

Abstract
Visceral primary afferents enter the CNS at the caudal solitary tract nucleus (NTS), and activate central pathways key to autonomic and homeostatic regulation. Excitatory transmission from primary solitary tract (ST)-afferents consists of multiple contacts originating from single axons that offer a remarkably high probability of glutamate release and high safety factor for ST afferent excitation. ST afferent activation sometimes triggers polysynaptic GABAergic circuits, which feedback onto second-order NTS neurons. Although inhibitory transmission is observed at second-order neurons, much less is known about the organization and mechanisms regulating GABA transmission. Here, we used a focal pipette to deliver minimal stimulus shocks near second-order NTS neurons in rat brainstem slices and directly activated single GABAergic axons. Most minimal focal shocks activated low jitter EPSCs from single axons with characteristics resembling ST afferents. Much less commonly (9% of sites), minimal focal shocks activated monosynaptic IPSCs at fixed latency (low jitter) that often failed (30%) and had no frequency-dependent facilitation or depression. These GABA release characteristics contrasted markedly to the unfailing, large amplitudes for glutamate released during ST-EPCSs recorded from the same neurons. Surprisingly, unitary GABAergic IPSCs were only weakly calcium dependent. In some neurons, strong focal shocks evoked compound IPSCs indicating convergent summation of multiple inhibitory axons. Our studies demonstrate that second-order NTS neurons receive GABAergic transmission from a diffuse network of inhibitory axons that rely on an intrinsically less reliable and substantially weaker release apparatus than ST excitation. Effective inhibition depends on co-activation of convergent inputs to blunt excitatory drive.
Key points
Successful transmission of information to the brain relies on a balance of excitation and inhibition, and this balance is different across different brain regions.
In the brain areas responsible for initiating homeostatic reflex control of visceral organs, excitation is known to be particularly powerful whereas inhibition is often less obvious.
Using minimal focal activation of single axons, this study shows that elementary inhibitory transmission in this brainstem region is founded on an intrinsically weak process with limited transmitter release that is surprisingly prone to failures.
Strong inhibitory transmission requires multi-axon convergence of modestly reliable synapses, whereas primary afferent excitation arises from single, multi-contact axons with highly reliable neurotransmitter release.
The results suggest that the entry of primary afferent information along cranial nerves enjoys a high safety factor in part due to the fundamental weakness of inhibitory transmission at these initial central neurons.
Introduction
Homeostatic reflexes rely on primary afferent updates to constantly match life support systems to behavioural, emotional and executive demands (Loewy, 1990; Andresen & Paton, 2011). Primary afferents in the solitary tract (ST) drive reflex regulation of the respiratory, circulatory and gastrointestinal systems. The solitary tract nucleus (nucleus tractus solitarii, NTS) in the CNS serves as a nexus of interaction as second-order neurons directly receive primary afferent terminals as well as an extraordinarily rich array of other CNS inputs (Andresen & Paton, 2011). Translation of afferent input into activation of NTS second-order neurons is a key step in the broadcast of visceral information to the CNS. Some of these regulatory pathways are as short as a loop with as few as two central neurons before returning to modify peripheral organ function. For example, cardiac vago-vagal reflexes exhibit full loop latencies averaging ∼50 ms from afferent activation (Kunze, 1972; Mendelowitz, 1999; Travagli et al. 2006).
Most neurons in the medial NTS are second order (McDougall et al. 2009), and a minority is classified as higher order with distinct input and output characteristics (Bailey et al. 2006a; Fernandes et al. 2011). The afferent limb of these pathways (ST to NTS) features exceptionally strong excitatory glutamate drive. This ST glutamate is delivered to the second-order neurons in NTS by a basic structure with limited numbers of ST afferents (generally one–three axons) in which each afferent axon supplies multiple contacts (∼20 active zones) that rarely fail to release glutamate (Andresen & Peters, 2008; McDougall et al. 2009; Peters et al. 2011). The functional result is a very high safety factor for excitatory transmission (Andresen & Yang, 1995; Liu et al. 2000; Bailey et al. 2002). ST afferent terminals and their glutamate release machinery are commonly targeted for modulation by descending inputs (e.g. forebrain) that act presynaptically on ST afferents (Bailey et al. 2006b; Peters et al. 2008a; Michelini & Stern, 2009). This strong excitatory afferent drive sometimes is countered by GABAergic inhibition, which shapes primary afferent integration (Mifflin & Felder, 1988). However, much less is known about the fundamental architecture of the inhibitory synaptic network and the mechanisms governing GABA transmission in NTS.
GABA transmission can strongly inhibit NTS performance (McDougall et al. 2008; Herman et al. 2012), and all neurons have spontaneous miniature IPSCs (in TTX) indicating ubiquitous GABA terminals, albeit at very low frequencies of release (Jin et al. 2004). GABAergic neurons are found throughout caudal NTS (Chan & Sawchenko, 1998; Bailey et al. 2008; Austgen et al. 2009), although the nature of functional connections is less obvious. A prominent relationship appears to be that most local GABAergic interneurons are themselves second order to ST primary afferents (Bailey et al. 2008). Much of what is known about GABAergic transmission in the NTS derives from in vivo observations (Mifflin et al. 1988; Mifflin & Felder, 1988), in which GABA actions are accentuated by general anaesthetics (Bailey et al. 2008; McDougall et al. 2008; Peters et al. 2008b). Despite studies on spontaneous IPSPs or electrically evoked compound events that include indirectly evoked IPSCs (Kawai & Senba, 1996), little is known about the fundamental mechanisms of unitary GABA transmission in the NTS. Here we directly activated individual GABA axons by probing in close proximity to second-order neurons using minimal intensity focal shocks. Our chief findings include: GABA axons were present at lower density than excitatory axons as we rarely detected monosynaptic GABA fibres in medial NTS. These single GABA axons unreliably evoked low-amplitude IPSCs despite invariant monosynaptic latencies. Variance–mean testing indicated a low probability of GABA release and a mechanism with relatively low calcium sensitivity. In some neurons, strong shocks evoked long-lasting IPSCs indicating convergence of GABA axons reaching second-order NTS neurons. Thus, fundamentally weak GABAergic transmission relies on convergent inputs in contrast to more robust excitatory drive from single primary afferent axons within the NTS.
Methods
Ethical approval
Animals were maintained and experimental procedures were performed under the supervision and approval of the Institutional Animal Care and Use Committee at the Oregon Health & Science University, and conform to the National Institutes of Health publication ‘Guide for the Care and Use of Laboratory Animals’.
Horizontal brainstem slice
Brainstem slices were prepared from adult (>180 g, average weight 320 ± 11 g, n= 45) Sprague–Dawley rats (Charles River Laboratories, Wilmington, MA, USA) as previously described (Doyle & Andresen, 2001). Briefly, rats were placed in a Plexiglas container, deeply anaesthetized with 3% isoflurane and the medulla removed. A 250 μm horizontal brainstem slice was cut, which contained ST axons together with caudal NTS (Delaware Diamond Knives, Wilmington, DE, USA; and VT1000S; Leica Microsystems, Bannockburn, IL, USA). External artificial cerebrospinal fluid contained (in mm): NaCl, 125; KCl, 3; KH2PO4, 1.2; MgSO4, 1.2; NaHCO3, 25; dextrose, 10; CaCl2, 2 (300 mosmol l−1), bubbled with 95% O2–5% CO2, perfused at 34°C.
Whole-cell recordings
Pipettes (2.5–3.5 MΩ) were visually guided to neurons in the medial subnucleus of the caudal NTS (Zeiss, Thornwood, NJ, USA; Hamamatsu Photonic Systems, Bridgewater, NJ, USA). Intracellular solution contained (in mm): NaCl, 10; KCl, 40; potassium gluconate, 90; EGTA, 11; CaCl2, 1; MgCl2, 1; Hepes, 10; Na2ATP, 2; Na2GTP, 0.2 (pH 7.3, 296 mosmol l−1). This internal/external solution combination at VH=−60 mV yields inward EPSCs and inward IPSCs (ECl=−25 mV) in open, whole-cell voltage-clamp configuration (Multiclamp 700B; Molecular Devices, Sunnyvale, CA, USA). Signals were sampled at 30 kHz (filtered at 10 kHz), displayed traces were filtered with a low-pass Bessel (eight-pole) to 3 kHz. Liquid junction potentials were uncorrected (9.5 mV at 34°C).
Evoked synaptic currents with the medial subnucleus – ST and focal
In principle, low-intensity shocks activate axons nearest to the stimulating electrode, and each axon will be excited in an all-or-none manner (Andresen & Yang, 1995; Doyle & Andresen, 2001). To selectively activate ST afferent axons, a concentric bipolar electrode (50 μm inner core and 200 μm outer diameters; Frederick Haer, Bowdoinham, ME, USA) was placed on the visible ST at a distance of 1–3 mm from recorded neurons minimizing activation of non-ST axons or local neurons (Doyle et al. 2004; Bailey et al. 2008). Such shocks never activated monosynaptic IPSCs even at the highest intensities. To intercept local axons, a pipette (1.0–1.5 MΩ) filled with external solution was placed <400 μm from the recorded soma and focal shock intensity (0.1 ms duration) in graded fashion at each location.
Shock intensity-recruitment profiles
To assess focally intercepted inputs, responses to graded shocks (AMPI, Jerusalem, Israel) were discriminated by arrival time and waveform (McDougall et al. 2009). Recruitment profiles plotted event amplitudes or integrated currents against shock intensity. At each focal intensity (10–800 μA, ST; 10–200 μA, focal), one to five shock bursts (50 Hz) were repeated every 6 s for ≥10 consecutive trials.
Discriminating synaptic events: order and glutamatergic/GABAergic
Detailed analyses was performed to characterize the first synaptic events triggered each 6 s, i.e. EPSC1 or IPSC1 from the train of five shocks and the latency and jitter calculated over 40 trials (jitter = standard deviation of latency). Jitters of <200 μs were considered monosynaptic, but >200 μs were polysynaptic (Doyle & Andresen, 2001; Bailey et al. 2006a). At successful sites, any trial in which shocks failed to evoke characteristic PSCs were counted as a synaptic failure for calculating failure rates over 40 trials. Only second-order NTS neurons were included in this study. If focal shocks evoked no response, the focal electrode was moved to a different location. Events were initially identified kinetically – fast decaying as EPSCs and slow decaying as IPSCs (Jin et al. 2004). Pharmacological antagonists tested glutamatergic non-NMDA (2,3-dioxo-6-nitro-1,2,3,4-tetrahydro-benzo[f]quinoxaline-7-sulphonamide, NBQX), NMDA (2-amino-5-phosphonopentanoic acid, AP-5), metabotropic glutamate (LY314495) and/or GABAA receptors (gabazine, GBZ; Tocris, Ellisville, MO, USA).
Data analysis and statistics
Evoked PSC1 latency, jitter, amplitude and failure rates were not normally distributed, so that comparisons between groups used Kruskal–Wallis one-way or two-way ANOVA on ranks with Student–Newman–Keuls post hoc tests (SigmaStat, Systat Software Inc., Chicago, IL, USA). During high failure rates, the first two sequentially successful events generated paired-pulse ratios (PPRs) regardless of position within a shock train and compared with the ratio 1.0 (no depression/facilitation) by Student's paired t test. Data were expressed as means ± SEM, and were considered statistically significant if P < 0.05.
Results
Diffuse GABAergic network in medial NTS
An estimated three-quarters of the neurons within the medial subregion of the caudal NTS are directly contacted by ST afferents (i.e. second order) as distant ST stimulation activated monosynaptic EPSCs (McDougall et al. 2009) with invariant latencies (Fig. 1). Once a low jitter ST-EPSC identified the recorded neuron as second order, minimal focal shocks tested for additional input axons in the near vicinity of the neuron. In some cases, a distinct slowly decaying synaptic event was triggered at constant latency and classified as a monosynaptic IPSC (Fig. 1A and B). A striking characteristic of monosynaptic IPSCs was the presence of intermittent failures in which no event followed identical shocks (focal; Fig. 1B). Focal shocks required higher intensities to activate synaptic events than for ST activation but, as with ST-EPSCs, increments in focal shock intensity above threshold produced constant amplitude responses (Fig. 1C) – evidence that a single axon was responsible. This procedure defined the minimal intensity for an effective shock to trigger the synaptic event at each test site. The kinetics of evoked IPSCs (Fig. 1B) effectively discriminated these events as longer lasting GABA-mediated synaptic events (Jin et al. 2004) – classifications that were confirmed by receptor-specific antagonists. ST shocks always activated monosynaptic EPSCs (jitter <200 μs). Many focally evoked IPSCs had similar jitters and were considered monosynaptic despite the frequent failures (Fig. 1B). Although focal evoked, low jitter IPSCs were rarely intercepted at lower shock intensities than EPSCs, successful IPSCs had distinctly high failure rates that were not relieved by increases in shock intensity – observations that suggest that inhibitory transmission was intrinsically unreliable.
Figure 1. Focal pipette activated axons near representative second-order NTS neurons.
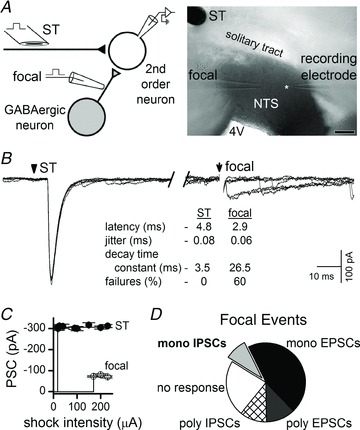
A, schematic illustrates the orientation of the solitary tract (ST) activation of primary afferent axon and focal pipette intercepting an inhibitory axon close to the neuron. The photograph shows example locations of stimulating (ST and focal) and recording electrodes. 4V, fourth ventricle. Scale bar: 200 μm. B, ST-evoked EPSCs identified neurons as second order to ST (left, 5 traces with artefacts blanked, shock at arrowhead), and were large-amplitude EPSCs with invariant latencies (80 μs in this example). Minimal shocks from the focal pipette (right, arrowhead) placed 200 μm from recorded soma evoked IPSCs in this same neuron. Focal shocks commenced 800 ms following ST shocks (break in traces). Focally evoked IPSCs had brief but invariant latencies, slower decay kinetics and higher failures (flat line traces) than ST-EPSCs. C, ST-EPSCs and focal-IPSCs (same neuron as in A and B) exhibited all-or-none stimulus-recruitment profiles, indicating minimal shocks activated single axons. D, across a total of 69 second-order solitary tract nucleus (NTS) neurons and 128 focal sites tested, nearly a quarter of focal sites failed to activate synaptic events (no response). The majority of successful events were monosynaptic (mono) EPSCs, but a large percentage were initiated through more complex pathways (polysynaptic – poly EPSCs and poly IPSCs). Only 9% of sites activated monosynaptic IPSCs, which were the major focus of the remainder of the studies.
Spontaneously released miniature IPSCs are commonly present in NTS neurons in action potential-free (TTX) conditions (Jin et al. 2004), and shocks to ST axons often activate polysynaptic IPSCs at NTS neurons (Mifflin & Felder, 1988; Andresen & Yang, 1995; Jin et al. 2004; McDougall et al. 2008). We attempted to detect inhibitory axons converging on second-order neurons by serially relocating the focal pipette to test additional sites surrounding the neurons. The focal pipette was placed generally rostral and/or medial (50–400 μm) from the soma of the recorded neuron. Across 128 sites surrounding 69 second-order NTS neurons, shocks failed to recruit any synaptic response. This suggests that, despite delivering shocks at substantially increased intensities (200 μA), about one-quarter of focal sites lacked axons that contacted the recorded neuron (Fig. 1D). However, moving the focal pipette as little as 25 μm from an unsuccessful site generally intercepted an afferent axon that could be activated at low intensity. Together such spatial results indicated a generally limited (20–30 μm) effective spread of focal current at high shock intensities from these unipolar focal stimulation pipettes. At nearly three-quarters of all test locations (≥10 trials per site), minimal intensity focal shocks activated an axon that triggered a synaptic response of some variety (Fig. 1D). Quite uncommonly (9% of sites), shocks activated low jitter IPSCs conspicuous for their long decay times and indicated the activation of monosynaptic GABAergic axons contacting the recorded neuron. The most common outcome for focal shocks, however, was the interception of incoming excitatory axons that predominantly (45% of sites) were monosynaptic EPSCs (mono EPSCs; Fig. 1D), with the remainder of sites activating polysynaptic PSCs (poly EPSCs and poly IPSCs; Fig. 1D). Thus, combining the number of sites at which focal shocks initiated polysynaptic synaptic events (poly IPSCs + poly EPSCs) together with the number of sites evoking monosynaptic EPSCs suggested that excitatory axons traversed 67% of regions near these neurons. Therefore, 88% of all the axons successfully activating synaptic responses (i.e. excluding ‘no response’ sites) were excitatory in origin. Using our approach of focal activation, we found disproportionately fewer GABAergic axons connected directly to second-order NTS neurons. Overall, the functional predominance of excitatory focal sites is consistent with dense anatomical trajectories and synaptic varicosities of ST visceral afferent axons within the NTS (Kubin et al. 2006).
Unreliable GABA release
In our studies, the striking paucity of focally activated, low jitter IPSCs is consistent with a low density of GABAergic axons in the vicinity of the recorded neurons that were directly connected (Fig. 1D). Successfully evoked, monosynaptic GABAA mediated IPSCs were long lasting, slowly relaxing events with amplitudes that fluctuated from shock to shock (Fig. 2A and B). Monosynaptic IPSCs were unaffected by non-NMDA antagonists, but were blocked by GBZ (Fig. 2A and B). In many neurons, increases in intensity of focal shocks activated compound synaptic events (Fig. 2A). These compound synaptic events displayed multiple latencies for different components within each event, but often contained kinetically slow currents similar to low jitter IPSCs. Application of ionotropic glutamate blockers (NBQX and AP-5) eliminated overlying, lower threshold EPSCs as well as ST-EPSCs, but monosynaptic IPSCs persisted (Fig. 2A). The remaining focal-IPSCs had consistent arrival times (low jitter) characteristic of monosynaptic IPSCs (Fig. 2A, lower right). Note that an analysis using narrow time–amplitude discrimination detected IPSCs before and after glutamatergic receptor block, which had similar latency distributions (insets, Fig. 2A, right). Failures were common even in these pharmacologically dissected IPSCs, despite their monosynaptic character (Fig. 2A–C). In other cases, minimal intensity focal shocks evoked high jitter, slow kinetic events that were completely eliminated by NBQX, indicating that GABA release for the IPSC was the final component of a polysynaptic pathway with an intervening non-NMDA glutamatergic link (polysynaptic IPSCs; Fig. 3). This was a surprising result considering the very close proximity of the focal electrode to the recorded soma. Equally surprising was the finding that focally activated polysynaptic IPSCs were somewhat more common than monosynaptic IPSCs using the minimal intensity criteria (11%vs. 9% respectively; Fig. 1D). Despite their polysynaptic network path, these polysynaptic events typically had unitary thresholds (constant amplitude responses with increases in shock intensity; Fig. 3) Together, these findings are consistent with a low density of GABA axons reaching recorded neurons.
Figure 2. Focal shocks often activated monosynaptic IPSCs as a component of complex synaptic responses in second-order neurons.
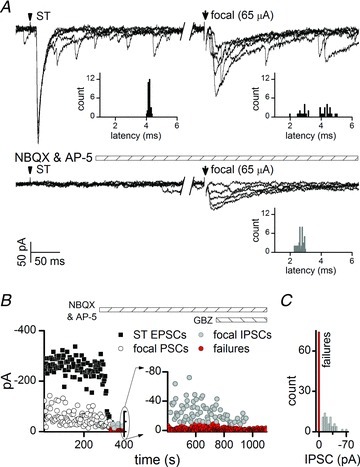
A, in a representative neuron, supra-minimal intensity focal shocks evoked compound events that included components with short and long decay times and variable latencies (focal, right). Glutamate receptor antagonists (2-amino-5-phosphonopentanoic acid (AP-5) 100 μm and 2,3-dioxo-6-nitro-1,2,3,4-tetrahydro-benzo[f]quinoxaline-7-sulphonamide (NBQX) 20 μm) blocked EPSCs, but unmasked a single, focally activated IPSC at fixed, low jitter latency (177 μs, 5 traces, 800 ms break between solitary tract (ST) and focal shocks, artefacts blanked). Note that glutamatergic block failed to alter the recruitment threshold for the IPSC (65 μA). IPSC amplitudes were highly variable and failures occurred frequently (defined as no response after a shock). B, AP-5 and NBQX blocked both ST (filled squares) and focal-EPSCs (open circles), but low jitter IPSCs remained until addition of GABAA blocker, gabazine (GBZ, 3 μm; focal-IPSCs, grey circles; synaptic failures, red circles). C, the amplitude variability of focal-IPSCs was high and failures (zero amplitude, red bar) were common.
Figure 3. Focal shocks near second-order NTS neurons just as often recruited polysynaptic IPSCs as monosynaptic IPSCs.
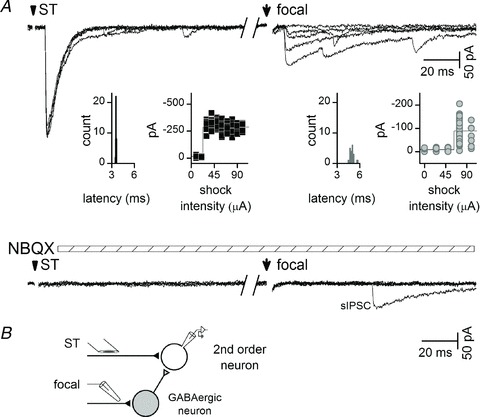
A, in a representative second-order NTS neuron, focal shocks activated high jitter IPSCs with a sharp intensity threshold and no increment in amplitude with higher intensities (focal, right, intensity-recruitment profile insert). Glutamate antagonists (AP-5 100 μm and 2,3-dioxo-6-nitro-1,2,3,4-tetrahydro-benzo[f]quinoxaline-7-sulphonamide (NBQX) 20 μm) blocked solitary tract (ST)-EPSC and focal-IPSCs (note persisting spontaneous IPSC). B, a minimal network representation to account for the evidence suggests that focal shocks activated an excitatory axon that initiated activation of a GABAergic neuron resulting in polysynaptic IPSCs arriving indirectly.
Across neurons with monosynaptic IPSCs (n= 20), focally activated IPSCs (n= 9 with; n= 11 without glutamate block) exhibited jitter values considered monosynaptic, but had an average jitter that was significantly higher than for ST-EPSCs (Fig. 4; P= 0.002, one-way ANOVA). The greater jitter hinted that GABA release properties might be unique. Focal, monosynaptic IPSCs arrived substantially earlier than ST-EPSCs to these same neurons presumably reflecting a much shorter conduction path. Threshold (minimum) focal shock intensities were uniformly similar with no differences across all focal synaptic response types (overall average of 58 ± 6 μA, P= 0.942, one-way ANOVA; see Fig. 1D for classes, mono EPSC, n= 57; poly EPSC, n= 15; mono IPSC, n= 12; poly IPSC, n= 14). Unlike ST-EPSCs, however, monosynaptic focal-IPSCs displayed a nearly threefold greater amplitude variability (Cv) compared with ST-EPSCs (Fig. 4), and IPSCs failed in >25% of trials (Figs 2C and 4). Despite the tightly time-locked synchrony of monosynaptic IPSCs, the elevated amplitude variability, greater jitter and frequent failures suggested that GABA release properties might fundamentally differ from ST glutamate release.
Figure 4. On average (23 second-order NTS neurons), monosynaptic focal-IPSCs had similar latencies and jitters whether isolated by glutamate blockers or not.
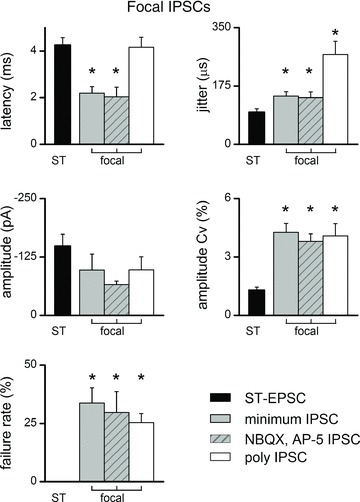
Monosynaptic focal-IPSC jitters were higher than solitary tract (ST)-EPSCs (*P < 0.05, one-way ANOVA). Coefficient of amplitude variation (Cv) and failures for all IPSCs exceeded ST-EPSCs to the same cells (*P < 0.05, one-way ANOVA). Activation threshold intensities (not shown, see Results) were not different across the focal groups. AP-5, 2-amino-5-phosphonopentanoic acid; NBQX, 2,3-dioxo-6-nitro-1,2,3,4-tetrahydro-benzo[f]quinoxaline-7-sulphonamide.
Unitary GABAergic transmission in NTS
The mechanisms underlying GABA release in NTS are poorly understood. The intrinsic characteristics of synaptic transmission can differ substantially between brain areas and neurotransmitters (Zucker & Regehr, 2002). To better define synaptic release of GABA in the NTS, we focally activated monosynaptic IPSCs with trains (five shocks at 25 Hz; Fig. 5A). Calculated IPSC PPRs averaged near 1.0 – i.e. indicating little depression or facilitation and substantially greater PPR than for ST-EPSCs to the same neurons (Fig. 5B). For ST-EPSCs (Fig. 5C), synaptic failures increased with repeated shocks as previously reported (Mifflin & Felder, 1988; Andresen & Yang, 1995). In contrast, failure rates of monosynaptic IPSCs were uniformly high (∼30%; Fig. 5C) and, at 25 Hz, failure rate did not differ by position within the shock train (Fig. 5C). Together, such evidence suggests that the GABA release process functions at lower frequencies and likely relies on a cascade of release machinery that is quite different than for primary afferent glutamate release.
Figure 5. Frequency-dependent depression was absent in monosynaptic focal-IPSCs suggesting unique release characteristics.
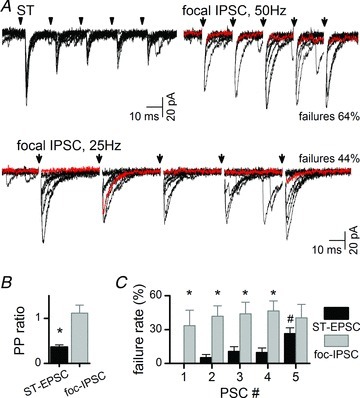
A, in a representative neuron (5 shocks, traces for 10 trials overlaid), solitary tract (ST)-EPSCs (upper) depressed at 50 Hz by the second shock in a train without failures at EPSC1 (upper, shock artefacts blanked), whereas focal-IPSCs (lower, NBQX 20 μm and AP-5 100 μm) often failed (red trace highlights failure within 10 successive traces overlaid). Focal-IPSCs exhibited no frequency-dependent changes in amplitude at 25 Hz. B, on average (n= 5 neurons), the paired-pulse (PP) ratio (PSP2/PSP1) for focal-IPSCs was not different from 1.0, whereas ST-EPSCs on average showed greater depression to 0.36 (P < 0.05, paired t test). C, failure rates were higher for focal-IPSCs compared with ST-EPSCs within these same neurons (*P < 0.05 focal vs. ST-EPSC, two-way ANOVA). # indicates late shock failure rate greater than the first ST-EPSC failure rate (P < 0.05, two-way ANOVA).
To test the GABA release process more directly, we assessed the relationship between extracellular calcium and GABA release probability using mean-variance analysis of IPSC amplitudes (Fig. 6). As expected, decreasing external calcium [Ca2+]o decreased both the mean amplitude as well as the amplitude variance Cv and increased the failure rate – consistent with decreases in the release probability of GABA (Fig. 6A and B). However, increases in [Ca2+]o only modestly augmented both the evoked IPSC amplitudes and their variance across the group (Fig. 6C). Thus, in this supra-physiological range of [Ca2+]o, only the ascending limb (increased release probability) of the GABA release relation could be discerned, and the expected descending limb of the parabolic relation where maximal release occurs was not detected. As a result, the probability of GABA release must be substantially <50% even at 2 mm[Ca2+]o (Fig. 6C). In stark contrast, ST glutamate release consistently averages ∼90% in identical conditions with sharply lower amplitude variance as the ST-EPSC reaches near maximal levels at 2 mm[Ca2+]o (Bailey et al. 2006b; Andresen & Peters, 2008; Peters et al. 2008a). Overall, GABA release at second-order NTS neurons exhibited very low probability release characteristics even at supra-maximal calcium, a finding consistent with a unique release machinery controlling GABA vesicle fusion.
Figure 6. Variance–mean (V–M) analysis of focal IPSC amplitudes indicated surprisingly low calcium sensitivity of evoked GABA release.
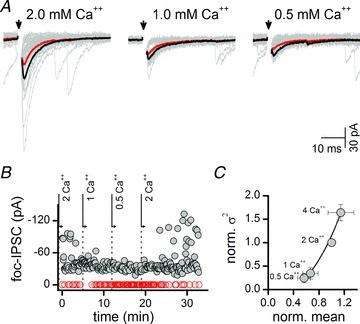
A, focal shocks (arrows) evoked IPSCs that fluctuated in amplitude, and both the mean and variance decreased when external calcium (Ca2+) decreased from the basal levels (2 mm). NBQX (20 μm), AP-5 (100 μm) and the metabotropic glutamate antagonist LY314495 (50 μm) were present throughout to block glutamate responses. Individual trial responses (40 light grey traces) were superimposed on IPSCs averaged excluding failures (black) or averaged IPSC including failures (red) as per V–M analysis. B, same cell as in A, focal-IPSC amplitudes reversibly decreased and failures increased (red circles) with reductions in [Ca2+]o. C, on average across second-order NTS neurons (n= 5), the V–M relation indicated that amplitude and variance monotonically increased for focal-IPSCs with increasing calcium. The result suggests that GABA release was relatively Ca2+ insensitive, and that release probability was <50% at all [Ca2+]o tested. Not that maximal release had not been reached by raising [Ca2+]o.
Convergence of GABAergic transmission
Our approach of minimal intensity focal shocks to evoke unitary GABA inputs to NTS neurons allowed us to assess most directly the characteristics of GABA transmission. To test if larger numbers of direct GABA inputs could be recruited, we utilized higher intensity focal shocks in the presence of non-NMDA antagonist. The glutamate block eliminated polysynaptic GABA responses (i.e. indirect) so that we could examine the response characteristics of GABA fibre ensembles, i.e. compound IPSCs. In some neurons, increasing intensity at the most sensitive focal pipette locations recruited substantially larger and more complicated compound synaptic events (Fig. 7). Under our recording conditions, IPSCs were inward, but most of the early compound synaptic current was contributed by activation of local glutamatergic axons (Fig. 7A). Addition of NBQX and AP-5 eliminated these early currents and only much slower kinetic IPSCs remained. Focusing on neurons with compound IPSCs, incrementing focal shocks recruited a larger current that often had a shorter latency and earlier peak than the minimum intensity focal IPSC (Fig. 7B). The stimulus-recruitment profile indicated that a constant amplitude was found over an interval of intensities before further increments resulted in larger and often earlier peaks in the compound IPSCs – clear evidence of recruitment of additional GABA axons and the development of fused compound IPSCs. Using bursts of focal shocks (five shocks at 25 Hz), we first observed that increasing focal intensity evoked increased IPSC amplitudes and that compound IPSCs exhibited little frequency-dependent depression in common with unitary minimal intensity focal evoked IPSCs (Fig. 7C and D, left). In addition, compound IPSCs showed substantially lower failure rates across each shock position as focal intensity was increased and additional inhibitory inputs were recruited (Fig. 7C and D, right). Thus, co-activation of redundant inputs greatly increased overall transmission reliability, but this redundancy masked the underlying unreliable release process.
Figure 7. Intense focal shocks revealed multiple, convergent inhibitory inputs to some second-order neurons.
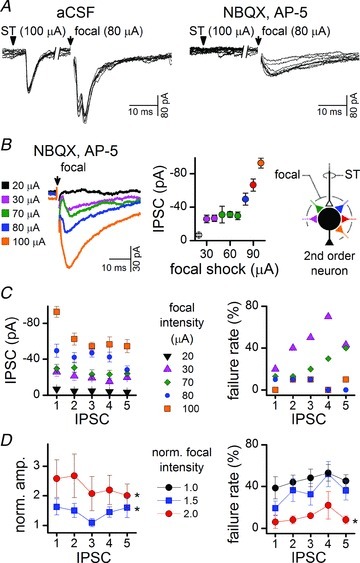
A, solitary tract (ST) shocks (arrow) evoked low jitter EPSCs defining neurons as second order. Focal shocks (arrow) at some locations generated variable and complex synaptic responses with high focal intensities. Early large glutamatergic EPSCs obscured GABAergic transmission and some responses might have been polysynaptic. Application of glutamate ionotropic blockers (2-amino-5-phosphonopentanoic acid (AP-5) 100 μm and 2,3-dioxo-6-nitro-1,2,3,4-tetrahydro-benzo[f]quinoxaline-7-sulphonamide (NBQX) 20 μm) revealed that focal shocks activated slow kinetic IPSCs (8 traces). Note that 800 ms break in time axis between ST and focal shocks has collapsed the true time relationship (stimulus artefacts blanked). B, in the same neuron as represented in A, increasing intensity under glutamate blockade recruited successively larger compound IPSCs. The added GABA axons triggered fused compound IPSCs that had a shorter latency, peaked earlier but had a much longer time course than the minimal focal event at 30 μA (average trace of 10 IPSCs per shock intensity, failures excluded). Note that the stimulus-recruitment profile showed an initial ‘all-or-none’ interval above the threshold (30–40 μA) over which amplitude did not increase. As the plots register the peak amplitude of the compound event, they represent the collective response and not single components. At higher intensities (>50 μA), the peak amplitude increased further and/or failure rate decreased, indicating additional inhibitory axons contributed to the summed, compound response. A diagrammatic representation of inhibitory inputs this neuron was shown to receive. C, in the same neuron as for A and B, conventional assays of frequency-dependent responses to bursts (5 shocks at 25 Hz) of focal stimuli evoked IPSCs with similar amplitudes at each position (1–5). The failure rate of IPSCs decreased markedly with stronger shocks. Note that failures decreased dramatically from 30 to 70 μA shocks despite a relatively small change in amplitude over the same range. D, across the group (n= 9), focal shocks 1.5× and 2× the minimal intensity resulted in significantly larger IPSC amplitudes (*P < 0.05, minimal shock IPSC amplitudes, normalized to 1 here, two-way ANOVA, failures excluded). Similarly, focal shocks twice the intensity of minimal threshold shocks significantly decreased failure rate as more inhibitory axons were recruited (*P < 0.05, two-way ANOVA minimal shock IPSC failure rate, black filled circles). aCSF, artificial cerebrospinal fluid.
Discussion
Cranial visceral afferents communicate with NTS neurons through a glutamate vesicular release mechanism with uniformly high probability. This glutamate transmission initiates activation of central regulatory pathways with a very high safety factor with reliable vesicle release and activation of postsynaptic action potentials (Andresen & Kunze, 1994; Andresen et al. 2004). GABAergic transmission provides an inhibitory influence balancing excitation but has been more difficult to study. Absent well defined fibre tracts, we turned to focal shocks to assess the fundamental characteristics of inhibitory transmission in the NTS. Minimal intensity focal shocks activated single local axons that converged onto second-order neurons detected as IPSCs. Activation of elemental inhibitory transmission to second-order neurons suggests the following new findings: (1) inhibitory axons to single cells were uncommonly encountered (9% of sites) in medial NTS; (2) most focal sites near cells were unexcitable or contained only excitatory axons that either directly or indirectly contacted second-order NTS neurons; (3) unitary GABAergic axons supported an intrinsically low probability of release of modest amplitude but with substantial failure rates (∼30%) and no frequency-dependent facilitation; (4) intense shocks at some focal sites recruited multiple inhibitory axons that converged on single neurons to effectively decrease IPSC transmission failures in the form of fused compound IPSCs. Combined, these findings reveal a synaptic organization for NTS second-order neurons that is dominated by powerful ST excitatory fibres with less effective GABAergic contacts. These results provide new insight into the contrasting amino acid release mechanisms and axon patterning in medial NTS. Whereas excitation occurs through highly effective, unfailing unitary transmission from ST primary afferents, in contrast inhibitory transmission is remarkably weak and unreliable but becomes more dependable with convergent activation of like fibres.
Hierarchy – primary excitatory afferent transmission drives inhibitory input in the NTS
Although tightly synchronized extracellular spikes indicated excitatory responses, only with the advent of intracellular recordings in the NTS could the later arriving, polysynaptic inhibitory responses to peripheral afferent stimulation be identified clearly (Miura, 1975). Second-order NTS neurons in vivo displayed primarily simple, single-latency EPSPs with surprisingly limited direct convergence either from within or across multiple nerve trunks (Ciriello & Calaresu, 1981; Donoghue et al. 1981, 1985; Mifflin, 1996). However, in approximately 30–50% of neurons, IPSPs were triggered by afferent ST activation and these were loosely synchronized, a finding consistent with an indirect pathway triggered by afferent activation (Mifflin, 1996). Most often IPSCs arrived after initial EPSPs, an EPSC–IPSC sequence, which is also consistent with local presence of GABAergic interneurons within or nearby the NTS neuron (Mifflin, 1996). In isolated brain slices, ST activation sometimes triggered similar EPSP–IPSP sequences (Andresen & Yang, 1995), and transgenic mouse models confirm the substantial numbers of local GABAergic neurons are monosynaptically contacted by ST primary afferent (Bailey et al. 2008). Interestingly, the substantial numbers of fluorescently labelled GAD-67 caudal NTS neurons in transgenic mice (Bailey et al. 2008) motivated us to use paired recordings to study connections from inhibitory neurons to nearby non-fluorescent neurons, but we failed to detect such connections (unpublished results). This effort suggested that functioning local synaptic connections were less likely than we might assume simply from the distribution of GABA-synthesizing neurons. In studies of spontaneous transmission in rats, miniature EPSC rates of glutamate release were generally 2- to 10-fold higher than the miniature IPSC rates of GABA release (in TTX) in identified second-order NTS neurons (Jin et al. 2004; Zhang & Mifflin, 2007; McDougall et al. 2008; Peters et al. 2008b; Fernandes et al. 2011). Together, these previous reports suggested that excitatory events at second-order NTS neurons were clearly more common than evoked inhibitory transmission.
Minimal focal shocks intercept single axons
To study inhibitory transmission directly, a means of evoking monosynaptic responses needed to be established. Indirect, polysynaptic IPSCs or compound IPSCs reveal little about the fundamental aspects of evoked inhibitory transmission. Our approach to test the basis of inhibitory transmission in medial NTS was grounded in two simple premises that: (1) axons in direct contact with a neuron would be most likely to be encountered close to the neuron; (2) weak electrical shocks placed close to an axon could activate that single axon selectively without activating other afferents to the same neuron. Our results suggest that both premises can be supported. Careful gradations in focal shock intensity allowed recruitment of very discrete responses – both EPSCs and IPSCs – and these titrations of intensity uncovered the activation of individual axons with higher intensities added to substantial numbers of mixed inputs converging on individual NTS neurons. This discrete recruitment protocol together with quantitative analysis of the latencies and amplitudes of these synaptic events distinguished directly coupled events (i.e. monosynaptic) from polysynaptic events as well as allowed an orderly dissection of compound events. This degree of control of focally activated events also provided discrete tests of potential interactions between focally activated GABA release and ST transmission to the same neuron – tests that are not possible in vivo or using distal ST shocks alone. All threshold responses were consistent with dependence on activation of single axons. Thus, augmentation of near minimal intensities did not change synaptic event amplitudes, latency or dynamics over a range of stimulus shock magnitudes. Moving the focal pipette to a different location recruited new inputs with different characteristics consistent with activation of different afferent axons depending on the location of the stimulus shock. From such results we conclude that minimal focal shocks detected a limited network of inputs converging onto single neurons. It is possible that focal shocks may have activated only limited branches from a common GABAergic source axon to the recorded neuron. However, the lack of recruitment or change in event dynamic with increased shock intensity and the equivalent amplitudes of focally activated monosynaptic IPSCs compared with polysynaptic IPSCs (e.g. activated through action potentials driving the GABAergic axon) would argue that focal shocks activated whole GABAergic inputs. From this perspective, monosynaptic responses had invariant latencies (jitters of <200 μs) that were not different whether the axon was glutamatergic or GABAergic. To our surprise, minimal focal shocks often activated axons that were indirectly connected to the recorded neuron – i.e. their high jitter indicated polysynaptic responses (EPSCs or IPSCs). However, even polysynaptic IPSCs had sharp, all-or-nothing thresholds. Thus, all results with minimal shocks were consistent with activation of a single axon along a pathway (mono- or polysynaptic) that ultimately contacted the recorded NTS neuron.
Focal shocks were highly localized
Moving the stimulus pipette to different locations suggested that effective shocks were highly localized with minimal to moderate intensities. Thus, moving the stimulating pipette even 25 μm resulted in either the loss of evoked responses and/or recruitment of new events with different thresholds and waveform characteristics. This technical approach was well suited to discrete recruitment of single axons, and allowed assessment of crisp latencies and jitters that is not possible with other methodologies such as release of caged glutamate or optogenetic activation (Katz & Dalva, 1994; Brill & Huguenard, 2009). The focused nature of the stimuli suggested that the pipette tip needed to be very close to an axon for successful activation. However, even at >10× higher intensities, many focal sites (23%) were unresponsive, whereas re-location of the focal pipette to an adjacent site successfully recruited synaptic inputs. The findings suggest highly heterogeneous distribution of axons at relatively low spatial density. However, our site sampling with focal electrodes was not comprehensive as access was obstructed by the recording pipette and this means that our studies offer no cumulative estimate of the total number of inhibitory inputs to these second-order neurons. Our results reinforce the conclusion that NTS neurons tend to be relatively synaptically isolated from near neighbours. This present finding is consistent with results from paired recordings of medial NTS neurons that indicated that two-thirds of these second-order neurons received only one ST-driven input despite intense shocks, and no synaptic interconnections or common ST inputs were found between close neighbour NTS neurons (McDougall et al. 2009). During glutamate block, high-intensity shocks at some locations recruited multiple and complex IPSCs that likely reflected local positioning of multiple convergent axons at some locations.
Our application of focal pipette stimulation used a remote current return path that functioned to attenuate stimulus shock intensity away from the pipette tip and limit the recruitment of fibres distant from the pipette tip. This stimulus configuration likely affected the recruitment characteristics and contrasts substantially from other stimulation electrode approaches. We used a concentric bipolar metal electrode for ST stimulation, and this electrode has a large contact area (200 μm diameter) and a configuration that passes current from the central pole to the outer annulus. In recruitment of ST axons, the concentric electrode effectively recruits one or more ST axons converging onto single NTS neurons (Doyle & Andresen, 2001; Andresen & Peters, 2008; McDougall et al. 2009; Peters et al. 2011) with the same characteristics as a pair of tungsten wires straddling the ST (Mendelowitz et al. 1995). The current spread pattern, selectivity and recruitment characteristics were quite different with a metal electrode pair placed close to the recorded neurons (Brooks et al. 1992; Aylwin et al. 1997; Kawai & Senba, 2000). Activation of compound IPSCs reflects multiple inputs and would have obscured key aspects of the intrinsic mechanisms of GABA transmission that were so apparent with minimal focal shocks used here, such as failure rates.
Low density of inhibitory axons to second-order NTS neurons
Minimal focal stimuli intercepted GABAergic axons to second-order NTS neurons relatively rarely (<10% of focal sites). High-intensity shocks in some locations recruited additional IPSCs above minimal thresholds presumably by current spread. Because IPSCs were often evident within the experimental context of mixed compound events with EPSCs, we had more success recruiting direct IPSCs under pharmacological isolation and studied most GABA release under non-NMDA blockers. We detected no electrical threshold difference between IPSCs and EPSCs so that electrode proximity to the convergent axon was likely the most important factor. The overall data were consistent with a particularly diffuse network of GABA inputs to any single second-order neuron in medial NTS. Thus, the universal presence of spontaneous IPSCs (McDougall et al. 2008) must arise from a limited number of GABAergic axons with a lower number of total GABA release sites on single second-order neurons compared with glutamatergic terminals. We concentrated on the medial subnucleus, and other regions of NTS may differ in this respect.
Low-fidelity GABAergic transmission
Focally evoked IPSCs failed frequently even with the very first shock – a rested state in which the readily releasable pool should be primed and maximal responsiveness favoured (Neher & Sakaba, 2008). Under similar conditions, ST fibres almost never failed (<0.1%). Furthermore, the lack of influence of frequent activation (shock bursts) suggested a surprising underlying GABA neurotransmission process. Tests indicated that GABA release was quite insensitive to external Ca2+ and the low slope of the variance–mean relation was consistent with a low release probability. Although similar release characteristics exist in other CNS regions (Nusser, 2002; Kravchenko et al. 2006), NTS GABA release starkly contrasts to the highly reliable ST glutamate process at these same neurons (Bailey et al. 2006b; Peters et al. 2008a). Most inhibitory neurons in NTS are second order (Bailey et al. 2008), and our findings indicate they have a limited GABA terminal distribution to other individual second-order neurons. Secondly, these inhibitory second-order neurons likely exhibit a uniform, low-pass filtering in translating primary ST sensory signals into IPSCs postsynaptically. An important factor in the range of inhibitory transmission is the phenotype of the postsynaptic neuron that is likely diverse in NTS and may include expression of different postsynaptic GABA receptors (Kasparov et al. 2001; Saha et al. 2001; Milligan et al. 2004; Herman et al. 2012). Though we do not know the source of inhibitory axons activated by focal shocks, the uniformity of evoked IPSC characteristics suggests that GABA transmission from local (Bailey et al. 2008) as well as nearby (Stornetta & Guyenet, 1999) or descending inhibitory projections into NTS (Saha et al. 2000) may be similarly unreliable (Jin & Andresen, 2011).
The relative numbers of convergent axons were remarkably low relative to the spatial/focal recruitment observed in other central neurons. In many regions outside the NTS, excitatory inputs often consist of low-release axons that augment with recruitment of multiple convergent inputs to create nearly continuously incrementing amplitudes in compound synaptic currents (Clark & Cull-Candy, 2002). In the NTS, the low density of GABA axons raises fundamental questions about pathway organization of the NTS and the relative numbers of inhibitory inputs contacting second-order neurons that this study is among the first to assess. All evidence suggests that the medial subnucleus of the NTS appears to tilt substantially to predominance by afferent excitatory inputs in excess of inhibitory inputs. GABA transmission provides important targets for anaesthetics on autonomic control mechanisms and an example of how this balance can shift (Lee et al. 2002; McDougall et al. 2008; Peters et al. 2008b). Thus, the ratio between excitatory and inhibitory recruitment within the NTS indicates a network arrangement in which second-order neurons are primed for signal relay at a high safety factor.
Acknowledgments
This work was supported by grants from the National Institutes of Health, HL-41119 (M.C.A.), HL-105703 (M.C.A.), and National Health and Medical Research Council of Australia, Overseas Training C J Martin Fellowship no. 400405 (S.J.M.). The content is solely the responsibility of the authors, and does not necessarily represent the official views of the National Heart, Lung and Blood Institute or the NIH.
Glossary
- AP-5
2-amino-5-phosphonopentanoic acid
- GBZ
gabazine
- NBQX
2,3-dioxo-6-nitro-1,2,3,4- tetrahydro-benzo[f]quinoxaline-7-sulphonamide
- NTS
nucleus of the solitary tract
- PPR
paired-pulse ratio
- ST
solitary tract
Author contributions
The study was designed by S.J.M. and M.C.A. Experiments were conducted at the Oregon Health and Science University by S.J.M. Data analysis was conducted by S.J.M. The paper was written by S.J.M. and M.C.A. All authors approved the final version of the manuscript. Conflicts of interest: none.
References
- Andresen MC, Doyle MW, Bailey TW, Jin Y-H. Differentiation of autonomic reflex control begins with cellular mechanisms at the first synapse within the nucleus tractus solitarius. Braz J Med Biol Res. 2004;37:549–558. doi: 10.1590/s0100-879x2004000400012. [DOI] [PubMed] [Google Scholar]
- Andresen MC, Kunze DL. Nucleus tractus solitarius: gateway to neural circulatory control. Annu Rev Physiol. 1994;56:93–116. doi: 10.1146/annurev.ph.56.030194.000521. [DOI] [PubMed] [Google Scholar]
- Andresen MC, Paton JF. The nucleus of the solitary tract: processing information from viscerosensory afferents. In: Llewellyn-Smith IJ, Verberne AJ, editors. Central Regulation of Autonomic Functions. London: Oxford; 2011. pp. 23–46. [Google Scholar]
- Andresen MC, Peters JH. Comparison of baroreceptive to other afferent synaptic transmission to the solitary tract nucleus. Am J Physiol Heart Circ Physiol. 2008;295:H2032–H2042. doi: 10.1152/ajpheart.00568.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andresen MC, Yang M. Dynamics of sensory afferent synaptic transmission in aortic baroreceptor regions of nucleus tractus solitarius. J Neurophysiol. 1995;74:1518–1528. doi: 10.1152/jn.1995.74.4.1518. [DOI] [PubMed] [Google Scholar]
- Austgen JR, Fong AY, Foley CM, Mueller PJ, Kline DD, Heesch CM, Hasser EM. Expression of Group I metabotropic glutamate receptors on phenotypically different cells within the nucleus of the solitary tract in the rat. Neuroscience. 2009;159:701–716. doi: 10.1016/j.neuroscience.2008.09.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aylwin ML, Horowitz JM, Bonham AC. NMDA receptors contribute to primary visceral afferent transmission in the nucleus of the solitary tract. J Neurophysiol. 1997;77:2539–2548. doi: 10.1152/jn.1997.77.5.2539. [DOI] [PubMed] [Google Scholar]
- Bailey TW, Appleyard SM, Jin YH, Andresen MC. Organization and properties of GABAergic neurons in solitary tract nucleus (NTS) J Neurophysiol. 2008;99:1712–1722. doi: 10.1152/jn.00038.2008. [DOI] [PubMed] [Google Scholar]
- Bailey TW, Hermes SM, Andresen MC, Aicher SA. Cranial visceral afferent pathways through the nucleus of the solitary tract to caudal ventrolateral medulla or paraventricular hypothalamus: target-specific synaptic reliability and convergence patterns. J Neurosci. 2006a;26:11893–11902. doi: 10.1523/JNEUROSCI.2044-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TW, Jin Y-H, Doyle MW, Andresen MC. Vanilloid sensitive afferents activate neurons with prominent A-type potassium currents in nucleus tractus solitarius. J Neurosci. 2002;22:8230–8237. doi: 10.1523/JNEUROSCI.22-18-08230.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TW, Jin Y-H, Doyle MW, Smith SM, Andresen MC. Vasopressin inhibits glutamate release via two distinct modes in the brainstem. J Neurosci. 2006b;26:6131–6142. doi: 10.1523/JNEUROSCI.5176-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brill J, Huguenard JR. Robust short-latency perisomatic inhibition onto neocortical pyramidal cells detected by laser-scanning photostimulation. J Neurosci. 2009;29:7413–7423. doi: 10.1523/JNEUROSCI.6098-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks PA, Glaum SR, Miller RJ, Spyer KM. The actions of baclofen on neurones and synaptic transmission in the nucleus tractus solitarii of the rat in vitro. J Physiol. 1992;457:115–129. doi: 10.1113/jphysiol.1992.sp019367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan RKW, Sawchenko PE. Organization and transmitter specificity of medullary neurons activated by sustained hypertension: implications for understanding baroreceptor reflex circuitry. J Neurosci. 1998;18:371–387. doi: 10.1523/JNEUROSCI.18-01-00371.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciriello J, Calaresu FR. Projections from buffer nerves to the nucleus of the solitary tract: an anatomical and electrophysiological study in the cat. J Auton Nerv Syst. 1981;3:299–310. doi: 10.1016/0165-1838(81)90071-0. [DOI] [PubMed] [Google Scholar]
- Clark BA, Cull-Candy SG. Activity-dependent recruitment of extrasynaptic NMDA receptor activation at an AMPA receptor-only synapse. J Neurosci. 2002;22:4428–4436. doi: 10.1523/JNEUROSCI.22-11-04428.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donoghue S, Felder RB, Gilbey MP, Jordan D, Spyer KM. Post-synaptic activity evoked in the nucleus tractus solitarius by carotid sinus and aortic nerve afferents in the cat. J Physiol. 1985;360:261–273. doi: 10.1113/jphysiol.1985.sp015616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donoghue S, Fox RE, Kidd C, Koley BN. The distribution in the cat brain stem of neurones activated by vagal non-myelinated fibres from the heart and lungs. Q J Exp Physiol Cogn Med Sci. 1981;66:391–404. doi: 10.1113/expphysiol.1981.sp002582. [DOI] [PubMed] [Google Scholar]
- Doyle MW, Andresen MC. Reliability of monosynaptic transmission in brain stem neurons in vitro. J Neurophysiol. 2001;85:2213–2223. doi: 10.1152/jn.2001.85.5.2213. [DOI] [PubMed] [Google Scholar]
- Doyle MW, Bailey TW, Jin Y-H, Appleyard SM, Low MJ, Andresen MC. Strategies for cellular identification in nucleus tractus solitarius slices. J Neurosci Methods. 2004;37:37–48. doi: 10.1016/j.jneumeth.2004.02.007. [DOI] [PubMed] [Google Scholar]
- Fernandes LG, Jin YH, Andresen MC. Heterosynaptic crosstalk: GABA-glutamate metabotropic receptors interactively control glutamate release in solitary tract nucleus. Neuroscience. 2011;174:1–9. doi: 10.1016/j.neuroscience.2010.11.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herman MA, Gillis RA, Vicini S, Dretchen KL, Sahibzada N. Tonic GABAA receptor conductance in medial subnucleus of the tractus solitarius neurons is inhibited by activation of Mu-opioid receptors. J Neurophysiol. 2012;107:1022–1031. doi: 10.1152/jn.00853.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin YH, Andresen MC. GABAB restrains release from singly-evoked GABA terminals. Neuroscience. 2011;193:54–62. doi: 10.1016/j.neuroscience.2011.07.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Y-H, Bailey TW, Andresen MC. Cranial afferent glutamate heterosynaptically modulates GABA release onto second order neurons via distinctly segregated mGluRs. J Neurosci. 2004;24:9332–9340. doi: 10.1523/JNEUROSCI.1991-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasparov S, Davies KA, Patel UA, Boscan P, Garret M, Paton JFR. GABA(A) receptor epsilon-subunit may confer benzodiazepine insensitivity to the caudal aspect of the nucleus tractus solitarii of the rat. J Physiol. 2001;536:785–796. doi: 10.1111/j.1469-7793.2001.00785.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz LC, Dalva MB. Scanning laser photostimulation: a new approach for analyzing brain circuits. J Neurosci Methods. 1994;54:205–218. doi: 10.1016/0165-0270(94)90194-5. [DOI] [PubMed] [Google Scholar]
- Kawai Y, Senba E. Organization of excitatory and inhibitory local networks in the caudal nucleus of tractus solitarius of rats revealed in in vitro slice preparation. J Comp Neurol. 1996;373:309–321. doi: 10.1002/(SICI)1096-9861(19960923)373:3<309::AID-CNE1>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- Kawai Y, Senba E. Electrophysiological and morphological characteristics of nucleus tractus solitarii neurons projecting to the ventrolateral medulla. Brain Res. 2000;877:374–378. doi: 10.1016/s0006-8993(00)02701-3. [DOI] [PubMed] [Google Scholar]
- Kravchenko MO, Moskalyuk AO, Fedulova SA, Veselovsky NS. Calcium-dependent changes of paired-pulse modulation at single GABAergic synapses. Neurosci Lett. 2006;395:133–137. doi: 10.1016/j.neulet.2005.10.070. [DOI] [PubMed] [Google Scholar]
- Kubin L, Alheid GF, Zuperku EJ, McCrimmon DR. Central pathways of pulmonary and lower airway vagal afferents. J Appl Physiol. 2006;101:618–627. doi: 10.1152/japplphysiol.00252.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunze DL. Reflex discharge patterns of cardiac vagal efferent fibres. J Physiol. 1972;222:1–15. doi: 10.1113/jphysiol.1972.sp009784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JS, Andresen MC, Morrow D, Chang KSK. Isoflurane depresses baroreflex control of heart rate in decerebrate rats. Anesthesiology. 2002;96:1214–1222. doi: 10.1097/00000542-200205000-00026. [DOI] [PubMed] [Google Scholar]
- Liu Z, Chen CY, Bonham AC. Frequency limits on aortic baroreceptor input to nucleus tractus solitarii. Am J Physiol Heart Circ Physiol. 2000;278:H577–H585. doi: 10.1152/ajpheart.2000.278.2.H577. [DOI] [PubMed] [Google Scholar]
- Loewy AD. Central autonomic pathways. In: Loewy AD, Spyer KM, editors. Central Regulation of Autonomic Functions. New York: Oxford University Press; 1990. pp. 88–103. [Google Scholar]
- McDougall SJ, Bailey TW, Mendelowitz D, Andresen MC. Propofol enhances both tonic and phasic inhibitory currents in second-order neurons of the solitary tract nucleus (NTS) Neuropharmacology. 2008;54:552–563. doi: 10.1016/j.neuropharm.2007.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDougall SJ, Peters JH, Andresen MC. Convergence of cranial visceral afferents within the solitary tract nucleus. J Neurosci. 2009;29:12886–12895. doi: 10.1523/JNEUROSCI.3491-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendelowitz D. Advances in parasympathetic control of heart rate and cardiac function. News Physiol Sci. 1999;14:155–161. doi: 10.1152/physiologyonline.1999.14.4.155. [DOI] [PubMed] [Google Scholar]
- Mendelowitz D, Yang M, Reynolds PJ, Andresen MC. Heterogeneous functional expression of calcium channels at sensory and synaptic regions in nodose neurons. J Neurophysiol. 1995;73:872–875. doi: 10.1152/jn.1995.73.2.872. [DOI] [PubMed] [Google Scholar]
- Michelini LC, Stern JE. Exercise-induced neuronal plasticity in central autonomic networks: role in cardiovascular control. Exp Physiol. 2009;94:947–960. doi: 10.1113/expphysiol.2009.047449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mifflin SW. Convergent carotid sinus nerve and superior laryngeal nerve afferent inputs to neurons in the NTS. Am J Physiol Regul Integr Comp Physiol. 1996;271:R870–R880. doi: 10.1152/ajpregu.1996.271.4.R870. [DOI] [PubMed] [Google Scholar]
- Mifflin SW, Felder RB. An intracellular study of time-dependent cardiovascular afferent interactions in nucleus tractus solitarius. J Neurophysiol. 1988;59:1798–1813. doi: 10.1152/jn.1988.59.6.1798. [DOI] [PubMed] [Google Scholar]
- Mifflin SW, Spyer KM, Withington-Wray DJ. Baroreceptor inputs to the nucleus tractus solitarius in the cat: modulation by the hypothalamus. J Physiol. 1988;399:369–387. doi: 10.1113/jphysiol.1988.sp017086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan CJ, Buckley NJ, Garret M, Deuchars J, Deuchars SA. Evidence for inhibition mediated by coassembly of GABAA and GABAC receptor subunits in native central neurons. J Neurosci. 2004;24:9241–9250. doi: 10.1523/JNEUROSCI.1979-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miura M. Postsynaptic potentials recorded from nucleus of the solitary tract and its subjacent reticular formation elicited by stimulation of the carotid sinus nerve. Brain Res. 1975;100:437–440. doi: 10.1016/0006-8993(75)90497-7. [DOI] [PubMed] [Google Scholar]
- Neher E, Sakaba T. Multiple roles of calcium ions in the regulation of neurotransmitter release. Neuron. 2008;59:861–872. doi: 10.1016/j.neuron.2008.08.019. [DOI] [PubMed] [Google Scholar]
- Nusser Z. Release-independent short-term facilitation at GABAergic synapses in the olfactory bulb. Neuropharmacology. 2002;43:573–583. doi: 10.1016/s0028-3908(02)00158-2. [DOI] [PubMed] [Google Scholar]
- Peters JH, McDougall SJ, Fawley JA, Andresen MC. TRPV1 marks synaptic segregation of multiple convergent afferents at the rat medial solitary tract nucleus. PLoS One. 2011;6:e25015. doi: 10.1371/journal.pone.0025015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters JH, McDougall SJ, Kellett DO, Jordan D, Llewellyn-Smith IJ, Andresen MC. Oxytocin enhances cranial visceral afferent synaptic transmission to the solitary tract nucleus. J Neurosci. 2008a;28:11731–11740. doi: 10.1523/JNEUROSCI.3419-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters JH, McDougall SJ, Mendelowitz D, Koop DR, Andresen MC. Isoflurane differentially modulates inhibitory and excitatory synaptic transmission to the solitary tract nucleus. Anesthesiology. 2008b;108:675–683. doi: 10.1097/ALN.0b013e318167af9a. [DOI] [PubMed] [Google Scholar]
- Saha S, Batten TF, Henderson Z. A GABAergic projection from the central nucleus of the amygdala to the nucleus of the solitary tract: a combined anterograde tracing and electron microscopic immunohistochemical study. Neuroscience. 2000;99:613–626. doi: 10.1016/s0306-4522(00)00240-2. [DOI] [PubMed] [Google Scholar]
- Saha S, Sieghart W, Fritschy JM, McWilliam PN, Batten TF. Gamma-aminobutyric acid receptor (GABA(A)) subunits in rat nucleus tractus solitarii (NTS) revealed by polymerase chain reaction (PCR) and immunohistochemistry. Mol Cell Neurosci. 2001;17:241–257. doi: 10.1006/mcne.2000.0919. [DOI] [PubMed] [Google Scholar]
- Stornetta RL, Guyenet PG. Distribution of glutamic acid decarboxylase mRNA-containing neurons in rat medulla projecting to thoracic spinal cord in relation to monoaminergic brainstem neurons. J Comp Neurol. 1999;407:367–380. [PubMed] [Google Scholar]
- Travagli RA, Hermann GE, Browning KN, Rogers RC. Brainstem circuits regulating gastric function. Annu Rev Physiol. 2006;68:279–305. doi: 10.1146/annurev.physiol.68.040504.094635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Mifflin SW. Modulation of synaptic transmission to second-order peripheral chemoreceptor neurons in caudal nucleus tractus solitarius by {alpha}1-adrenoreceptors. J Pharmacol Exp Ther. 2007;320:670–677. doi: 10.1124/jpet.106.114033. [DOI] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG. Short-term synaptic plasticity. Annu Rev Physiol. 2002;64:355–405. doi: 10.1146/annurev.physiol.64.092501.114547. [DOI] [PubMed] [Google Scholar]