and analysed their dendritic and axonal aspects (Fig. 1A). We observed that these neurons had round cell bodies and short, sparsely spiny dendrites arranged in a stellate fashion around the soma. The axon branched profusely mostly close to the soma, displaying frequent, small en passant varicosities. It also produced a dense local plexus that we found was the landmark of these cells (see reconstruction in Fig. 1A). The axon and dendrites were contained within the BLA, suggesting an interneuron-like, local function. The soma appearance as well as the dendritic and axonal patterns of our recorded cells closely resembled NGFCs reported by anatomical studies of amygdala in other species (Tombol & Szafranska-Kosmal, 1972; McDonald & Culberson, 1981; McDonald, 1982), and we will refer to them as such.
Figure 1. Dendritic, axonal and firing patterns of NGFCs in the BLA.
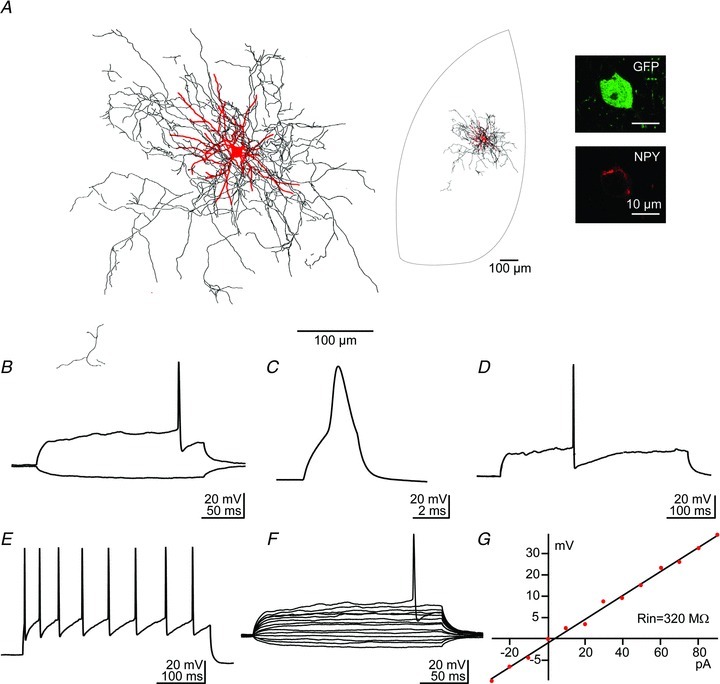
A, light microscopic reconstruction (63×) of a biocytin-labelled neuropeptide Y (NPY)+ cell (red, soma and dendrites; black, axon). The location of the reconstructed cell within the BLA boundaries is also illustrated. Note that this recorded neuron evoked a GABAA,slow IPSC in postsynaptic PN (not shown). Confocal immunofluorescence micrographs showing a green fluorescent protein (GFP) and NPY+ cell from a NPY-hrGFP BAC perfused transgenic mouse. B–G, passive and active electrophysiological responses from another NPY+ NGFC. B, voltage responses to injected current pulses (−30 pA, +70 pA, 200 ms), note the late firing. C and D, action potentials evoked by a short (C, current pulse: +100 pA, 3 ms) and a longer (D, +120 pA, 400 ms) depolarizing current pulse, note AHP in D. E, action potential frequency adaptation (current pulse: +280 pA, 400 ms). F, voltage responses to hyerpolarizing-depolarizing current pulses (range: −30/+90 pA, 400 ms) used to construct the I–V plot shown in G and to determine the value of Rin. All traces were recorded at the resting membrane potential of −65 mV from the same NPY+ NGFC.
The injection of hyperpolarizing-depolarizing current pulses from the resting membrane potential revealed some intrinsic functional aspects of these neurons (n= 13; Fig. 1B–G; Table 1). In particular, NGFCs exhibited a relatively broad action potential, pronounced spike AHP, a relatively low input resistance (Fig. 1C, D and G), and a delayed firing in response to a depolarizing current pulse of rheobase intensity (Fig. 1B, D and F). Larger depolarizing current steps elicited accommodating spike trains (Fig. 1E). Next, we performed whole-cell patch-clamp recordings of pairs between presynaptic NPY-GFP+ cells and postsynaptic PNs of the BLA. These experiments were usually performed in the presence of kynurenic acid, an antagonist for the ionotropic glutamate receptor (3 mm, at Vh=−65 mV) to prevent excitatory network activity, but identical results were obtained in experiments performed in control ACSF. An action current in a NPY+ cell elicited a uIPSC in a PN in most cases (postsynaptic uIPSC, 98/128 cases; Fig. 2A). This protocol could also elicit an inward current in the presynaptic cell occurring shortly after the action current (therefore referred to as an autaptic uIPSC, 86/98 cases; Fig. 2B). The reconstruction of a representative connected pair is shown in Fig. 2E. On average, the postsynaptic and autaptic uIPSCs peak amplitudes were 143.1 ± 17.6 pA (n= 58) and 135.7 ± 14.0 pA (n= 51); the 20–80% rise times were 3.00 ± 0.15 ms (n= 58) and 4.02 ± 0.55 ms (n= 37); and the single exponential fitted decay time constants were 41.60 ± 2.02 ms (n= 58) and 74.80 ± 6.00 ms (n= 45), respectively (Fig. 2C). When the decay of the IPSCs was fitted with a double exponential, the weighted decay (τw) values were: 41.51 ± 2.83 ms and 71.66 ± 5.73 ms, for postsynaptic and autaptic uIPSCs, respectively. The decay of autaptic IPSCs was significantly slower than that of postsynaptic IPSCs (P < 0.001), but the rise time and the peak amplitude did not significantly differ (P > 0.05). The rise time and decay time constant of the IPSCs were positively correlated (Pearson's correlation, R= 0.63 and 0.68 for autaptic and postsynaptic IPSCs, respectively, P < 0.001; Fig. 2D). Because the kinetics of postsynaptic and autaptic IPSCs matched closely that described in the literature as GABAA,slow (Capogna & Pearce, 2011), we refer to them as such or as ‘slow uIPSCs’. Notably, presynaptic action currents always evoked slow uIPSCs without failures (Fig. 2A and B). Both currents were purely mediated by the GABAA receptor, as they were abolished by the GABAA receptor antagonist SR95531 (5 μm), and no other synaptic component was detected (n= 17; Fig. 2A and B). The remaining outward current seen in the presynaptic cell (Fig. 2B) was an AHP mediated by Ca2+-dependent voltage gated K+ channels, as this current reversed at Vh of −90 mV (n= 8, data not shown). We tested the potential influence of the AHP outward current on the autaptic uIPSC kinetics. The inclusion of the slow Ca2+ chelator EGTA (10 mm, 5–10 min) into the pipette internal solution blocked the AHP outward current, as expected (Faber & Sah, 2002). In the presence of EGTA, the rise time and decay of the autaptic uIPSC was not significantly different from that recorded with control patch solution (n= 5, P > 0.05 for all tested parameters, data not shown). Moreover, no electrical synapses were detected in synaptically coupled cells (n= 6, data not shown), as a hyperpolarizing current pulse injected in a NPY+ cell did not evoke a detectable voltage deflection in a PN. Our data demonstrate that NGFCs of the BLA evoked slow uIPSCs solely mediated by GABAA receptors. In contrast, NGFCs of the neocortex (Tamas et al. 2003) or hippocampus (Price et al. 2005) elicit slow uIPSCs mediated by both GABAA and GABAB receptors.
Table 1.
Summary of passive and active membrane responses of NPY-GFP+ NGFCs
| Parameter | AVG ± SEM (n= 13) |
|---|---|
| Rin (MΩ) | 287.9 ± 23.3 |
| AP width (ms) | 0.92 ± 0.06 |
| AP amplitude (mV) | 64.9 ± 4.1 |
| AHP amplitude (mV) | 33.9 ± 1.8 |
| Adaptation index | 0.27 ± 0.04 |
| Membrane τ (ms) | 13.6 ± 1.4 |
| Cm (nF) | 0.05 ± 0.01 |
Data shown as means ± SEM. AHP, afterhyperpolarization; AP, action potential; Cm, membrane capacitance; Rin, input resistance.
Figure 2. NGFCs evoke postsynaptic and autaptic slow unitary inhibitory postsynaptic currents (uIPSCs).
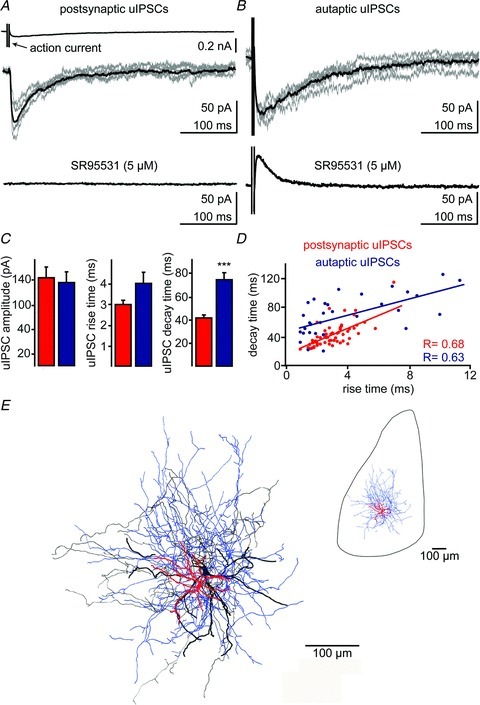
A, short square depolarizing voltage pulse applied to a presynaptic NPY-GFP+ cell elicited an action current (top trace), which evoked a slow uIPSC in a postsynaptic PN of the BLA (middle trace) and an autaptic uIPSCs in the NPY-GFP+ cell. B, individual uIPSCs are shown in grey; the average of five traces is shown in black. Bath application of the GABAA receptor antagonist SR95531 (5 μm) abolished postsynaptic (A, lower trace) and autaptic uIPSCs (B, lower trace). The outward current (B) is mediated by voltage gated K+ channels. C, quantification of 20–80% rise time, decay time and peak amplitude of postsynaptic and autaptic uIPSCs, ***P < 0.005, Mann–Whitney test. Data shown as mean ± SEM. D, scatter plot of decay time versus rise time of autaptic and postsynaptic uIPSCs (Pearson correlation, R= 0.63 and 0.68, respectively, P < 0.001). E, light microscopic reconstruction (63× magnification) of biocytin-labelled presynaptic NPY+ and postsynaptic PN cells. Soma and dendrites (thick) of the presynaptic cell are shown in red, the axon (thin) is shown in blue; soma and dendrites (thick), axon (thin) of the postsynaptic cell are shown in black. The insert shows the location of the reconstructed cells within the BLA boundaries.
Additional experiments confirmed the lack of GABAB receptor activation by NGFCs in spite of the presence of this receptor in PNs of the BLA (Pape & Pare, 2010). Firstly, in the presence of SR95531 (5 μm) and the vesicular GABA transporter (VGAT) 1 antagonist SKF89976A (25 μm) to increase GABA spillover, stimulation of a NGFC did not elicit any detectable current. The subsequent application of the GABAB receptor antagonist CGP35348 (5 μm) had no effect (n= 5, data not shown). Secondly, the GABAB receptor agonist baclofen (10 μm) evoked a clear-cut outward current (78 ± 10 pA, n= 3, data not shown) in PNs of the BLA voltage-clamped at −65 mV, demonstrating that GABAB receptor-mediated currents could be detected in our preparation.
As previously mentioned, some presynaptic NPY+ cells did not evoke uIPSCs in PNs (30/128 cases). We noticed that in a few cases (n= 8, data not shown), some passive and active membrane responses in these NPY+ cells were different from those observed in NGFCs. Specifically, the Rin was higher (514 ± 37 MΩ,P < 0.001), the membrane τ was higher (38.7 ± 7.9 ms, P < 0.001), and the adaptation index was higher (0.54 ± 0.08, P < 0.01) compared with NGFC data shown in Table 1. This suggests that these NPY+ cells were not NGFCs, consistent with the heterogeneity of NPY+ cells observed in other brain areas (Karagiannis et al. 2009; Ibanez-Sandoval et al. 2011). However, because the aim of our study was to identify the cell type and the mechanisms involved in GABAA,slow in the amygdala, we have not further examined this issue.
NGFCs of the BLA fire phase-locked to hippocampal theta oscillations in vivo
To understand further the function of NGFCs in the BLA, we studied their firing patterns in vivo. Neurons were recorded extracellularly in urethane-anaesthetized rats and subsequently labelled using the juxtacellular method (Bienvenu et al. 2012). Two neurons were identified as NGFCs on the basis of their axonal and dendritic patterns, and the expression of NPY (tjx54a and 80a). These cells co-expressed VGAT, as well as somatostatin (Fig. 3B) and calbindin (CB). A third cell (tjx76a) was classified as a NGFC for its axonal and dendritic arborizations exhibiting all features of the cells we recorded in vitro in the mouse; this cell was CB+. The anatomical section containing its soma was lost during processing, and immunoreactivity for NPY could not be assessed. All cells were negative for parvalbumin (PV; Supplementary Fig. 1). Immunohistochemistry results are recapitulated in Supplementary Table 2.
Figure 3. In vivo firing pattern of NGFCs of the basolateral amygdala complex (BLA).
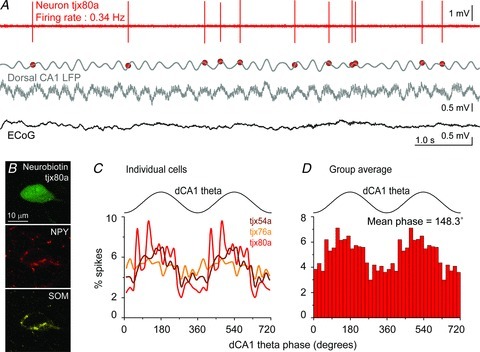
A, firing of neuron tjx80a recorded in a urethane-anaesthetized adult rat. The red trace shows single-unit activity (spikes) of tjx80a during a period of firing. Spiking occurred preferentially around the late ascending phase of theta oscillations recorded in the dorsal hippocampus (CA1 stratum pyramidale). Local field potential (LFP) is shown in grey: bottom, raw LFP; top, band-pass filtered theta oscillations 3–6 Hz. Red symbols illustrate the occurrence of tjx80a spikes relative to CA1 theta oscillations. Theta oscillations are observed during the cortical ‘activated state’, as seen in the surface electrocorticogram (ECoG; black trace). B, tjx80a was juxtacellularly filled with Neurobiotin, and was found to express neuropeptide Y (NPY) and somatostatin (SOM) using immunohistochemistry. Confocal single optical slice of 1.1 μm thickness. Note that the same channel was used to test PV immunoreactivity (negative, see Supplementary Fig. 1) as to test NPY subsequently. C, NGFCs recorded in vivo in the BLA fired preferentially during the ascending phase of hippocampal theta oscillations. Phase histograms showing the normalized distribution of cells’ spikes across the CA1 theta oscillation cycle. The theta cycle was duplicated for clarity. D, consistent firing modulation of the three NGFCs with hippocampal theta resulted in a significant modulation at the population level. Average phase histogram showing the preference of NGFC firing for the late ascending phase of CA1 theta.
In vivo NGFCs fired at low frequencies and irregularly (mean rate = 0.91 Hz, mean coefficient of variation = 1.73, n= 3; Fig. 3A; Supplementary Table 3). Single action potentials had a peak to trough width of 0.4 ± 5.3−5 ms, a value comparable to other interneuron types and shorter than that recorded in PNs with the juxtacellular method in vivo (Bienvenu et al. 2012).
Synchronization of hippocampal and BLA activities at theta frequencies is thought to be important for fear memory (Pape & Pare, 2010). We investigated the spike timing relationship of NGFCs with theta oscillations recorded in dorsal CA1 (Bienvenu et al. 2012). The three NGFCs displayed a marked firing preference for the ascending phase of hippocampal theta (Fig. 3A and C; Supplementary Table 3). As a result of precise timing across neurons, NGFCs as a group were modulated in phase with CA1 theta oscillations (population mean phase = 148.3 deg and modulation depth r= 0.14; P < 0.05, Moore test; Fig. 3D).
Thus, NGFCs of BLA fire phase-locked with theta oscillations in addition to evoking GABAA,slow with a time course consistent with a theta cycle, suggesting this cell type could be a key local contributor to this rhythm in the BLA.
Low concentration of extracellular GABA causes slow NGFC-IPSC
The kinetics of slow uIPSC after stimulation of NGFCs of the BLA was much slower than uIPSCs evoked by any type of interneuron of the BLA tested so far (Woodruff & Sah, 2007; Spampanato et al. 2011). What causes such prolonged synaptic inhibition? We hypothesized that the spatiotemporal concentration profile of GABA at the synapse determines the IPSC kinetics. To test this idea and assess the transmitter concentration at the postsynaptic receptors of PNs, first we compared the effect of the low-affinity, fast unbinding (∼0.6 ms; Jones et al. 2001) competitive GABAA receptor antagonist TPMPA (75 μm) on the slow uIPSC and on fast sIPSCs recorded from PNs. This antagonist should be more effective on IPSCs evoked by lower extracellular GABA concentration (Barberis et al. 2011). As expected, bath application of TPMPA reduced the amplitude of slow uIPSC (n= 16, decrease to 41.9% of initial value, P < 0.05; Fig. 4A and C), but produced a non-significant decrease on the amplitude of fast sIPSCs (n= 5, decrease to 81.6% of initial value, P > 0.05; Fig. 4B and C). Moreover, TPMPA had no significant effect on the rise time and decay time of slow uIPSC or fast sIPSC (all P values > 0.05; Fig. 4C), although it tended to decrease the decay of slow uIPSC, a result consistent with transmitter spillover (Overstreet & Westbrook, 2003). A summary of the data obtained with TPMPA is presented in Table 2.
Figure 4. NGFCs release low non-saturating concentration of GABA.
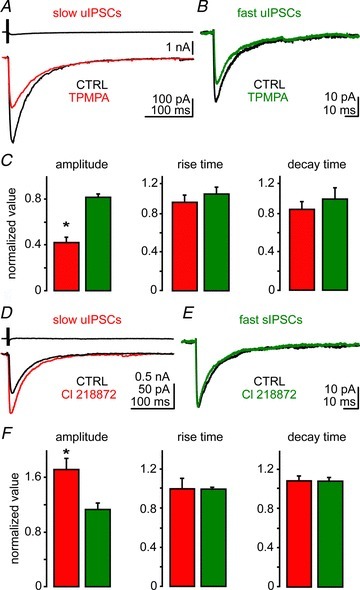
A, action current in a presynaptic NPY+ cell (upper trace) evoked a unitary inhibitory postsynaptic current (uIPSC) in a postsynaptic PN (black trace); the same current upon application of the fast unbinding GABAA receptor competitive antagonist (1,2,5,6-tetrahydropyridin-4-yl)methylphosphinic acid (TPMPA; 75 μm, red trace). B, effect of TPMPA (green trace) on fast spontaneous (s)IPSCs. Each trace is the average of five sweeps in A, and 48 events in B. C, quantification of the effects of TPMPA on amplitude, rise and decay time of slow uIPSCs (n= 16) and fast sIPSCs (n= 5). Mann–Whitney test, *P < 0.05. D, an action current in a presynaptic NPY+ cell (upper trace) evoked a uIPSC in a PN in control (CTRL; black trace) and during the application of the GABAA receptor α1 subunit agonist C1218872 (1 μm, red trace). E, effect of C1218872 (green trace) on fast sIPSCs. Each trace is the average of five sweeps in D, and 61 events in E. F, quantification of drug effect on amplitude, rise and decay time of slow uIPSC (n= 14) and fast sIPSCs (n= 5). Wilcoxon's test, *P < 0.05.
Table 2.
Quantification of drugs effect on amplitude, rise and decay time of slow uIPSC and fast sIPSCs
| Slow uIPSCs | Fast sIPSCs | |||||
|---|---|---|---|---|---|---|
| Amplitude (pA) | Rise time (ms) | Decay time (ms) | Amplitude (pA) | Rise time (ms) | Decay time (ms) | |
| Pre-TPMPA | 220.7 ± 38.8 | 2.76 ± 0.43 | 44.35 ± 5.65 | 35.5 ± 4.9 | 1.25 ± 0.19 | 12.17 ± 1.95 |
| TPMPA | 108.5 ± 30.1* | 2.35 ± 0.30 | 38.68 ± 7.17 | 28.8 ± 3.9 | 1.22 ± 0.13 | 10.92 ± 1.62 |
| n= 16 | n= 5 | |||||
| Pre-C1218872 | 173.6 ± 49.2 | 2.93 ± 0.36 | 34,93 ± 3,65 | 38.2 ± 7.6 | 1.40 ± 0.15 | 14.00 ± 1.46 |
| C1218872 | 229.1 ± 48.6* | 2.62 ± 0.41 | 39.14 ± 4.15 | 36.6 ± 1.5 | 1.37 ± 0.16 | 147.5 ± 1.57 |
| n= 14 | n= 5 | |||||
| Pre-dextran | 183.9 ± 40.8 | 2.55 ± 0.39 | 45.95 ± 12.9 | 35.1 ± 4.2 | 1.32 ± 0.11 | 14.45 ± 1.79 |
| Dextran | 292.7 ± 68.8* | 2.53 ± 0.40 | 41.79 ± 5.38 | 33.1 ± 2.2 | 1.32 ± 0.13 | 13.66 ± 1.35 |
| n= 15 | n= 6 | |||||
| Pre-L655708 | 109.7 ± 25.9 | 4.47 ± 0.69 | 61.31 ± 7.84 | – | – | – |
| L655708 | 106.6 ± 22.4 | 4.42 ± 0.66 | 58.30 ± 8.26 | – | – | – |
| n= 11 | ||||||
| Pre-R04938581 | 85.8 ± 19.9 | 2.01 ± 0.49 | 50.55 ± 6.54 | – | – | – |
| R04938581 | 78.2 ± 22.8 | 2.43 ± 0.61 | 61.50 ± 12.85 | – | – | – |
| n= 7 | ||||||
Data shown as means ± SEM. Wilcoxon's test, *P < 0.05. sIPSC, spontaneous IPSC; uIPSC, unitary inhibitory postsynaptic current; TPMPA, (1,2,5,6-tetrahydropyridin-4-yl)methylphosphinic acid.
The GABAA receptor α1 subunit agonists potentiate the IPSC amplitude to various extents depending on the degree of receptor occupancy (Hajos et al. 2000). If NGFCs generate a lower concentration of transmitter compared with other interneurons, then slow IPSCs should be more sensitive to α1 agonists than fast IPSCs. To test this idea we used the selective α1 agonist Cl218872, as it displays a greater selectivity for GABAA receptors containing the α1 subunit than the more widely used zolpidem (Korpi et al. 2002). The application of Cl218872 (1 μm) significantly increased the amplitude of slow uIPSCs without significantly affecting their kinetics (n= 14, P < 0.05; Fig. 4D and F). In contrast, this drug had no effect on the amplitude of fast sIPSCs recorded from PNs (n= 5, P > 0.05; Fig. 4E and F). A summary of the data obtained with Cl218872 is presented in Table 2. These results indicate that GABAA receptors were not saturated by GABA released from NGFCs, and also suggest that the α1 subunit is present at these synapses.
Taken together, these results suggest that compared with other interneurons of the BLA, transmitter release from NGFCs results in lower GABA concentration exposure of the postsynaptic receptors.
GABA spillover contributes to the slow IPSC kinetics
Next, we tested whether spillover of GABA contributes to the slow kinetics of the IPSCs evoked by NGFCs. Firstly, we used the high molecular weight molecule dextran to increase the viscosity and impair the diffusion of the neurotransmitter across the synaptic cleft (Min et al. 1998). We expected that dextran should increase intra-cleft GABA concentration and decrease spillover and activation of extrasynaptic receptors. This should affect the amplitude of slow uIPSC rather than fast sIPSC. As predicted, the addition of dextran (1.25 mm, MW: 40,000) to the ACSF significantly increased the amplitude (n= 15, increase to 183.1% of initial value, P < 0.01; Fig. 5A and C) and tended to accelerate the decay time constant of slow uIPSCs (although this effect did not reach significance, P > 0.05). In contrast, dextran had no significant effect on fast sIPSC amplitude and kinetics (n= 6, 98.0% of initial value, P > 0.05; Fig. 5B and C). These results indicate that GABA released from NGFC, but not from other interneurons of the BLA, diffuses away from the releasing sites. A summary of the data obtained with dextran is shown in Table 2.
Figure 5. NGFCs elicit GABA spillover: actions of dextran and α5 GABAA receptor ligands.
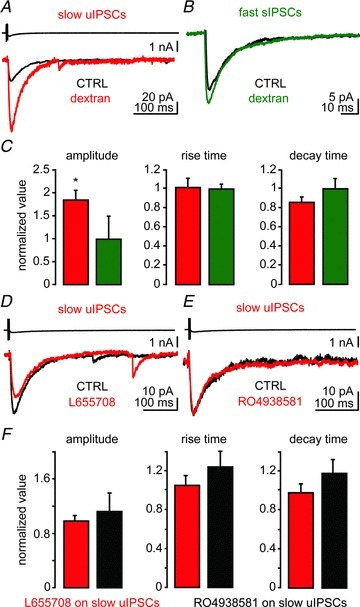
A, action current in presynaptic NPY+ cell (upper trace) evoked a unitary inhibitory postsynaptic current (uIPSC) in a postsynaptic PN (black trace); the same current upon application of dextran (1.25 mm, red trace). B, effect of dextran (green trace) on fast spontaneous (s)IPSCs. Each trace is the average of five sweeps in A, and 75 events in B. C, quantification of the effect of dextran on the amplitude, rise and decay time of slow uIPSCs (n= 15) and fast IPSCs (n= 6). Mann–Whitney test, *P < 0.05. C and D, action of GABAA receptor α5 subunit inverse agonists L655708 (D, 25 nm, red trace) or RO4938581 (E, 500 nm, red trace) on slow uIPSCs. Each trace is the average of five sweeps. F, quantification of drug effect on amplitude, rise and decay time of slow uIPSC and fast sIPSCs (n= 11, 7). Wilcoxon's test, *P < 0.05. CTRL, control.
There is evidence for the presence of high-affinity extrasynaptic GABAA receptors on the target cells for neocortical and hippocampal NGFCs (Olah et al. 2009; Karayannis et al. 2010), likely activated by low GABA concentrations. Therefore, we tested the presence of the high-affinity α5 subunit in PNs activated by NGFCs. We used two different ligands: L655708, an inverse agonist selective for the α5 subunit up to 25 nm (Vargas-Caballero et al. 2010), and RO4938581, a highly selective α5 subunit inverse agonist (Ballard et al. 2009). None of these ligands significantly changed the amplitude or kinetics of slow uIPSCs (n= 11, 25 nm, L655708; Fig. 5D and F; or 500 nm, RO4938581, n= 7; Fig. 5E and F; all P values > 0.05). A summary of the data obtained with these drugs is shown in Table 2. Therefore, GABA released from NGFCs does not appear to activate extrasynaptic α5 subunits, in contrast to hippocampal NGFCs (Karayannis et al. 2010). This is perhaps because their expression in the BLA is too low to mediate detectable changes in cell excitability upon their inhibition (Fritschy & Mohler, 1995).
Moreover, presynaptic GABAB receptors are located at extrasynaptic sites of neocortical (Tamas et al. 2003) and hippocampal NGFCs (Price et al. 2005). If GABA escapes the synaptic cleft, spillover of GABA should activate these receptors. To test this hypothesis, we stimulated presynaptic NGFCs at theta frequency (5 Hz). This protocol elicited a transient short-term depression of IPSC amplitude (n= 10; Fig. 6A and C). Application of the GABAB receptor antagonist CGP35348 (5 μm) attenuated the synaptic depression (P < 0.05 for 2nd, 3rd and 4th stimulation, n= 10; Fig. 6A and C). The same protocol was repeated by recording the postsynaptic cell with caesium-containing patch solution, to block postsynaptic GABAB receptors (Gahwiler & Brown, 1985). Under these conditions, CGP35348 produced a similar increase of uIPSCs as in control conditions (P > 0.05 for 2nd, 3rd and 4th stimulation, n= 6; Fig. 6B and D), suggesting the presynaptic location of GABAB receptors. These data indicate that GABA released by NGFCs can effectively activate presynaptic GABAB receptors located outside release sites.
Figure 6. NGFCs elicit GABA spillover: presynaptic GABAB receptor attenuates synaptic depression.
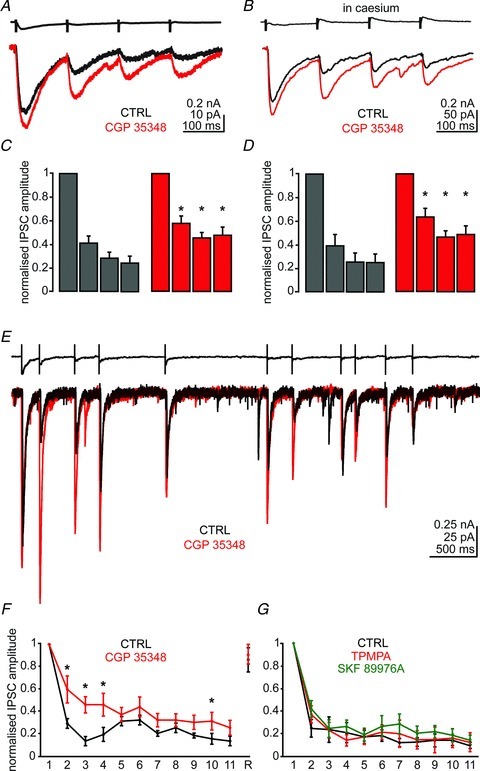
A, action current in a presynaptic NPY+ cell (5 Hz stimulation, upper trace) evoked unitary inhibitory postsynaptic currents (uIPSCs) in a PN (lower trace, in black) that depressed over repeated activation. Application of the GABAB receptor antagonist CGP35348 (50 μm) increased the amplitude of the uIPSCs and attenuated synaptic depression (lower trace, in red). Each trace is the average of five sweeps. B, as in A, but the PN cell was recorded with a caesium methanesulphonate-containing solution to block postsynaptic GABAB receptors. Each trace is the average of five sweeps. C, histograms show the amplitude of each IPSC recorded during 5 Hz stimulation in control (black) and in the presence of CGP35348 (red) normalized to the amplitude of the first uIPSC (n= 10). D, as in C, in the presence of caesium (n= 6). E, action current in presynaptic NPY+ cell (in vivo train of action potentials, upper trace) evoked uIPSCs in PN (lower trace, in black). Application of CGP35348 (50 μm) increased the amplitude of the uIPSCs and attenuated synaptic depression (lower trace, in red). F, graph shows the amplitude of each IPSC evoked by in vivo firing in control and in the presence of CGP35348 normalized to the amplitude of the first uIPSC (n= 8; the numbers on the x-axis refer to the action potential occurrence in the train). The IPSC amplitude recorded 2 min after the end of the train stimulation is also shown (R, recovery). Asterisks mark significant differences with respect to the pre-drug condition; Wilcoxon test, *P < 0.05. G, same graph as in F, in control (CTRL), and during the application in the same cells of (1,2,5,6-tetrahydropyridin-4-yl) methylphosphinic acid (TPMPA; 75 μm) or SKF89976A (25 μm; n= 6; P > 0.05 for all comparisons, Wilcoxon's test).
We also injected into NPY+-NGFC the firing from a NGFC recorded in vivo as described above. The application in vitro of a representative series of 11 action potentials (5 s duration) recorded during hippocampal theta activity in vivo elicited a robust synaptic depression that fully reversed after 2 min (n= 8; Fig. 6E and F). Consistent with the theta frequency stimulation experiments described above, the application of CGP35348 partially rescued the synaptic depression (n= 8; P < 0.05 for 2nd, 3rd, 4th and 10th slow uIPSC; Fig. 6E and F). These data thus confirm that GABA released by NGFCs spills over and activates extrasynaptic receptors. To further test the mechanisms involved in this short-term synaptic plasticity, this protocol of stimulation was repeated in the presence of the weak GABAA receptor antagonist TPMPA, or the VGAT-1 GABA transporter antagonist SKF89976A, to reduce or increase, respectively, the concentration and hence modify the time GABA stays bound to postsynaptic receptors. In these experiments, SKF89976A (25 μm) increased the size of slow uIPSCs (amplitude: 131% of control, P < 0.05; decay time: 273% of control, n= 6, P < 0.05), consistent with GABA spillover and the lack of GABAA receptor saturation (Szabadics et al. 2007; Karayannis et al. 2010). We observed that neither manipulation significantly changed the pattern of the synaptic depression (n= 6, P > 0.05; Fig. 6G). Thus, this short-term synaptic plasticity was likely due to presynaptic mechanisms, such as activation of GABAB autoreceptors and functional synaptic vesicles depletion.
NGFC axon terminals form non-synaptic appositions
The data illustrated so far suggest that the spatiotemporal concentration profile of GABA released from axon terminals is the major factor that determines the kinetics of slow uIPSC evoked by NGFCs of the BLA. Does this have a structural correlate? We tested this issue by using EM. Slices were analysed in which NPY+ cells filled with biocytin evoked slow uIPSCs. Consecutive ultrathin sections from three to five slices (at least five grids from each slice, each containing 10–20 consecutive ultrathin sections) were examined for each cell. Each biocytin-labelled axon terminal was scanned in every ultrathin section (even in consecutive grids, if required), to determine whether it was forming conventional synapses. Strikingly, the majority of the visualized 40 terminals of NPY-GFP+ cells formed non-synaptic appositions (n= 31, 77.5): on somata (n= 5; Fig. 7A); on dendrites (n= 14; Fig. 7B–D); on another terminal (n= 2); the remaining 10 appositions were formed on unidentified structures (Fig. 7E). We noticed encapsulation of targets by presynaptic terminals (Fig. 7B), consistent with NGFC terminals of the neocortex (Olah et al. 2009). Furthermore, we observed clear non-synaptic appositions between a terminal of NPY-GFP+ cell and one of its dendrite (Fig. 7F), representing a potential structural substrate of autaptic uIPSC. We also identified nine synapses formed on: dendrites (n= 6); soma (n= 1); another terminal (n= 1); and on a glial cell (n= 1). It is likely that the ratio between synaptic and non-synaptic appositions formed on targeted cells influences the kinetics of the uIPSCs. These results are consistent with the idea that GABA can diffuse away from the release sites and reach receptors sensitive to a low concentration of GABA on various membrane compartments of nearby cells.
Figure 7. Non-synaptic appositions are formed by NGFC terminals.
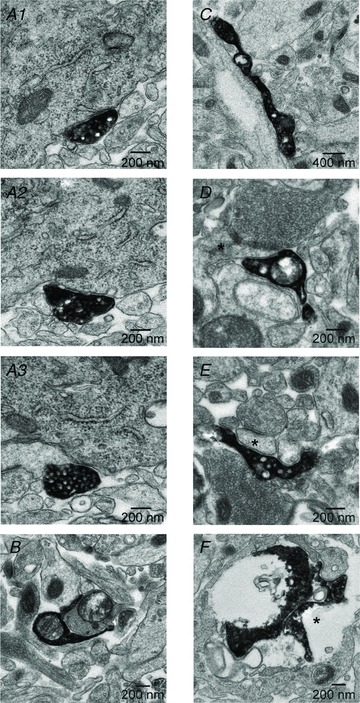
A, biocytin labelled terminal of a NPY+ cell making close apposition with the soma or the initial part of a dendrite. B, labelled terminal making close apposition with a dendritic profile; note the encapsulation of the target dendrite. C, labelled terminal making close apposition with a medium sized dendrite. D, labelled terminal making close apposition with a spine that receives a type I synapse (asterisk). E, another labelled terminal making close apposition with another postsynaptic structure that receives a type I synapse (asterisk). F, labelled terminal (asterisk) making close apposition with a dendrite of the presynaptic cell (autapse). Note that all appositions are not classical synaptic structures. Each panel is a representative image(s) from several consecutive ultrathin sections, namely: 11 (A), 15 (B), 22 (C), 9 (D), 12 (E), 25 (F).
Discussion
This work reports a novel type of NPY+, local inhibitory cell of the BLA and demonstrates that this cell type is a source of slow phasic inhibition to PNs.
The anatomical aspect of these NPY+ cells closely resembles NGFC-like class III neurons observed in BLA Golgi preparations in rat and opossum (McDonald & Culberson, 1981; McDonald, 1982) and Golgi IIc cells described in the cat (Tombol & Szafranska-Kosmal, 1972). Therefore, we have termed them NGFC of the amygdala. We found that these neurons are positive for NPY and somatostatin, consistent with previous observations (McDonald, 1989), and they are also likely to express nitric oxide synthase (McDonald et al. 1993). Future studies should further characterize the immunohistochemical markers of NPY+ cells and also test for the presence of different subpopulations in the BLA. In the hippocampus, bistratified cells, NGFCs and Ivy cells can express NPY (Klausberger et al. 2004; Price et al. 2005; Fuentealba et al. 2008). In the striatum, two types of NPY+ GABAergic cells have been recently reported: NGFC and plateau-depolarization low-threshold spike interneuron (Ibanez-Sandoval et al. 2011).
Several types of interneurons have so far been studied in the BLA. This classification is based mainly on the expression of calcium-binding proteins and neuropeptides, single-cell RT-PCR, and electrophysiological characterization of anatomically identified neurons in vitro (Ehrlich et al. 2009; Pape & Pare, 2010; Spampanato et al. 2011). We recently fully characterized four types of interneurons of the rat BLA recorded in vivo (Bienvenu et al. 2012). The present work combines the in vitro and in vivo approaches, and defines the NGFC type in the BLA. The kinetics of IPSCs evoked by NGFCs of the BLA is much slower than any other interneuron type previously studied in the amygdala, such as PV+ cells (Woodruff & Sah, 2007). It closely matches the kinetics of the IPSCs evoked by NGFCs of the neocortex, hippocampus and striatum (Tamas et al. 2003; Price et al. 2008; Ibanez-Sandoval et al. 2011). The discovery of slow GABAA receptor-mediated IPSC in the amygdala and the identification of NGFC as the cell type mediating it provides additional evidence for the wide presence and significance of this signal in the CNS (Capogna & Pearce, 2011).
What mechanisms underlie the slow NGFC-IPSC? Because NGFCs of cortex and hippocampus target the distal dendrites of pyramidal cells (Vida et al. 1998; Tamas et al. 2003; Price et al. 2008), dendritic filtering could in principle explain the slow kinetics of the synaptic currents. However, we observed with EM that the terminals of NGFCs in the amygdala are apposed not only to dendritic profiles but also to somatic membranes. This renders this possibility unlikely. The subunit composition of the GABAA receptors can determine the kinetics of the IPSCs at CNS synapses (Farrant & Kaila, 2007). For example, slow IPSC evoked in CA1 pyramidal cells are impaired in the hippocampus of GABAA receptor β3 subunit null mice (Hentschke et al. 2009). It has been suggested that neocortical and hippocampal NGFC synapses express both low- and high-affinity GABAA receptors, at both synaptic and extrasynaptic sites (Capogna & Pearce, 2011). The present results indicate the involvement of the GABAA receptor α1 subunit at BLA-NGFC synapses. Because this subunit usually confers fast gating (Farrant & Kaila, 2007), it is an unlikely determinant of the IPSC slowness. A third causative factor could be the spatiotemporal concentration profile of GABA. Our results obtained with the use of TPMPA and an α1 subunit agonist demonstrate that the GABA released from NGFC terminals reaches a relatively low concentration and does not saturate postsynaptic receptors. This result is in agreement with the low concentration and the prolonged presence of transmitter found at neocortical and hippocampal NGFC synapses (Szabadics et al. 2007; Karayannis et al. 2010). Such prolonged exposure of receptors to the transmitter favours receptor desensitization (Karayannis et al. 2010). The present results suggest that presynaptic inhibition mediated by GABAB receptors and functional depletion of presynaptic vesicles are the most likely causes of the IPSC depression observed in the BLA. On note: the firing frequency of NGFCs of the BLA (here) was lower than that observed in NGFCs of the hippocampus (Fuentealba et al. 2010). For this reason we injected fewer action potentials in NGFCs of the BLA compared with the ones applied to NGFCs in the hippocampus (Karayannis et al. 2010). The different protocol used here might have contributed to shift the locus of this short-term synaptic plasticity towards the presynaptic site.
The results obtained with dextran and a GABAB receptor antagonist suggest spillover of GABA to extrasynaptic sites. Indeed, we observed that the terminals of NGFCs of the BLA form non-synaptic appositions more often than classical synaptic contacts on target neurons. This might allow diffusion of GABA from the cleft and its action as a volume transmitter onto extrasynaptic sites. Furthermore, a lack of clearly defined postsynaptic elements favours the diffusion of the transmitter and can influence the concentration of GABA reaching postsynaptic receptors. This view is consistent with ultrastructural observations made on neocortical (Olah et al. 2009) and hippocampal NGFCs (Karayannis et al. 2010).
Several observations made in NGFCs of the BLA reported here were similar to the ones detected in NGFCs of the neocortex or hippocampus. However, some of the features of NGFCs in the BLA were distinct. Firstly, slow uIPSCs elicited by NGFCs of the amygdala are entirely mediated by the GABAA receptor and lack a GABAB receptor component typically detected in the neocortex or the hippocampus (Tamas et al. 2003; Price et al. 2005). Secondly, NGFCs make autapses more frequently in the BLA than in the hippocampus (∼90% reported here versus∼50% reported in Karayannis et al. 2010), suggesting a more robust self-regulation of activity in the former. Thirdly, appositions between NGFC terminals and the soma of PNs were observed in the BLA, whereas NGFCs selectively target the dendrites of pyramidal cells in the neocortex and CA1 hippocampus (Tamas et al. 2003; Price et al. 2008). Fourthly, NGFC-evoked IPSCs in the amygdala are not sensitive to GABAA receptor α5 inverse agonists, in contrast to the neocortex and hippocampus (Szabadics et al. 2007; Karayannis et al. 2010). This difference might be explained by the lower level of expression of that subunit in the BLA compared with the neocortex or hippocampus (Fritschy & Mohler, 1995). This result is consistent with the idea that the subunit composition of the GABAA receptor is not the major determinant shaping kinetics of slow IPSCs. Finally, NGFCs of the BLA and hippocampus both fire close to the peak of hippocampal theta (Fuentealba et al. 2010), but the former discharge at a lower fire rate. On note, Ivy cells of the hippocampus recorded in anaesthetized (Fuentealba et al. 2008) or freely-moving (Lapray et al. 2012) rats preferentially fire at the through and ascending phase of theta cycle, respectively.
What is the physiological role of NGFCs of the BLA? NGFCs’ firing preference for the late ascending phase of CA1 theta oscillations is virtually identical to that of CB+ dendrite-targeting cells (Bienvenu et al. 2012). The present EM analysis indicates that NGFCs of the BLA release GABA mainly around dendritic domains. Thus, we provide further evidence for rhythmic inhibition of PN dendrites in phase with hippocampal theta oscillations, in basal conditions. This, together with the other observation that this cell type elicits IPSCs with kinetics near to one theta cycle, suggests a possible function in the coordination of hippocampal-amygdala theta oscillations that emerge during fear memory retrieval (Seidenbecher et al. 2003). Distinct temporal profiles of inhibition may explain the need for several types of cells firing at similar times and releasing GABA onto dendrites. NGFCs and other dendrite-targeting cells of the BLA might cooperatively be instrumental for the formation and recall of emotional memories via rhythmic inhibition.
Furthermore, it is estimated that the dense axonal arborization of one cortical NGFC provides potential release sites equivalent to six basket cells (Olah et al. 2009). The GABA released from NGFCs inhibits the release of glutamate or GABA from axon terminals, which are located at some distance from NGFC release sites (Olah et al. 2009). Because action potentials of NGFCs evoke IPSCs without failures, they might have a profound effect on excitatory inputs to nearby cells and be very effective in silencing spontaneous activity of PNs in the BLA (Gaudreau & Pare, 1996). Moreover, series of action potentials generated by NGFCs lead to the depression of slow synaptic events and disinhibition of postsynaptic targets favouring long-term plasticity of excitatory inputs. GABA released from NGFCs can acts as a volume transmitter (Olah et al. 2009; Karayannis et al. 2010, present results). Therefore, the activity of NGFCs can provide an important source of accumulated extrasynaptic GABA that activates presynaptic GABAB receptors and mediates suppression of glutamatergic input to PNs of the BLA (Pan et al. 2009).
We show here that NGFCs in the amygdala can express NPY. This peptide has an anxiolytic action, as demonstrated by anxiety-related behavioural tests (Sajdyk et al. 1999). Application of NPY decreases the excitability of most PNs of the lateral nucleus via activation of Y1 receptors and subsequently G-protein coupled inwardly rectifying potassium channels (Sosulina et al. 2008). Although NPY can be found in PNs, local GABAergic cells are likely to be the main source of NPY in the BLA (McDonald & Mascagni, 2002). Our data suggest that NGFCs are the most abundant NPY-containing type of neurons within the BLA. Therefore they might be a source of this peptide. Growing evidence shows that local GABAergic cells of the BLA are critically involved in the acquisition, manifestation and extinction of fear (Ehrlich et al. 2009; Pape & Pare, 2010). Identification of cell types and their connectivity within the amygdala is crucial for understanding how emotional information is processed. We propose that slow phasic inhibition evoked by NGFCs is an essential component of the amygdala network actively contributing to the maintenance and modulation of its high inhibitory tone.
Acknowledgments
We thank Dr Robert Stewart for the Matlab program used in the protocol of in vivo firing injection into in vitro recorded neurons. We also thank Dr Damien Lapray for help in cell reconstruction, Linda Katona for help with anatomical analysis of cell tjx54a, and Katharine Whitworth for excellent technical aid. Dr Colin Akerman, Prof. Francesco Ferraguti and Prof. Dmitri Rusakov are acknowledged for their comments on a previous version of the manuscript. This work was supported by the Medical Research Council, UK (MRC award U138197106). Mirosława Mańko is in receipt of an MRC studentship. Thomas Bienvenu is funded by an MRC DPhil studentship, and is a fellow of Ecole de l' Inserm Liliane Bettencourt MD-PhD Program, France.
Glossary
- ACSF
artificial cerebrospinal fluid
- AHP
afterhyperpolarization
- BAC
bacterial artificial chromosome
- BLA
basolateral amygdala complex
- CB
calbindin
- DAB
diaminobenzidine
- ECl
equilibrium potential for Cl− ions
- ECoG
electrocorticogram
- EM
electron microscopy
- GFP
green fluorescent protein
- hrGFP
humanized Renilla green fluorescent protein
- IPSC
inhibitory postsynaptic current
- NGFC
neurogliaform cell
- NPY
neuropeptide Y
- PB
phosphate buffer
- PN
principal neuron
- PV
parvalbumin
- Rin
input resistance
- sIPSC
spontaneous IPSC
- τ
membrane time constant
- TPMPA
(1,2,5,6-tetrahydropyridin-4-yl)methylphosphinic acid
- uIPSC
unitary inhibitory postsynaptic current
- Vh
holding potential
- VGAT
vesicular GABA transporter
Author contributions
M.C. ideated and supervised the project; M.M., Y.D. and M.C. designed and performed the in vitro experiments, analysed and interpreted the data; T.B. performed and analysed in vivo experiments, and drafted the relative text; M.M. and M.C. drafted the article; M.M., T.B. and M.C. revised it critically. All authors approved the final version.
Author's present address
T. C. M. Bienvenu: INSERM U862, Neurocentre Magendie, University of Bordeaux, 146 rue Léo Saignat, 33077 Bordeaux, France.
Supplementary material
Supplementary Table 1
Supplementary Table 2
Supplementary Table 3
Supplementary Fig. 1
References
- Armstrong C, Szabadics J, Tamas G, Soltesz I. Neurogliaform cells in the molecular layer of the dentate gyrus as feed-forward gamma-aminobutyric acidergic modulators of entorhinal-hippocampal interplay. J Comp Neurol. 2011;519:1476–1491. doi: 10.1002/cne.22577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballard TM, Knoflach F, Prinssen E, Borroni E, Vivian JA, Basile J, Gasser R, Moreau JL, Wettstein JG, Buettelmann B, Knust H, Thomas AW, Trube G, Hernandez MC. RO4938581, a novel cognitive enhancer acting at GABAA alpha5 subunit-containing receptors. Psychopharmacology (Berl) 2009;202:207–223. doi: 10.1007/s00213-008-1357-7. [DOI] [PubMed] [Google Scholar]
- Banks MI, Li TB, Pearce RA. The synaptic basis of GABAA,slow. J Neurosci. 1998;18:1305–1317. doi: 10.1523/JNEUROSCI.18-04-01305.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barberis A, Petrini EM, Mozrzymas JW. Impact of synaptic neurotransmitter concentration time course on the kinetics and pharmacological modulation of inhibitory synaptic currents. Front Cell Neurosci. 2011;5:6. doi: 10.3389/fncel.2011.00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartos M, Vida I, Frotscher M, Meyer A, Monyer H, Geiger JR, Jonas P. Fast synaptic inhibition promotes synchronized gamma oscillations in hippocampal interneuron networks. Proc Natl Acad Sci U S A. 2002;99:13222–13227. doi: 10.1073/pnas.192233099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bienvenu TC, Busti D, Magill PJ, Ferraguti F, Capogna M. Cell-type-specific recruitment of amygdala interneurons to hippocampal theta rhythm and noxious stimuli in vivo. Neuron. 2012;74:1059–1074. doi: 10.1016/j.neuron.2012.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buhl EH, Halasy K, Somogyi P. Diverse sources of hippocampal unitary inhibitory postsynaptic potentials and the number of synaptic release sites. Nature. 1994;368:823–828. doi: 10.1038/368823a0. [DOI] [PubMed] [Google Scholar]
- Capogna M, Pearce RA. GABA A,slow: causes and consequences. Trends Neurosci. 2011;34:101–112. doi: 10.1016/j.tins.2010.10.005. [DOI] [PubMed] [Google Scholar]
- Crowley JJ, Fioravante D, Regehr WG. Dynamics of fast and slow inhibition from cerebellar golgi cells allow flexible control of synaptic integration. Neuron. 2009;63:843–853. doi: 10.1016/j.neuron.2009.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond GB. Reporting ethical matters in the Journal of Physiology: standards and advice. J Physiol. 2009;587:713–719. doi: 10.1113/jphysiol.2008.167387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrlich I, Humeau Y, Grenier F, Ciocchi S, Herry C, Luthi A. Amygdala inhibitory circuits and the control of fear memory. Neuron. 2009;62:757–771. doi: 10.1016/j.neuron.2009.05.026. [DOI] [PubMed] [Google Scholar]
- English DF, Ibanez-Sandoval O, Stark E, Tecuapetla F, Buzsaki G, Deisseroth K, Tepper JM, Koos T. GABAergic circuits mediate the reinforcement-related signals of striatal cholinergic interneurons. Nat Neurosci. 2012;15:123–130. doi: 10.1038/nn.2984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faber ES, Sah P. Physiological role of calcium-activated potassium currents in the rat lateral amygdala. J Neurosci. 2002;22:1618–1628. doi: 10.1523/JNEUROSCI.22-05-01618.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrant M, Kaila K. The cellular, molecular and ionic basis of GABA(A) receptor signalling. Prog Brain Res. 2007;160:59–87. doi: 10.1016/S0079-6123(06)60005-8. [DOI] [PubMed] [Google Scholar]
- Fritschy JM, Mohler H. GABAA-receptor heterogeneity in the adult rat brain: differential regional and cellular distribution of seven major subunits. J Comp Neurol. 1995;359:154–194. doi: 10.1002/cne.903590111. [DOI] [PubMed] [Google Scholar]
- Fuentealba P, Begum R, Capogna M, Jinno S, Marton LF, Csicsvari J, Thomson A, Somogyi P, Klausberger T. Ivy cells: a population of nitric-oxide-producing, slow-spiking GABAergic neurons and their involvement in hippocampal network activity. Neuron. 2008;57:917–929. doi: 10.1016/j.neuron.2008.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuentealba P, Klausberger T, Karayannis T, Suen WY, Huck J, Tomioka R, Rockland K, Capogna M, Studer M, Morales M, Somogyi P. Expression of COUP-TFII nuclear receptor in restricted GABAergic neuronal populations in the adult rat hippocampus. J Neurosci. 2010;30:1595–1609. doi: 10.1523/JNEUROSCI.4199-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gahwiler BH, Brown DA. GABAB-receptor-activated K+ current in voltage-clamped CA3 pyramidal cells in hippocampal cultures. Proc Natl Acad Sci U S A. 1985;82:1558–1562. doi: 10.1073/pnas.82.5.1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaudreau H, Pare D. Projection neurons of the lateral amygdaloid nucleus are virtually silent throughout the sleep–waking cycle. J Neurophysiol. 1996;75:1301–1305. doi: 10.1152/jn.1996.75.3.1301. [DOI] [PubMed] [Google Scholar]
- Hajos N, Nusser Z, Rancz EA, Freund TF, Mody I. Cell type- and synapse-specific variability in synaptic GABAA receptor occupancy. Eur J Neurosci. 2000;12:810–818. doi: 10.1046/j.1460-9568.2000.00964.x. [DOI] [PubMed] [Google Scholar]
- Hentschke H, Benkwitz C, Banks MI, Perkins MG, Homanics GE, Pearce RA. Altered GABAA,slow inhibition and network oscillations in mice lacking the GABAA receptor beta3 subunit. J Neurophysiol. 2009;102:3643–3655. doi: 10.1152/jn.00651.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herry C, Ciocchi S, Senn V, Demmou L, Muller C, Luthi A. Switching on and off fear by distinct neuronal circuits. Nature. 2008;454:600–606. doi: 10.1038/nature07166. [DOI] [PubMed] [Google Scholar]
- Huntsman MM, Porcello DM, Homanics GE, DeLorey TM, Huguenard JR. Reciprocal inhibitory connections and network synchrony in the mammalian thalamus. Science. 1999;283:541–543. doi: 10.1126/science.283.5401.541. [DOI] [PubMed] [Google Scholar]
- Ibanez-Sandoval O, Tecuapetla F, Unal B, Shah F, Koos T, Tepper JM. A novel functionally distinct subtype of striatal neuropeptide Y interneuron. J Neurosci. 2011;31:16757–16769. doi: 10.1523/JNEUROSCI.2628-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones MV, Jonas P, Sahara Y, Westbrook GL. Microscopic kinetics and energetics distinguish GABA(A) receptor agonists from antagonists. Biophys J. 2001;81:2660–2670. doi: 10.1016/S0006-3495(01)75909-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapur A, Pearce RA, Lytton WW, Haberly LB. GABAA-mediated IPSCs in piriform cortex have fast and slow components with different properties and locations on pyramidal cells. J Neurophysiol. 1997;78:2531–2545. doi: 10.1152/jn.1997.78.5.2531. [DOI] [PubMed] [Google Scholar]
- Karagiannis A, Gallopin T, David C, Battaglia D, Geoffroy H, Rossier J, Hillman EM, Staiger JF, Cauli B. Classification of NPY-expressing neocortical interneurons. J Neurosci. 2009;29:3642–3659. doi: 10.1523/JNEUROSCI.0058-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karayannis T, Elfant D, Huerta-Ocampo I, Teki S, Scott RS, Rusakov DA, Jones MV, Capogna M. Slow GABA transient and receptor desensitization shape synaptic responses evoked by hippocampal neurogliaform cells. J Neurosci. 2010;30:9898–9909. doi: 10.1523/JNEUROSCI.5883-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klausberger T, Marton LF, Baude A, Roberts JDB, Magill P, Somogyi P. Spike timing of dendrite-targeting bistratified cells during hippocampal network oscillations in vivo. Nat Neurosci. 2004;7:41–47. doi: 10.1038/nn1159. [DOI] [PubMed] [Google Scholar]
- Klausberger T, Somogyi P. Neuronal diversity and temporal dynamics: the unity of hippocampal circuit operations. Science. 2008;321:53–57. doi: 10.1126/science.1149381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korpi ER, Grunder G, Luddens H. Drug interactions at GABA(A) receptors. Prog Neurobiol. 2002;67:113–159. doi: 10.1016/s0301-0082(02)00013-8. [DOI] [PubMed] [Google Scholar]
- Kuo SP, Bradley LA, Trussell LO. Heterogeneous kinetics and pharmacology of synaptic inhibition in the chick auditory brainstem. J Neurosci. 2009;29:9625–9634. doi: 10.1523/JNEUROSCI.0103-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapray D, Lasztoczi B, Lagler M, Viney TJ, Katona L, Valenti O, Hartwich K, Borhegyi Z, Somogyi P, Klausberger T. Behavior-dependent specialization of identified hippocampal interneurons. Nat Neurosci. 2012;215:1265–1271. doi: 10.1038/nn.3176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makkar SR, Zhang SQ, Cranney J. Behavioral and neural analysis of GABA in the acquisition, consolidation, reconsolidation, and extinction of fear memory. Neuropsychopharmacology. 2010;35:1625–1652. doi: 10.1038/npp.2010.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markwardt SJ, Dieni CV, Wadiche JI, Overstreet-Wadiche L. Ivy/neurogliaform interneurons coordinate activity in the neurogenic niche. Nat Neurosci. 2011;14:1407–1409. doi: 10.1038/nn.2935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald AJ. Neurons of the lateral and basolateral amygdaloid nuclei: a Golgi study in the rat. J Comp Neurol. 1982;212:293–312. doi: 10.1002/cne.902120307. [DOI] [PubMed] [Google Scholar]
- McDonald AJ. Coexistence of somatostatin with neuropeptide Y, but not with cholecystokinin or vasoactive intestinal peptide, in neurons of the rat amygdala. Brain Res. 1989;500:37–45. doi: 10.1016/0006-8993(89)90297-7. [DOI] [PubMed] [Google Scholar]
- McDonald AJ, Culberson JL. Neurons of the basolateral amygdala: a Golgi study in the opossum (Didelphis virginiana) Am J Anat. 1981;162:327–342. doi: 10.1002/aja.1001620404. [DOI] [PubMed] [Google Scholar]
- McDonald AJ, Mascagni F. Immunohistochemical characterization of somatostatin containing interneurons in the rat basolateral amygdala. Brain Res. 2002;943:237–244. doi: 10.1016/s0006-8993(02)02650-1. [DOI] [PubMed] [Google Scholar]
- McDonald AJ, Payne DR, Mascagni F. Identification of putative nitric oxide producing neurons in the rat amygdala using NADPH-diaphorase histochemistry. Neuroscience. 1993;52:97–106. doi: 10.1016/0306-4522(93)90185-i. [DOI] [PubMed] [Google Scholar]
- Min MY, Rusakov DA, Kullmann DM. Activation of AMPA, kainate, and metabotropic receptors at hippocampal mossy fiber synapses: role of glutamate diffusion. Neuron. 1998;21:561–570. doi: 10.1016/s0896-6273(00)80566-8. [DOI] [PubMed] [Google Scholar]
- Nusser Z, Naylor D, Mody I. Synapse-specific contribution of the variation of transmitter concentration to the decay of inhibitory postsynaptic currents. Biophys J. 2001;80:1251–1261. doi: 10.1016/S0006-3495(01)76101-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olah S, Fule M, Komlosi G, Varga C, Baldi R, Barzo P, Tamas G. Regulation of cortical microcircuits by unitary GABA-mediated volume transmission. Nature. 2009;461:1278–1281. doi: 10.1038/nature08503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olah S, Komlosi G, Szabadics J, Varga C, Toth E, Barzo P, Tamas G. Output of neurogliaform cells to various neuron types in the human and rat cerebral cortex. Front Neural Circuits. 2007;1:4. doi: 10.3389/neuro.04.004.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overstreet LS, Westbrook GL. Synapse density regulates independence at unitary inhibitory synapses. J Neurosci. 2003;23:2618–2626. doi: 10.1523/JNEUROSCI.23-07-02618.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan BX, Dong Y, Ito W, Yanagawa Y, Shigemoto R, Morozov A. Selective gating of glutamatergic inputs to excitatory neurons of amygdala by presynaptic GABAb receptor. Neuron. 2009;61:917–929. doi: 10.1016/j.neuron.2009.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pape HC, Pare D. Plastic synaptic networks of the amygdala for the acquisition, expression, and extinction of conditioned fear. Physiol Rev. 2010;90:419–463. doi: 10.1152/physrev.00037.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pare D, Collins DR. Neuronal correlates of fear in the lateral amygdala: multiple extracellular recordings in conscious cats. J Neurosci. 2000;20:2701–2710. doi: 10.1523/JNEUROSCI.20-07-02701.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearce RA. Physiological evidence for two distinct GABAA responses in rat hippocampus. Neuron. 1993;10:189–200. doi: 10.1016/0896-6273(93)90310-n. [DOI] [PubMed] [Google Scholar]
- Pinault D. A novel single-cell staining procedure performed in vivo under electrophysiological control: morpho-functional features of juxtacellularly labeled thalamic cells and other central neurons with biocytin or Neurobiotin. J Neurosci Methods. 1996;65:113–136. doi: 10.1016/0165-0270(95)00144-1. [DOI] [PubMed] [Google Scholar]
- Prenosil GA, Schneider Gasser EM, Rudolph U, Keist R, Fritschy JM, Vogt KE. Specific subtypes of GABAA receptors mediate phasic and tonic forms of inhibition in hippocampal pyramidal neurons. J Neurophysiol. 2006;96:846–857. doi: 10.1152/jn.01199.2005. [DOI] [PubMed] [Google Scholar]
- Price CJ, Cauli B, Kovacs ER, Kulik A, Lambolez B, Shigemoto R, Capogna M. Neurogliaform neurons form a novel inhibitory network in the hippocampal CA1 area. J Neurosci. 2005;25:6775–6786. doi: 10.1523/JNEUROSCI.1135-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price CJ, Scott R, Rusakov DA, Capogna M. GABA(B) receptor modulation of feedforward inhibition through hippocampal neurogliaform cells. J Neurosci. 2008;28:6974–6982. doi: 10.1523/JNEUROSCI.4673-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sajdyk TJ, Vandergriff MG, Gehlert DR. Amygdalar neuropeptide Y Y1 receptors mediate the anxiolytic-like actions of neuropeptide Y in the social interaction test. Eur J Pharmacol. 1999;368:143–147. doi: 10.1016/s0014-2999(99)00018-7. [DOI] [PubMed] [Google Scholar]
- Schofield CM, Huguenard JR. GABA affinity shapes IPSCs in thalamic nuclei. J Neurosci. 2007;27:7954–7962. doi: 10.1523/JNEUROSCI.0377-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidenbecher T, Laxmi TR, Stork O, Pape HC. Amygdalar and hippocampal theta rhythm synchronization during fear memory retrieval. Science. 2003;301:846–850. doi: 10.1126/science.1085818. [DOI] [PubMed] [Google Scholar]
- Sosulina L, Schwesig G, Seifert G, Pape HC. Neuropeptide Y activates a G-protein-coupled inwardly rectifying potassium current and dampens excitability in the lateral amygdala. Mol Cell Neurosci. 2008;39:491–498. doi: 10.1016/j.mcn.2008.08.002. [DOI] [PubMed] [Google Scholar]
- Spampanato J, Polepalli J, Sah P. Interneurons in the basolateral amygdala. Neuropharmacology. 2011;60:765–773. doi: 10.1016/j.neuropharm.2010.11.006. [DOI] [PubMed] [Google Scholar]
- Suzuki N, Bekkers JM. Microcircuits mediating feedforward and feedback synaptic inhibition in the piriform cortex. J Neurosci. 2012;32:919–931. doi: 10.1523/JNEUROSCI.4112-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabadics J, Tamas G, Soltesz I. Different transmitter transients underlie presynaptic cell type specificity of GABAA,slow and GABAA,fast. Proc Natl Acad Sci U S A. 2007;104:14831–14836. doi: 10.1073/pnas.0707204104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamas G, Lorincz A, Simon A, Szabadics J. Identified sources and targets of slow inhibition in the neocortex. Science. 2003;299:1902–1905. doi: 10.1126/science.1082053. [DOI] [PubMed] [Google Scholar]
- Tombol T, Szafranska-Kosmal A. A golgi study of the amygdaloid complex in the cat. Acta Neurobiol Exp (Wars) 1972;32:835–848. [PubMed] [Google Scholar]
- Vargas-Caballero M, Martin LJ, Salter MW, Orser BA, Paulsen O. alpha5 Subunit-containing GABA(A) receptors mediate a slowly decaying inhibitory synaptic current in CA1 pyramidal neurons following Schaffer collateral activation. Neuropharmacology. 2010;58:668–675. doi: 10.1016/j.neuropharm.2009.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vida I, Halasy K, Szinyei C, Somogyi P, Buhl EH. Unitary IPSPs evoked by interneurons at the stratum radiatum-stratum lacunosum-moleculare border in the CA1 area of the rat hippocampus in vitro. J Physiol. 1998;506:755–773. doi: 10.1111/j.1469-7793.1998.755bv.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodruff AR, Sah P. Networks of parvalbumin-positive interneurons in the basolateral amygdala. J Neurosci. 2007;27:553–563. doi: 10.1523/JNEUROSCI.3686-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wozny C, Williams SR. Specificity of synaptic connectivity between layer 1 inhibitory interneurons and layer 2/3 pyramidal neurons in the rat neocortex. Cereb Cortex. 2011;21:1818–1826. doi: 10.1093/cercor/bhq257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zar JH. Biostatistical Analysis. Upple Saddle River, NJ: Prentice Hall; 1999. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.