

Abstract
The Topoisomerase II (topo II) DNA incision and ligation cycle can be poisoned (e.g following treatment with cancer chemotherapeutics) to generate cytotoxic DNA double strand breaks (DSBs) with topo II covalently conjugated to DNA. Tyrosyl-DNA phosphodiesterase 2 (Tdp2) protects genomic integrity by reversing 5′-phosphotyrosyl (5′-Y) linked topo II-DNA adducts. Here, X-ray structures of mouse Tdp2-DNA complexes reveal that a Tdp2 β-2-helix-β DNA damage binding “grasp”, helical “cap”, and DNA lesion binding elements fuse to form an elongated protein-DNA conjugate substrate interaction groove. The Tdp2 DNA binding surface is highly tailored for engagement of 5′-adducted ssDNA ends, and restricts non-specific endonucleolytic or exonucleolytic processing. Structural, mutational and functional analyses support a single-metal ion catalytic mechanism for the endonuclease–exonuclease–phosphatase (EEP) nuclease superfamily, and establish a molecular framework for targeted small molecule blockade of Tdp2-mediated resistance to anti-cancer topoisomerase drugs.
To relieve DNA topological strain and facilitate cellular DNA and DNA/RNA transactions, type II topoisomerases metabolize DNA topoisomers by incising DNA, gating passage of a second DNA duplex through a topo II-linked DSB, and re-ligating the DNA break. The reversibility of topo II DNA cleavage reactions is facilitated by the formation of covalent enzyme-phosphotyrosyl linkages between the 5′-phosphate ends of the incised duplex and an active site topo II tyrosine. Although topo II-DSB intermediates are transient, genetic and environmental perturbations can accelerate topo II DNA cleavage, or stall topoisomerase re-ligation3,4,5, shifting DNA cleavage and ligation equilibrium towards production of excessive DSBs that retain topoisomerase subunits covalently adducted to the DSB 5′ termini via their active site tyrosine residue1,2. Left un-processed, such protein-adducted DNA ends are expected to block DNA double strand break repair. Widely prescribed and potent anticancer chemotherapeutic topo II poisons such as the anthracyclines (e.g. Adriamycin) and etoposide pharmacologically exploit this topoII mechanistic vulnerability to create genomic instability and cell death6,7.
Vertebrate tyrosyl-DNA phosphodiesterase Tdp2 (also known as TTRAP or EapII) processes topo II-adducts to 5′-phosphorylated DNA termini via direct reversal of the 5′-phosphotyrosyl linkage8 (Fig. 1a). The turnover of stalled type II topoisomerase covalent complexes proceeds via a ubiquitin proteasome degradation pathway, so Tdp2 may remove degraded Topo2 peptides covalently linked to the 5′ terminus 9,10,11. Targeted RNAi knockdown of Tdp2 sensitizes A549 lung cancer cells to etoposide, and increases formation of nuclear γH2AX foci, a marker of DSBs8, supporting the notion that Tdp2 is an important component in enabling cellular repair of topoII-adducted DSBs. Tdp2 is also overexpressed in lung cancers, and transcriptionally up-regulated in mutant p53 cells12. Thus, it is hypothesized that Tdp2 functions in cellular topo II drug resistance13 and mediates mutant p53 gain of function phenotypes, including acquisition of therapy resistance during cancer progression12. However, the molecular basis underlying Tdp2 topo II-DNA adduct repair activities remains unclear in the absence of protein structural information for any Tdp2 homolog.
Figure 1. Tdp2 catalytic activity.
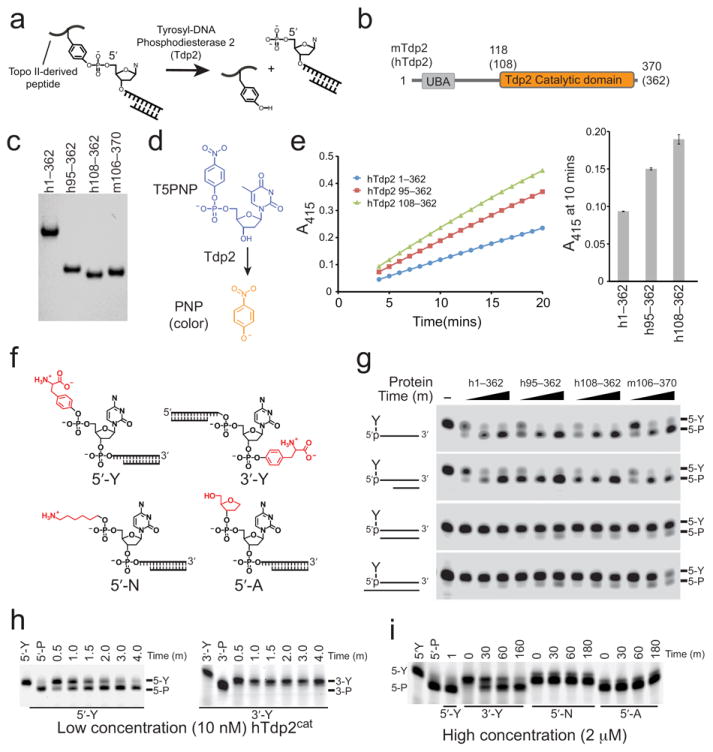
(a) Poisoned topo II results in a tyrosine covalently linked to the 5′-phosphate of a dsDNA break with 5′overhangs. The 5′-Y bond is cleaved by Tdp2. (b) Domain structure of mammalian Tdp2 homologs showing mouse and human (bracketed) amino acid domain boundaries. (c) SDS-PAGE of purified human (h) and mouse (m) Tdp2 proteins used in structural and activity assays. (d) Tdp2 hydrolyzes T5PNP to produce p-nitrophenolate (PNP). (e) Catalytic activity of Tdp2 proteins assayed using the T5PNP reagent. Error bars indicate the standard deviation from three independent measurements. (f) Structures of assayed substrates with varied 5′ and 3′ modifications. (g) Catalytic activity of Tdp2 on 5′-Y substrates in the context of indicated secondary structures analyzed by denaturing gel electrophoresis. (h) 5′- vs 3′-phosphotyrosine cleavage assayed using 1 μM synthetic oligo containing a phosphotyrosine at the indicated terminus with 10 nM hTdp2cat, analyzed as in panel g. (i) Tdp2cat activity assayed on sub-optimal substrates using 1 μM synthetic oligo containing the indicated terminal phosphate modification with 2 μM hTdp2cat analyzed as in panel g.
Tdp2 is a two-domain DNA repair protein with an N-terminal ubiquitin associated (UBA) domain that may link Tdp2 to cellular signaling and stress responses9, and a carboxyl terminal exonuclease-endonuclease-phosphatase (EEP) catalytic domain (Fig. 1b). EEP domain nucleases cleave DNA and RNA backbones and have diverse cellular functions including RNA processing (eg. the CNOT6L poly-A deadenylase14), and DNA repair (Tdp2 and Ape1)8,15. Through use of a common enzymatic scaffold, EEP phosphoesterases have evolved very diverse substrate specificities. Tdp2 is particularly intriguing and distinct in that it processes protein-DNA conjugates. This raises the question of how Tdp2 identifies its substrates, and how Tdp2 discriminates 5′-terminal DNA adducts from polynucleotides to prevent inappropriate endo- or exonucleolytic cleavages, and how this specificity and activity might be regulated. To clarify Tdp2 functions in genomic maintenance and cellular cancer therapeutic resistance, we report combined structural and functional characterization of Tdp2 catalytic activity and enzymatic selectivity.
RESULTS
Tdp2 domain mapping and catalytic activity
We used limited trypsin proteolysis (Supplementary Fig. 1), truncation mutagenesis, sequence analysis (Supplementary Fig. 2), and small angle X-ray scattering (SAXS) coupled to measurement of Tdp2 5′ tyrosyl-phosphodiesterase activity (Figs. 1c–e) to identify the minimal catalytically active domain (referred to as Tdp2cat hereafter) from human (hTdp2cat, residues 108–362) and murine (mTdp2cat, residues 118–370) Tdp2 (Supplementary Fig. 2). Analysis of the SAXS electron pair distribution function and solution scattering parameters (Supplementary Fig. 3, and Supplementary Table 1) shows that full length hTdp2 (hTdp2FL) adopts an elongated structure (maximum particle dimension, 102 Å) that is susceptible to trypsin protease digestion (Supplementary Fig. 1). In contrast, a truncated, trypsin-stable hTdp2cat fragment is globular based on the SAXS analysis (Supplementary Fig. 3). Thus, the Tdp2 N-terminal UBA domain (Fig. 1b), that may interface with the cellular signaling apparatus and ubiquitination machinery16,17, appears to be flexibly linked to the folded C-terminal EEP domain core.
Purified hTdp2cat and mTdp2cat proteins (Fig. 1c) have Mg2+-dependent (Supplementary Fig. 4a) activity on 5′-tyrosylated (5′-Y) termini of single-stranded DNA or on duplex substrates with 1–4 nt 5′-overhangs, as well as the small molecule 5′-Y mimic thymidine 5′-monophosphate p-nitrophenyl ester (T5PNP) (Figs. 1d–g, Supplementary Fig. 4b). Blunt or 5′-recessed 5′-Y termini (Fig. 1g), and 3′-Y ssDNAs are comparatively poor substrates (Figs. 1h,i). In addition, even at high enzyme concentrations in excess of substrate, Tdp2 displays no detectable endo- or exonuclease activity, showing that Tdp2 is highly specific for a 5′-Y terminus. Furthermore, Tdp2 displays no detectable activity on a 5′-adducted alkylamine (5′-N) adduct, nor can it remove a 5′-tetrahydrofuran ring abasic site mimic despite its similarity in size to a tyrosine (Figs. 1f,i). This suggests that in addition to recognizing the size and shape of the 5′ adduct, Tdp2 may also exploit the aromatic character of the tyrosine leaving group to promote catalysis. Consistent with published work8, Tdp2 processes 3′-Y adducts, albeit only at high protein concentration with extended incubation (Fig. 1i). Thus, mammalian Tdp2 homologs display enzymatic preferences for the secondary structure of the DNA end (single-stranded 5′-overhangs), the polarity of phosphotyrosine adduction (5′ ≫ 3′), and for the chemical structure of the 5′ adduct. The Tdp2 specificity for 5′-Y within 5′ overhangs is most consistent with the role for Tdp2 in promoting type II topoisomerase protein-adduct repair. Deletion of the Tdp2 N-terminal domain does not alter substrate preferences, but confers a ~1.5–2 fold increase of Tdp2 catalytic activity on T5PNP and the 5′ tyrosylated polynucleotide substrates (Fig. 1e,g). Thus, the flexibly linked Tdp2 N-terminus may regulate Tdp2 activity.
Crystallization of Tdp2–DNA complexes
Mouse Tdp2cat X-ray crystal structures were determined in 4 distinct states, including 3 DNA–protein complexes (see online Methods): 1) DNA complex I, an mTdp2cat–5′-N-DNA 5′-adduct substrate analog complex (at 2.1 Å, in a Mg2+ free form), 2) DNA complex II, a mTdp2cat–5′-PO4-DNA Mg2+ catalytic product complex (at 1.5 Å), 3) DNA complex III, an mTdp2cat–DNA–Mg2+ complex, with ssDNA excluded from the active site (at 1.85 Å), and 4) An mTdp2cat–Mg2+–5′-dAMP complex (see Table 1). An 8 base-pair DNA duplex with 1nt overhangs derived from a self-complimentary oligonucleotide (Supplementary Table 2) was utilized for crystallization of the non-hydrolyzable substrate analog 5′-N adduct (complex I) and the 5′-PO4 reaction product complex (complex II) (Fig. 2 and Supplementary Fig. 5). Importantly, Tdp2 is catalytically active on 1nt overhanging substrates (Supplementary Fig. 4b), though activity is decreased, probably due to reduced accessibility of the 5′ terminus in these substrates, as compared to preferred 2 or 4 nucleotide overhangs. In all three DNA complex structures Tdp2 utilizes conserved protein side chains to extensively engage the terminal nucleotides of the DNA 5′-adducted strand (Fig. 2a–c, blue DNA strands, and Supplementary Fig. 6). In contrast, there is a dearth of DNA–protein contacts made to the 3′ end of the complementary strand of the duplex DNA (gray DNA strands, Figures 2a–c) employed as a vehicle for protein–DNA co-complex crystallization. Thus, the use of two-ended crystallization substrates has trapped protein–DNA interactions relevant to Tdp2 DNA processing and specificity for 5′-strand overhanging ends (Fig. 1g), consistent with a biological role for Tdp2 processing of TopoII linked double strand breaks8.
Table 1.
Data collection and refinement statistics
| mTdp2cat–Mg2+–dAMP | mTdp2cat–DNA complex I | mTdp2cat–DNA complex II | mTdp2cat–DNA complex III | Se–Met mTdp2cat–Ba2+–dAMP | |
|---|---|---|---|---|---|
| Data collection | |||||
| Space group | P212121 | P212121 | P212121 | P21 | P212121 |
| Cell dimensions | |||||
| a, b, c (Å) | 85.47, 169.60, 185.51 | 114.72, 118.79, 160.47 | 54.60, 68.60, 166.03 | 60.48, 42.97, 107.95 | 85.68, 170.93, 184.09 |
| α, β, γ (°) | 90, 90, 90 | 90, 90, 90 | 90, 90, 90 | 90, 95.89, 90 | 90, 90, 90 |
|
Peak
|
|||||
| Wavelength (Å) | 0.9795 | ||||
| Resolution (Å) | 50–2.55 (2.64–2.55) | 50–2.10 (2.18–2.10) | 20-1.5 (1.55–1.50) | 50–1.85 (1.92–1.85) | 50–2.9 (3.0–2.9) |
| Rsym or Rmerge | 0.105 (0.510) | 0.079 (0.535) | 0.054 (0.518) | 0.119 (0.404) | 0.128 (0.468) |
| I / σI | 13.5 (2.8) | 13.0 (2.1) | 22.1 (2.6) | 10.0 (2.0) | 9.1 (2.0) |
| Completeness (%) | 97.7 (92.1) | 97.9 (99.0) | 98.9 (99.0) | 97.4 (93.6) | 99.9 (99.5) |
| Redundancy | 5.3 (4.7) | 3.0 (2.9) | 4.4 (4.5) | 3.1 (2.3) | 3.8 (3.5) |
| Refinement | |||||
| Resolution (Å) | 50–2.55 | 50–2.10 | 50–1.50 | 50–1.85 | |
| No. reflections | 84882 | 130977 | 105826 | 46631 | |
| Rwork /Rfree | 0.191/0.220 | 0.160/0.193 | 0.145/0.175 | 0.205/0.245 | |
| No. atoms | |||||
| Protein | 17574 | 12056 | 4140 | 4017 | |
| Ligand/ion | 149 | 1691 | 388 | 490 | |
| Water | 397 | 1041 | 472 | 437 | |
| B-factors | |||||
| Protein | 52.1 | 42.6 | 30.7 | 25.5 | |
| Ligand/ion | 70.1 | 61.2 | 51.9 | 40.7 | |
| Water | 40.8 | 52.5 | 45.8 | 35.1 | |
| R.m.s. deviations | |||||
| Bond lengths (Å) | 0.004 | 0.005 | 0.009 | 0.010 | |
| Bond angles (°) | 0.6 | 0.9 | 1.3 | 1.0 |
Each dataset was collected from a single crystal. * Values in parentheses are for highest-resolution shell.
Figure 2. Structures of the mTdp2cat–DNA complexes.
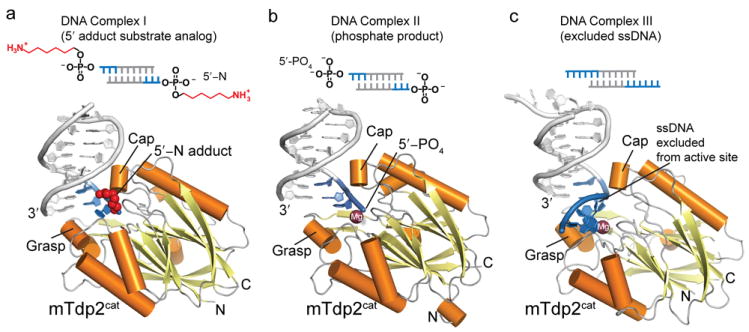
DNA crystallization constructs are diagramed showing the regions of DNA that contacts Tdp2 (blue). Extensive DNA contacts made by conserved Tdp2 side chains are made to the 3 terminal 5′ nucleotides (Complex I and II) or 6 terminal 5′ nucleotides Complex III (a) DNA 5′-N substrate analog complex (Complex I) displayed with 5′-N in red, and a cartoon representation with bound DNA (blue), Tdp2 helices (orange) and β-strands (yellow). (b) Tdp2-DNA product complex (Complex II). (c) Excluded ssDNA complex (Complex III).
Architectures of Tdp2 DNA damage recognition complexes
The 28.5 kDa mTdp2cat catalytic domain adopts a compacted globular α-β fold with a central 12-stranded β-sandwich surrounded by 8 helical elements (Fig. 2a, Supplementary Fig. 2). Approximate 2-fold topological pseudo-symmetry relates the two halves of the Tdp2cat β-sandwich. Tdp2 directly engages and directs the 5′ terminus into the active site with 14 residues mediating sequence-independent DNA contacts to the three terminal nucleotides of 5′ strand (blue DNA in complexes I and II, Figs. 2a–c, Supplementary Fig. 6). By comparison, direct protein contacts to the 3′ end of the complementary strand are limited to a DNA base stacking interaction with the exposed 3′ terminus and are mediated by non-conserved amino acids (Fig. 1g and Supplementary Fig. 6), suggesting these contacts are not relevant to Tdp2–DNA interactions in solution.
Eight highly conserved Tdp2 motifs (M1–M8, Fig. 3a–c, Supplementary Figs. 2 and 6) define a contiguous surface of the protein mediating: 1) Mg2+ coordination and active site chemistry (Motifs M1, M2, M5, M6, and M8); 2) DNA binding (Motifs M5, M6, and M7); and 3) A hydrophobic 5′-adduct recognition groove (Motifs M1, M3 and M4). Regions M5–M7 assemble to form the Tdp2 DNA binding groove. M7 adopts a previously unrecognized β-(2-helix)-β (β2Hβ) DNA-binding fold that projects outward from the Tdp2 catalytic core to envelop the exposed 5′ DNA end. An extended network of conserved hydrophobic residues (Trp307, Leu315, Ile317, Pro318, Tyr321, and Phe325) fuse and form a platform for DNA base-stacking and deoxyribose sugar interactions that “grasp” the DNA 5′-terminus (Fig. 3a, Fig 3c, and Supplementary Fig. 6). Proximal to the active site, three hydrophobic side chains (Phe325, Leu315 and Trp307) form a groove that makes van der Waals contacts to the 5′ terminal sugar, and interact non-specifically with the Cyt1 pyrimidine base (Fig. 3c).
Figure 3. Tdp2 DNA recognition motifs.
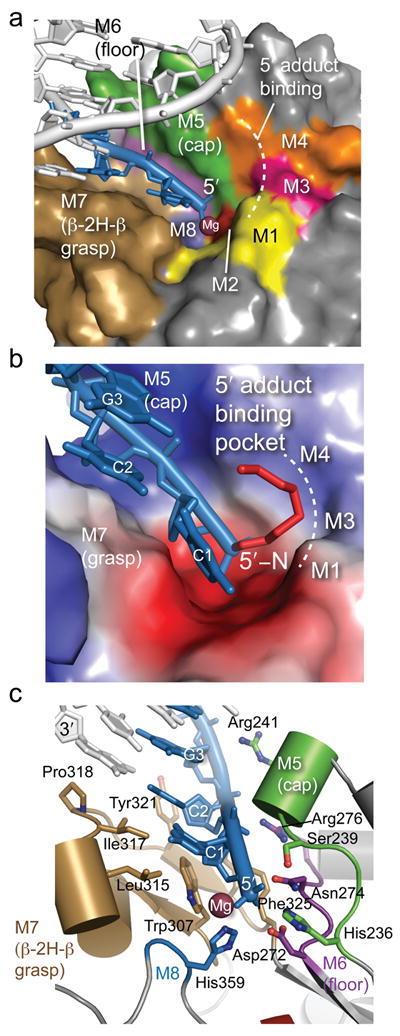
(a) DNA binding motifs (M1–M8) of Tdp2cat. The DNA is shown in blue and the Mg ion is shown in purple. (b) Surface charge representation of mTdp2cat (Blue = positive, red = negative, gray = neutral or hydrophobic) shows a positively charged groove for binding the 5′ end of the DNA and a hydrophobic patch for binding the 5′-N adduct of complex I. (c) The DNA 5′ terminus is bound by the β2Hβ (M7 motif, tan) and helical Cap (M5 motif, green) and floor (Purple) DNA binding elements.
Along with the β2Hβ-grasp, motif M5 forms a helical DNA binding “cap” on the opposite side of the cleft to orient the 5′ terminus for its approach into the active site. M5-cap interactions from Arg241 and the cap 310- helix main chain amide directly bind to the DNA sugar-phosphate backbone of the approaching 5′ strand (Fig. 3c). Between the grasp and the cap, three residues from motif M6 (Arg 276, Asn274 and Asp 272) form the “floor” of this highly conserved DNA interaction groove (Figs. 3a–c). In the DNA substrate analog complex, regions M1, M3 and M4 form a pocket binding the 5′-N alkylamine adduct (Figs. 3a,b).
5′ DNA end binding motifs
To test roles for Tdp2–DNA interfaces observed in our mTdp2cat structures, we evaluated the impact of amino acid substitutions on the strictly conserved DNA binding groove on human Tdp2cat catalytic activity, using three substrates, 1) p-nitrophenyl phosphate (PNPP), 2) thymidine 5′-monophosphate p-nitrophenyl ester (T5PNP), and 3) the DNA substrate 4nt-5′-Y (Fig. 4a). Our goal in analyzing the activity of mutant hTdp2cat proteins (Supplementary Fig. 7) on this set of chemically related substrates was to dissect structure–activity relationships of the Tdp2 DNA binding surface (Fig. 4a–c). We hypothesized that mutations impacting the basic DNA binding groove would impair catalysis on DNA substrates but not significantly alter processing of T5PNP. In support of roles for orienting the 5′ terminus of the 4nt-5′-Y substrates, substitution of two conserved cap and floor DNA binding arginine side chains R266EhTdp2 (R276mTdp2) and R231EhTdp2 (R241mTdp2) have reduced activity on 4nt-5′-Y (Fig. 4c: compare lanes 3–5 to lanes 20–22 and 37–39), but had modest impacts on hydrolysis of T5PNP (Fig. 4b).
Figure 4. Structure-based mutagenesis analysis of Tdp2 DNA interaction elements and active site residues.
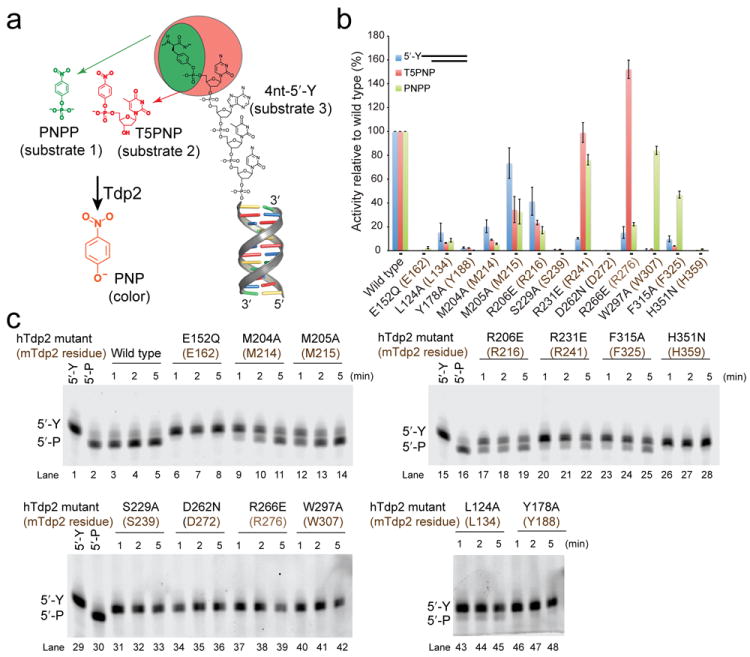
(a) Nested substrate approach for assaying catalytic activity of Tdp2 mutants. A phosphotyrosine analog (PNPP, green, substrate 1) is a minimal substrate. A phosphotyrosine mononucleotide analog (T5PNP, orange, substrate 2) is a more complex substrate bearing a 5′ nucleoside. T5PNP and PNPP are processed by Tdp2 to generate p-nitrophenol (PNP, see online methods). Substrate 3 is the preferred 4nt-5′-Y synthetic oligonucleotide with 5′-Y in a 4nt overhang. (b) Bar graph displaying the activity of wildtype and mutant human Tdp2cat-MBP fusion proteins (MBP-hTdp2cat) on the 3 substrates in panel “a”. Quantification of activity is the mean activity of 3 replicates expressed as a fraction of wild type with error bars indicating the standard deviation. For substrate 3, a quantification of activity at the 2-minute timepoint of panel “c” is shown. (c) 15% TBE-Urea PAGE analysis of Tdp2cat mutants for processing of substrate 3.
At the DNA 5′ terminus, two β2Hβ-motif hydrophobic residues (Trp307 and Phe325) cup the terminal deoxyribose sugar. Consistent with important roles for orienting the DNA terminus for catalysis, the substitution of these β2Hβ hydrophobic side chains severely impairs hydrolysis (< 5% of wildtype activity) of both 4nt-5′-Y and T5PNP (Fig. 4b,c lanes 23–25 and 40–42). Interestingly the F315AhTdp2 (F325mTdp2) and W279AhTdp2 (W307mTdp2) mutants both retain >45% of wildtype activity on PNPP, a substrate lacking a 5′ nucleoside (Fig. 4a,b). Overall, observations from mutagenesis and activity studies are consistent with the polarity of DNA binding in the Tdp2 DNA interaction groove, and support the structural observations that sequence non-specific DNA binding contacts from the cap, floor, and grasp orient the 5′ terminus to promote catalysis with combined substrate sugar-phosphate binding.
5′Adduct binding site
The position of the entire 5′-N alkylamine adduct is clearly visible in 5 of the 6 Tdp2 molecules in the crystallographic asymmetric unit, and well defined for first three methyl groups in the remaining molecule (Fig. 5). The overall trajectory of the C1 and C2 methyl carbons is consistent. Structural overlays of the six DNA–protein complexes show that while the flexible adduct adopts variable structures, they all occupy a hydrophobic groove created by Leu134 (Motif M1), Tyr188 (Motif M3), and Met214 (Motif M4) (Fig. 5a).
Figure 5. Tdp2 active site and catalytic mechanism.
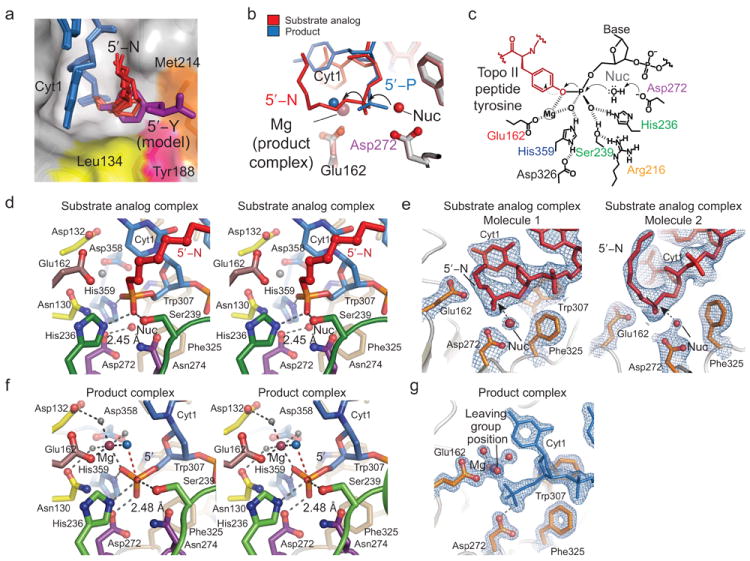
(a) Six positions of the 5′-N adduct in the crystallographic asymmetric unit are displayed showing the position of the 5′-adduct binding site. Proposed position of a 5′-Y substrate (model) is shown as a purple stick representation. (b) Structural overlay of product (blue DNA) and substrate analog (red DNA) complexes illustrating inversion of configuration about the 5′-phosphorous during the reaction. (c) Proposed Tdp2 structure-based catalytic mechanism. Tdp2 residues are colored as in Fig. 2a. (d) Stereo view of the substrate analog active site with 5′-N displayed in red. Tdp2 residues are colored as in Fig. 2a. (e) A 2.1 Å resolution σ-A weighted 2Fo–Fc map (blue mesh, contoured at 1.0 σ), is displayed overlaid on the substrate analog structure (red). “Nuc” indicates the position of the proposed water nucleophile. (f) Stereo view of the product complex active site. Interactions important for magnesium ion coordination and catalytic activity are indicated with gray dashed lines. The trajectory of the hydrolyzed bond between the 5′-phosphorous atom (orange) and the water which occupies the position of the leaving group (blue) is indicated with a dashed red line. (g) Experimental electron density of the product complex active site. Final 1.5 Å σ–A weighted 2Fo–Fc map displaying density for the DNA (blue, contoured at 1.7 σ), active site residues of mTdp2 (orange), and the magnesium ion (purple) with its octahedrally coordinated waters (gray, red).
To better understand how Tdp2 might bind a cognate 5′-Y substrate, we assessed possible Tyr binding positions in this hydrophobic pocket based on binding of 5′-N. We positioned the tyrosine hydroxyl to be anchored at bridging oxygen position, and posit 5′-Y could bind analogous to the alkyl chains occupying the hydrophobic 5′-N binding groove (Fig. 5a). Modeling of the 5′-Y into this pocket reveals van der Waals complementarily between the protein surface and the tyrosine aromatic ring. Consistent with this model, alanine substitutions of any of the hTdp2cat pocket residues, or of an adjacent residue stabilizing the conformation of Met 214 (Met 215) (hTdp2cat mutations L124A, M204A, M205A, L124A, and Y178A) all confer catalytic defects for processing of 5′-Y (Fig. 4c lanes 9–11, 12–14, 43–35, and 46–48), as well as the small molecule substrates PNPP and T5PNP (Fig. 4b).
Tdp2 catalytic mechanism
The Tdp2 substrate analog and product complex structures provide a high-resolution view of the Tdp2 active site, in the absence of mutagenic perturbation. A structural superposition of DNA end bound conformations in complexes I and II highlights a trajectory for a reaction coordinate for the adduct hydrolysis reaction that is characterized by inversion of configuration about the 5′ -phosphorous (Figs. 5b and 5c). In the substrate analog structure (a Mg2+ free crystal form), a bound water molecule (labeled “Nuc”, Fig. 5b–e) is positioned appropriately for in-line nucleophilic attack ~180° opposite of the P-O bond of the 5′-N adduct. Although 5′-Y substrates were included in the crystallization solution of the product complex (a Mg2+ bound crystal form), enzymatic processing occurred in the crystallization drop, and the DNA of complex II is unambiguously bound as a 5′-PO4 reaction product (Fig. 5f and 5g). No electron density is visible for the cleaved tyrosine moiety, suggesting it does not remain bound to Tdp2 after phosphotyrosine bond hydrolysis. In the product state, the single bound Mg2+ ion coordinates a water molecule (blue sphere, Fig. 5b and 5f) occupying a position analogous to the bridging oxygen of the 5′ adduct leaving group. Similarly, the proposed nucleophile in the substrate analog complex occupies the approximate position of the 5′-PO4 product oxygen, generated by phosphate inversion (Fig. 5d).
Based on geometry of the bound substrates and products we propose a catalytic mechanism for Tdp2 DNA adduct processing (Fig. 5c). In both the product and substrate complexes, Asp272 is best positioned to act as a catalytic base to activate a water molecule for nucleophilic attack in an SN2 displacement reaction, and forms close hydrogen bonds (average distance of 2.45 Å for the 6 monomers in the crystallographic ASU) to the proposed water nucleophile, or to the 5′-PO4 product oxygen (average distance 2.48 Å for the two monomers in the crystallographic ASU). Magnesium is absent from our substrate analog complex, however Mg2+ directly interacts with the 5′-PO4 in the product structure. Bound Mg2+, along with three additional conserved residues (His236, His359 and Ser239) binding 5′-PO4, are positioned to interact with the substrate phosphate moiety, and stabilize a pentacovalent reaction transition state in this scheme (Fig. 5c,d and f). Supporting their predicted roles in tyrosyl-phosphodieseter hydrolysis, mutation of the presumed conserved catalytic base Asp272 (Fig. 4c lanes 34–36) or transition state stabilization ligands Ser239 or His359 impairs catalytic activity on all 3 substrates (Fig. 4b, 4c lanes 31–33 and 26–28). Based on structural and mutagenesis data, our data are most consistent with a single metal ion mechanism (Fig. 5c) that is similar to a mechanism originally proposed for the related EEP nuclease Ape1 (ref.15.)
Tdp2 DNA processing specificity
Overall, the architecture of the Tdp2 DNA binding groove formed by the M6–M8 motifs explains the exquisite in vitro Tdp2 DNA structure-dependant DNA substrate specificity (Fig. 1, g,h,i). Consistent with the substrate cleavage preference for 5′-Y overhangs, the dimensions of the substrate-binding groove proximal to the active center appear too small to efficiently accommodate dsDNA without distortion of the enzyme active site and/or terminal DNA base pair unwinding, providing a possible explanation for reduced efficiency of processing on blunt or recessed 5′ termini. In addition, Tdp2 displays a marked preference for 5′-Y over 3′-Y substrates (Fig. 1h,i and ref. 8). Based on the structures and mutagenesis, substrate interactions with the DNA 5′-end are facilitated by van der Waals contacts from the β2Hβ grasp (Trp307, Phe325) to the terminal C4′ and C5′ methyl groups (Fig. 6a). We posit substrate interactions may be suboptimal for binding a 3′-Y DNA end where the phosphate is more closely juxtaposed to the deoxyribose sugar (Fig. 6b).
Figure 6. Determinants of Tdp2 substrate specificity.
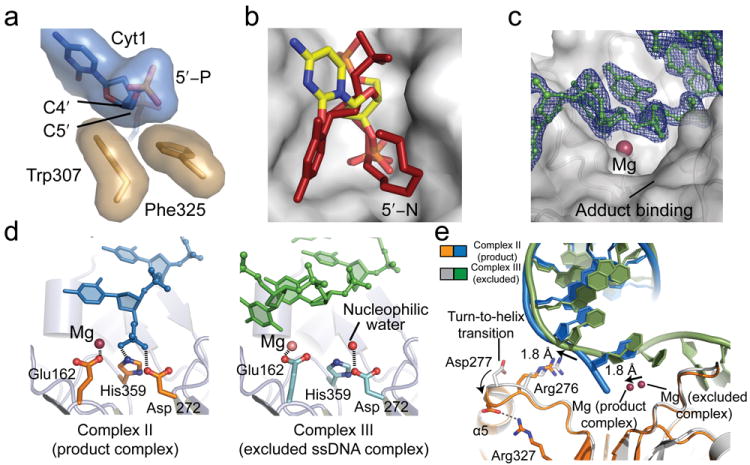
(a) Trp307 and Phe325 of the β2Hβ (M7 motif, tan) recognize the 5′ DNA end through van der Waals interactions with C4′ and C5. (b) 5′ vs. 3′ adduct processing. The substrate binding pocket with the 5′-N adduct (red) is displayed on the Tdp2 surface (gray). A model of DNA in the reverse orientation with a 3′ phosphate in the active site is shown in yellow. (c) Experimental electron density of the excluded DNA complex. A 2Fo–Fc map contoured at 1.5 σ shows DNA (green), which is distant from the catalytic Mg2+ ion (purple). (d) Details of active site geometry. Left panel: In the product complex, the DNA (blue) is bound in the active site residues of Tdp2 (orange) and Mg2+ ion (purple). Right panel: In the excluded ssDNA complex the DNA (green) phosphate backbone is far from the active site (cyan) or Mg ion (pink). In the absence of substrate bound in the active site, the putative active site water nucleophile (red) is hydrogen-bonded to Asp 272. (e) Structural comparison of Tdp2–DNA complex 2 and 3 structures reveals conformational changes in Tdp2.
While Tdp2 rapidly processes 5′-Y, a single-stranded 5′-end of the DNA is not further processed nucleolytically by Tdp2 in solution, even at high protein concentration (Fig. 1h), underscoring the precise selectivity of the Tdp2 active site for 5′-Y reversal. Relevant to this selectivity, we determined an additional structure of DNA bound Tdp2 in which the single-stranded 5′ terminus is found excluded from the active site (DNA complex III). In this complex, which we term the “Excluded ssDNA” complex (Fig. 6c–e), the ssDNA is bound outside of the Tdp2 active site recess, and hydrophobic base stacking interactions meditated by Leu134 stabilize this active-site excluded conformation (Fig 5c, Supplemental Fig. 6). We hypothesize this third, non-productively DNA bound conformation reflects a mode of Tdp2 substrate encounter with intact ssDNA, and a basis for understanding protection against Tdp2 mediated endonucleolytic cleavage.
Comparisons of the excluded DNA conformation to the active site occupied complex structures I and II, reveal conformational differences involving the Asp277 and Arg276 (floor), and the Mg2+ binding site (Fig. 6d and 6e). A ~2Å shift in the DNA appears coupled to motion of Arg276. Movement of Arg276 is also coincident with a turn-to-helix transition at the N-terminal end of helix α5, and to the formation of a salt-bridging contact between Asp277 and Arg327 (Fig. 6e). Interestingly, the active site Mg also moves inward 1.8 Å in the product state (relative to the excluded ssDNA complex). We speculate that these conformational rearrangements may reflect induced fit assembly of the Tdp2 active site upon productive interaction with the ssDNA terminus in the active site, and might provide an additional level of substrate selectivity.
DISCUSSION
Overall, our results define the molecular basis for Tdp2 recognition and hydrolytic processing of 5′-tyroslyated protein-DNA conjugates, and support a testable structure-based single magnesium ion mediated catalytic mechanism for the EEP superfamily of nucleases. Tdp2 architecture is closely related to Ape115, but shares only 16% sequence identity with Ape1 over the EEP domain (Fig. 7). Ape1 is an endonuclease, and recognizes and cleaves the 5′-terminal side of abasic sites to facilitate DNA base excision repair15. However, DNA and 5′-adduct recognition elements make Tdp2 distinct from Ape1, and Tdp2 excludes an intact phosphodiester backbone from its active site to ensure selectivity and restrict endo- or exonucleolytic processing. Superposition of the Ape1 and Tdp2 DNA complex structures indicates that although the central beta sheet and active site chemistry is preserved, the DNA substrate interactions and secondary structure elements engaging DNA damage are structurally diverged and distinct. Interestingly, the internal topological pseudo 2-fold symmetry of the EEP proteins breaks down with respect to protein surface loops defining substrate binding. For Tdp2, one half of the binding cleft has evolved to recognize the 5′ adduct (Motifs M1, M3 and M4), and on the opposing side, the M5–M7 surface motifs define binding to the deoxy-polynucleotide. The analogous regions from Ape1 also participate in substrate recognition, but bear little resemblance to Tdp2 (Fig. 7). However, despite global differences in DNA binding mode, the approach of the scissile DNA-phosphotyrosyl linkage (Tdp2), or DNA phosphodiester linkage (Ape1) within the EEP active site is quite similar. Collectively, these observations highlight that divergent EEP surface DNA interacting recognition elements have evolved for tailored protein-DNA adduct processing (Tdp2) (Fig. 8) and DNA damage (Ape1) recognition with a common hydrolytic enzymatic processing mechanism.
Figure 7. DNA damage recognition by EEP domains.
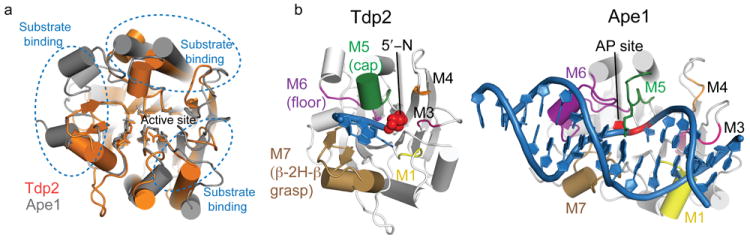
(a) Structural superposition of Tdp2 (orange) and human Ape1 (gray, RCSB accession 1DE8 (ref. 15)). The core EEP fold β–sandwich and active site residues are conserved, but DNA substrate interaction loops (blue dotted lines) are structurally divergent. (b) Comparison of DNA–substrate interaction modes by Tdp2 and human Ape1.
Figure 8. Model for removal of 5′-phosphotyrosine linked topo II adducts from DNA by Tdp2.
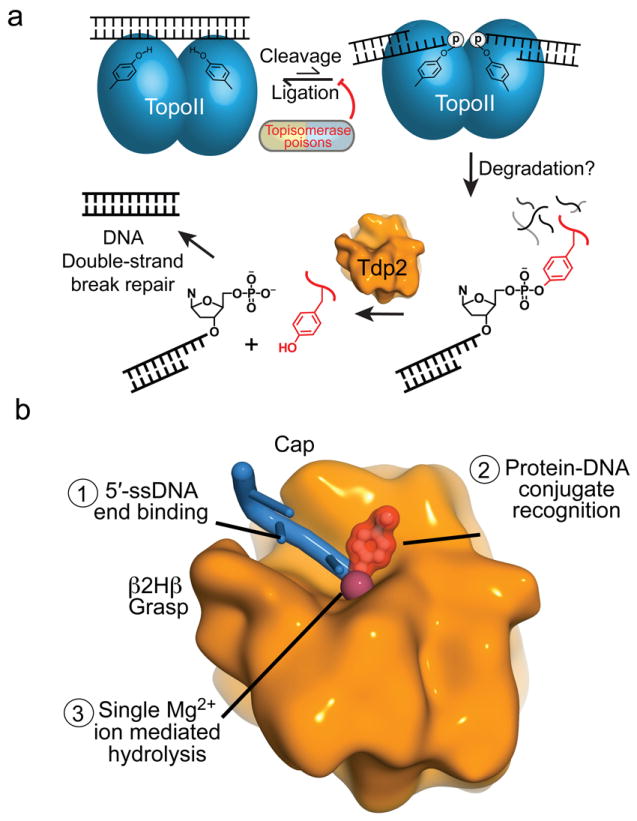
(a) Stalled topo II cleavage complexes are degraded in a proteasome-dependent manner. Tdp2 encounters and removes the Topo II peptides by hydrolyzing the 5′-phosphotyrosine bond, yielding 5′ phosphorylated DNA termini that are repaired by the cellular DSB repair machinery. (b) Substrate interactions by Tdp2 involves structure-specific recognition of 5′-phosphotyrosine linked protein-adducted DNA termini, and Tdp2 employs single metal ion catalysis to reverse DNA damage.
Structures of several DNA repair end-cleansing enzymes are now available, including those for DNA polynucleotide kinase–phosphatase (PNK)18,19,20,21, Aptx22, and the Flap endonculease Fen1 (ref. 23). A common feature of DNA end binding recognition in these enzymes is the deployment of hydrophobic α-helical DNA base platforms to sequester exposed DNA termini. Our X-ray structures further expand the repertoire of DNA end recognition mechanisms to include the β2Hβ grasp and cap combination of Tdp2. By engulfing DNA termini, these DNA end damage-processing enzymes may protect exposed damage termini from inappropriate 5′ or 3′ exonucleolytic degradation. The observation that Tdp2 extensively engages the 5′ terminal 3 nt is substrate has additional implications for understanding Tdp2 activity on topo II adducts. Structural analysis of an etoposide trapped human topo IIβ24, and of the S. cerevisiae topo II covalent cleavage complex25 show how type II topoisomerases extensively engage the DNA 5′ end. Limited accessibility of 5′-topo II adducted DNA ends suggest that large conformational changes, and possibly unfolding of the topo II Toprim catalytic domain, would be required for Tdp2 to access the substrate (Fig. 8a). Thus Tdp2–DNA complex structures imply that Tdp2 DNA end-processing might be regulated by access, and Tdp2 catalysis may require proteolysis of topoII isoforms prior to de-tyrosylation and DNA end joining10,11.
The DNA binding polarity defined here by structures and mutagenesis, the mode of 5′-adduct recognition, and the Tdp2 enzymatic preference for model 5′-Y substrates in vitro are all consistent with cellular roles for Tdp2 in promoting repair of topoII linked DNA damage (Fig. 8)8,13. Evidence in yeast26 and mammalian cells27 shows processing of topo II adducts, as well as removal of the related Spo11 adduct that is an obligate intermediate in meiotic recombination28, can also involve endonucleolytic processing by the multifunctional DSB sensing and processing Mre11–Rad50–Nbs1–Ctp1(CtIP,Sae2) complex (MRN–CtIP)29. The choice of MRN–CtIP or Tdp2 for resolution of 5′-Y adducted termini might thus dictate DNA double strand break repair pathway choice, with the former channeling breaks to repair by homologous recombination, while it has been hypothesized Tdp2 generates ends appropriate for repair by non-homologous end joining8,13. Functional redundancy of 5′ adduct processing and use of a Tdp2-dependant direct reversal repair pathway may be particularly critical in terminally differentiated cells, where repair by homologous recombination may not be possible.
Tdp2 was originally identified in screens for factors encoding cellular resistance to camptothecin, an anti-cancer agent that generates 3′-phosphotyrosyl linked Topoisomerase I (TopoI) DNA adducts8. Accordingly, Tdp2 also exhibits activity towards 3′-Y in vitro (Fig. 1h, I and ref. 8), but this activity is inefficient relative to 5′-Y hydrolysis, or when compared to the 3′-Y hydrolysis activity of Tdp1 — a phospholipase 2 superfamily enzyme that catalyzes the metal-independent reversal of 3′ phosphotyrosyl linkages30. Tdp1 is structurally unrelated to Tdp2 (ref. 30). Thus, structures defined here also highlight convergent evolutionary solutions of eukaryotic cells for coping with topoisomerase protein-DNA adducts. Tdp1 deficiency is linked to the neurological disease spinocerebellar ataxia with axonal neuropathy (SCAN1)31,32 and Tdp2 activity on 3′-linked topoisomerase adducts appears important in the absence of Tdp1 (ref. 33). So, Tdp2 mediated processing of 3′-Y linked TopoI adducts may partly compensate for loss of Tdp1 activity in SCAN1.
Finally, given Tdp2 variation in the human population34, Tdp2–DNA complex structure activity studies described here have implications for understanding roles for Tdp2 polymorphisms in modulation of cancer chemotherapy, and susceptibility to environmentally linked topo II poisons. Given that Tdp2 siRNA knockdown sensitizes lung cancer A549 cells to etoposide8, and that Tdp2 is overexpressed in mutant p53 cells and human lung cancers12, Tdp2 inhibitors may synergize or potentiate cytotoxic effects of current anticancer treatments targeting topo II. The molecular determinants of Tdp2 DNA-protein recognition and catalysis defined here will likely enable unique opportunities for development of Tdp2 inhibitors.
ONLINE METHODS
Expression and purification of Tdp2 proteins
Human Tdp2 (hTdp2) and mouse Tdp2 (mTdp2) were expressed at 17° C from pMCSG935 in BL21 Rosetta2 E. coli (EMD Biosciences). Following lysis by sonication in lysis buffer (20 mM Tris pH 7.5, 500 mM NaCl, 1 mM tris(2-carboxyethyl)phosphine (TCEP), and 1 mM phenylmethylsulfonyl fluoride with the addition of 0.1 mg ml-1 lysozyme), maltose-binding protein (MBP) - tagged protein affinity purified on amylose resin (New England Biolabs), and eluted in lysis buffer plus 10 mM maltose. MBP-Tdp2 fusion proteins were purified by size exclusion chromatography on a 16/60 S-200 column (GE Life Sciences) in size-exclusion column buffer (20 mM Tris pH 7.5, 300 mM NaCl, 1 mM TCEP). For biochemical assays, the MBP-tagged proteins were concentrated and used without further purification. For mTdp2cat crystallization, the MBP tag was removed by TEV protease digestion, and mTdp2cat was purified further with butyl-sepharose hydrophobic interaction chromatography (GE Life Sciences), followed by dialysis into size-exclusion buffer.
Small angle X-ray scattering
For SAXS studies, Tdp2 variants were buffer exchanged into SAXS buffer (15mM Tris pH 7.5, 300 mM NaCl, 1mM TCEP, 2mM MgCl2, and 1% glycerol), and concentrated in Amicon 3K 0.5mL centrifugal concentrators (Fisher), immediately prior to SAXS data collection. SAXS data collection was performed at at 3, 1.5, and 0.75 mg mL-1 protein concentration, and SAXS data was analyzed using the ATSAS suite of SAXS data analysis tools (http://www.embl-hamburg.de/biosaxs/software.html).
Crystallization of Tdp2 and Tdp2–DNA complexes
Crystals of Tdp2–DNA complexes were grown by sitting drop vapor diffusion by mixing 200 nL of precipitant with 200nL of Tdp2 protein–DNA complex. mTdp2cat nucleotide and DNA complexes contained a final protein concentration of 8 mg ml-1 and, 1) for Se-Met AMP complex crystals, 10 mM dAMP, 2) for DNA complex I, a 1.2-fold molar excel of 5N-9SA oligonucleotide and 1 mM MgCl2, 3) for DNA complex II, a 1.2-fold molar excess of 5Y-9SA oligonucleotide and 1 mM MgCl2, or 4) for DNA complex III, a 1.2-fold molar excess of 12SA oligonucleotide (Supplementary Table 4) and 1 mM MgCl2. For the DNA-bound complexes I, II, and III, protein-DNA complex crystallization at 2:1 protein/DNA stoichiometry was promoted with the use of oligonucleotide substrates that are self-complimentary through an 8 base pair duplex region, and bear double-ended overhangs (Supplementary Fig. 5, Supplementary Table 2). To facilitate observation of Tdp2 bound to a 5′-adducted substrate analog without mutation of the Tdp2 active site, we used the hydrolysis resistant 5′-N adducted substrate (Fig. 1i, Fig. 2a). Selenomethione-derivatized mTdp2cat–dAMP complex crystals were grown with a precipitant containing 100 mM MES pH 6.5, 20 mM BaCl2, and 10% PEG-20,000, then cryoprotected in liquid nitrogen in the crystallization mother liquor supplemented by 25% PEG-20,000, 8% glycerol, and 5% glucose. Crystals of mTdp2cat bound to a 5′-N DNA (complex I) were grown with a precipitant containing 100 mM sodium acetate pH 4.5, 5% PEG1000, and 50% ethylene glycol. Crystals of mTdp2cat bound to a product DNA (complex II) were grown with a precipitant containing 100 mM HEPES pH 7.5, 20% PEG3350, 200 mM sodium acetate, and 10mM MgCl2, then cryoprotected in the crystallization mother liquor plus 25% PEG-3350, 8% glycerol, and 5% glucose. DNA Complex III crystals mTdp2cat were grown with a precipitant containing 100 mM Tris pH 8.5, 25% PEG3350, 250 mM magnesium formate, then cryoprotected in the crystallization condition with 8% glycerol, and 5% glucose.
X-ray diffraction data collection, phasing and refinement
X-ray data (Table 1) were collected at 105 K on beamline 22-ID of the Advanced Photon Source at a wavelength of 1.0000 Å for the mTdp2cat–Mg2+–5′-dAMP complex, Tdp2–DNA complex II and Tdp2–DNA complex III. Tdp2–DNA complex I was collected at 0.8266Å and the Se–Met mTdp2cat–Ba2+–dAMP dataset was collected at the experimentally determined Se-K edge of 0.9795Å. X-ray diffraction data was processed and scaled using the HKL2000 suite36.
The SeMet-derivatized mTdp2cat–dAMP complex crystals were phased using single-wavelength anomalous dispersion (SAD) dataset (Table 1). Positions of the 36 Se atoms of the 9 Tdp2 monomers in the crystallographic asymmetric unit were located and refined with SOLVE37,38. An initial model built with RESOLVE38,39 was improved by iterative rounds of manual fitting in Coot and refinement against the mTdp2cat–Mg2+–dAMP dataset using the program PHENIX38. Chain A of this model was used as the search model for determination of the mTdp2cat–DNA complexes I, II, and III by molecular replacement in PHASER40. All crystallographic refinement and data analysis was performed in PHENIX37. The final models display excellent geometry. MOLPROBITY38 Ramachandran statistics for the mTdp2cat–Mg2+–dAMP complex: 96.9% favored, 0.0% outliers, mTdp2cat–5′-N DNA complex I: 98.4% favored, 0.0% outliers, mTdp2cat–product DNA complex II: 98.9% favored, 0.0% outliers, and mTdp2cat–excluded DNA complex III: 97.2% favored, 0.0% outliers.
T5PNP and PNPP cleavage assays
Enzymatic reactions were performed in clear 96-well plates in assay buffer containing 100 mM NaCl, 20 mM Tris pH 7.5, and 2 mM MgCl2. Reactions consisted of 2 mM T5PNP or PNPP and 1μM TDP2 proteins. Increase in absorbance at 415 nm was monitored for 60 minutes in a POLARStar Omega plate reader (BMG Labtech). Data was analyzed in Microsoft Excel.
Preparation of oligonucleotide substrates
Oligonucleotide substrates and construction are summarized in Supplementary Tables 2–4. 5′ or 3′ modified oligonucleotides were purchased from Midland Certified Reagent Company, and unlabeled DNA oligonucleotides were purchased from Integrated DNA. Oligonucleotides were re-suspended in distilled water and diluted to a working concentration in buffer (10mM Tris pH 7.5, 50 mM NaCl, 0.1 mM EDTA) and either used directly in reaction assays or crystallization trials, or annealed to a 1.2-fold excess of the indicated complimentary nucleotide in buffer (10mM Tris pH 7.5, 50 mM NaCl, 0.1 mM EDTA), heated to 70° C, and slowly cooled to room temperature before using in reaction assays.
Tdp2 oligonucleotide-substrate assays
Purified Tdp2 proteins (10 nM) were incubated with 1 μM FITC-labeled DNA substrates in reaction buffer containing 100 mM NaCl, 20 mM Tris pH 7.5, and 2 mM MgCl2. Samples were removed at the indicated timepoints, and the reaction was stopped by adding a 4-fold excess of Novex TBE-Urea loading buffer (Invitrogen). Reactions were resolved on Novex 15% TBE-Urea PAGE gels (Invitrogen). The FITC-labeled oligonucleotides were imaged using a Typhoon 9000 imager with an excitation wavelength of 488 nm and a band pass emission filter set at 520 nm (GE Healthcare Life Sciences). ImageQuant software was used to quantify band intensities.
Supplementary Material
Acknowledgments
This research was supported by the intramural research program of the US National Institutes of Health (NIH), National Institute of Environmental Health Sciences (NIEHS) grant 1Z01ES102765-01 (R.S.W.) and by NIH grant CA 084442 (D.A.R.). We thank T. Kunkel and members of the Williams Lab for discussions and critical reading of the manuscript, L. Pedersen of the NIEHS Collaborative crystallography group, the Advanced Light Source beamline 12.3.1 (SIBYLS) staff and the Advanced Photon Source (APS) Southeast Regional Collaborative Access Team (SER-CAT) staff for assistance with SAXS and crystallographic data collection, and J. Williams of the NIEHS Protein Microcharacterization Core Facility for mass spectrometry analysis.
Footnotes
AUTHOR CONTRIBUTIONS
All authors performed experiments. M.J.S., D.R. and R.S.W. designed experiments, analyzed results, and wrote the manuscript.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
ACCESSION CODES
Coordinates have been submitted to the RCSB protein data bank for the mTdp2cat–Mg2+–5′-dAMP complex (accession number 4GYZ), mTdp2cat–DNA complex I (accession number 4GZ0), mTdp2cat–DNA complex II (accession number 4GZ1), and mTdp2cat–DNA complex III (accession number 4GZ2).
References
- 1.Nitiss JL. DNA topoisomerase II and its growing repertoire of biological functions. Nat Rev Cancer. 2009;9:327–37. doi: 10.1038/nrc2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nitiss JL. Targeting DNA topoisomerase II in cancer chemotherapy. Nat Rev Cancer. 2009;9:338–50. doi: 10.1038/nrc2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Deweese JE, Osheroff N. The DNA cleavage reaction of topoisomerase II: wolf in sheep’s clothing. Nucleic Acids Res. 2009;37:738–48. doi: 10.1093/nar/gkn937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wilstermann AM, Osheroff N. Base excision repair intermediates as topoisomerase II poisons. J Biol Chem. 2001;276:46290–6. doi: 10.1074/jbc.M105733200. [DOI] [PubMed] [Google Scholar]
- 5.Bandele OJ, Osheroff N. (-)-Epigallocatechin gallate, a major constituent of green tea, poisons human type II topoisomerases. Chem Res Toxicol. 2008;21:936–43. doi: 10.1021/tx700434v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hande KR. Etoposide: four decades of development of a topoisomerase II inhibitor. Eur J Cancer. 1998;34:1514–21. doi: 10.1016/s0959-8049(98)00228-7. [DOI] [PubMed] [Google Scholar]
- 7.Baldwin EL, Osheroff N. Etoposide, topoisomerase II and cancer. Curr Med Chem Anticancer Agents. 2005;5:363–72. doi: 10.2174/1568011054222364. [DOI] [PubMed] [Google Scholar]
- 8.Cortes Ledesma F, El Khamisy SF, Zuma MC, Osborn K, Caldecott KW. A human 5’-tyrosyl DNA phosphodiesterase that repairs topoisomerase-mediated DNA damage. Nature. 2009;461:674–8. doi: 10.1038/nature08444. [DOI] [PubMed] [Google Scholar]
- 9.Bahmed K, Nitiss KC, Nitiss JL. UnTTrapping the ends: a new player in overcoming protein linked DNA damage. Cell Res. 2010;20:122–3. doi: 10.1038/cr.2010.17. [DOI] [PubMed] [Google Scholar]
- 10.Mao Y, Desai SD, Ting CY, Hwang J, Liu LF. 26 S proteasome-mediated degradation of topoisomerase II cleavable complexes. J Biol Chem. 2001;276:40652–8. doi: 10.1074/jbc.M104009200. [DOI] [PubMed] [Google Scholar]
- 11.Fan JR, et al. Cellular processing pathways contribute to the activation of etoposide-induced DNA damage responses. DNA Repair (Amst) 2008;7:452–63. doi: 10.1016/j.dnarep.2007.12.002. [DOI] [PubMed] [Google Scholar]
- 12.Do PM, et al. Mutant p53 cooperates with ETS2 to promote etoposide resistance. Genes Dev. 2012;26:830–45. doi: 10.1101/gad.181685.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zeng Z, Cortes-Ledesma F, El Khamisy SF, Caldecott KW. TDP2/TTRAP is the major 5’-tyrosyl DNA phosphodiesterase activity in vertebrate cells and is critical for cellular resistance to topoisomerase II-induced DNA damage. J Biol Chem. 2011;286:403–9. doi: 10.1074/jbc.M110.181016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang H, et al. Crystal structure of the human CNOT6L nuclease domain reveals strict poly(A) substrate specificity. EMBO J. 2010;29:2566–76. doi: 10.1038/emboj.2010.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mol CD, Izumi T, Mitra S, Tainer JA. DNA-bound structures and mutants reveal abasic DNA binding by APE1 and DNA repair coordination. Nature. 2000;403:451–6. doi: 10.1038/35000249. [DOI] [PubMed] [Google Scholar]
- 16.Pype S, et al. TTRAP, a novel protein that associates with CD40, tumor necrosis factor (TNF) receptor-75 and TNF receptor-associated factors (TRAFs), and that inhibits nuclear factor-kappa B activation. J Biol Chem. 2000;275:18586–93. doi: 10.1074/jbc.M000531200. [DOI] [PubMed] [Google Scholar]
- 17.Varady G, Sarkadi B, Fatyol K. TTRAP is a novel component of the non-canonical TRAF6-TAK1 TGF-beta signaling pathway. PLoS One. 2011;6:e25548. doi: 10.1371/journal.pone.0025548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Garces F, Pearl LH, Oliver AW. The structural basis for substrate recognition by Mammalian polynucleotide kinase 3’ phosphatase. Mol Cell. 2011;44:385–96. doi: 10.1016/j.molcel.2011.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schellenberg MJ, Williams RS. DNA end processing by polynucleotide kinase/phosphatase. Proc Natl Acad Sci U S A. 2011;108:20855–6. doi: 10.1073/pnas.1118214109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bernstein NK, et al. The molecular architecture of the mammalian DNA repair enzyme, polynucleotide kinase. Mol Cell. 2005;17:657–70. doi: 10.1016/j.molcel.2005.02.012. [DOI] [PubMed] [Google Scholar]
- 21.Coquelle N, Havali-Shahriari Z, Bernstein N, Green R, Glover JN. Structural basis for the phosphatase activity of polynucleotide kinase/phosphatase on single- and double-stranded DNA substrates. Proc Natl Acad Sci U S A. 2011;108:21022–7. doi: 10.1073/pnas.1112036108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tumbale P, et al. Structure of an aprataxin-DNA complex with insights into AOA1 neurodegenerative disease. Nat Struct Mol Biol. 2011;18:1189–95. doi: 10.1038/nsmb.2146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tsutakawa SE, et al. Human flap endonuclease structures, DNA double-base flipping, and a unified understanding of the FEN1 superfamily. Cell. 2011;145:198–211. doi: 10.1016/j.cell.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wu CC, et al. Structural basis of type II topoisomerase inhibition by the anticancer drug etoposide. Science. 2011;333:459–62. doi: 10.1126/science.1204117. [DOI] [PubMed] [Google Scholar]
- 25.Schmidt BH, Burgin AB, Deweese JE, Osheroff N, Berger JM. A novel and unified two-metal mechanism for DNA cleavage by type II and IA topoisomerases. Nature. 2010;465:641–4. doi: 10.1038/nature08974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hartsuiker E, Neale MJ, Carr AM. Distinct requirements for the Rad32(Mre11) nuclease and Ctp1(CtIP) in the removal of covalently bound topoisomerase I and II from DNA. Mol Cell. 2009;33:117–23. doi: 10.1016/j.molcel.2008.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nakamura K, et al. Collaborative action of Brca1 and CtIP in elimination of covalent modifications from double-strand breaks to facilitate subsequent break repair. PLoS Genet. 2010;6:e1000828. doi: 10.1371/journal.pgen.1000828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Neale MJ, Pan J, Keeney S. Endonucleolytic processing of covalent protein-linked DNA double-strand breaks. Nature. 2005;436:1053–7. doi: 10.1038/nature03872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Williams RS, Williams JS, Tainer JA. Mre11-Rad50-Nbs1 is a keystone complex connecting DNA repair machinery, double-strand break signaling, and the chromatin template. Biochem Cell Biol. 2007;85:509–20. doi: 10.1139/O07-069. [DOI] [PubMed] [Google Scholar]
- 30.Davies DR, Interthal H, Champoux JJ, Hol WG. The crystal structure of human tyrosyl-DNA phosphodiesterase, Tdp1. Structure. 2002;10:237–248. doi: 10.1016/s0969-2126(02)00707-4. [DOI] [PubMed] [Google Scholar]
- 31.El-Khamisy SF, et al. Defective DNA single-strand break repair in spinocerebellar ataxia with axonal neuropathy-1. Nature. 2005;434:108–13. doi: 10.1038/nature03314. [DOI] [PubMed] [Google Scholar]
- 32.Takashima H, et al. Mutation of TDP1, encoding a topoisomerase I-dependent DNA damage repair enzyme, in spinocerebellar ataxia with axonal neuropathy. Nature genetics. 2002;32:267–72. doi: 10.1038/ng987. [DOI] [PubMed] [Google Scholar]
- 33.Zeng Z, et al. TDP2 promotes repair of topoisomerase I-mediated DNA damage in the absence of TDP1. Nucleic acids research. 2012 doi: 10.1093/nar/gks622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sherry ST, et al. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;29:308–11. doi: 10.1093/nar/29.1.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stols L, et al. A new vector for high-throughput, ligation-independent cloning encoding a tobacco etch virus protease cleavage site. Protein Expr Purif. 2002;25:8–15. doi: 10.1006/prep.2001.1603. [DOI] [PubMed] [Google Scholar]
- 36.Otwinowski Z, Minor W. Processing of X-ray Diffraction Data Collected in Oscillation Mode. In: Carter CW Jr, Sweets RM, editors. Methods in Enzymology. Vol. 276. Academic Press; New York: 1997. pp. 307–326. [DOI] [PubMed] [Google Scholar]
- 37.Terwilliger TC, Berendzen J. Automated MAD and MIR structure solution. Acta Crystallogr D Biol Crystallogr. 1999;55(Pt 4):849–61. doi: 10.1107/S0907444999000839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Adams PD, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–21. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Terwilliger TC. Maximum-likelihood density modification. Acta Crystallogr D Biol Crystallogr. 2000;56(Pt 8):965–72. doi: 10.1107/S0907444900005072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McCoy AJ, RW G-K, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Cryst. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.