

Abstract
Recombinant immunotoxins (RITs) are chimeric proteins that are being developed for cancer treatment. We have produced RITs that contain PE38, a portion of the bacterial protein Pseudomonas exotoxin A. Because the toxin is bacterial, it often induces neutralizing antibodies, which limit the number of treatment cycles and the effectiveness of the therapy. Because T cells are essential for antibody responses to proteins, we adopted an assay to map the CD4+ T-cell epitopes in PE38. We incubated peripheral blood mononuclear cells with an immunotoxin to stimulate T-cell expansion, followed by exposure to overlapping peptide fragments of PE38 and an IL-2 ELISpot assay to measure responses. Our observation of T-cell responses in 50 of 50 individuals correlates with the frequency of antibody formation in patients with normal immune systems. We found a single, highly immunodominant epitope in 46% (23/50) of the donors. The immunodominant epitope is DRB1-restricted and was observed in subjects with different HLA alleles, indicating promiscuity. We identified two amino acids that, when deleted or mutated to alanine, eliminated the immunodominant epitope, and we used this information to construct mutant RITs that are highly cytotoxic and do not stimulate T-cell responses in many donors.
Keywords: deimmunization, immunogenicity, protein engineering, antidrug antibodies
Recombinant immunotoxins (RITs) are chimeric proteins that are being developed for the targeted therapy of cancer. Our laboratory has produced RITs composed of the variable fragment (Fv) of an antibody specific for a tumor-associated cell-surface antigen, joined to a 38-kDa fragment of Pseudomonas exotoxin A (PE38) that kills the target cell (1). We are currently developing RITs that target CD22 for B-cell malignancies (HA22, also known as “Moxetumomab Pasudotox”) and mesothelin for mesothelioma and other epithelial malignancies (SS1P). In a recently completed phase 1 trial in refractory hairy cell leukemia, HA22 had a response rate of 86%, with 46% complete remissions (2). HA22 also has produced complete remissions in several children with acute lymphoblastic leukemia (3). Although PE38 is a bacterial protein, HA22 does not frequently induce the formation of neutralizing antibodies in patients with hematological malignancies, because their immune systems are suppressed by chemotherapy and by the malignant cells, which proliferate in the bone marrow. This suppression usually allowed HA22 to be given for many cycles, contributing to the high response rate (4). In contrast, the response rate to SS1P was much lower in patients with mesothelioma, who have normal immune systems that rapidly produce antibodies to PE38. Therefore, most patients only received a single cycle of treatment (4, 5).
The formation of neutralizing antibodies is a common occurrence when foreign proteins are used as therapeutic agents in humans (6, 7), and the more foreign the protein, the more likely it is that a rapid immune response will be generated (8–10). To deimmunize PE38 and thus allow more treatment cycles with RITs to be given, we initially focused on identifying and removing B-cell epitopes. We initially identified the B-cell epitopes in PE38 that are responsible for the mouse immune response and used this information to make mutant immunotoxins that can be given to mice for many cycles without inducing antibody production (11, 12). We have extended these studies to identify and remove human B-cell epitopes (13). T cells play a pivotal role in the ability to elicit an antibody response. One of the early events in the development of antibodies is the antigen-specific activation of CD4+ T-helper cells (14). CD4+ T-cell support is initiated by antigen-presenting cells (APCs), which display peptide fragments derived from foreign proteins on MHC class II molecules that bind T-cell receptors (14, 15). Several studies have identified human-specific T-cell epitopes in therapeutic proteins (16–18), and in some cases proteins were produced by mutating amino acids within the protein and were shown to be less immunogenic using mouse models (19–22).
The goal of the current study was to identify and remove human T-cell epitopes in immunotoxins containing PE38. Depending on the type of assay used, it is possible to identify peptides that result in T-cell activation, but these peptides might never be formed in vivo. To ensure that the epitopes we identified were naturally produced by APCs, we adapted an assay developed by Sette and colleagues (23) in which we first incubated donor peripheral blood mononuclear cells (PBMCs) with RITs for 14 d to allow the immunotoxin to be processed by donor APCs and relevant peptides to be presented to T cells. We subsequently exposed the activated T cells to overlapping synthetic peptides from the sequence of PE38 and used an ELISpot assay for IL-2 to measure T-cell activation. We analyzed samples from 50 normal donors with no recorded previous exposure to PE38 and with a broad distribution of HLA alleles and found that all donors showed a significant response to at least one peptide, as would be expected from a highly immunogenic foreign protein. We also found one immunodominant epitope that was HLA class II DRB1-restricted and promiscuous because of the diversity of donors that responded to it. Using alanine-scanning mutagenesis we identified single amino acids within PE38 that were responsible for the epitope and constructed highly active mutant RITs targeting CD22 in which the T-cell response was abolished in 34% of donors and diminished in an additional 42%.
Results
The structure of an RIT is shown in Fig. 1A. It is composed of an Fv fused to a 38-kDa portion of PE38. The Fv binds to the cancer cell and brings PE38 into the cell. PE38 is made up of two domains; domain II (amino acids 253–364) carries out toxin processing, and domain III (amino acids 395–613) contains the ADP ribosylating activity. Amino acids 365–394 are not needed for activity and are not present in PE38. Because of the bacterial origin of PE38, we focused on that portion of the RIT.
Fig. 1.

Structural model of RIT variants. (A) Structural model of Moxetumomab Pasudotox. The VL is in cyan, and the VH is in magenta. Domain II of the toxin is in gray, and domain III is in yellow. (B) HA22-LR. The linker containing the furin cleavage sequences is in gray. Image courtesy of Byungkook Lee (National Cancer Institute, NIH, Bethesda, MD).
Identification of T-Cell Epitopes.
To identify T-cell epitopes in the PE38, we used an assay originally developed to identify peptides responsible for human T-cell responses to a hay fever allergen (23). We stimulated PBMCs from 50 donors with intact RIT, followed by restimulation with 111 15-mer overlapping peptides spanning the entire sequence of PE38 (Table S1) and an ELISpot assay for IL-2 to measure T-cell response (Fig. S1).
Fig. 2A shows an example of a screen of pools for one of the 50 donors. Only a single pool (pool 3) had a response that met the three criteria we established for a positive response [≥85 spot-forming cells (SFCs) per 106 cells, more than three times the number in the negative control, and ≥10% of all of the spots for that donor]. The response to pool 14 fulfilled two of the three criteria (the response was ≥85 SFC per 106 cells and was greater than three times that of the negative control); however, the spots made up only 8% of the total spots, and therefore the response not considered positive. We subsequently performed a fine screen of pool 3 with the individual peptides (Fig. 2B) and found that only peptides 14 and 15 gave positive responses.
Fig. 2.

Representative pattern of CD4+ T-cell IL-2 secretion in response to PE38-derived peptides in a single donor. (A) IL-2 secretion in response to stimulation with 22 peptide pools from PE38. (B) IL-2 response to the five peptides that comprise pool 3. (C) IL-2 response of isolated CD4+ T cells to the five peptides that comprise pool 3.
Pool 3 contains an immunodominant epitope.
We screened a total of 50 donors and found that all 50 gave positive responses, as would be expected for a highly immunogenic protein such as PE38 (4). Fig. 3A presents the positive and negative responses of the 50 donors in a heatmap format. The strongest positive response for each donor is shown in black, weaker responses in gray, and the absence of a response in white. We found 108 positive responses and 992 negative responses (white). Pools 4, 5, 13, 21, and 22 gave no responses. In 16 of the 50 donors a single pool induced a response. Fifteen donors had two responses, 14 donors had three responses, and 5 donors had four responses. Fig. 3A also shows that pool 3 produced the most responses (23 of 50), and for nine of these donors it was the only pool that induced a response. We also found that the second most frequent responses were in pool 16 in domain III, where 16 of 50 donors responded.
Fig. 3.

CD4+ T-cell IL-2 secretion in response to PE38-derived peptide pools. Samples from 50 normal donors were stimulated with PE38-containing immunotoxin for 14 d and were restimulated with the overlapping peptides pools. Each pool was tested in quadruplicate, and SFC/1E6 cells were calculated. (A) Visual illustration of the strongest (black), weak (gray), and negative (white) responses. Donors were clustered using an automatic sorting based on the responsiveness of the pools. (B) Relative responses to 22 pools (n = 50).
Fig. 3B summarizes the responses of the 50 donors to stimulation by the 22 pools. As previously shown (24, 25), individual donors responded variably; to correct for these variations, the values were normalized to the total number of spots for each donor. Pool 3 elicited a positive response in 23 donors, which is extraordinarily strong. The pool 3 responses were statistically greater than all other pools (P < 0.0001 in Friedman’s test). Out of the entire screen for all of the donors and all of the pools (106,998 SFC per106 cells), pool 3 in domain II had more than 21% of all of the spots in the screen (22,850 SFC per106 cells), and pool 16 in domain III produced the second largest number of responses (8.2% of all of the spots in the screen).
To identify individual peptides within pool 3 that caused a response in 23 donors, samples were screened with individual peptides (Fig. 4A). Peptides 14 and 15 (which have a core sequence of LVALYLAARLSW) had a significantly stronger response than the other peptides in pool 3 (P < 0.0001, Mann–Whitney–Wilcoxon test). This epitope does not correspond to any human B-cell epitopes (13).
Fig. 4.

CD4+ T-cell response to single peptides that compose pool 3 and HLA II restriction. (A) Response to overlapping peptides that compose pool 3 (n = 23). (B) HLA class II restriction. Expanded PBMC were incubated with antibodies against HLA DR, DP, and DQ, a combination of the three (All), or class I (Pan). The responses to stimulation with peptide 15 were compared with the positive control (peptide 15 with no antibody), and the relative response was calculated for each donor (n = 9).
In addition to a fine screen of the peptides in pool 3, we identified the HLA-DRB1 allotypes of the donors. The samples used for the fine screen of pool 3 contained donors with many different HLA-DRB1 allotypes, including homozygosity for DRB1_04 and DRB1_15 (Table S2), indicating that the two immunodominant peptides comprise a promiscuous epitope.
Response is CD4+ T-cell specific.
To confirm that the epitopes are specific for CD4+ T cells, we performed the ELISpot assay using CD4+ cells isolated from the expanded PBMCs. Following the identification of peptides containing an epitope in a particular donor, CD4+ cells were isolated by negative MACS selection and were combined with irradiated (4,000 rad), freshly thawed autologous PMBCs in an ELISpot plate. Irradiated PBMCs were plated alone to confirm that those cells did not contribute to the response, and the CD4-fractionated cells also were plated to confirm that CD8+ or other PBMCs were not responsible for the response. A typical result from one of the 10 donors tested is shown in Fig. 2C. We found that only peptides 14 and 15 produced a significant response and that the strongest response was observed with CD4+ T cells. The response from CD4-depleted cells was very low and probably was attributable to contaminating CD4+ cells in the depleted population. There was no response from irradiated PBMCs. We conclude that the T-cell responses both are specific for peptides and are limited to the CD4+ subset.
HLA restriction.
To determine which HLA molecules are required for the T-cell response, we measured the ability of antibodies specific for HLA-DR, DP, or DQ to inhibit the response (Fig. 4B). The responses of all nine donors were inhibited by the anti-DR antibody but not by antibodies against either DP or DQ. A mixture of all three antibodies likewise inhibited the response. Antisera to HLA class I did not block the response. These data show that the responses are restricted by class II HLA-DR.
HLA types within the donor pool.
To determine if the responses occurred in donors with different HLA alleles, we analyzed the HLA-DRB1 status of all of the donors and compared this result with the frequency of HLA-DRB1 alleles in the human population. We found that the representation of HLA-DRB1 alleles in our cohort is not dramatically different from the worldwide distribution of human HLA (www.allelefrequencies.net) (26) (Fig. S2). Our cohort is enriched for HLA-DR08 and HLA-DR15 and lacks DR09, but the frequencies of these alleles are well within the reported ranges of some major ethnic groups (27). We conclude that the distribution of our donors is representative of the population that will be treated with RITs containing PE38.
Identification of amino acids required for T-cell activation.
To identify amino acid variants that reduced the CD4+ T cell responses in the immunodominant peptide, we synthesized a set of variant peptides for the sequence shared by peptides 14 and 15 (LVALYLAARLSW). To keep the peptide in a 15-mer format, we used the scaffold of peptide 15, because more donors responded to peptide 15 (21/23) than to peptide 14 (20/23). Each variant had one amino acid replaced with alanine, and in the place of alanine we used glycine. The sequences for the variants are shown in Table 1. The peptides were tested on 21 donors who were positive for peptide 15 in the IL-2 ELISpot assay. The number of spots relative to the variant peptides was normalized to the parent peptide, so that peptide 15 had a response of 100% for each run.
Table 1.
CD4+ T-cell response to alanine-variant peptides
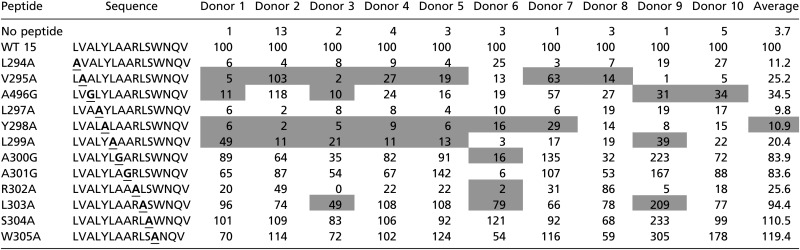 |
Data are shown as percent of response to WT peptide. Gray shading indicates a response of <10% to WT peptide. Boldface and underlining indicate the amino acid residues that were changed.
Representative responses of 10 of the 21 donors to each variant peptide are shown in Table 1. The average relative responses of the 21 donors to the mutant peptides L294A, L297A, Y298A, and R302A were below 20%. The mutant peptides L297A and Y298A had even more pronounced effects, with average relative responses below 11%. The responses to other variant peptides, such as S304A and W305A, were not greatly affected by the alanine mutations, with average relative scores of 110.5% and 119.4%, respectively.
Mutant immunotoxins.
Because the responses to peptides containing L297A and Y298A in domain II of PE38 were very low, we incorporated these mutations into HA22, making HA22-L297A and HA22-Y298A. We also were able to delete the entire region containing the epitope. In a previous study focused on eliminating B-cell epitopes in immunotoxin HA22-LR, we deleted most of domain II of PE38 (amino acids 253–364, corresponding to peptides 1–38) but maintained excellent cytotoxic activity. HA22-LR retains the 11 amino acids making up the furin site (in pool 2) (Fig. 1B); based on the in silico prediction algorithm (28) these 11 residues were not predicted to make up a T-cell epitope. We were able to purify the mutant RITs to near homogeneity, as shown in Fig. 5B. The proteins with point mutations and HA22 have the expected size of 63 kDa (lanes 1, 2 and 4), whereas HA22-LR has the expected molecular weight of 51 kDa (lane 5).
Fig. 5.

Purity and activity of mutant immunotoxins. (A) Tris⋅glycine gel (4–20%) run under nonreducing conditions shows the purity of the variant immunotoxins. Lane 1, HA22-L297A; lane 2, HA22-Y298A; lane 4, WT HA22; lane 5, HA22-LR. (B) Representative WST8 cytotoxicity assay in a CD22+ cell line (Daudi) after 3-d incubation with either HA22 or mutant immunotoxins.
Properties of mutant immunotoxins.
The cytotoxic activities of the mutant and WT RITs were measured in four cell lines, as previously described (29). Fig. 5A shows a representative assay using Daudi cells, and Table 2 shows the average EC50 values for each cell line. All three mutant immunotoxins (HA22-L297A, HA22-Y298A, and HA22-LR) were very cytotoxic, although somewhat less so than the parent molecule, HA22. Compared with HA22, HA22-L297A showed relative activities >50% in all four cell lines and was more active than the other two mutants in three of the four cell lines. We would not expect the differences in the relative activities to be clinically significant, because all mutants were very active.
Table 2.
Summary of EC50 and relative activity of parent and variant molecules on four CD22+ cell lines
| Protein | CA46 (n = 5) |
Raji (n = 5) |
Daudi (n = 4) |
HAL-01 (n = 4) |
||||
| EC50 ± SD (ng/mL) | Relative activity (%) | EC50 ± SD (ng/mL) | Relative activity | EC50 ± SD (ng/mL) | Relative activity (%) | EC50 ± SD (ng/mL) | Relative activity (%) | |
| HA22 | 0.18 ± 0.09 | 100 | 0.15 ± 0.09 | 100 | 0.1 ± 0.05 | 100 | 1.14 ± 0.43 | 100 |
| HA22-LR | 0.41 ± 0.27 | 45 | 0.45 ± 0.18 | 33 | 0.11 ± 0.05 | 85 | 1.5 ± 0.45 | 76 |
| HA22-L297A | 0.32 ± 0.15 | 58 | 0.27 ± 0.13 | 54 | 0.12 ± 0.05 | 83 | 2.25 ± 0.61 | 51 |
| HA22-Y298A | 0.79 ± 0.52 | 23 | 0.47 ± 0.34 | 32 | 0.34 ± 0.15 | 28 | 6.7 ± 1.16 | 17 |
The stability of the mutant immunotoxins was investigated by heating them for 15 min at various temperatures. We found that all four proteins were completely stable up to 50 °C. We found that L297A lost 50% of its activity at 51 °C, LR at 56 °C, Y298A at 57 °C, and HA22 at 58 °C (Fig. S3). Thus, the introduction of the point mutations had very little effect on thermal stability.
To test the capacity of the mutant RITs HA22-L297A and HA22-Y298A to stimulate a T-cell response, we synthesized mutant peptides containing the alanine mutations present in the mutant immunotoxins for all of the overlapping peptides that contain position L297A and Y298A (peptides 12, 13, 14, 15, and 16). We also examined the HA22- LR deletion mutant using the WT peptides. We pooled the variant peptides and used them to evaluate the immunogenicity of the variant RITs. PBMCS from donors known to have a positive response in pool 3 were cultured with HA22, HA22-L297A, HA22-Y298A, or HA22-LR and were restimulated with the appropriate peptide pools or the whole protein. A representative response to the four proteins is shown in Fig. 6. PBMCs that underwent in vitro expansion with WT HA22 and were restimulated with WT peptides on day 14 had a response of 713 SFC per106 cells in pool 3, which is statistically significant over the no-peptide control (P < 0.0001 in a Student t test). The same donors’ PBMCs were stimulated with the mutant proteins and then were restimulated with the mutant peptide pools (or WT peptides for LR). These cells had an extremely low response of <15 SFC per 106 cells, similar to that in the negative control. PBMCs that were stimulated with mutant proteins and restimulated with WT peptides showed no significant response. Furthermore, the deletion and removal of the amino acids in domain II (pools 1–8) did not induce the formation of new epitopes corresponding to the PE38 peptides. Restimulation using the whole protein did not give any response.
Fig. 6.

Mutant immunotoxins do not create new T-cell epitopes. Representative responses to 22 peptide pools after stimulation with (A) HA22, (B) HA22-LR, (C) HA22-L297A, or (D) HA22-Y298A and restimulation with appropriate mutant peptides.
Discussion
In this study we report the identification, characterization, and elimination of an immunodominant T-cell epitope in PE38. Our approach was to stimulate donor PBMCs with intact RIT, followed by restimulation with overlapping peptides and an ELISpot assay for IL-2 to measure T-cell response. We identified two overlapping peptides that contain an immunodominant, DR-restricted, promiscuous epitope and several subdominant epitopes. The immunodominant epitope, located in domain II of PE, stimulated substantial T-cell responses in 23 of 50 normal donors. We then used alanine-scanning mutagenesis to identify the amino acids responsible for establishing the epitope and used this information to construct mutant RITs that did not stimulate T cells but retained excellent cytotoxic activity.
In Vitro Expansion.
Previously, human helper T-cell epitopes have been identified by exposure of PBMCs or dendritic cells to peptides, followed by measurement of T-cell proliferation by thymidine incorporation. We initially tried variations of this assay and found, as previously described (30), that many donors did not respond to peptide stimulation and that often the magnitude of the response was low. The lack of a response in some donors was unexpected, because immunotoxins containing PE38 are highly immunogenic in humans with a normal immune system (4, 5). In an attempt to improve the number of responses, we adopted the assay developed by Sette and colleagues (23) to identify epitopes in a hay fever allergen. In this approach PBMCs are incubated with whole protein to allow the generation and presentation of normally processed peptides that initiate the expansion of responsive T cells. This first incubation is followed by a second one in which cells are stimulated with synthetic peptides and by an ELISpot assay to measure IL-2 production. The addition of the in vitro expansion step resulted in very strong and reproducible responses, ranging from hundreds to thousands of SFC per 106 cells in 100% of the donors. The background response level was very low, with a median of only 10 SFC per 106 cells. Because of the wide interest in deimmunizing proteins, we believe this approach could be of general use.
We initially evaluated IL-4, IFN-γ, and IL-2 to measure T-cell stimulation and found that they gave very similar responses. We used IL-2 because it supports T-cell activation, differentiation, and memory and is a less specialized cytokine than IL-4 or IFN-γ (31). In addition, it was shown previously in vaccination studies that IL-2 is a reliable indicator of CD4 T-cell activation, whereas IFN-γ was more variable (32).
In vitro expansion with whole protein is useful for evaluating the immunogenicity of mutant RITs, in which specific T-cell epitopes have been abolished. Evaluating T-cell activation with peptides alone is useful for showing that a known epitope is abolished (20), but it is insufficient for demonstrating that new epitopes have not been created. For instance, mutations could alter antigen processing of the protein and potentially generate new epitopes. This assay also allows one to evaluate the emergence of subdominant epitopes after the removal of an immunodominant epitope.
Previous Efforts to Map PE38 T-Cell Epitopes.
An earlier study investigating PE38 for the presence of CD4+ T-cell epitopes used a 3H-thymidine incorporation assay to monitor peptide activation of dendritic cells (30). This study identified three epitopes; the major one corresponds to peptides 75 and 76 (in pools 15 and 16), and the two others correspond to peptides 15 and 65 (in pools 3 and 13, respectively). The response rates were 23% for the major epitope and 14% for the other two. Peptides 75, 76, and 15 also were identified in our assay (peptides 75 and 76 were particularly responsive peptides in the fine screens of pools 15 and 16), although with markedly different dominance. Peptide 65, however, which was found to be equally as immunogenic as peptide 15 by dendritic cell presentation and T-cell proliferation, was not found in our study. We predict that this discrepancy is a result of antigen processing and presentation of the full RIT, such that peptide 65 is irrelevant in a native setting.
HLA II Restriction and Promiscuity.
To investigate the molecular basis of the immunodominant epitope, we evaluated the HLA restriction of the responsive donors and found that responses were restricted to class II and specifically to HLA DR. Examination of the HLA DRB1 composition of the donors that responded to peptides 14 and 15 shows a wide range of haplotypes, including homozygosity and numerous combinations of different DR haplotypes (Table S2). This result indicates that this epitope is promiscuous among several HLA class II haplotypes. Moreover, in silico prediction using Propred (28), which predicts the binding of peptides to HLA class II molecules, supports our conclusion. The peptides corresponding to the immunodominant epitope are predicted to bind in the top third percentile to 48 of the 51 DR molecules covered by the software, indicating a strong, promiscuous epitope. We therefore expect that removal of this single epitope will affect a broad and diverse population and will allow many patients to benefit from the deimmunization of the RIT.
Southwood et al. (33) described an HLA-DR supertype that is characterized by a large group of DR molecules that promiscuously bind to an overlapping peptide repertoire. The supertype includes DRB1_0101, DRB1_0401, and DRB1_0701, which share a binding motif characterized by a large aromatic or hydrophilic residue in position 1 and a small noncharged residue in position 6. The promiscuous epitope identified in our study, LVALYLAARLSW, matches this description, with the aromatic tyrosine being at P1 and the small leucine at P6.
Deletion of the Immunodominant Epitope.
In this study we demonstrate that the epitope can be destroyed by point mutations that presumably interfere with binding to the HLA molecule or to the T-cell receptor. Alternatively, it can be destroyed by a deletion of the epitope as part of a major deletion of a large portion of PE38 (Fig. 1A) (34). Weldon et al. (29) have reported that most of the amino acids in domain II, except for an 11-residue sequence comprising the furin cleavage site, can be deleted from HA22 without major loss of cytotoxic activity (Fig. 1B). Deletion of the entire epitope should be more beneficial than the point mutations, because it eliminates both the immunodominant epitope in the pool and the subdominant epitopes in pools 1 and 2. Based on the heatmap in Fig. 3A that shows that 18 of the 50 donors had epitopes only in domain II, the LR deletion should eliminate 36% of responses completely.
New Epitopes Are Not Created.
One potential concerns surrounding the elimination of T-cell epitopes are that new epitopes will appear as a consequence of the mutation or that cryptic epitopes will emerge that were suppressed by the stronger epitopes (35). We used alanine mutagenesis because alanine substitutions reduce the binding of a peptide to an HLA molecule (36, 37). We examined the mutant immunotoxins and did not find new epitopes, probably because the alanine substitution destroyed the epitope, either by disrupting peptide–HLA binding or, less likely, by disrupting the binding to the T-cell receptor. This result is in agreement with findings by Yeung et al. (38), who identified murine T-cell epitopes in human IFN-β and found that eliminating the immunodominant epitopes did not result in a response directed at the subdominant epitope.
In summary, we have identified the T-cell epitopes in PE38, including a promiscuous immunodominant epitope that can be eliminated without a major loss in activity or stability. Immunotoxin HA22-LR, with a deletion of almost all the amino acids in domain II, was not immunogenic in 34% of donors and was less immunogenic in an additional 42% of the donors. We now plan to combine the domain II deletion with point mutations that remove epitopes in domain III. These deletions should produce an RIT with very low immunogenicity that can be investigated in animal models. We anticipate that immunotoxins containing mutations that eliminate T-cell epitopes can be given for several cycles to patients with normal immune systems, allowing better antitumor responses to be achieved.
Materials and Methods
Peptide Synthesis.
A series of 15-mer peptides, which overlap by 12 amino acids and span the entire sequence of PE38, were synthesized by American Peptides. Peptides were purified to >95% homogeneity by HPLC, and their composition was confirmed by mass spectrometry. Peptides were dissolved in DMSO at 10 mM and stored at −20 °C. For initial screening, peptides were pooled into groups of five consecutive peptides, with the exception of pool 22, which contained six peptides (Table S1).
Human Donor PBMC Samples.
Apheresis samples from healthy volunteer donors at the National Institutes of Health (NIH) blood bank were used for all experiments. Specimens were collected under research protocols approved by the NIH Institutional Review Board (99-CC-0168) and were obtained after informed consent. PBMCs were isolated by Ficoll-Hypaque (GE Healthcare) density-gradient separation according to manufacturer’s instructions, and the buffy coat was collected and washed three times with Dulbecco's PBS without Ca and Mg. PBMCs were cryopreserved in liquid nitrogen at a concentration of 1–3 × 107 cells/mL. Class I and II HLA typing was performed using PCR sequence-specific primers/sequence-specific oligonucleotides–based tissue typing by the HLA typing unit in the Warren G. Magnuson Clinical Center, NIH.
In Vitro Expansion of PE38-Specific T Cells.
In vitro expansion was carried out as previously described (23). PBMCs were stimulated with an RIT containing PE38 (typically LMB9) (39), because we had a large amount of this protein available in a highly purified state at a final concentration of 5 μg/mL or 75 mM. Medium alone was used as a negative control, and CEFT (5 µg/mL of each peptide in the peptide mixture) (Axxura) was used as a positive control. On day 14, cells were harvested, washed once, and screened for reactivity against peptide pools. On day 17, the screen was repeated along with fine screens for the individual peptides.
ELISpot Assays.
The secretion of IL-2 following stimulation with RIT was analyzed in an IL-2 ELISpot assay according to manufacturer’s recommendations (Mabtech). Negative controls were incubated with medium, and positive controls were incubated with CEFT peptides or phytohemagglutinin (Sigma). Spots were counted by computer-assisted image analysis (Immunospot 5.0; Cellular Technology Limited). Each assay was performed in quadruplicate. Positive pools were fine screened to identify the individual immunogenic peptides by testing individual peptides from the pool. The threshold for a positive response was based on an initial analysis of the results from the screening of five donors and included three factors: A response was considered positive if (i) the value was ≥85 SFCs per 106 cells, (ii) the value was more than three times that of the negative control, and (iii) the spots in the pool made up more than 10% of all of the spots for that donor. This threshold gave reproducible responses for all donors.
CD4+ T-Cell Isolation.
CD4+ T cells were purified by negative selection from frozen aliquots of PBMCS using the MACS CD4+ T-cell isolation kit and LS separation column (Miltenyi Biotec) according to the manufacturer’s protocol. CD4+ T-cell populations were >80% pure, as determined by flow cytometry, and were >90% viable as judged by Trypan blue exclusion.
HLA Class II Restriction.
Antibody inhibition assays were performed to determine the HLA restriction of the immunodominant epitope as previously described (23). The responses of nine donors from various HLA groups to peptide 15 when inhibited with HLA antibodies was measured. The responses were normalized to the untreated control.
Construction, Expression, and Purification of RIT.
HA22 and mutant RITs thereof are composed of the heavy-chain Fv fused to PE38 (VH-PE38) disulfide-linked to the light-chain Fv (VL). The mutations L297A and Y298A were introduced into the parent expression plasmid (HA22 VH-PE38) using PCR overlap extension (40). The resulting PCR products were cloned back into the parent plasmid, and the mutations were confirmed by DNA sequencing. All RITs were purified by a standard protocol (41).
WST8 Assay.
Cytotoxicity assays were performed on CD22+ human Burkitt lymphoma cell lines (CA46, Raji, and Daudi) and an acute lymphoblastic leukemia cell line (HAL-01). The assay was performed as previously described (29).
Thermal Stability.
RITs were heated for 15 min at various temperatures as previously described (42).
Statistical Analysis.
A nonparametric Friedman’s test was used to compare the screen results of the 22 pools for 50 donors. P < 0.01 was considered statistically significant. The Mann–Whitney–Wilcoxon test was used to compare the alanine-variant peptides and positive single peptides within the pools.
Supplementary Material
Acknowledgments
We thank Dr. Byungkook Lee for providing the figures showing the structures of the immunotoxins, Dr. Paul Robins for immunological advice, Susan L. Strobl for help in technical troubleshooting, Dr. Alexandro Sette and Carla Ossehof for sharing their epitope-mapping protocol, Dr. Onda Massanori for providing useful reagents, and Dr. Jay Berzofsky, Dawn Walker and Dr. Jon Yewdell for providing helpful comments. This research was supported by the Intramural Research Program of the Center for Cancer Research, National Cancer Institute, National Institutes of Health and by a Cooperative Research and Development Agreement with MedImmune, LLC.
Footnotes
The authors declare no conflict of interest.
See Author Summary on page 20790 (volume 109, number 51).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1218138109/-/DCSupplemental.
References
- 1.FitzGerald DJ, Wayne AS, Kreitman RJ, Pastan I. Treatment of hematologic malignancies with immunotoxins and antibody-drug conjugates. Cancer Res. 2011;71(20):6300–6309. doi: 10.1158/0008-5472.CAN-11-1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kreitman RJ, et al. Phase I trial of anti-CD22 recombinant immunotoxin moxetumomab pasudotox (CAT-8015 or HA22) in patients with hairy cell leukemia. J Clin Oncol. 2012;30(15):1822–1828. doi: 10.1200/JCO.2011.38.1756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wayne AS, et al. A novel anti-CD22 immunotoxin, moxetumomab pasudotox: Phase I study in pediatric acute lymphoblastic leukemia. Blood. 2011;118:1317a. doi: 10.1182/blood-2017-02-749101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hassan R, et al. Phase I study of SS1P, a recombinant anti-mesothelin immunotoxin given as a bolus I.V. infusion to patients with mesothelin-expressing mesothelioma, ovarian, and pancreatic cancers. Clin Cancer Res. 2007;13(17):5144–5149. doi: 10.1158/1078-0432.CCR-07-0869. [DOI] [PubMed] [Google Scholar]
- 5.Kreitman RJ, Hassan R, Fitzgerald DJ, Pastan I. Phase I trial of continuous infusion anti-mesothelin recombinant immunotoxin SS1P. Clin Cancer Res. 2009;15(16):5274–5279. doi: 10.1158/1078-0432.CCR-09-0062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wolbink GJ, Aarden LA, Dijkmans BA. Dealing with immunogenicity of biologicals: Assessment and clinical relevance. Curr Opin Rheumatol. 2009;21(3):211–215. doi: 10.1097/bor.0b013e328329ed8b. [DOI] [PubMed] [Google Scholar]
- 7.Krieckaert CL, Bartelds GM, Wolbink GJ. Therapy: Immunogenicity of biologic therapies-we need tolerance. Nat Rev Rheumatol. 2010;6(10):558–559. doi: 10.1038/nrrheum.2010.153. [DOI] [PubMed] [Google Scholar]
- 8.Nagata S, Pastan I. Removal of B cell epitopes as a practical approach for reducing the immunogenicity of foreign protein-based therapeutics. Adv Drug Deliv Rev. 2009;61(11):977–985. doi: 10.1016/j.addr.2009.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.van den Berg HA, Rand DA. Foreignness as a matter of degree: The relative immunogenicity of peptide/MHC ligands. J Theor Biol. 2004;231(4):535–548. doi: 10.1016/j.jtbi.2004.07.008. [DOI] [PubMed] [Google Scholar]
- 10.Shankar G, Shores E, Wagner C, Mire-Sluis A. Scientific and regulatory considerations on the immunogenicity of biologics. Trends Biotechnol. 2006;24(6):274–280. doi: 10.1016/j.tibtech.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 11.Onda M, et al. Characterization of the B cell epitopes associated with a truncated form of Pseudomonas exotoxin (PE38) used to make immunotoxins for the treatment of cancer patients. J Immunol. 2006;177(12):8822–8834. doi: 10.4049/jimmunol.177.12.8822. [DOI] [PubMed] [Google Scholar]
- 12.Hansen JK, et al. A recombinant immunotoxin targeting CD22 with low immunogenicity, low nonspecific toxicity, and high antitumor activity in mice. J Immunother. 2010;33(3):297–304. doi: 10.1097/CJI.0b013e3181cd1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu W, et al. Recombinant immunotoxin engineered for low immunogenicity and antigenicity by identifying and silencing human B-cell epitopes. Proc Natl Acad Sci USA. 2012;109(29):11782–11787. doi: 10.1073/pnas.1209292109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Parker DC. T cell-dependent B cell activation. Annu Rev Immunol. 1993;11:331–360. doi: 10.1146/annurev.iy.11.040193.001555. [DOI] [PubMed] [Google Scholar]
- 15.Parker DC. The functions of antigen recognition in T cell-dependent B cell activation. Semin Immunol. 1993;5(6):413–420. doi: 10.1006/smim.1993.1047. [DOI] [PubMed] [Google Scholar]
- 16.Harding FA, et al. A beta-lactamase with reduced immunogenicity for the targeted delivery of chemotherapeutics using antibody-directed enzyme prodrug therapy. Mol Cancer Ther. 2005;4(11):1791–1800. doi: 10.1158/1535-7163.MCT-05-0189. [DOI] [PubMed] [Google Scholar]
- 17.Tangri S, et al. Rationally engineered therapeutic proteins with reduced immunogenicity. J Immunol. 2005;174(6):3187–3196. doi: 10.4049/jimmunol.174.6.3187. [DOI] [PubMed] [Google Scholar]
- 18.Cantor JR, et al. Therapeutic enzyme deimmunization by combinatorial T-cell epitope removal using neutral drift. Proc Natl Acad Sci USA. 2011;108(4):1272–1277. doi: 10.1073/pnas.1014739108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.De Groot AS, Knopp PM, Martin W. De-immunization of therapeutic proteins by T-cell epitope modification. Dev Biol (Basel) 2005;122:171–194. [PubMed] [Google Scholar]
- 20.Moise L, et al. Effect of HLA DR epitope de-immunization of Factor VIII in vitro and in vivo. Clin Immunol. 2012;142(3):320–331. doi: 10.1016/j.clim.2011.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jones TD, et al. The development of a modified human IFN-alpha2b linked to the Fc portion of human IgG1 as a novel potential therapeutic for the treatment of hepatitis C virus infection. J Interferon Cytokine Res. 2004;24(9):560–572. doi: 10.1089/jir.2004.24.560. [DOI] [PubMed] [Google Scholar]
- 22.Warmerdam PA, et al. Elimination of a human T-cell region in staphylokinase by T-cell screening and computer modeling. Thromb Haemost. 2002;87(4):666–673. [PubMed] [Google Scholar]
- 23.Oseroff C, et al. Molecular determinants of T cell epitope recognition to the common Timothy grass allergen. J Immunol. 2010;185(2):943–955. doi: 10.4049/jimmunol.1000405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Deenadayalan A, et al. Immunoproteomic identification of human T cell antigens of Mycobacterium tuberculosis that differentiate healthy contacts from tuberculosis patients. Mol Cell Proteomics. 2010;9(3):538–549. doi: 10.1074/mcp.M900299-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Khan N, et al. T cell recognition patterns of immunodominant cytomegalovirus antigens in primary and persistent infection. J Immunol. 2007;178(7):4455–4465. doi: 10.4049/jimmunol.178.7.4455. [DOI] [PubMed] [Google Scholar]
- 26.Gonzalez-Galarza FF, Christmas S, Middleton D, Jones AR. Allele frequency net: A database and online repository for immune gene frequencies in worldwide populations. Nucleic Acids Res. 2011;39(Database issue):D913–D919. doi: 10.1093/nar/gkq1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Marsh SGE, Parham P, Barber LD. The HLA Factsbook. San Diego: Academic; 2000. [Google Scholar]
- 28.Singh H, Raghava GP. ProPred: Prediction of HLA-DR binding sites. Bioinformatics. 2001;17(12):1236–1237. doi: 10.1093/bioinformatics/17.12.1236. [DOI] [PubMed] [Google Scholar]
- 29.Weldon JE, et al. A protease-resistant immunotoxin against CD22 with greatly increased activity against CLL and diminished animal toxicity. Blood. 2009;113(16):3792–3800. doi: 10.1182/blood-2008-08-173195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Harding FA. 2008. MedImmune L US 2008/0193976.
- 31.Tassignon J, et al. Monitoring of cellular responses after vaccination against tetanus toxoid: Comparison of the measurement of IFN-gamma production by ELISA, ELISPOT, flow cytometry and real-time PCR. J Immunol Methods. 2005;305(2):188–198. doi: 10.1016/j.jim.2005.07.014. [DOI] [PubMed] [Google Scholar]
- 32.De Rosa SC, et al. Vaccination in humans generates broad T cell cytokine responses. J Immunol. 2004;173(9):5372–5380. doi: 10.4049/jimmunol.173.9.5372. [DOI] [PubMed] [Google Scholar]
- 33.Southwood S, et al. Several common HLA-DR types share largely overlapping peptide binding repertoires. J Immunol. 1998;160(7):3363–3373. [PubMed] [Google Scholar]
- 34.Allured VS, Collier RJ, Carroll SF, McKay DB. Structure of exotoxin A of Pseudomonas aeruginosa at 3.0-Angstrom resolution. Proc Natl Acad Sci USA. 1986;83(5):1320–1324. doi: 10.1073/pnas.83.5.1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu Z, Williams KP, Chang YH, Smith JA. Immunodominance: A single amino acid substitution within an antigenic site alters intramolecular selection of T cell determinants. J Immunol. 1993;151(4):1852–1858. [PubMed] [Google Scholar]
- 36.Allen PM, et al. Identification of the T-cell and Ia contact residues of a T-cell antigenic epitope. Nature. 1987;327(6124):713–715. doi: 10.1038/327713a0. [DOI] [PubMed] [Google Scholar]
- 37.Johansen BH, Vartdal F, Eriksen JA, Thorsby E, Sollid LM. Identification of a putative motif for binding of peptides to HLA-DQ2. Int Immunol. 1996;8(2):177–182. doi: 10.1093/intimm/8.2.177. [DOI] [PubMed] [Google Scholar]
- 38.Yeung VP, et al. Elimination of an immunodominant CD4+ T cell epitope in human IFN-beta does not result in an in vivo response directed at the subdominant epitope. J Immunol. 2004;172(11):6658–6665. doi: 10.4049/jimmunol.172.11.6658. [DOI] [PubMed] [Google Scholar]
- 39.Kuan CT, Pastan I. Improved antitumor activity of a recombinant anti-Lewis(y) immunotoxin not requiring proteolytic activation. Proc Natl Acad Sci USA. 1996;93(3):974–978. doi: 10.1073/pnas.93.3.974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Horton RM, Cai ZL, Ho SN, Pease LR. Gene splicing by overlap extension: Tailor-made genes using the polymerase chain reaction. Biotechniques. 1990;8(5):528–535. [PubMed] [Google Scholar]
- 41.Pastan I, Beers R, Bera TK. Recombinant immunotoxins in the treatment of cancer. Methods Mol Biol. 2004;248:503–518. doi: 10.1385/1-59259-666-5:503. [DOI] [PubMed] [Google Scholar]
- 42.Liu W, et al. A recombinant immunotoxin engineered for increased stability by adding a disulfide bond has decreased immunogenicity. Protein Eng Des Sel. 2012;25(1):1–6. doi: 10.1093/protein/gzr053. [DOI] [PMC free article] [PubMed] [Google Scholar]
