

Abstract
Huntington's disease (HD) is a heritable neurodegenerative disorder, with heart disease implicated as one major cause of death. While the responsible mechanism remains unknown, autonomic nervous system (ANS) dysfunction may play a role. We studied the cardiac phenotype in R6/1 transgenic mice at early (3 months old) and advanced (7 months old) stages of HD. While exhibiting a modest reduction in cardiomyocyte diameter, R6/1 mice had preserved baseline cardiac function. Conscious ECG telemetry revealed the absence of 24-h variation of heart rate (HR), and higher HR levels than wild-type littermates in young but not older R6/1 mice. Older R6/1 mice had increased plasma level of noradrenaline (NA), which was associated with reduced cardiac NA content. R6/1 mice also had unstable R–R intervals that were reversed following atropine treatment, suggesting parasympathetic nervous activation, and developed brady- and tachyarrhythmias, including paroxysmal atrial fibrillation and sudden death. c-Fos immunohistochemistry revealed greater numbers of active neurons in ANS-regulatory regions of R6/1 brains. Collectively, R6/1 mice exhibit profound ANS-cardiac dysfunction involving both sympathetic and parasympathetic limbs, that may be related to altered central autonomic pathways and lead to cardiac arrhythmias and sudden death.
Key points
Heart disease has been attributed as a major cause of death in patients with Huntington's disease (HD). While little is known about cardiac complication(s) in HD, dysfunction of the autonomic nervous system (ANS) may play a role.
Using a mouse model of HD, we demonstrated that even from an early HD phase, there was enhanced sympathetic and parasympathetic nervous activities, leading to an unstable heart beat, various arrhythmias and, ultimately, sudden arrhythmic death.
Greater numbers of active neurons were found in brain regions important for autonomic regulation in HD mice, suggesting a centrally mediated mechanism that was underlying the ANS-cardiac dysfunction.
Our findings provide insight into a likely neurocardiac cause of death in HD, and warrant further clinical investigation.
Introduction
Huntington's disease (HD) is a neurodegenerative disorder caused by a CAG repeat expansion within the huntingtin gene, resulting in an abnormal elongation of the poly-glutamine (poly-Q) tract of the huntingtin protein. The pathological consequences include neuronal dysfunction and formation of protein aggregates containing ubiquitinated forms of the mutant protein (Bates, 2005). Patients with HD progressively present with symptoms of clinical psychiatric disorders, cognitive deficits, dementia and physical disability (Munoz-Sanjuan & Bates, 2011). There is no cure or effective treatment for HD, and death typically occurs 15–20 years after the onset of HD motor symptoms.
Recent research on the abnormalities of peripheral organs or tissues in HD has extended our understanding of HD pathophysiology and provided clues for the design of new reparative therapies (Sassone et al. 2009; van der Burg et al. 2009). While HD is primarily a disease of the CNS, early epidemiological reviews of mortality of patients with HD indicated pneumonia (about 40%) and heart disease (about 30%) as leading causes of death (Chiu & Alexander, 1982; Lanska et al. 1988; Sorensen & Fenger, 1992). The nature of cardiac abnormalities in patients with HD remains completely unknown. Aberrant activity of the autonomic nervous system (ANS), specifically sympathetic nervous activation together with parasympathetic withdrawal, has been reported in patients with HD by several groups based on heart rate variability (HRV) as a marker of ANS function (Den Heijer et al. 1988; Sharma et al. 1999; Andrich et al. 2002; Kobal et al. 2004; Bar et al. 2008). These studies, however, did not examine potential cardiac consequences of ANS dysfunction, despite the fact that strong evidence exists for a causal role of ANS dysfunction and onset of arrhythmias or sudden cardiac death (Du et al. 1999; Billman, 2006; Chen et al. 2007).
R6 mice were the first models of HD that contained the 5′-end of the human HD gene and consequently expressed mutant forms of the protein (Mangiarini et al. 1996; Bates et al. 1997). Two transgenic lines, R6/1 and R6/2, have been commonly used for HD research (Mangiarini et al. 1996; Bates et al. 1997). Both transgenic lines develop cognitive deficits followed by motor disorders that closely model HD, as well as progressive dysfunction of the cerebral cortex and striatum, and molecular abnormalities (Mangiarini et al. 1996; Gil & Rego, 2009). Unlike R6/2 mice that develop motor symptoms at about 2 months old and survive up to 4 months old, the R6/1 mice develop adult-onset depressive-like abnormalities and cognitive deficits by 3 months old and motor symptoms at about 4 months old, and have a lifespan of 10 months (Mangiarini et al. 1996; Gil & Rego, 2009; Pang et al. 2009). In the current study, we have examined the neurocardiac phenotype in R6/1 mice from 3 to 7 months old, covering early to advanced HD phases.
Methods
Ethical approval
All experimental procedures were approved by the local Alfred Medical Research and Education Precinct Animal Ethics Committee, and were in accordance with the Australian Government National Health and Medical Research Council's Code of Practice for the Care and Use of Animals for Scientific Purposes (7th edn, 2004).
Animals
Male R6/1 mice (CAG repeat length of 134–138) and wild-type (WT) littermates of CBA×C57Bl/6 background were used. Animals were housed together with three to six per cage (unless otherwise specified), and maintained on a 12:12 h light–dark cycle with water and food at libitum.
Assessment of cardiac function and histology
Cardiac function was assessed using echocardiography and micromanometry (Du et al. 2000; Pretorius et al. 2009). Mice were maintained under light isoflurane anaesthesia (∼1.7%) for non-invasive serial echocardiography tests. Left ventricular dimension at diastole (LVDd), fractional shortening (FS) and heart rate (HR) were determined. Mice fully recovered after removal of the anaesthesia at the end of the test.
For micromanometry experiments, mice were anaesthetised with isoflurane (∼2–3%). Surgical anaesthesia depth was confirmed with the absence of pedal reflex. Using a 1.4F Millar catheter, systolic and diastolic aortic pressures (SAP, DAP), LV systolic pressure (LVSP), LV end-diastolic pressure (LVEDP), and the maximal rates of rise or fall of LV pressures (dP/dtmax or dP/dtmin) were determined.
At the end of the micromanometry experiment, isoflurane was increased to 4%, and the depth of anaesthesia was confirmed with the absence of pedal reflex. The chest cavity was cut open and the heart removed. Wet weights of atria and ventricles and body weight were obtained, and the tibial length was measured.
LVs were either frozen for biochemical assays or fixed in 10% formalin in phosphate-buffered saline, and tissue sections with Masson's trichrome staining were prepared. Diameters of about 40 cardiomyocytes per LV were measured in a blinded fashion, and the average was calculated.
Telemetry ECG recording
Mice were anaesthetised with isoflurane (∼2–3%), and carprofen (5 mg kg−1 s.c.) was administered for analgesia. Surgical anaesthesia depth was confirmed with the absence of pedal reflex. The body of the ETA-F10 ECG radio telemetry transmitter [Data Sciences International (DSI), St Paul, MN, USA] was positioned in the abdominal cavity, and the two biopotential leads were anchored subcutaneously on the lower left of the chest wall and over the right pectoral muscle, respectively (Pretorius et al. 2009). Telemetry probes were implanted in both young and older mice (3 and 6 months old, respectively). Following a 2 week recovery period, telemetry transmitters were turned on and mice were housed individually with cages placed on separate receiver pads (model RPC-1, DSI), allowing simultaneous recording of ECG and locomotor activity. Signals from the receiver were passed through an analog output adapter (Option R08, DSI) and converted by an analog-to-digital acquisition card (PC plus, National Instruments 6024E) using software written in Labview (National Instruments, Austin, TX, USA). Data were also converted into European Data Format files, which were imported into Chart v5.5.6 (ADInstruments, Bella Vista, NSW, Australia) for analyses.
Twenty-four hour HR changes and HR responses to stress or drugs in conscious mice
To assess HR and activity changes over a 12:12 h light–dark cycle, telemetry recordings were made continuously for a period of 24 h while mice were in their home cage and left undisturbed. To determine the incidence of different types of arrhythmias, we manually scrolled through 24 h baseline telemetry ECG recordings. For each animal, confirmation of the presence and type of arrhythmias was made by three persons (X.J.D., H.K. and N.L.J.).
Using the same batch of mice under telemetry ECG monitoring, we examined, on alternative days, changes in HR variation in the presence of the muscarinic antagonist atropine (1.2 mg kg−1, i.p.), and HR response to the β-adrenergic agonist isoproterenol (4 μg kg−1, i.p.). HR was determined prior to and after isoproterenol injection.
Furthermore, we examined HR response to shake stress during the inactive period (lights-on). As previously described (Davern et al. 2010), the home cage containing a mouse was placed onto the platform of an orbital mixer (Ratek Instruments, Boronia, VIC, Australia) set at 100 rpm (0.18 g) for 5 min. Recordings were made for a period of 5 min prior to, during and after the shaking, respectively.
HR responses to drugs in anaesthetised mice
HR effects of the drugs were studied in a separate experiment in mice anaesthetised with isoflurane. The standard lead-II ECG was recorded, and HR changes in response to atropine, (1.2 mg kg−1, i.p.) or isoproterenol (4 μg kg−1, i.p.) were determined. Body temperature was monitored during anaesthesia using a rectal probe, and maintained at 36–37°C using a heat lamp to warm the animal. While anaesthesia is commonly associated with alteration of ANS activity, isoflurane anaesthesia has less interference relative to other anaesthetics on the ANS activity or baseline HR (Tan et al. 2003).
Biochemical assays
RNA was extracted from the LV tissues and expression levels of various genes were determined, along with housekeeping gene (18S), by quantitative real-time RT-PCR. Plasma levels of noradrenaline (NA) and 3,4-dihydroxyphenylglycol (DHPG), and cardiac content of NA were determined by HPLC, as previously described (Lambert & Jonsdottir, 1998; Du et al. 1999).
Brain immunohistochemistry
Brains were collected 4 h prior to lights-off when animals were in the inactive phase of a 24 h cycle. Mice were anaesthetised within 1 min using a mixture of ketamine (100 mg kg−1, i.p.) and xylazine (20 mg kg−1, i.p.). After anaesthesia depth was confirmed with the absence of pedal reflex, the chest cavity was cut open and mice were perfused transcardially with 20 ml 0.9% saline and 70 ml 4% paraformaldehyde in phosphate buffer (0.1 mm, pH 7.2). Brains were postfixed in 4% paraformaldehyde in phosphate buffer for 1 h and stored in 20% sucrose in phosphate buffer at 4°C. As previously described (Davern & Head, 2007; Davern et al. 2009), coronal brain cryostat sections (40 μm) were cut, and c-Fos was immunohistochemically stained using anti-c-Fos primary antibody (sc-52, Santa Cruz Biotechnology, Santa Cruz, CA, USA). Sections were then washed and incubated in avidin-biotin peroxidase complex (1:100, Vector Laboratories, Inc., Burlingame, CA, USA). After washes, sections were incubated in 50 mm Tris buffer containing 0.08% nickel ammonium sulphate and 0.1% 3’diaminobenzidine hydrochloride (both from Sigma-Aldrich, Castle Hill, NSW, Australia) for 10 min. Subsequently, 30 μl of 30% hydrogen peroxide was added for a further 6 min. After the final wash, sections were mounted on gelatin-coated slides. The density of c-Fos-positive neurons of four to five sections per brain per region was counted by an investigator blinded to animal diagnosis, as described previously (Davern & Head, 2007; Davern et al. 2009).
The brain regions selected for analyses included the central and medial nucleus of the amygdala (CeAM and MeAM, respectively), paraventricular nucleus of the hypothalamus (PVN) and in the medulla, the nucleus tractus solitarii (NTS), raphe pallidus and rostral ventrolateral medulla (RVLM). These regions are important for autonomic regulation (Dampney, 1994).
Statistics
Results are presented as means ± SEM, unless otherwise specified. Statistical analyses were performed using Student's t test or ANOVA and Bonferroni post hoc t test. Measures from the same animals before and after an intervention were compared by paired Student's t test. Fisher's exact test was used to compare the incidence of arrhythmias. A value of P < 0.05 was considered as statistically significant.
Results
Gross and cardiac phenotypes of R6/1 mice
Male R6/1 mice were studied from 3 to 7 months old. HD phenotypes were fully developed during this period, manifested by a progressive development of motor abnormalities and loss in body mass (Fig. 1A and B). Using non-invasive echocardiography and invasive micromanometry, we observed that parameters of cardiac function in R6/1 mice at both ages were comparable to that of WT mice, except for a lower blood pressure (Table 1). R6/1 mice had a significant and age-dependent reduction in absolute heart weight (P < 0.05 by two-way ANOVA), which was in proportion to the reduced body mass (Table 1). R6/1 mice had no change in the weight of atria when normalised by body weight (Table 1). There were no histological signs of cardiomyocyte disarray or overt fibrosis (Fig. 1C), although LV cardiomyocyte diameter was smaller in R6/1 mice at both 3 and 7 months old (Fig. 1D).
Figure 1. Gross phenotype and cardiac histology of R6/1 mice at early HD (3 months old) and advanced HD (7 months old) stages.
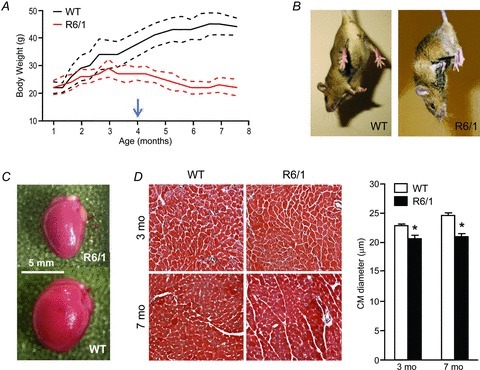
A, growth curves (mean ± SD) showing cessation of body weight gain in R6/1 mice prior to the age when they exhibit typical HD motor symptoms (arrow). B, when suspended by the tail, R6/1 mice showed a typical hindlimb clasping, whereas wild-type (WT) mice spread their four limbs. C, reduced heart size in male R6/1 mice at 7 months old. D, there were no overt histological abnormalities of the LV myocardium (×20, Masson's trichrome), except for a 10–15% reduction in cardiomyocyte (CM) diameter at both 3 and 7 months old. Data are means ± SEM, *P < 0.05 vs. age-matched WT (two-way ANOVA); n= 4 per group.
Table 1.
Cardiac function, ECG parameters, and heart weight in R6/1 and WT mice at two ages
| 3 months | 7 months | |||
|---|---|---|---|---|
| WT | R6/1 | WT | R6/1 | |
| Haemodynamics | ||||
| HR (bpm) | ND | ND | 363 ± 15 (6) | 399 ± 23 (6) |
| SAP (mmHg) | ND | ND | 100 ± 6 | 85 ± 4 |
| DAP (mmHg) | ND | ND | 72 ± 4 | 57 ± 3* |
| LVSP (mmHg) | ND | ND | 98 ± 6 | 84 ± 3 |
| LVEDP (mmHg) | ND | ND | 5.5 ± 0.2 | 4.5 ± 0.6 |
| dP/dtmax (mmHg s−1) | ND | ND | 6210 ± 710 | 6910 ± 504 |
| dP/dtmin (mmHg s−1) | ND | ND | 6568 ± 730 | 6048 ± 480 |
| Echocardiography | ||||
| HR (bpm) | 525 ± 14 (5) | 431 ± 34 (5)† | 519 ± 10 (9) | 437 ± 21 (9)† |
| LVDd (mm) | 3.98 ± 0.05 | 3.80 ± 0.14 | 4.51 ± 0.15‡ | 3.49 ± 0.12† |
| FS (%) | 33 ± 4 | 33 ± 2 | 27 ± 1 | 28 ± 2 |
| ECG | ||||
| R–R (ms) | 145 ± 3 (5) | 145 ± 3 (5) | 157 ± 4 (6) | 158 ± 10 (5) |
| P–R interval (ms) | 39.2 ± 1.3 | 37.0 ± 0.4 | 38.3 ± 1.1 | 39.2 ± 0.7 |
| QRS interval (ms) | 7.7 ± 0.5 | 8.2 ± 0.7 | 10.4 ± 0.5 | 9.6 ± 0.7 |
| Morphometrics | ||||
| Body weight (g) | 30 ± 1 (14) | 25 ± 1† (8) | 38 ± 2‡ (10) | 22 ± 1† (9) |
| Tibial length (mm) | 17.8 ± 0.2 | 17.7 ± 0.2 | 18.4 ± 0.2‡ | 18.4 ± 0.1‡ |
| Heart (mg) | 126 ± 3 | 108 ± 3† | 140 ± 8‡ | 89 ± 4†‡ |
| Atria (mg) | 10 ± 1 | 10 ± 1 | 12 ± 1 | 7 ± 1†‡ |
| HW/TL (mg mm−1) | 7.1 ± 0.2 | 6.1 ± 0.3† | 7.6 ± 0.4 | 4.8 ± 0.2†‡ |
| Atria/TL (mg mm−1) | 0.57 ± 0.03 | 0.56 ± 0.03 | 0.64 ± 0.03 | 0.37 ± 0.03†‡ |
| HW/BW (mg g−1) | 4.2 ± 0.1 | 4.3 ± 0.1 | 3.7 ± 0.1‡ | 4.1 ± 0.1† |
| Atria/BW (mg g−1) | 0.34 ± 0.02 | 0.40 ± 0.03 | 0.31 ± 0.01 | 0.32 ± 0.03 |
Group size indicated in parentheses. Abbreviations: BW, body weight; DAP, diastolic aortic pressure; dP/dtmax, maximal rate of the rise of left ventricular pressure; dP/dtmin, maximal rate of the fall of left ventricular pressure; FS, fractional shortening; HR, heart rate; HW, heart weight; LVDd, left ventricular dimension at diastole; LVEDP, left ventricular end-diastolic pressure; LVSP, left ventricular systolic pressure; ND, not determined; SAP, systolic aortic pressure; TL, tibial length; WT, wild-type. *P < 0.05 vs. age-matched WT (Student's unpaired t test). †P < 0.05 vs. age-matched WT. ‡P < 0.05 vs. 3-month-old group of same genotype (two-way ANOVA).
At the mRNA level, LV expression of the genes studied was mostly similar between age-matched R6/1 and WT mice, including those important for myocardial structure, function and energy metabolism, extracellular matrix proteins, and fetal genes (Table 2). Compared with the 3-month-old R6/1 group, expression of sarcoplasmic reticulum calcium ATPase-2a (SERCA2a) and β-myosin heavy chain (MHC) was increased, and that of pro-collagen III was lower in 7-month-old R6/1 mouse hearts (P < 0.05, Table 2).
Table 2.
Expression levels of genes in LV from R6/1 and WT mice
| 3 months | 7 months | |||
|---|---|---|---|---|
| WT (8) | R6/1 (8) | WT (7) | R6/1 (7) | |
| α-MHC | 1.00 ± 0.08 | 0.83 ± 0.04 | 1.00 ± 0.16 | 1.06 ± 0.15 |
| SERCA2a | 1.00 ± 0.13 | 0.88 ± 0.08 | 1.00 ± 0.20 | 1.22 ± 0.14† |
| ANP | 1.00 ± 0.16 | 0.72 ± 0.20 | 1.00 ± 0.22 | 0.53 ± 0.07 |
| β-MHC | 1.00 ± 0.22 | 0.59 ± 0.14 | 1.00 ± 0.21 | 2.13 ± 0.65† |
| β1-AR | 1.00 ± 0.08 | 0.89 ± 0.08 | 1.00 ± 0.21 | 1.19 ± 0.20 |
| CPT-1β | 1.00 ± 0.11 | 1.07 ± 0.06 | 1.00 ± 0.18 | 1.08 ± 0.14 |
| Pro-Col I | ND | ND | 1.00 ± 0.15 | 0.57 ± 0.11* |
| Pro-Col III | 1.00 ± 0.12 | 0.98 ± 0.15 | 1.00 ± 0.24 | 0.46 ± 0.09† |
| Glut 4 | 1.00 ± 0.05 | 1.12 ± 0.05 | 1.00 ± 0.14 | 1.08 ± 0.12 |
| VEGF | ND | ND | 1.00 ± 0.17 | 0.92 ± 0.08 |
| PGC-1 | 1.00 ± 0.12 | 0.72 ± 0.03* | ND | ND |
Abbreviations: ANP, atrial natriuretic peptide; AR, adrenergic receptor; CPT, carnitine palmitoyltransferase; Glut, glucose transporter; MHC, myosin heavy chain; ND, not determined; PGC-1, peroxisome proliferator activated receptor γ coactivator 1-α; Pro-Col, pro-collagen; SERCA, sarcoplasmic reticulum calcium ATPase; VEGF, vascular endothelial growth factor; WT, wild-type. Gene expression levels were normalised to 18S. *P < 0.05 vs. age-matched WT (Student's unpaired t test). †P < 0.05 vs. 3-month-old R6/1. Group size indicated in parentheses.
Changes in HR and physical activity in conscious mice across 24 h
The HR in WT mice of both ages studied showed a 24 h variation, with hourly averaged conscious HR increasing from approximately 500 bpm during the lights-on phase to, at times, over 600 bpm during the lights-off phase (effect of time P < 0.001, repeated measures ANOVA, Fig. 2A and B). Such a 24 h HR variation pattern was absent in R6/1 mice at both ages studied (effect of time P > 0.05, Fig. 2A and B). Furthermore, the HR in 3.5-month-old R6/1 mice was higher than age-matched WT mice for most of the 24 h period (Fig. 2A), whereas at 6.5 months old the HR level was lower in R6/1 than age-matched WT controls during the lights-off phase (active period) (Fig. 2B). These findings are summarised by bar graphs showing 12 h averaged HR for the lights-on and lights-off phases (Fig. 2A and B). R6/1 and WT mice had a similar physical activity pattern across 24 h at both ages studied (Fig. 2A and B).
Figure 2. Changes in conscious heart rate (HR) and physical activity across 24 h in R6/1 and wild-type (WT) mice.
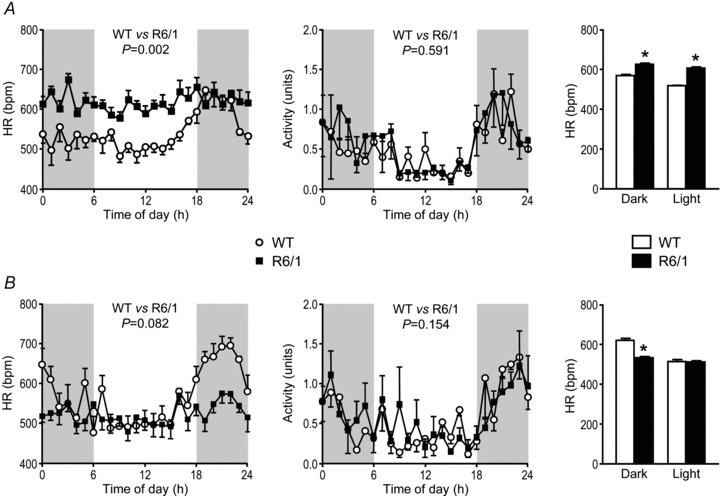
Plots of hourly averaged conscious HRs across 24 h for mice at 3.5 (A) and 6.5 (B) months old show a variation with time of day in WT (n= 4–6/group, effect of time P < 0.001 both ages) but not in R6/1 mice (n= 5–6/group, effect of time P > 0.05 both ages). HR levels over the 24 h period were significantly higher in the 3.5-month-old R6/1 than age-matched WT mice (P= 0.002), but not in the older mice (P= 0.082). Notably, HR was lower in old compared with young R6/1 mice (P= 0.001). Activity was not significantly different between the groups at both ages (P= 0.591 and 0.154, respectively). Shaded areas represent lights-off periods of the day; 24 h data were analysed using ANOVA for repeated measures. Bar graphs illustrate the HR averaged over the 12 h light and 12 h dark periods, and show that compared with age-matched WT mice, young R6/1 mice have a higher HR throughout the day; whereas, older R6/1 mice have a lower HR during the dark phase (*P < 0.05, Student's unpaired t test).
R–R intervals in conscious or anaesthetised mice, and the effects of atropine
To further explore the phenotype of autonomic nervous activity in R6/1 mice, we analysed beat-to-beat HR variation using ECG data from mice during their inactive period. R6/1 mice exhibited unstable HR compared with WT littermates, manifested visually by the chaotic HR rhythm and irregular beat-to-beat R–R intervals (Figs 3A–C and 4A–E), and quantified by assessing the standard deviation of normal R–R intervals (SDNN) in conscious (P < 0.05, Fig. 4C and D) or isoflurane-anaesthetised states (P < 0.05, Fig. 4E). Treatment with atropine markedly reduced the extent of disparity of R–R intervals seen in R6/1 mice (Fig. 4C–E), suggesting that the erratic beat-to-beat variation in R6/1 mice is likely due to an enhanced but variable parasympathetic tone. In WT mice, treatment with atropine reduced SDNN in the conscious state, but had no effect when tested under isoflurane anaesthesia (Fig. 4C–E).
Figure 3. Variation of cardiac rhythm in conscious mice.
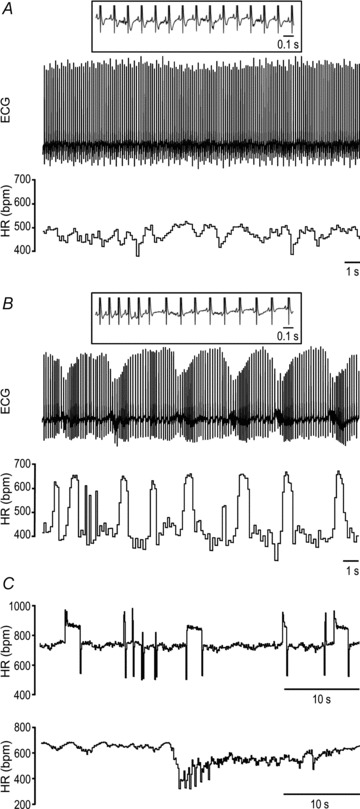
Telemetry ECG and/or ECG-derived heart rate (HR) from a typical wild-type (A) and R6/1 mouse at 3 months old (B and C), with the latter showing marked instability in HR. Insets show P waves coupled with QRS.
Figure 4. Heart rate (HR) variation and effect of the muscarinic antagonist atropine in conscious and isoflurane-anaesthetised mice.
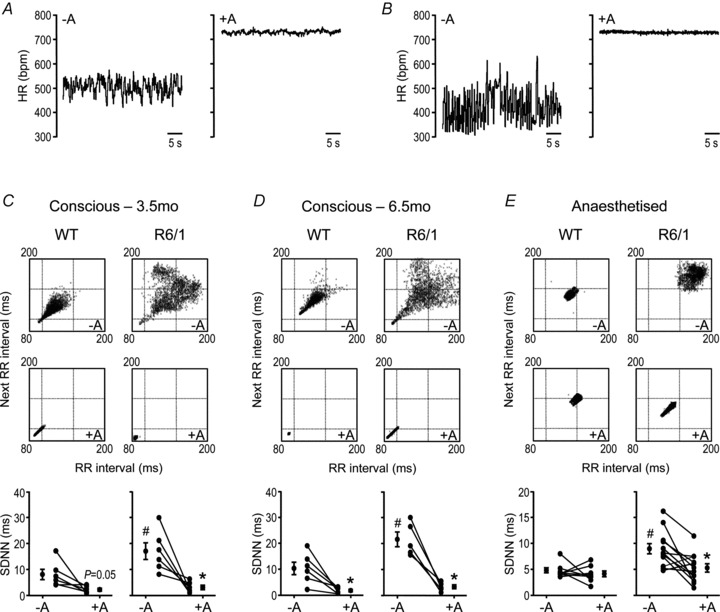
A and B, ECG-derived HR in representative wild-type (WT) (A) and R6/1 (B) mice immediately before and 10 min after atropine (1.2 mg kg−1 i.p.). C–E, Poincaré plots (upper, no atropine, and middle, with atropine) showing beat-to-beat HR variation in conscious (C, at 3.5 months old, and D, 6.5 months old) and isoflurane anaesthesia (E, at 4–5 months old) R6/1 and WT representative mice, with each R–R interval plotted against the preceding R–R interval, derived from 5 min recordings (∼2000–3500 beats). HR variation was attenuated by atropine. The findings are summarised in the graphs (bottom) showing standard deviation of normal R–R intervals (SDNN), with the mean ± SEM plotted either side of data from individual mice (circles + lines). At baseline (no atropine), SDNN was significantly higher for R6/1 versus its age-matched WT group (#P < 0.05, Student's unpaired t test). Following atropine, SDNN was attenuated compared with the corresponding pre-atropine values, and this was more clearly evident in R6/1 mice (*P < 0.05, Student's paired t test). There were no group differences in SDNN in the presence of atropine (P > 0.05, Student's unpaired t test). n= 5–12; –A, no atropine; +A, with atropine.
HR increase in response to isoproterenol
We also examined the HR response to the β-adrenergic agonist isoproterenol in mice under conscious state (at 3.5 and 6.5 months old) or isoflurane anaesthesia (4–5 months old). Isoproterenol was tested at 4 μg kg−1 (i.p.), a dose known to stimulate close to maximal cardiac functional responses. In conscious 3.5-month-old mice, the maximum HR in response to isoproterenol was similar between genotypes, though the change in HR from baseline was lower in R6/1 mice (P= 0.018, Fig. 5A). HR response to isoproterenol was attenuated in 6.5-month-old R6/1 compared with age-matched WT mice (P < 0.05, Fig. 5B). In conscious mice, treatment with atropine markedly increased HR (Fig. 4A and B). Therefore, the potential influence of atropine on isoproterenol-induced HR increase was studied in mice under isoflurane anaesthesia. Administration of isoproterenol markedly increased HR in WT, but the chronotropic response was blunted in R6/1 mice (Fig. 5C and D), and pre-treatment for 10 min with atropine (1.2 mg kg−1, i.p.) restored the HR response in R6/1 mice to that of WT mice (Fig. 5D).
Figure 5. Heart rate (HR) response to the β-adrenergic agonist isoproterenol in conscious and isoflurane-anaesthetised mice.

A and B, absolute HR and change in HR (Δ HR) following injection with isoproterenol (indicated by filled triangle, 4 μg kg−1, i.p.), in conscious 3.5- (A) and 6.5-month-old (B) R6/1 and wild-type (WT) mice (n= 5–6). Group differences were more prominent at 6.5 months old. *P < 0.05, ANOVA for repeated measures. C, representative traces from R6/1 and WT anaesthetised 4–5-month-old mice showing a blunted HR response to isoproterenol (filled triangle, 4 μg kg−1, i.p.) and the effect of pre-treatment with atropine (1.2 mg kg−1 i.p.). D, grouped data show that the blunted HR response to isoproterenol (filled triangle) in R6/1 mice was normalised by atropine pre-treatment (n= 3–5 per group). Symbols and faint lines represent means ± SEM. –A, no atropine; +A, with atropine.
HR response to shake stress test
In 3.5-month-old mice, the peak HR response to the shake stress test of 5 min duration was no different between the genotypes, but R6/1 mice showed a slower restoration of HR towards baseline during the 5 min period (P= 0.013, Fig. 6A). At 6.5 months old, there was a tendency for the peak change in HR in response to the shake stress to be reduced and a slower recovery towards baseline in R6/1 compared with that of WT littermates (Fig. 6B).
Figure 6. Heart rate (HR) response to shake stress test.
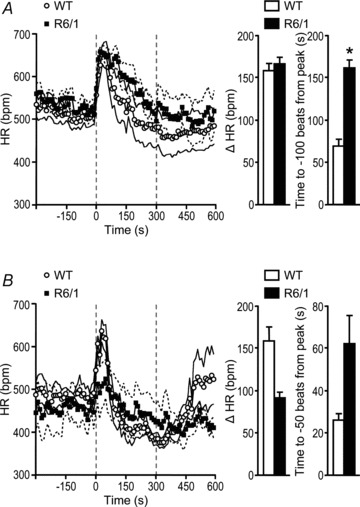
Animals that were 3.5 (A, n= 6–7) and 6.5 (B, n= 4) months old were subjected to the shake stress test for 5 min (indicated by the period between the dashed vertical lines), and HR was derived from telemetry ECG recordings. Plotted data are means ± SEM (the latter indicated by the accompanying faint lines). A, compared with wild-type (WT) mice, R6/1 mice at 3.5 months old showed a similar peak HR (P= 0.804, Student's unpaired t test) but a slower HR recovery to baseline (*P= 0.013, Student's unpaired t test). B, at 6.5 months old, although there was a tendency for a decrease in the peak HR level and a slower recovery towards baseline in response to the shake stress test in R6/1 compared with WT mice, these parameters were not different between the two groups (P= 0.112 and 0.134, respectively, Student's unpaired t test).
Catecholamine measures
Plasma levels of NA were similar between R6/1 and WT mouse hearts at 3 months old, but were sixfold higher in 7-month-old R6/1 versus WT groups (Fig. 7). DHPG levels were similar between R6/1 and WT mice at both ages studied. NA content was similar in the LV of R6/1 and WT mice at 3 months old, but was 40% lower in R6/1 than WT mice at 7 months old (P < 0.05 by two-way ANOVA with age and genotype as factors, Fig. 7), indicating an excessive neuronal release.
Figure 7. Plasma levels of noradrenaline (NA) and 3,4-dihydroxyphenylglycol (DHPG), and cardiac content of NA.
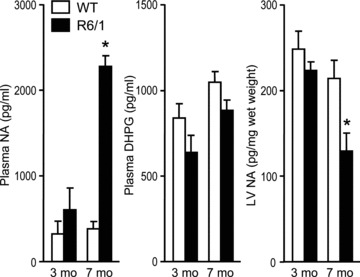
Plasma NA was significantly increased and LV NA decreased in 7-month-old R6/1 compared with age-matched wild-type (WT) mice. These changes were not evident at 3 months old. There were no significant changes in plasma DHPG levels between R6/1 and WT mice at 3 or 7 months old. *P < 0.05 vs. age-matched WT and 3-month-old R6/1 mice (two-way ANOVA). n= 4–6 per group.
Arrhythmia phenotype in R6/1 mice
The ECG parameters at stable basal conditions were comparable between R6/1 and WT mice (Table 1). However, telemetry ECG recording of conscious R6/1 mice at 3.5 and 6.5 months old revealed a variety of arrhythmias, including atrial–ventricular conduction blockade (mostly occurring as a single episode without consistent change in preceding P–R intervals, Fig. 8A), sinus arrhythmia (Fig. 8A), atrial flutter (Fig. 8B), atrial fibrillation (AF) (Fig. 8C), supra-ventricular and ventricular premature beats (Fig. 8D and E), and short episodes of ventricular tachycardia (Fig. 8F). Whilst AF occurred in one WT mouse as a single episode during a 24 h period, R6/1 mice had an average of four episodes/24 h (1–17 episodes). Table 3 summarises the prevalence of different types of arrhythmias detected over a 24 h period from 3.5- and 6.5-month-old mice combined, showing a significantly higher incidence of arrhythmias in R6/1 compared with WT mice. There was no evidence at autopsy of heart failure or gross cardiac pathology in these mice.
Figure 8. Different types of arrhythmias detected by telemetry ECG in conscious R6/1 mice.
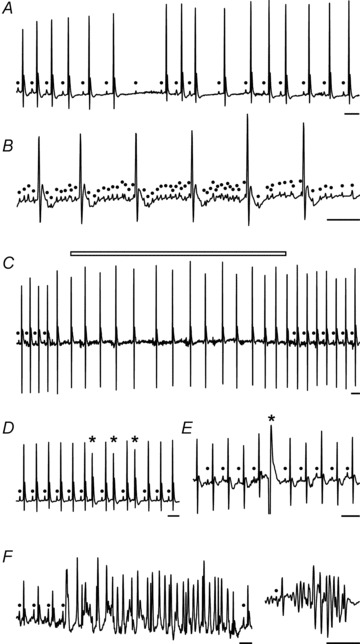
A, sinus arrhythmias and atrial–ventricular conduction blockade (note the absence of consistent change in P–R duration). B, atrial flutter. C, paroxysmal AF (open bar) evidenced by loss of P waves, unstable baseline and irregularity of R–R intervals. D, supra- ventricular ectopic beats. E, ventricular premature beat. F, episodes of ventricular tachycardia; closed circles indicates P wave; asterisk indicates ectopic beats; scale bars: 0.1 s. Group data shown in Table 3.
Table 3.
Incidence of various types of arrhythmias over 24 h in conscious R6/1 and WT mice
| WT | R6/1 | P value | |
|---|---|---|---|
| Atrial–ventricular blockade | 6/10 | 6/12 | 0.691 |
| Supra-ventricular premature beat | 5/10 | 3/12 | 0.378 |
| Ventricular premature beat | 2/10 | 8/12 | 0.043 |
| Atrial flutter | 4/10 | 10/12 | 0.074 |
| Proximal atrial fibrillation | 1/10 | 10/12 | 0.002 |
| Transient ventricular tachycardia | 0/10 | 7/12 | 0.005 |
Data are numbers of animals at both 3 and 7 months old combined. P values were calculated by Fisher's exact test. Abbreviation: WT, wild-type.
Of 16 R6/1 mice that were under investigation, three died at 6–7 months old. One R6/1 mouse died during conscious ECG telemetry monitoring, and Fig. 9 shows the onset of AF immediately followed by complete atrial–ventricular dissociation (AVD) and a slow ventricular rhythm all occurring within seconds prior to death. Sudden cardiac death was also indicated by the lack of findings at autopsy that might indicate an alternative cause of death.
Figure 9. Telemetry ECG from a 7-month-old R6/1 mouse revealing development of arrhythmias and sudden death.
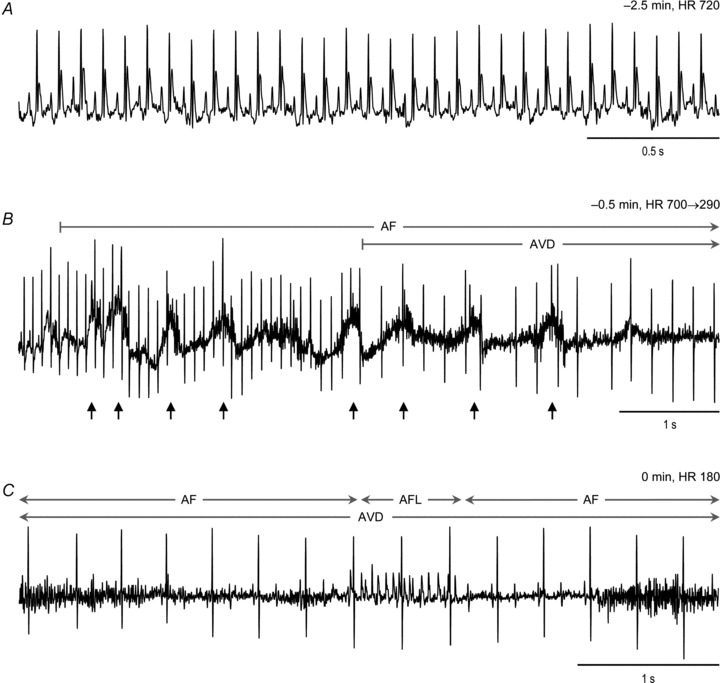
A and B, ECG traces starting from 2.5 min prior to death show the time course from normal cardiac rhythm (A) to development of atrial fibrillation (AF) immediately followed by complete atrial–ventricular disassociation (AVD) (B). Small arrows indicate changes in ECG baseline likely due to hypoxia-driven gasps just prior to death. C, finally, sustained AF interrupted by a short episode of atrial flutter (AFL), together with a slow and regular ventricular rhythm as the terminal presentation of ECG. HR, heart rate.
c-Fos positive neurons in the forebrain and hindbrain regions
We determined the number of c-Fos positive neurons in the several brain regions known to be relevant to regulating the ANS (Horiuchi et al. 2004). Animals were deeply anaesthetised 4 h before ‘lights-off’, when mice normally would be in the inactive phase of a 24 h cycle (Fig. 2). In all brain regions examined, counts of c-Fos positive neurons were mostly twofold greater in R6/1 mice compared with corresponding regions in WT brains (Fig. 10A and B). These included the PVN (1.9-fold, P < 0.001), MeAM (2.1-fold, P < 0.001), CeAM (2.5-fold, P= 0.02), NTS (2.2-fold, P < 0.001), RVLM (2.2-fold, P= 0.003) and the raphe pallidus (3.4-fold, P < 0.001).
Figure 10. Density of active neurons in various brain regions controlling autonomic nervous activity.
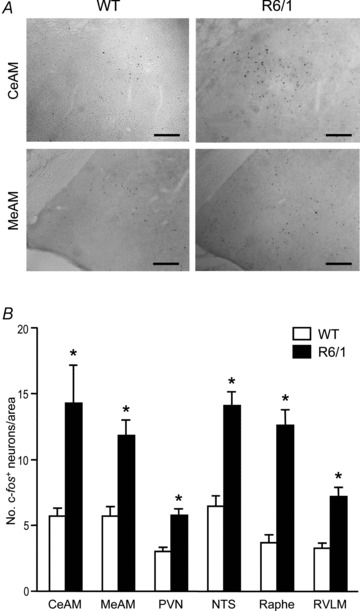
A, images of brain sections showing active neurons (black dots) detected by immunohistochemistry for c-Fos. Photomicrographs of coronal sections through the central (CeAM) and medial (MeAM) nuclei of the amygdala in 5.5-month-old wild-type (WT) and R6/1 mouse brains. Scale bars: 100 μm. B, bar graph showing grouped data of active neurons in brain regions that are known autonomic nervous centres (n= 4/group). CeAM and MeAM, central and medial nuclei of the amygdala, respectively; PVN, paraventricular nucleus of the hypothalamus; NTS, nucleus tractus solitarii; Raphe, raphe pallidus; RVLM, rostral ventrolateral medulla. *P < 0.05 vs. WT (ANOVA).
Discussion
We aimed to provide experimental insights into cardiac-associated abnormalities in HD. Our thorough investigation of R6/1 mice has led to several novel findings. First, R6/1 mice have normal baseline cardiac function and largely unchanged cardiac gene expression or histology, except for cardiomyocyte atrophy. Second, our results of HR monitoring, drug validation and catecholamine measurement suggest that from the early HD phase, R6/1 mice exhibited ANS-cardiac dysregulation manifested as enhanced sympathetic and parasympathetic nervous activities, leading to unstable and chaotic cardiac rhythm as well as a lack of the normal nocturnal variation in HR. Third, R6/1 mice developed a variety of arrhythmias, including paroxysmal AF and sudden arrhythmic death. Finally, ANS dysfunction in R6/1 mice may be related to central mechanisms indicated by greater numbers of active neurons in the brain regions that are known to regulate ANS activity.
There is evidence for cardiac abnormalities owing to poly-Q aggregates in cardiomyocytes. Pattison et al. (2008) generated transgenic mice with cardiomyocyte-restricted expression of polyglutamine encoding transgene leading to cardiac dilatation, hypertrophy (with an upregulated fetal gene profile), fibrosis and heart failure death. Using the R6/2 model, Mihm et al. (2007) reported LV dilatation and contractile dysfunction by 8 weeks old. The authors also observed the presence of poly-Q aggregates in R6/2 hearts (Mihm et al. 2007). Even at the advanced HD phase, we found that R6/1 mice neither show cardiac dysfunction nor histopathology (such as fibrosis, cardiomyocyte apoptosis or hypertrophy), although a moderate degree of reduction in the heart mass and a lower arterial blood pressure were detected. The cardiac phenotype seen in the R6/1 mice in comparison to that of the R6/2 or poly-Q transgenic mouse models (Mihm et al. 2007; Pattison et al. 2008) implies that cardiac pathophysiology seen in the latter two models might be due to very high levels of poly-Q aggregates and may therefore less closely mimic what occurs in human patients with HD.
HD is well known to progress with ageing. In the present study, R6/1 mice were studied at 3 and 7 months old, representing early and advanced HD phases. Except for ageing associated body mass reduction, we observed age-dependent development of certain neurocardiac phenotypes, including: (1) loss of diurnal variation of HR; (2) attenuated HR response to shake stress or to a β-adrenergic agonist; (3) age-dependent changes in a few genes studied; and (4) elevation of plasma NA levels in association with reduced myocardial content. These findings would improve our understanding on HD pathophysiology beyond the brain, an important area of research that was recently reviewed (Sassone et al. 2009; van der Burg et al. 2009). A moderate extent of cardiac atrophy was observed in R6/1 mice given the reduction in heart weight and cardiomyocyte diameter. The reason for this phenotype remains unclear. Skeletal striated muscle atrophy is well documented in patients with HD and animal models of HD, and has been attributed to disruption of gene expression and cellular function associated with the presence of the mutant huntingtin gene (Sassone et al. 2009; van der Burg et al. 2009).
Our study implies significant cardiac ANS dysregulation in the R6/1 mice. Although plasma and cardiac NA levels were not altered at 3 months old, a sustained enhancement of sympathetic nervous activity is indicated by higher HR levels together with the loss of a typical nocturnal 24 h HR variation in 3-month-old R6/1 mice, in which the parasympathetic tone was elevated rather than reduced. Likewise, episodes of sinus tachycardia or tachyarrhythmias are likely the consequence of elevated cardiac sympathetic drive. A long-term sustained cardiac sympathetic activation is strongly indicated by the finding in 7-month-old R6/1 mice of a marked increase in plasma levels of NA together with a 40% reduction in cardiac NA content. Meanwhile, plasma levels of DHPG, an intraneuronal metabolite of NA, were not significantly different between age-matched R6/1 and WT controls, indicating a comparable degree of neuronal NA reuptake. These changes are very similar to those seen in the setting of heart failure where cardiac sympathetic drive and release of NA remain higher (Himura et al. 1993; Kaye et al. 1994; Eisenhofer et al. 1996) despite a reduced tissue content due to excessive release (Liang et al. 1991; Himura et al. 1993).
Twenty-four hour HR levels by telemetry were higher at 3.5 months old and lower at 6.5 months old in R6/1 compared with age-matched WT mice, and there was a tendency for a reduced peak HR response to the shake stress test. All these differences are likely due in part to the development of desensitisation of cardiac β-adrenergic signalling following a long-term exposure to elevated NA owing to sympathetic overdrive, although β1-adrenoceptor gene expression was unaltered. This was in keeping with our finding of attenuated HR response to isoproterenol in the older R6/1 mice. In addition, another HD mouse model (N171–82Q) showed an attenuated HR response to restraint stress (Griffioen et al. 2012). Our findings also suggest activation of the parasympathetic nervous system, as evidenced by atropine reducing the marked variation in R–R intervals, measured as SDNN, in the conscious and anaesthetised states. Unstable HR with episodes of single atrial–ventricular blockade or sinus bradycardia in R6/1 mice is likely evoked by an augmented parasympathetic tone, as indicated by a robust reduction in R–R variation following atropine treatment. We observed a blunted HR response to isoproterenol in R6/1 mice under isoflurane anaesthesia. The improvement by atropine pre-treatment of the HR response to isoproterenol in anaesthetised R6/1 mice implies tonic postsynaptic inhibition of β-adrenergic responses by the parasympatho-muscarinic signalling under our experimental conditions. Such muscarinic inhibition is well known to be enhanced in the setting of an enhanced sympatho-β-adrenergic activity (Morady et al. 1988; Furukawa et al. 1996; Olshansky et al. 2008; Brack et al. 2009). Parasympathetic hyperactivity is especially evident in patients with the juvenile or rigid forms of HD (Stober et al. 1983).
Very recently, a few teams have reported ANS dysfunction in other HD models (Kudo et al. 2011; Schroeder et al. 2011; Griffioen et al. 2012). In the BACHD mouse model of HD, a blunted baroreceptor reflex under anaesthetised conditions was observed (Schroeder et al. 2011). Using telemetry, two groups reported abnormalities in sleep patterns and HR changes over a 12:12 h light–dark cycle, and in circadian rhythm recorded during 12:12 h dark–dark conditions in HD models including BACHD and R6/2 (Morton et al. 2005; Kudo et al. 2011). Our 24 h HR telemetry findings are in line with these reports. Interestingly, we did not observe differences in the 24 h activity pattern between R6/1 and WT mice. Our experimental protocol cannot exclude the possibility that mice were periodically waking up during the inactive period but were not moving around the cage (and therefore not registered on the telemetry activity channel). In fact, using video technology to monitor R6/2 mice in their home cage, Steele et al. (2007) revealed that, compared with WT mice, the HD mice displayed 2.5-fold increased awakening events, increased twitching (during rest) and hypergrooming, but decreased distance travelled and activities such as stretching, hanging vertically and jumping. Studies on patients with HD also have shown nocturnal sleep disturbances together with daytime somnolence (Morton et al. 2005; Aziz et al. 2009, 2010; Videnovic et al. 2009).
Clinical studies have consistently reported sympathetic predominance and parasympathetic withdrawal in patients with HD relative to healthy controls (Den Heijer et al. 1988; Sharma et al. 1999; Andrich et al. 2002; Kobal et al. 2004; Bar et al. 2008). In these studies, ANS activity was assessed using the HRV technique. However, these studies might suffer from some limitations. The source of raw data was from a very brief period (1–10 min) of ECG recording in patients known to have an unstable psychological state. There was lack of confirmative validation or specificity, and it was difficult to ascertain the onset or timing of ANS dysfunction along the course of HD. Importantly, these studies did not relate ANS abnormalities to cardiac consequences and/or excluded patients with arrhythmias (Andrich et al. 2002; Kobal et al. 2004; Bar et al. 2008). Thus, further clinical research is required, and methods such as 24 h Holter monitoring of ECG should be adopted.
c-Fos expression as a marker of activated neurons has been shown to be increased 40–60 min after a stressor (Dampney et al. 1995; Davern & Head, 2007; Davern et al. 2009). Our protocol of brain preparation excluded the influence of acute stress-associated changes on neuronal activity, but revealed ‘constitutive’ activity of brain neurons at the time when animals are normally asleep. A twofold increase in the number of active neurons was noticed in various forebrain and hindbrain regions of R6/1 brains. These regions (PVN, NTS, RVLM) are well known in integrating cardiovascular responses to arousal, acute stress or anxiety, and in governing ANS regulation of the cardiovascular system (Davern & Head, 2007; Davern et al. 2010). The midline raphe region is of particular interest, and is known to influence cardiac sympathetic activity (Horiuchi et al. 2004). These findings would be consistent with the view that HD mice have more wakeful events as the amygdala is a region integrating the emotional and cardiovascular responses to arousal stimuli (Davern & Head, 2011). The very marked activation of the raphe pallidus is known to relay cardiac sympathetic activation due to arousal (Horiuchi et al. 2004). This pattern may well explain the lack of reduction in HR during the light phase, which is normally seen in a nocturnal species. Thus, the ANS dysregulation of the heart is due most likely to abnormalities in the function of brain centres that govern ANS activity.
Early mortality surveys indicated that heart disease constituted the second major cause of death for patients with HD (Chiu & Alexander, 1982; Lanska et al. 1988; Sorensen & Fenger, 1992). The nature of cardiac pathophysiology associated with HD remains unknown. An arrhythmogenic role of the activated sympatho-β-adrenergic system has been established in settings of heart diseases such as ischaemia or chronic myocardial infarction (Schwartz et al. 1992; Du et al. 1999; Chen & Tan, 2007; Tan et al. 2008), or in transgenic interventions leading to an enhanced cardiac β-adrenergic activity (Du et al. 2000). In R6/1 mice, while cardiac function was largely preserved, unstable baseline HR and arrhythmias do occur. AF reported in various mouse models is typically paroxysmal and usually associated with cardiac pathology as arrhythmic substrates (Sah et al. 1999; Xiao et al. 2004; Saba et al. 2005; Pretorius et al. 2009). Of interest is our finding of various types of arrhythmias that were short-lasting in nature in the R6/1 mice free of overt cardiac pathology. There is evidence that on the background of an enhanced sympathetic nervous activity, parasympathetic activation would enhance the chance of supra-ventricular arrhythmias (Tan et al. 2008). Further, enhanced sympathetic as well as parasympathetic tone has been shown to induce paroxysmal AF, for example in patients with obstructive sleep disorder (Leung, 2009). Denervation of cardiac parasympathetic nerves by catheter ablation is also effective in preventing vagal activation-induced AF both experimentally and clinically (Schauerte et al. 2000; Arora et al. 2008). Our findings provide invaluable clues for likely cardiac causes of death in patients with HD, and for further clinical studies in patients with HD on ANS function, potential development of arrhythmias and therapeutic interventions.
While our experimental results suggest a central role of the ANS dysfunction in the development of HR instability and arrhythmias, we cannot exclude the possibility that a chronically altered autonomic tone and/or cardiomyocyte dysfunction due to mutant huntingtin gene might ultimately induce cardiac abnormalities, and that a combination of these factors contributed to the observed arrhythmia. To confirm this, a detailed electrophysiological study is necessary to identify potential cardiac factors that make the HD heart vulnerable to arrhythmias in the setting of altered function of the ANS.
In conclusion, our findings in the R6/1 mice suggest a centrally mediated dysregulation by the ANS of cardiac activity with resultant absence of typical 24 h HR changes and neurogenic arrhythmias leading to sudden cardiac death.
Acknowledgments
This work was supported by the National Heart Foundation of Australia (grant number G10M5126). A.J.H. was Future Fellow of the Australian Research Council; G.L., G.A.H. and X.J.D. were fellows of the National Health and Medical Research Council of Australia; X.D. was a recipient of a Melbourne University Research Scholarship. This study was supported in part by the Victorian Government's Operational Infrastructure Support Program.
Glossary
- AF
atrial fibrillation
- ANS
autonomic nervous system
- AVD
atrial–ventricular dissociation
- CeAM
central nucleus of the amygdala
- DAP
diastolic aortic pressure
- DHPG
3,4-dihydroxyphenylglycol
- dP/dtmax
maximal rate of rise of LV pressure
- dP/dtmin
maximal rate of fall of LV pressure
- FS
fractional shortening
- HD
Huntington's disease
- HR
heart rate
- HRV
heart rate variability
- LV
left ventricle or left ventricular
- LVDd
LV dimension at diastole
- LVEDP
LV end-diastolic pressure
- LVSP
LV systolic pressure
- MeAM
medial nucleus of the amygdala
- MHC
myosin heavy chain
- NA
noradrenaline
- NTS
nucleus tractus solitarii
- poly-Q
poly-glutamine
- PVN
paraventricular nucleus of the hypothalamus
- RVLM
rostral ventrolateral medulla
- SAP
systolic aortic pressure
- SDNN
standard deviation of normal R–R intervals
- SERCA
sarcoplasmic reticulum calcium ATPase
- WT
wild-type
Author contributions
H.K. and N.L.J.: conception, design of experiments, collection, analysis and interpretation of data, drafting of manuscript; P.D. and G.L.: conception, collection, analysis and interpretation of data, critically revising the manuscript; Y.S., T.P., X.D. and L.L.: collection, analysis and interpretation of data; G.A.H.: conception and design, interpretation of data, critically revising the manuscript; A.J.H.: conception and design, providing transgenic mice, interpretation of data, critically revising the manuscript; X.J.D: conception and design of the study, obtaining grant, analysis and interpretation of data, drafting of manuscript. The major part of the work was done in the laboratory of X.J.D. at Baker IDI Heart and Diabetes Institute. All authors approved the final version for publication.
References
- Andrich J, Schmitz T, Saft C, Postert T, Kraus P, Epplen JT, Przuntek H, Agelink MW. Autonomic nervous system function in Huntington's disease. J Neurol Neurosurg Psychiatry. 2002;72:726–731. doi: 10.1136/jnnp.72.6.726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arora R, Ulphani JS, Villuendas R, Ng J, Harvey L, Thordson S, Inderyas F, Lu Y, Gordon D, Denes P, Greene R, Crawford S, Decker R, Morris A, Goldberger J, Kadish AH. Neural substrate for atrial fibrillation: implications for targeted parasympathetic blockade in the posterior left atrium. Am J Physiol Heart Circ Physiol. 2008;294:H134–H144. doi: 10.1152/ajpheart.00732.2007. [DOI] [PubMed] [Google Scholar]
- Aziz NA, Anguelova GV, Marinus J, van Dijk JG, Roos RA. Autonomic symptoms in patients and pre-manifest mutation carriers of Huntington's disease. Eur J Neurol. 2010;17:1068–1074. doi: 10.1111/j.1468-1331.2010.02973.x. [DOI] [PubMed] [Google Scholar]
- Aziz NA, Pijl H, Frolich M, Schroder-van der Elst JP, van der Bent C, Roelfsema F, Roos RA. Delayed onset of the diurnal melatonin rise in patients with Huntington's disease. J Neurol. 2009;256:1961–1965. doi: 10.1007/s00415-009-5196-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar KJ, Boettger MK, Andrich J, Epplen JT, Fischer F, Cordes J, Koschke M, Agelink MW. Cardiovagal modulation upon postural change is altered in Huntington's disease. Eur J Neurol. 2008;15:869–871. doi: 10.1111/j.1468-1331.2008.02173.x. [DOI] [PubMed] [Google Scholar]
- Bates GP. History of genetic disease: the molecular genetics of Huntington disease – a history. Nat Rev Genet. 2005;6:766–773. doi: 10.1038/nrg1686. [DOI] [PubMed] [Google Scholar]
- Bates GP, Mangiarini L, Mahal A, Davies SW. Transgenic models of Huntington's disease. Hum Mol Genet. 1997;6:1633–1637. doi: 10.1093/hmg/6.10.1633. [DOI] [PubMed] [Google Scholar]
- Billman GE. A comprehensive review and analysis of 25 years of data from an in vivo canine model of sudden cardiac death: implications for future anti-arrhythmic drug development. Pharmacol Ther. 2006;111:808–835. doi: 10.1016/j.pharmthera.2006.01.002. [DOI] [PubMed] [Google Scholar]
- Brack KE, Coote JH, Ng GA. Vagus nerve stimulation inhibits the increase in Ca2+ transient and left ventricular force caused by sympathetic nerve stimulation but has no direct effects alone – epicardial Ca2+ fluorescence studies using fura-2 AM in the isolated innervated beating rabbit heart. Exp Physiol. 2009;95:80–92. doi: 10.1113/expphysiol.2009.048215. [DOI] [PubMed] [Google Scholar]
- Chen LS, Zhou S, Fishbein MC, Chen PS. New perspectives on the role of autonomic nervous system in the genesis of arrhythmias. J Cardiovasc Electrophysiol. 2007;18:123–127. doi: 10.1111/j.1540-8167.2006.00590.x. [DOI] [PubMed] [Google Scholar]
- Chen PS, Tan AY. Autonomic nerve activity and atrial fibrillation. Heart Rhythm. 2007;4:S61–S64. doi: 10.1016/j.hrthm.2006.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu E, Alexander L. Causes of death in Huntington's disease. Med J Aust. 1982;1:153. doi: 10.5694/j.1326-5377.1982.tb132224.x. [DOI] [PubMed] [Google Scholar]
- Dampney RA. Functional organization of central pathways regulating the cardiovascular system. Physiol Rev. 1994;74:323–364. doi: 10.1152/physrev.1994.74.2.323. [DOI] [PubMed] [Google Scholar]
- Dampney RA, Li YW, Hirooka Y, Potts P, Polson JW. Use of c-fos functional mapping to identify the central baroreceptor reflex pathway: advantages and limitations. Clin Exp Hypertens. 1995;17:197–208. doi: 10.3109/10641969509087065. [DOI] [PubMed] [Google Scholar]
- Davern PJ, Chen D, Head GA, Chavez CA, Walther T, Mayorov DN. Role of angiotensin II Type 1A receptors in cardiovascular reactivity and neuronal activation after aversive stress in mice. Hypertension. 2009;54:1262–1268. doi: 10.1161/HYPERTENSIONAHA.109.139741. [DOI] [PubMed] [Google Scholar]
- Davern PJ, Head GA. Fos-related antigen immunoreactivity after acute and chronic angiotensin II-induced hypertension in the rabbit brain. Hypertension. 2007;49:1170–1177. doi: 10.1161/HYPERTENSIONAHA.106.086322. [DOI] [PubMed] [Google Scholar]
- Davern PJ, Head GA. Role of the medial amygdala in mediating responses to aversive stimuli leading to hypertension. Clin Exp Pharmacol Physiol. 2011;38:136–143. doi: 10.1111/j.1440-1681.2010.05413.x. [DOI] [PubMed] [Google Scholar]
- Davern PJ, Jackson KL, Nguyen-Huu TP, La Greca L, Head GA. Cardiovascular responses to aversive and nonaversive stressors in Schlager genetically hypertensive mice. Am J Hypertens. 2010;23:838–844. doi: 10.1038/ajh.2010.69. [DOI] [PubMed] [Google Scholar]
- Den Heijer JC, Bollen WL, Reulen JP, van Dijk JG, Kramer CG, Roos RA, Buruma OJ. Autonomic nervous function in Huntington's disease. Arch Neurol. 1988;45:309–312. doi: 10.1001/archneur.1988.00520270087025. [DOI] [PubMed] [Google Scholar]
- Du XJ, Cox HS, Dart AM, Esler MD. Sympathetic activation triggers ventricular arrhythmias in rat heart with chronic infarction and failure. Cardiovasc Res. 1999;43:919–929. doi: 10.1016/s0008-6363(99)00139-x. [DOI] [PubMed] [Google Scholar]
- Du XJ, Gao XM, Wang B, Jennings GL, Woodcock EA, Dart AM. Age-dependent cardiomyopathy and heart failure phenotype in mice overexpressing β2-adrenergic receptors in the heart. Cardiovasc Res. 2000;48:448–454. doi: 10.1016/s0008-6363(00)00187-5. [DOI] [PubMed] [Google Scholar]
- Eisenhofer G, Friberg P, Rundqvist B, Quyyumi AA, Lambert G, Kaye DM, Kopin IJ, Goldstein DS, Esler MD. Cardiac sympathetic nerve function in congestive heart failure. Circulation. 1996;93:1667–1676. doi: 10.1161/01.cir.93.9.1667. [DOI] [PubMed] [Google Scholar]
- Furukawa Y, Hoyano Y, Chiba S. Parasympathetic inhibition of sympathetic effects on sinus rate in anesthetized dogs. Am J Physiol Heart Circ Physiol. 1996;271:H44–H50. doi: 10.1152/ajpheart.1996.271.1.H44. [DOI] [PubMed] [Google Scholar]
- Gil JM, Rego AC. The R6 lines of transgenic mice: a model for screening new therapies for Huntington's disease. Brain Res Rev. 2009;59:410–431. doi: 10.1016/j.brainresrev.2008.12.001. [DOI] [PubMed] [Google Scholar]
- Griffioen KJ, Wan R, Brown TR, Okun E, Camandola S, Mughal MR, Phillips TM, Mattson MP. Aberrant heart rate and brainstem brain-derived neurotrophic factor (BDNF) signaling in a mouse model of Huntington's disease. Neurobiol Aging. 2012;33:1481.e1–1481.e5. doi: 10.1016/j.neurobiolaging.2011.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Himura Y, Felten SY, Kashiki M, Lewandowski TJ, Delehanty JM, Liang CS. Cardiac noradrenergic nerve terminal abnormalities in dogs with experimental congestive heart failure. Circulation. 1993;88:1299–1309. doi: 10.1161/01.cir.88.3.1299. [DOI] [PubMed] [Google Scholar]
- Horiuchi J, McAllen RM, Allen AM, Killinger S, Fontes MA, Dampney RA. Descending vasomotor pathways from the dorsomedial hypothalamic nucleus: role of medullary raphe and RVLM. Am J Physiol Regul Integr Comp Physiol. 2004;287:R824–R832. doi: 10.1152/ajpregu.00221.2004. [DOI] [PubMed] [Google Scholar]
- Kaye DM, Lambert GW, Lefkovits J, Morris M, Jennings G, Esler MD. Neurochemical evidence of cardiac sympathetic activation and increased central nervous system norepinephrine turnover in severe congestive heart failure. J Am Coll Cardiol. 1994;23:570–578. doi: 10.1016/0735-1097(94)90738-2. [DOI] [PubMed] [Google Scholar]
- Kobal J, Meglic B, Mesec A, Peterlin B. Early sympathetic hyperactivity in Huntington's disease. Eur J Neurol. 2004;11:842–848. doi: 10.1111/j.1468-1331.2004.00894.x. [DOI] [PubMed] [Google Scholar]
- Kudo T, Schroeder A, Loh DH, Kuljis D, Jordan MC, Roos KP, Colwell CS. Dysfunctions in circadian behavior and physiology in mouse models of Huntington's disease. Exp Neurol. 2011;228:80–90. doi: 10.1016/j.expneurol.2010.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert GW, Jonsdottir IH. Influence of voluntary exercise on hypothalamic norepinephrine. J Appl Physiol. 1998;85:962–966. doi: 10.1152/jappl.1998.85.3.962. [DOI] [PubMed] [Google Scholar]
- Lanska DJ, Lavine L, Lanska MJ, Schoenberg BS. Huntington's disease mortality in the United States. Neurology. 1988;38:769–772. doi: 10.1212/wnl.38.5.769. [DOI] [PubMed] [Google Scholar]
- Leung RS. Sleep-disordered breathing: autonomic mechanisms and arrhythmias. Prog Cardiovasc Dis. 2009;51:324–338. doi: 10.1016/j.pcad.2008.06.002. [DOI] [PubMed] [Google Scholar]
- Liang CS, Frantz RP, Suematsu M, Sakamoto S, Sullebarger JT, Fan TM, Guthinger L. Chronic β-adrenoceptor blockade prevents the development of β-adrenergic subsensitivity in experimental right-sided congestive heart failure in dogs. Circulation. 1991;84:254–266. doi: 10.1161/01.cir.84.1.254. [DOI] [PubMed] [Google Scholar]
- Mangiarini L, Sathasivam K, Seller M, Cozens B, Harper A, Hetherington C, Lawton M, Trottier Y, Lehrach H, Davies SW, Bates GP. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell. 1996;87:493–506. doi: 10.1016/s0092-8674(00)81369-0. [DOI] [PubMed] [Google Scholar]
- Mihm MJ, Amann DM, Schanbacher BL, Altschuld RA, Bauer JA, Hoyt KR. Cardiac dysfunction in the R6/2 mouse model of Huntington's disease. Neurobiol Dis. 2007;25:297–308. doi: 10.1016/j.nbd.2006.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morady F, Kou WH, Nelson SD, de Buitleir M, Schmaltz S, Kadish AH, Toivonen LK, Kushner JA. Accentuated antagonism between β-adrenergic and vagal effects on ventricular refractoriness in humans. Circulation. 1988;77:289–297. doi: 10.1161/01.cir.77.2.289. [DOI] [PubMed] [Google Scholar]
- Morton AJ, Wood NI, Hastings MH, Hurelbrink C, Barker RA, Maywood ES. Disintegration of the sleep-wake cycle and circadian timing in Huntington's disease. J Neurosci. 2005;25:157–163. doi: 10.1523/JNEUROSCI.3842-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz-Sanjuan I, Bates GP. The importance of integrating basic and clinical research toward the development of new therapies for Huntington disease. J Clin Invest. 2011;121:476–483. doi: 10.1172/JCI45364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olshansky B, Sabbah HN, Hauptman PJ, Colucci WS. Parasympathetic nervous system and heart failure: pathophysiology and potential implications for therapy. Circulation. 2008;118:863–871. doi: 10.1161/CIRCULATIONAHA.107.760405. [DOI] [PubMed] [Google Scholar]
- Pang TY, Du X, Zajac MS, Howard ML, Hannan AJ. Altered serotonin receptor expression is associated with depression-related behavior in the R6/1 transgenic mouse model of Huntington's disease. Hum Mol Genet. 2009;18:753–766. doi: 10.1093/hmg/ddn385. [DOI] [PubMed] [Google Scholar]
- Pattison JS, Sanbe A, Maloyan A, Osinska H, Klevitsky R, Robbins J. Cardiomyocyte expression of a polyglutamine preamyloid oligomer causes heart failure. Circulation. 2008;117:2743–2751. doi: 10.1161/CIRCULATIONAHA.107.750232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pretorius L, Du XJ, Woodcock EA, Kiriazis H, Lin RC, Marasco S, Medcalf RL, Ming Z, Head GA, Tan JW, Cemerlang N, Sadoshima J, Shioi T, Izumo S, Lukoshkova EV, Dart AM, Jennings GL, McMullen JR. Reduced phosphoinositide 3-kinase (p110α) activation increases the susceptibility to atrial fibrillation. Am J Pathol. 2009;175:998–1009. doi: 10.2353/ajpath.2009.090126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saba S, Janczewski AM, Baker LC, Shusterman V, Gursoy EC, Feldman AM, Salama G, McTiernan CF, London B. Atrial contractile dysfunction, fibrosis, and arrhythmias in a mouse model of cardiomyopathy secondary to cardiac-specific overexpression of tumor necrosis factor-α. Am J Physiol Heart Circ Physiol. 2005;289:H1456–H1467. doi: 10.1152/ajpheart.00733.2004. [DOI] [PubMed] [Google Scholar]
- Sah VP, Minamisawa S, Tam SP, Wu TH, Dorn GW, 2nd, Ross J, Jr, Chien KR, Brown JH. Cardiac-specific overexpression of RhoA results in sinus and atrioventricular nodal dysfunction and contractile failure. J Clin Invest. 1999;103:1627–1634. doi: 10.1172/JCI6842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sassone J, Colciago C, Cislaghi G, Silani V, Ciammola A. Huntington's disease: the current state of research with peripheral tissues. Exp Neurol. 2009;219:385–397. doi: 10.1016/j.expneurol.2009.05.012. [DOI] [PubMed] [Google Scholar]
- Schauerte P, Scherlag BJ, Pitha J, Scherlag MA, Reynolds D, Lazzara R, Jackman WM. Catheter ablation of cardiac autonomic nerves for prevention of vagal atrial fibrillation. Circulation. 2000;102:2774–2780. doi: 10.1161/01.cir.102.22.2774. [DOI] [PubMed] [Google Scholar]
- Schroeder AM, Loh DH, Jordan MC, Roos KP, Colwell CS. Baroreceptor reflex dysfunction in the BACHD mouse model of Huntington's disease. PLoS Curr. 2011;3:RRN1266. doi: 10.1371/currents.RRN1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz PJ, La Rovere MT, Vanoli E. Autonomic nervous system and sudden cardiac death. Experimental basis and clinical observations for post-myocardial infarction risk stratification. Circulation. 1992;85:I77–I91. [PubMed] [Google Scholar]
- Sharma KR, Romano JG, Ayyar DR, Rotta FT, Facca A, Sanchez-Ramos J. Sympathetic skin response and heart rate variability in patients with Huntington disease. Arch Neurol. 1999;56:1248–1252. doi: 10.1001/archneur.56.10.1248. [DOI] [PubMed] [Google Scholar]
- Sorensen SA, Fenger K. Causes of death in patients with Huntington's disease and in unaffected first degree relatives. J Med Genet. 1992;29:911–914. doi: 10.1136/jmg.29.12.911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steele AD, Jackson WS, King OD, Lindquist S. The power of automated high-resolution behavior analysis revealed by its application to mouse models of Huntington's and prion diseases. Proc Natl Acad Sci U S A. 2007;104:1983–1988. doi: 10.1073/pnas.0610779104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stober T, Sen S, Burger L. Bradycardia and second-degree AV block: an expression of the dominance of cholinergic activity in the rigid form of Huntington's disease. J Neurol. 1983;229:129–132. doi: 10.1007/BF00313453. [DOI] [PubMed] [Google Scholar]
- Tan AY, Zhou S, Ogawa M, Song J, Chu M, Li H, Fishbein MC, Lin SF, Chen LS, Chen PS. Neural mechanisms of paroxysmal atrial fibrillation and paroxysmal atrial tachycardia in ambulatory canines. Circulation. 2008;118:916–925. doi: 10.1161/CIRCULATIONAHA.108.776203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan TP, Gao XM, Krawczyszyn M, Feng X, Kiriazis H, Dart AM, Du XJ. Assessment of cardiac function by echocardiography in conscious and anesthetized mice: importance of the autonomic nervous system and disease state. J Cardiovasc Pharmacol. 2003;42:182–190. doi: 10.1097/00005344-200308000-00005. [DOI] [PubMed] [Google Scholar]
- van der Burg JM, Bjorkqvist M, Brundin P. Beyond the brain: widespread pathology in Huntington's disease. Lancet Neurol. 2009;8:765–774. doi: 10.1016/S1474-4422(09)70178-4. [DOI] [PubMed] [Google Scholar]
- Videnovic A, Leurgans S, Fan W, Jaglin J, Shannon KM. Daytime somnolence and nocturnal sleep disturbances in Huntington disease. Parkinsonism Relat Disord. 2009;15:471–474. doi: 10.1016/j.parkreldis.2008.10.002. [DOI] [PubMed] [Google Scholar]
- Xiao HD, Fuchs S, Campbell DJ, Lewis W, Dudley SC, Jr, Kasi VS, Hoit BD, Keshelava G, Zhao H, Capecchi MR, Bernstein KE. Mice with cardiac-restricted angiotensin-converting enzyme (ACE) have atrial enlargement, cardiac arrhythmia, and sudden death. Am J Pathol. 2004;165:1019–1032. doi: 10.1016/S0002-9440(10)63363-9. [DOI] [PMC free article] [PubMed] [Google Scholar]