

Abstract
Precise regulation of the intracellular concentration of chloride [Cl-]i is necessary for proper cell volume regulation, transepithelial transport, and GABA neurotransmission. The Na-K-2Cl (NKCCs) and K-Cl (KCCs) cotransporters, related SLC12A transporters mediating cellular chloride influx and efflux, respectively, are key determinants of [Cl-]i in numerous cell types, including red blood cells, epithelial cells, and neurons. A common "chloride/volume-sensitive kinase", or related system of kinases, has long been hypothesized to mediate the reciprocal but coordinated phosphoregulation of the NKCCs and the KCCs, but the identity of these kinase(s) has remained unknown. Recent evidence suggests the WNK (with no lysine = K) serine-threonine kinases directly or indirectly via the downstream Ste20-type kinases SPAK/OSR1, are critical components of this signaling pathway. Hypertonic stress (cell shrinkage), and possibly decreased [Cl-]i, triggers the phosphorylation and activation of specific WNKs, promoting NKCC activation and KCC inhibition via net transporter phosphorylation. Silencing WNK kinase activity can promote NKCC inhibition and KCC activation via net transporter dephosphorylation, revealing a dynamic ability of the WNKs to modulate [Cl-]. This pathway is essential for the defense of cell volume during osmotic perturbation, coordination of epithelial transport, and the gating of sensory information in the peripheral system. Commiserate with their importance in serving these critical roles in humans, mutations in WNKs underlie two different Mendelian diseases, pseudohypoaldosteronism type II (an inherited form of salt-sensitive hypertension), and hereditary sensory and autonomic neuropathy type 2. WNKs also regulate ion transport in lower multicellular organisms, including Caenorhabditis elegans, suggesting their functions are evolutionarily-conserved. An increased understanding of how the WNKs regulate the Na-K-2Cl and K-Cl cotransporters may provide novel opportunities for the selective modulation of these transporters, with ramifications for common human diseases like hypertension, sickle cell disease, neuropathic pain, and epilepsy.
Introduction
Any mechanism that makes a homeostatic adjustment in response to a physiological perturbation requires sensors that detect the abnormality in the system and transducers that propagate the signal, once detected, to the appropriate effectors, which elicit the corrective response. Such mechanisms are often complex, with multiple components at each step, many times operating in parallel. Molecular genetics can define critical genes in such complex regulatory pathways in an unbiased way, paving the way for novel and unanticipated physiological insights. In 2001, a positional cloning effort performed by Lifton and colleagues in the quest for the genes underlying the monogenic form of hypertension with hyperkalemia named pseudohypoaldosteronism type II (PHAII; Online Mendelian Inheritance in Man (OMIM) no. 145260) resulted in the identification of the human genes encoding the human WNK kinases (1). Intronic deletions in WNK1 were found in affected patients of two different large PHAII families previously linked to the chromosome 12 PHAII locus, and were associated with a greater than 5-fold increase in WNK1 expression, suggesting mutations were gain-of-function. In other families previously linked to the chromosome 17 PHAII locus, four different charge-changing missense mutations in WNK4 were found, three of which clustered in a highly-conserved stretch of ten amino acids located distal to the molecule’s first coiled-coil domain, while the fourth mutation was located just beyond the second coiled-coil domain. These genetic findings revealed a completely novel pathway important for the regulation of blood pressure and electrolyte homeostasis in humans, and spawned investigation into the mechanism by which the WNKs achieve these crucial functions (1).
Physiological experiments in Xenopus laevis oocytes, mammalian cells, and mouse models have subsequently shown that WNK1 and WNK4 are multifunctional molecular switches with diverse actions on multiple targets of ion transport in the aldosterone-sensitive distal nephron, including the thiazide-sensitive Na-Cl cotransporter NCC (2). Through their diverse but coordinated actions on transporters and channels that mediate sodium, chloride, and potassium flux, achieved via multiple catalytic states of its kinase domain that is regulated by upstream stimuli like serum potassium, angiotensin II, and aldosterone, the WNK1 and WNK4 kinases regulate renal salt and potassium homeostasis. These studies opened new avenues into the understanding of kidney physiology, including the long-standing problem of the "aldosterone paradox", which refers to the question of how aldosterone can be at the same time a sodium-retaining and potassium-secreting hormone (3).
The WNK kinases (with no lysine=K), first discovered and cloned in rat, comprise a subfamily of protein serine-threonine kinases that lack a well-conserved lysine in β strand 3 of kinase subdomain II that is crucial for ATP-binding and the catalysis of phosphoryl transfer (4). The catalytic activation loop in WNK kinases is remodeled such that a lysine (Lys-233 in WNK1) in β strand 2 serves this role (5). Distant relatives of the WNK kinases are the mammalian Ste20-type serine-threonine kinases, named for their founding member that is a mitogen-activated protein kinase kinase kinase kinase (MAP4K) in the yeast MAPK signaling pathway (6). WNK1 was the first mammalian member of this kinase family to be cloned (4); in humans, rodents, and other mammals, a total of four WNK kinases exist (1, 7). In humans, WNK1 is encoded on chromosome 12, WNK2 on chromosome 9, WNK3 on the X chromosome, WNK4 on chromosome 17 (1, 7). WNK kinases appear to be expressed in most, if not all, multicellular organisms; for example, a single WNK ortholog exists in Drosophila melanogaster and Caenorhabditis elegans, while at least nine orthologs exist in Arabidopsis thaliana (7). Although WNK kinases have not been found in the yeast Saccharomyces cervesiae, the first reported WNK sequence was identified in the fungus Phycomyces, leaving unanswered the question as to whether WNK kinases exist in unicellular organisms.
Human WNK kinases exhibit high sequence similarity within their amino-terminal kinase domains, two coiled-coil domains, and an auto-inhibitory domain (1, 4, 7). WNK kinases probably exist as oligomers (possibly homo-tetramers); however, the kinase activity does not require oligomerization (8). Oligomerization may help to increase the scaffolding function of the large carboxy-terminal domain that contains multiple sites for protein-protein interactions (e.g., several PXXP motifs that can interact with SH3 domains on other proteins). While stable WNK-WNK heteroligomers have not been detected, specific members of the WNK kinase family (e.g., WNK1 and WNK4) have been shown to phosphorylate each other (8, 9).
At the time of their identification as human disease genes, little was known about WNK kinase function other than that the autophosphorylation of the rat homologue of the human WNK1 gene was increased in response to hypertonic stimuli like NaCl, mannitol, or sucrose (4). This finding was prescient, as several years later, the first report surfaced that demonstrated WNKs were not only important regulators of the renal-specific NCC, but also the genetically-related Na-K-2Cl and K-Cl cotransporters (10–12), which were known to be reciprocally activated and inhibited, respectively, by serine-threonine phosphorylation triggered by extracellular hypertonicity (13–17). Exploration into non-renal targets of the WNK kinases was in part spurned by the finding that they were shown to be discretely localized to epithelia such as bile ducts, pancreatic ducts, and the choroid plexus, as well as in non-epithelial cells like neurons (10, 18).
A critical link occurred when Delpire and colleagues found that the WNK kinases physically interacted with the Ste20-type kinases SPAK and OSR1, which were known to bind, phosphorylate, and stimulate the activity of NKCC1 (19, 20, 21). The first experiments to show that a WNK kinase functionally regulated a Na-K-2Cl (10) or K-Cl co-transporter (11, 22) were performed in oocytes, paving the way for additional studies that defined the mechanism of this regulation in oocytes and other model systems (12).
Subsequent work has established the current paradigm that different WNK kinase osmosensors, sensitive to changes in extracellular tonicity and possibly intracellular [Cl-]i, via their interaction with the associated downstream SPAK and OSR1 transducer kinases, reciprocally but coordinately regulate the effector Na-K-2Cl and K-Cl cotransporters to defend cell volume, control epithelial transport, and potentially regulate [Cl-]i in central and peripheral neurons to help determine the polarity of GABAergic neurotransmission (23, 24). Commiserate with their importance in serving these critical roles in humans, mutations in WNKs underlie two different Mendelian diseases, PHAII (1) and hereditary sensory and autonomic neuropathy type 2 (HSAN2; 25). Moreover, the WNK pathway is also important in regulating ion transport in lower multicellular organisms, including worms (26), and also plants (27), suggesting the WNKs are evolutionarily-selected to regulate osmotic homeostasis.
This review will attempt to synthesize the current molecular genetic, biochemical, and physiological data regarding the WNK kinases and their phosphoregulation of the Na-K-2Cl and K-Cl cotransporters, with a focus on their roles in cell volume regulation. We will also speculate on potential roles of the WNKs on GABAergic signaling in the brain and peripheral nervous system. Other recent papers have comprehensively covered the roles of the WNKs in regulating renal transepithelial transport and the pathogenesis of hypertension (e.g., 28, 29), along with the roles of the WNK-associated Ste20-type kinases SPAK and OSR1 (e.g., 24).
The Na-K-2Cl and K-Cl cotransporters: coordinated roles in cell volume regulation and GABAergic signaling
The Na-K-2Cl and K-Cl cotransporters are SLC12A cation/chloride cotransporters (CCCs) that mediate epithelial transport, maintain cellular volume, and regulate GABAergic neurotransmission (30). The Na-(K)-2Cl cotransporters execute the coupled movement of sodium and chloride, with or without potassium, into cells and include the bumetanide-sensitive NKCC1 (ubiquitously expressed), the furosemide-sensitive NKCC2 (expressed in the kidney's thick ascending limb), and the thiazide-sensitive NCC (expressed in the kidney's distal convoluted and connecting tubules) (17, 30). Mutations in NKCC2 and NCC, respectively, cause Bartter’s syndrome type I [OMIM no. 601678] and Gitelman’s syndrome (OMIM no. 263800), autosomal recessive diseases characterized by low blood pressure due to renal salt wasting, and hypokalemic alkalosis (31, 32). The K-Cl cotransporters (KCC1, KCC2, KCC3, and KCC4) transport chloride coupled with potassium, mediating net chloride transport out of cells (16). KCC1 is ubiquitously expressed (33), while KCC2 is expressed exclusively in neurons (34). KCC3 is highly-expressed in neurons and renal epithelia (35), and loss-of-function mutations in KCC3 cause a severe inherited peripheral neuropathy associated with agenesis of the corpus callosum termed Andermann’s syndrome (OMIM #218000; 36). KCC4 is highly-expressed in renal and inner ear epithelia, and in neurons and glia in various regions of the CNS (37).
Roles in cell volume regulation
The volume of mammalian cells (which lack cells walls) is incessantly challenged by changes in extracellular tonicity, the transport of osmotically-active substances across the cell membrane, and the formation or disappearance of intracellular osmoles (38). This is a particular problem for neurons, which experience large fluxes of ions during action potential generation; epithelial cells, which are responsible for transporting solute and water across their membranes to achieve the homeostasis of bodily fluids like blood, cerebrospinal fluid, pancreatic juice, bile, and seminal fluid; and red blood cells, which experience changes in extracellular osmolality, especially during their transit through blood vessels traversing the hypertonic interstitium of the renal medulla. Because alterations of cell volume can jeopardize the structural integrity and intracellular milieu of cells, the continued operation of cell volume regulatory mechanisms is required to maintain proper cell function and survival (38, 39). These mechanisms are complex, and contain multiple sensors, transducers, and effectors, often operating in parallel pathways (39).
Cells are able to rapidly respond to perturbations in cell volume by dynamically altering their level of intracellular solute (Figure 1). In hypotonic extracellular conditions, abrupt cell swelling results in a homeostatic counter-response termed regulatory volume decrease (RVD), which triggers a corrective release of K+ and Cl- via K-Cl cotransport through KCCs, and/or separate K+ and Cl- channels (38–40). Cell shrinkage triggers regulatory volume increase (RVI), which results in ion accumulation via Na-K-2Cl cotransport, Na+/H+ exchange, or Na+ channels (38–40). To prevent futile cycling and needless energy expenditure, the NKCCs and the KCCs are coordinately and reciprocally regulated during these cell volume regulatory mechanisms; RVD results in the activation of KCCs and inhibition of the NKCCs, while RVI results in the inhibition of the KCCs and activation of the NKCCs (41). This regulation is largely achieved by serine/threonine phosphorylation and dephosphorylation (16–17; 42–44). Evidence from kinetic studies, many done in red blood cells, first suggested that a common volume/chloride-sensitive kinase mediates the phosphoregulation of both pathways (13; 41; 45–48). In this model, cell shrinkage and/or decreases in intracellular chloride activate the kinase(s), which in turn phosphorylates the NKCCs and KCCs, promoting their activation and inactivation, respectively; cell swelling and/or increases in intracellular chloride inhibits kinase activity, promoting net dephosphorylation of both cotransporters, thereby resulting in the inhibition of NKCC and activation of KCC (13). Pathologic alterations in CCC activity have been shown to contribute to the altered cell volume regulation in red blood cells in sickle cell disease (49–51) and in neurons in neurodegenerative disorders (52). Despite this important homeostatic mechanism, the identity of the "chloride/volume-sensitive" kinase of the Na-K-2Cl and K-Cl cotransporters has been a matter of speculation, until recently.
Figure 1. Reciprocal phosphoregulation of the Na-K-2Cl and K-Cl cotransporters triggered by changes in extracellular tonicity or intracellular chloride concentration [Cl-]i.

In many cell types, including epithelial cells and red blood cells, hypertonic conditions and cell shrinkage stimulate cotransporter phosphorylation (P), resulting in NKCC1 activation and K-Cl cotransporter inhibition (shown), contributing to regulatory volume increase. Conversely, hypotonicity and cell swelling promotes cotransporter dephosphorylation, resulting in NKCC1 inhibition and K-Cl cotransporter inhibition (not shown), contributing to regulatory volume decrease. In GABAergic neurons, the balance between NKCC1 and K-Cl cotransporter activities sets [Cl-]i, which along with the membrane potential, determines the directionality of the cellular flux of Cl- and the neuronal response to GABA (whose receptor functions as a Cl- channel). With high NKCC1 activity and low KCC2 activity, GABA signaling is excitatory; the converse is true when NKCC1 activity is low, and KCC2 activity is high. A common Cl-/volume sensitive kinase has been proposed to reciprocally and coordinately phosphoregulate the NKCCs and the K-Cl cotransporters.
Roles in GABAergic neurotransmission
NKCC1 and the KCCs also play important role in establishing the level of intracellular Cl- [Cl-]i in neurons, which determines the polarity (excitatory versus inhibitory) of the response to the neurotransmitter γ-aminobutyric acid (GABA), because the GABAA receptor is coupled to a chloride channel (53, 54) (Figure 1). During early development, NKCC1 activity is high and KCC2 activity is low, resulting in a high [Cl-]I and excitatory GABAergic signaling (55–58). The developmental switch from excitatory to inhibitory GABA signaling occurs shortly after birth in the rat, but in other species exhibit heterogeneity in the timing of this switch (56, 57, 59). In the adult mammalian CNS, high KCC2 activity and low NKCC1 results in very a low [Cl-]i, and consequently, inhibitory GABAergic signaling in most neuronal populations (57). Interestingly, certain anatomic populations of neurons in the adult show cycled variation in their response to GABA from excitatory to inhibitory (60) due to the dynamic modulation of [Cl-]i secondary to variations in CCC activity (61). The mechanisms underlying the switches in GABAergic signaling are not all simply due to alterations in CCC expression, suggesting dynamic phosphorylation mechanisms might also play a role in this switch -- both in development, and in circadian rhythms (54). Pathologic alterations in NKCC1 and KCC activity have been shown to result in the alterations in [Cl-]i homeostasis and aberrant GABAergic neurotransmission that contributes to the hypo-excitability or hyper-excitability of neurons in neonatal seizures (54, 62), temporal lobe epilepsy (63, 64), and neuropathic pain (65, 66). However, the kinases that mediate these dynamic fluctuations in NKCC1 and KCC activity in the central and peripheral nervous systems are unknown.
The WNK kinase “osmosensor”
Hyperosmotic stress and cell shrinkage activates WNK1 by stimulating its phosphorylation at critical regulatory residues. Extracellular hyperosmotic conditions (e.g., sorbitol, NaCl, and KCl), but not other stresses (e.g., H2O2, anisomycin, phosphatase inhibitors, growth factors, cytokines, phorbol esters) rapidly (within 0.5 min) activate endogenous WNK1 in multiple mammalian cell types by increasing its phosphorylation at Ser382 within the T-loop of its kinase domain (4, 8, 67). Mutation of Ser382 to Ala prevents WNK1 activation by hypertonicity, whereas mutation of Ser382 to Glu, which mimics phosphorylation, increases WNK1 activity and prevents further activation by hypertonic conditions (67). Ser382 and the residues surrounding it are identical in all WNK isoforms, and conserved in the D. melanogaster and C. elegans WNK1 homologues (67). At present, is unclear how WNK1 is activated by the shrinkage-induced phosphorylation at Ser382, though evidence at present favors a trans autophosphorylation reaction, suggesting that WNK1 is itself an "osmosensor." When expressed in E. coli, wild-type but not kinase-inactive WNK1 is still phosphorylated at Ser382, while in mammalian cells, a recombinant kinase-inactive WNK1 mutant can still be normally phosphorylated at Ser382 in response to sorbitol, suggesting that endogenous WNK1 is able to transphosphorylate the kinase-inactive WNK1 mutant (67). Additional mechanisms that could explain these data include phosphorylation of WNK1 by another upstream kinase that is itself the osmosensor, or the inhibition of a protein phosphatase. Further studies are required to investigate these possibilities.
Hyperosmotic stress also activates SPAK and OSR1, interacting downstream kinases of the WNKs (see below) by stimulating their phosphorylation at critical regulatory residues in their T-loop and S-motif (Thr-233 and Ser-373 of SPAK) (67). Critically, these sites have been shown previously to be phosphorylated by WNK1 in vitro (68, 69), and genetic suppression of WNK1 decreases basal and sorbitol-induced activation of SPAK/OSR1 and phosphorylation (67, 70). These data suggest that SPAK and OSR1 are likely not themselves osmosensors. Sorbitol induces a dramatic re-localization of WNK1 from the cytosol to intracellular vesicles that co-localize with clathrin and AP-1, and partially with TGN46, suggesting that a considerable pool of WNK1 is localized to TGN/recycling endosomes after hyperosmotic stress (67). This effect of hypertonicity on WNK1 is mediated by its c-terminal non-catalytic region (67). This finding is compelling, given the fact that the over-expression of several different WNK kinases affects the trafficking of multiple ion channels and cotransporters between intracellular vesicles and the plasma membrane. For example, catalytically-inactive WNK1 or -4 decreases the membrane expression of the renal outer medullary potassium channel (ROMK) through a clathrin-dependent endocytosis (2). It is also interesting that SPAK has been shown to translocate from the cytoplasm to the cytoskeleton in response to hypertonicity (71).
Together, these data highlight the effects that hypertonicity and cell shrinkage have on WNK1 phosphorylation, activity, and cellular localization, as well the WNKs’ interaction with its downstream kinase partners SPAK and OSR1. At present, it is unclear whether WNK2, -3, and -4 isoforms are regulated in a manner similar to WNK1; however, the fact that Ser382 is conserved among all WNK isoforms in different species suggests a common mechanism may exist among the different WNKs. Future experiments will also be needed to explore whether the WNKs' regulation of ion channels and other cotransporters is mediated in part through its translocation of to the TGN/recycling endosomes upon exposure to hyperosmotic stress or changes in intracellular ion content.
WNK kinase regulation of Na-K-2Cl cotransport via the transducer SPAK/OSR1 kinases
The mammalian Ste(sterile)20-type kinases, named after their yeast homolog originally discovered in a genetic analysis of Saccharomyces cerevisiae mating, include the Ste20/Sps1-related proline/alanine-rich kinase (SPAK) and the oxidative-stress responsive kinase 1 (OSR1), genes orthologous to D. melanogaster Fray and the C. elegans GCK-3. SPAK and OSR1 are highly-expressed in neurons and chloride transporting epithelia, including the distal nephron (72, 73). Low intracellular chloride or extracellular hypertonicity activate SPAK and OSR1 in a manner that is dependent on WNK kinases, as it is abrogated by genetic ablation of WNK1 or WNK4 kinase activity (67, 68, 73, 74). SPAK and OSR1 are key substrates downstream of the WNK kinases in a signaling cascade that regulates the activity of the NKCCs. This model has been shaped by biochemical data showing that WNKs physically interact and phosphorylate SPAK/OSR1 (19, 20, 68, 69, 75), and functional data showing that an interaction between specific WNKs, SPAK/OSR1, and NKCCs is required for cotransporter activation (12, 76, 77).
Delpire and colleagues first demonstrated that SPAK and OSR1 physically interact with NKCC1 and KCCs via a specific conserved carboxy-terminal (CCT) domain (19, 20). This motif of OSR1 and SPAK recognizes a Arg-Phe-Xaa-Val (RFXV) domain harbored in the cytoplasmic amino terminal tails of NKCCs and the KCCs (19, 20, 69) The SPAK/OSR1 CCT domain also binds RFXV domains embedded within the carboxyl termini of WNK1 and WNK4 (12, 19, 20, 68, 69, 74). The physical binding of SPAK to WNK1/4 and NKCC1 is necessary for cotransporter activity under both baseline and hyperosmotic conditions (68, 69, 74, 76). Villa et al. have delved into the structural mechanism by which the CCT domain of SPAK/OSR1 interacts with the RFXV motif of the WNKs and NKCC1 by solving the crystal structure of the CCT domain of OSR1 in complex with a peptide of WNK4 containing the RFXV motif at a resolution of 1.95 Angstroms (78). These studies revealed OSR1’s CCT domain forms a novel protein fold that binds WNK4’s RFXV motif through a surface-exposed groove via a complex web of interactions that included a Thr residue (Thr 1008) positioned just after its RFXV motif. Delpire and colleagues had shown previously that RFXV motifs in WNK1/4 (and other known or putative SPAK/OSR1-binding proteins) are preceded by such serine or threonine residues (79). The structure of the WNK4-OSR1 complex suggests that phosphorylation of threonine 1008 in WNK4 would result in a steric clash with the backbone of the CCT domain, potentially preventing the binding of WNK4 to OSR1’s CCT domain (78). Interestingly, a biotinylated WNK4 RFXV-pT peptide, in which a threonine residue equivalent to threonine 1008 in WNK4 is phosphorylated, failed to interact with endogenously-expressed SPAK and OSR1 in cell lysates under conditions in which the dephosphorylated WNK4 peptide bound strongly to OSR1 (78). This result is particularly intriguing, because it suggests that the interaction of RFXV motifs might be dynamically regulated by phosphorylation of nearby serine/threonine residues on WNK1 and WNK4. Phosphorylation of this residue by another (as yet unknown) kinase could trigger the dissociation of WNK1 or WNK4 from SPAK and OSR1, allowing SPAK/OSR1 to interact with their RFXV-motif-containing targets including the cotransporters. Such an event could be triggered by changes in intracellular chloride and/or cell volume. These data establish that the CCT domain functions as a multipurpose docking site, enabling SPAK/OSR1 to interact with its downstream substrates (NKCC1) and upstream activators (WNK1/WNK4) (69).
In addition to physically interacting with SPAK/OSR1, the WNK isoforms activate SPAK and OSR1 by phosphorylating equivalent residues located within the T-loop of their catalytic domains (Thr233 in SPAK, Thr185 in OSR1) (68, 69). Alanine mutagenesis has shown that phosphorylation of these residues is essential for SPAK/OSR1's activation in response to hypertonicity and cell shrinkage, as well as their ability to physically interact with and phosphorylate NKCC1 (68, 69, 74, 75). WNKs phosphorylate other residues in SPAK/OSR1 (e.g., Thr243 in SPAK), but alanine mutagenesis of these residues does not affect SPAK/OSR1's activation in response to hypertonicity or its ability to activate NKCC1 (67, 75). Once activated, SPAK/OSR1 phosphorylates Thr199, Thr201, and Thr206 in NKCC1; phosphorylation at Thr206 is essential for NKCC1's activation by hypertonicity (75, 76). Binding of SPAK/OSR1 to NKCC1 is also necessary for cotransporter activity under both baseline and hyperosmotic conditions (76).
The functional interaction of WNKs and SPAK/OSR1 on NKCC1 was first demonstrated in oocytes, as has been since recapitulated in mammalian cell systems. While the activity of NKCC1 was not shown to be significantly affected by co-expression of SPAK in oocytes (21), the finding that a dominant-negative SPAK mutant dramatically reduced the regulatory phosphorylation and activity of NKCC1 in low intracellular chloride (activating) conditions suggested that SPAK might be a key element downstream of the chloride-sensing mechanism that regulates NKCC1 (21). This interaction was found to be of important functional significance, as co-expression of SPAK and WNK4 together, but not alone, robustly increased NKCC1 activity in a manner insensitive to conditions of external osmolarity (69, 74). This activation was dependent on the catalytic activities of SPAK and WNK4, and SPAK’s physical interaction with both WNK4 and NKCC1. OSR1 exhibited similar functional activation of NKCC1 when co-expressed with WNK1 (69). Genetically engineered SPAK knock-in mice in which the T-loop threonine residue (Thr243) was mutated to alanine to prevent activation by WNK isoforms exhibit markedly reduced phosphorylation of NCC and NKCC2 cotransporters at the residues phosphorylated by SPAK, and display significantly reduced salt-dependent blood pressure, consistent with a loss of function of NCC and NKCC2 (80).
Equally important to the activation of NKCC1 by hypertonicity and cell shrinkage is its deactivation in response to hypotonicity and cell swelling, which occurs secondary to cotransporter dephosphorylation by phosphatases. NKCC1 activity is stimulated by protein phosphatase 1 (PP1) inhibitors; however, it has been unclear how PP1 exerts its inhibitory effect on NKCC1 during hypotonicity: does PP1 act directly on the cotransporter, or indirectly by affecting an activating kinase like WNK or SPAK? Recently, Delpire and colleagues shed insight into the mechanism by which NKCC1 is inhibited by PP1, by showing that mutation of key residues in the PP1 binding motif located in the N-terminal tail of NKCC1 significantly reduces the inhibitory effect of PP1, and that PP1 dephosphorylates the cotransporter and SPAK in a time-dependent manner (81). Importantly, PP1's dephosphorylation of SPAK was significantly greater when SPAK and the N-terminal tail of NKCC1 were in a physical complex with one another in the reaction, indicating the necessity of scaffolding of PP1 and SPAK in proximity to one another. These data are consistent with a model where PP1 inhibits NKCC1 activity directly by dephosphorylating the cotransporter, and also indirectly by dephosphorylating its proximal-most activating kinase, SPAK (81). At present, it is unclear if PP1 dephosphorylates the WNK kinases. This will be an important question for future experiments, since WNK1/4 exists in a complex with SPAK in cells.
WNK kinase regulation of K-Cl cotransport
Exploration of WNK/Ste20-type kinase regulation of the K-Cl cotransporters has lagged behind that of the Na-K-2Cl cotransporters until recently. It seems that, in contrast to their effect on NKCC1, SPAK/OSR1 seem to play minor roles in the WNK-dependent regulation of the KCCs (82–84). A breakthrough study by Rinehart et al. (84), which utilized novel, titanium dioxide-based phosphopeptide enrichment followed by two techniques that allowed for the quantitative assessment of dynamic changes in KCC phosphorylation under hypotonic (activating) and hypertonic (inactivating) conditions (SILAC, or stable isotope labeling of amino acids in cell culture; and MRM, or multiple reactive monitoring, which measures the absolute percentage of phosphorylation at specific target sites) identified two essential sites of phosphorylation (T991 and T1048) in KCC3 that mediate its inactivation during hypertonic/cell shrinkage in both cell culture and in mature human erythrocytes. WNK1, but not SPAK or OSR1, is necessary for the phosphorylation of these residues of KCC3 (84). RNAi with multiple siRNAs against WNK1, WNK2, WNK3, WNK4, SPAK, and OSR1 in HEK-KCC3tetON cells, demonstrated that only WNK1 was necessary for the hypertonicity-induced phosphorylation and inhibition of KCC3 at T991 and T1048. Whether this effect is mediated by direct phosphorylation of KCC3 by WNK1, or via the activation of other kinases that act at these sites is unknown. Alternatively, WNK1 could negatively regulate phosphatase activity. These possibilities will require further experiments for elucidation. These regulatory sites on KCC3 are also present in other KCCs, including KCC2, and have been shown to be essential for KCC2's full activation in mammalian cells. Alanine substitutions introduced into KCC2 at positions Thr 906 and 1007 (the equivalent residues to T991 and T1048 in KCC3) resulted in a 4-fold increase in KCC2 activity compared with wild type KCC2 in HEK293 cells under isotonic conditions, as assayed by KCC-dependent 86 Rb uptake (84). Moreover, in the developing brain, the phosphorylation state of KCC2 at Thr 906 and 1007 parallels KCC2 activity, with essentially complete dephosphorylation in the adult, when KCC2 activity is highest. These are particularly intriguing results, suggesting that KCC2 phosphorylation at these sites may play a role in regulation of [Cl-]i in GABAergic neurons, in addition to other known mechanisms of KCC2 regulation in the CNS such as regulation of transcript and protein expression (85). Additional experiments are needed to assess the physiologic role of KCC2 phosphorylation in the brain and peripheral nervous system. Interestingly, the sequence context of T991 in the KCCs is also conserved in the amino-terminus of NKCC1, suggesting the same kinase could coordinately regulate the NKCCs and KCCs via opposing effects these two transport pathways.
WNK3: a brain-enriched WNK family member with reciprocal actions on the Na-K- 2Cl and K-Cl cotransporters
WNK3 deserves special mention, given its distinct expression profile and its opposing actions on the Na-K-2Cl and K-Cl cotransporters. WNK3 exists in several alternatively-spliced isoforms, including a brain-specific isoform that contains a unique exon (86). Like WNK1 and WNK4, WNK3 localizes to the intercellular junctions of chloride-transporting epithelia throughout the body (11); however, unlike WNK1 and WNK4, which are highly-expressed in the kidney, WNK3 is most highly expressed in the brain. In situ analysis has shown that WNK3 transcripts are robustly expressed in the several different cortical layers, the hippocampus, reticular formation, hypothalamic nuclei (including the supraoptic and suprachiasmatic nuclei), and the cerebellum, and undergoes a dynamic developmental shift in expression that parallels KCC2 in the hippocampus and cerebellum (11). Recently, NKCC1, several of the K-Cl cotransporters, and WNK3 have been shown to be co-expressed in vasoactive intestinal peptide, gastrin-releasing peptide, and vasopressin-releasing neurons in the suprachiasmatic nucleus (87), an interesting phenomenon because neurons within the SCN are capable of eliciting either an excitatory or inhibitory response to GABA, depending on the cell's level of [Cl-]i (60).
WNK3 modulates the balance between cellular chloride influx and efflux via reciprocal actions on the NKCCs and KCCs, and is able to override the normal effects of variation in extracellular osmolarity on cotransporter activity (11). Co-expression of wild-type WNK3 with NKCC1 in oocytes increases NKCC1 activity not only in hypertonic media (the normal condition for assaying NKCC1 activity), but also in hypotonic media (a condition in which NKCC1 is normally silenced) (11). Conversely, wild-type WNK3 inhibited each of the KCCs, even in hypotonic media (a condition in which KCCs are normally maximally active) (11). A catalytically-silent WNK3 mutant produced the mirror-image effects of wild-type WNK3 on the cotransporters, inhibiting NKCC1 but activating the KCCs (11). Interestingly, kinase-inactive WNK3’s stimulation of the KCCs in hypertonic conditions is abolished by calyculin A and cyclosporine A, implicating protein phosphatases PP1 and 2B in this effect (82). Wild type WNK3 increases the phosphorylation of the same Thr residues of NKCC1 phosphorylated by SPAK/OSR1, and kinase-inactive WNK3 prevented the phosphorylation of these essential residues, even in hypertonic (activating) conditions (11).
These data suggest that WNK3 can regulate [Cl-]i by dynamically regulating balance between NKCC1 and KCC activities, depending on whether WNK3 is itself “activated.” Presumably, this is achieved in response to changes in extracellular tonicity, cell volume, and intracellular chloride, though formal evidence is at present lacking. However, insight into the mechanism of how WNK3 regulates its targets was recently achieved by Ponce-Coria et al., who focused on the target NKCC2 (77). This group found that intracellular chloride depletion activates NKCC2 by promoting the phosphorylation of the cotransporter at homologous residues that are phosphorylated in NKCC1 by SPAK/OSR1. Ponce-Coria et al. demonstrated that the activation of NKCC2 in response to intracellular chloride depletion was dependent on an interaction between WNK3 and SPAK, where WNK3 is positioned upstream of SPAK and appears to be the chloride-sensitive kinase (77). Elimination of WNK3’s only SPAK-binding motif prevents its activation of NKCC2. A catalytically-inactive WNK3 mutant also completely prevents NKCC2 activation by intracellular chloride depletion (77). These data revealed a novel chloride-sensing mechanism that regulates NKCC2, and provided insight into the mechanism by which changes in the level of intracellular chloride can alter cotransporter activity (77). (Figure 2).
Figure 2. Proposed model for intracellular chloride concentration [Cl-]i, WNK3, and SPAK interaction in the control of Na-K-2Cl cotransporter activity.

Variations in [Cl-]i are likely associated with changes in the ratio of active versus inactive WNK3 kinase (W3R). Low [Cl-]i activates WNK3, which phosphorylates and activates SPAK, which then binds, phosphorylates, and activates the Na-K-2Cl cotransporters. See text for details. From Ponce-Coria 2008.
Evolutionary conservation of the WNK/Ste20-type kinase pathway
The ability to defend cell volume amidst changes in intracellular solute content or extracellular osmolality is a fundamental property common to cells in all species, since failure to do so would lead to osmotic lysis due to excessive cell swelling. Hence, it seems likely that common mechanisms of cell volume regulation would exist in different organisms through evolutionary conservation. Strange and colleagues have recently shown that the interaction between the WNK and Ste20-type kinases is an evolutionarily-conserved pathway that functions to regulate transepithelial ion transport and maintain systemic osmotic homeostasis in C. elegans (26, 82).
The SPAK worm homolog GCK-3 had been shown previously to bind and negatively regulate the C. elegans chloride channel CLH-3b, which is activated in response to cell swelling by serine/threonine dephosphorylation events mediated by the type 1 phosphatases GLC-7α and GLC-7β (88). CLH-3b is orthologous to the mammalian volume-sensitive and voltage-gated ClC-2 chloride channel, which is mutated in idiopathic generalized epilepsy, an inherited neurological disorder affecting about 0.4% of the world's population (89). By constructing genetically-engineered worms whose expression of GFP is driven by the endogenous gene’s promoter, Strange’s group demonstrated that both GCK-3 and the single WNK ortholog in worms, WNK-1, are expressed in the excretory cell (the worm-equivalent of the mammalian kidney), which plays a role in systemic osmoregulation (26). Worms genetically-engineered with tissue-specific RNAi of GCK-3 had a dramatically impaired systemic volume recovery and survival after hypertonic shrinkage. WNK-1 was shown to bind to GCK-3 in the yeast two-hybrid system, and knockdown of WNK-1 produced a phenotype similar to GCK-3 (RNAi) worms (26). Because knockdown of GCK-3 and WNK-1 together had no additive effect, these kinases were postulated to function in a common pathway in a manner similar to their mammalian homologs, with WNK-1 upstream of GCK-3 (26). Deletion of either of the worm WNK-1 or GCK-3 genes resulted in the Exc phenotype, which includes a defect in the tubular extension of excretory cells (90). Expression of the activated form of GCK-3 or the CLH-3 deletion mutation partly suppresses the phenotype of the WNK-1 or GCK-3 deletion mutants, suggesting that WNK-1, by activating GCK-3 and triggering the down-regulation of CIC channel activity, controls the tubular formation of excretory canals (90).
Phylogenetic analysis of the kinase domains of the Ste20-type and WNK kinases by Choe et al. suggest that the interaction between these two kinase families first evolved in the early metazoan lineage (26). Evolutionarily, this would have coincided with the emergence of multicellular organisms that contain different fluid compartments separated by epithelial cell sheets, and the need to precisely regulate the ionic and osmotic composition of these different fluids. The evidence provided by Strange and colleagues suggest the compelling possibility that the WNKs and the Ste20-type kinases arose early in animal evolution as a mechanism to regulate the epithelial transport processes of early metazoans, a function that is still crucial in humans today (91).
Closing comments
Since it was first hypothesized that a single "chloride/volume-sensitive kinase" regulates the Na-K-2Cl and K-Cl cotransporters, it has become increasingly clear that this model is far too simple. The current paradigm suggests that a more complex system, comprised of osmosensors like the WNK kinases, and associated downstream transducer kinases like SPAK/OSR1, work in concert to orchestrate this regulation. This system is important for maintaining cell volume during osmotic stress, executing transepithelial solute and water transport, and may be important for modulating the response (excitation versus inhibition) to GABA (Figure 3). Further complexity, not fully developed in this review, is added by the fact that WNKs interact with one another, and themselves are negative or positive regulators of one another's activity; for example, WNK1 phosphorylates both WNK2 and WNK4 and is a negative regulator of WNK4-mediated effects (e.g., 9), yet both kinases have been shown to activate SPAK/OSR1 (75). Moreover, for a given WNK, like WNK1, tissue-specific isoforms exist (e.g., both WNK1 and WNK4 have kidney-specific isoforms, and WNK1 and WNK3 have brain-specific isoforms) (1, 25, 86). Additionally, WNKs have been shown to regulate another serine-threonine kinase, SGK-1 (3), which has a known role in cell volume regulation (38). Given this complexity, it seems like unbiased genetic analyses, in combination with in vitro and in vivo physiological characterization, will continue to be useful tools to dissect out the details of this important, evolutionarily-conserved pathway and identify the prime targets for therapeutic drug development. For example, molecular genetic analysis has recently shown that autosomal recessive disease hereditary sensory and autonomic neuropathy type II (HSANII), an early-onset disorder characterized by loss of perception to pain, touch, and heat due to a loss of peripheral sensory nerves, results from mutations in the nervous system-specific exon of WNK1 (25). This finding, especially in light of the demonstrated ability of WNK1 to regulate both NKCC1 and the K-Cl cotransporters like KCC2 and KCC3, suggests that investigation into the neuronal-specific isoform of WNK1 might prove beneficial for targeting pain syndromes. Genetic investigation of the WNKs in model systems like D. melanogaster and C. elegans, which have single WNK homologs, may also be useful, given the tractability and established proteomic networks of these organisms. An increased understanding of how the WNKs regulate the Na-K-2Cl and K-Cl cotransporters may provide novel opportunities for the selective modulation of these transporters, with ramifications for common human diseases like hypertension, sickle cell disease, neuropathic pain, and epilepsy.
Figure 3. Model of WNK kinase phosphoregulation of the Na-K-2Cl and K-Cl cotransporters.
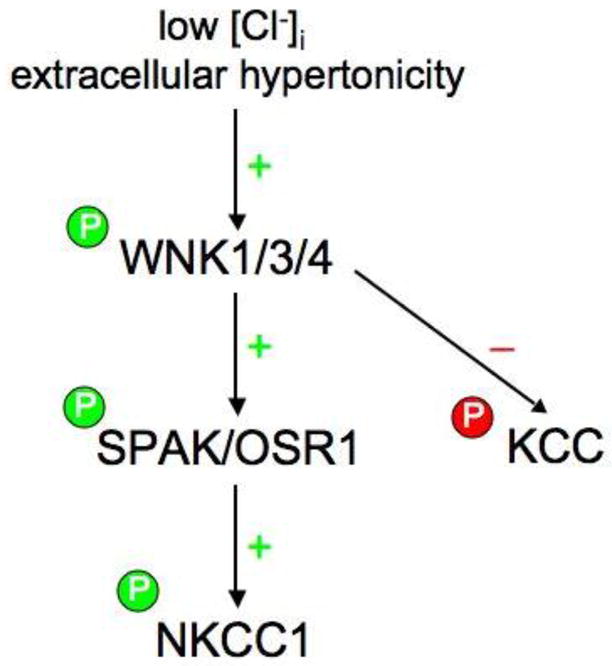
Low intracellular chloride concentration [Cl-]i and/or extracellular hypertonicity (resulting in cell shrinkage) activates WNK kinase(s) via autophosphorylation or activation by an unknown upstream kinase. Activated WNKs, in turn, bind, phosphorylate, and activate SPAK/OSR1, which then binds to NKCC1 and activates it by promoting its phosphorylation at key amino-terminal residues in its cytoplasmic tail. Under similar conditions, WNKs trigger the net phosphorylation and inhibition of the K-Cl cotransporters, either directly or indirectly via the inactivation of a phosphatase.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Wilson FH, Disse-Nicodeme S, Choate KA, Ishikawa K, Nelson-Williams C, Desitter I, Gunel M, Milford DV, Lipkin GW, Achard JM, Feely MP, Dussol B, Berland Y, Unwin RJ, Mayan H, Simon DB, Farfel Z, Jeunemaitre X, Lifton RP. Human hypertension caused by mutations in WNK kinases. Science. 2001;293:1107–1112. doi: 10.1126/science.1062844. [DOI] [PubMed] [Google Scholar]
- 2.Kahle KT, Wilson FH, Leng Q, Lalioti MD, O’Connell AD, Dong K, Rapson AK, MacGregor GG, Giebisch G, Hebert SC, Lifton RP. WNK4 regulates the balance between renal NaCl reabsorption and K+ secretion. Nat Genet. 2003;35:372–376. doi: 10.1038/ng1271. [DOI] [PubMed] [Google Scholar]
- 3.Kahle KT, Ring AM, Lifton RP. Molecular physiology of the WNK kinases. Annu Rev Physiol. 2008;70:329–55. doi: 10.1146/annurev.physiol.70.113006.100651. [DOI] [PubMed] [Google Scholar]
- 4.Xu B, English JM, Wilsbacher JL, Stippec S, Goldsmith EJ, Cobb MH. WNK1, a novel mammalian serine/threonine protein kinase lacking the catalytic lysine in subdomain II. J Biol Chem. 2000;275:16795–16801. doi: 10.1074/jbc.275.22.16795. [DOI] [PubMed] [Google Scholar]
- 5.Min X, Lee BH, Cobb MH, Goldsmith EJ. Crystal structure of the kinase domain of WNK1, a kinase that causes a hereditary form of hypertension. Structure. 2004;12(7):1303–11. doi: 10.1016/j.str.2004.04.014. [DOI] [PubMed] [Google Scholar]
- 6.Dan I, Watanabe NM, Kusumi A. The Ste20 group kinases as regulators of MAP kinase cascades. Trends Cell Biol. 2001;11:220–230. doi: 10.1016/s0962-8924(01)01980-8. [DOI] [PubMed] [Google Scholar]
- 7.Veríssimo F, Jordan P. WNK kinases, a novel protein kinase subfamily in multicellular organisms. Oncogene. 2001;20(39):5562–9. doi: 10.1038/sj.onc.1204726. [DOI] [PubMed] [Google Scholar]
- 8.Lenertz LY, Lee BH, Min X, Xu BE, Wedin K, Earnest S, Goldsmith EJ, Cobb MH. Properties of WNK1 and implications for other family members. J Biol Chem. 2005;280:26653–26658. doi: 10.1074/jbc.M502598200. [DOI] [PubMed] [Google Scholar]
- 9.Yang CL, Zhu X, Wang Z, Subramanya AR, Ellison DH. Mechanisms of WNK1 and WNK4 interaction in the regulation of thiazide-sensitive NaCl cotransport. J Clin Invest. 2005;115(5):1379–87. doi: 10.1172/JCI22452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kahle KT, Gimenez I, Hassan H, Wilson FH, Wong RD, Forbush B, Aronson PS, Lifton RP. WNK4 regulates apical and basolateral Cl- flux in extrarenal epithelia. Proc Natl Acad Sci USA. 2004;101:2064–2069. doi: 10.1073/pnas.0308434100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kahle KT, Rinehart J, de Los Heros P, Louvi A, Meade P, Vazquez N, Hebert SC, Gamba G, Gimenez I, Lifton RP. WNK3 modulates transport of Cl- in and out of cells: implications for control of cell volume and neuronal excitability. Proc Natl Acad Sci USA. 2005;102:16783–16788. doi: 10.1073/pnas.0508307102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gagnon KB, England R, Delpire E. Volume sensitivity of cation-chloride cotransporters is modulated by the interaction of two kinases: SPAK and WNK4. Am J Physiol Cell Physiol. 2006;290:C134–C142. doi: 10.1152/ajpcell.00037.2005. [DOI] [PubMed] [Google Scholar]
- 13.Parker JC. In defense of cell volume? Am J Physiol Cell Physiol. 1993;265:C1191–C1200. doi: 10.1152/ajpcell.1993.265.5.C1191. [DOI] [PubMed] [Google Scholar]
- 14.Lauf PK, Adragna NC. K-Cl cotransport: properties and molecular mechanism. Cell Physiol Biochem. 2000;10:341–354. doi: 10.1159/000016357. [DOI] [PubMed] [Google Scholar]
- 15.Flatman PW. Regulation of Na-K-2Cl cotransport by phosphorylation and protein-protein interactions. Biochim Biophys Acta. 2002;1566:140–151. doi: 10.1016/s0005-2736(02)00586-2. [DOI] [PubMed] [Google Scholar]
- 16.Adragna NC, Fulvio MD, Lauf PK. Regulation of K-Cl cotransport: from function to genes. J Membr Biol. 2004;201:109–137. doi: 10.1007/s00232-004-0695-6. [DOI] [PubMed] [Google Scholar]
- 17.Hebert SC, Mount DB, Gamba G. Molecular physiology of cation-coupled Cl- cotransport: the SLC12 family. Pflügers Arch. 2004;447:580–593. doi: 10.1007/s00424-003-1066-3. [DOI] [PubMed] [Google Scholar]
- 18.Choate KA, Kahle KT, Wilson FH, Nelson-Williams C, Lifton RP. WNK1, a kinase mutated in inherited hypertension with hyperkalemia, localizes to diverse Cl--transporting epithelia. Proc Natl Acad Sci USA. 2003;100:663–668. doi: 10.1073/pnas.242728499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Piechotta K, Lu J, Delpire E. Cation chloride cotransporters interact with the stress-related kinases Ste20-related prolinealanine-rich kinase (SPAK) and oxidative stress response 1 (OSR1) J Biol Chem. 2002;277:50812–50819. doi: 10.1074/jbc.M208108200. [DOI] [PubMed] [Google Scholar]
- 20.Plotkin MD, Snyder EY, Hebert SC, Delpire E. Expression of the Na-K-2Cl cotransporter is developmentally regulated in postnatal rat brains: a possible mechanism underlying GABA's excitatory role in immature brain. J Neurobiol. 1997;33(6):781–95. doi: 10.1002/(sici)1097-4695(19971120)33:6<781::aid-neu6>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 21.Dowd BF, Forbush B. PASK (prolinealanine-rich STE20-related kinase), a regulatory kinase of the Na-K-Cl cotrans-porter (NKCC1) J Biol Chem. 2003;278:27347–27353. doi: 10.1074/jbc.M301899200. [DOI] [PubMed] [Google Scholar]
- 22.Rinehart J, Kahle KT, de Los Heros P, Vazquez N, Meade P, Wilson FH, Hebert SC, Gimenez I, Gamba G, Lifton RP. WNK3 kinase is a positive regulator of NKCC2 and NCC, renal cation-Cl- cotransporters required for normal blood pressure homeostasis. Proc Natl Acad Sci USA. 2005;102:16777–16782. doi: 10.1073/pnas.0508303102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kahle KT, Rinehart J, Ring A, Gimenez I, Gamba G, Hebert SC, Lifton RP. WNK protein kinases modulate cellular Cl- flux by altering the phosphorylation state of the Na-K-Cl and K-Cl cotransporters. Physiology (Bethesda) 2006;21:326–35. doi: 10.1152/physiol.00015.2006. [DOI] [PubMed] [Google Scholar]
- 24.Delpire E, Gagnon KB. SPAK and OSR1: STE20 kinases involved in the regulation of ion homoeostasis and volume control in mammalian cells. Biochem J. 2008;409(2):321–31. doi: 10.1042/BJ20071324. [DOI] [PubMed] [Google Scholar]
- 25.Shekarabi M, Girard N, Rivière JB, Dion P, Houle M, Toulouse A, Lafrenière RG, Vercauteren F, Hince P, Laganiere J, Rochefort D, Faivre L, Samuels M, Rouleau GA. Mutations in the nervous system--specific HSN2 exon of WNK1 cause hereditary sensory neuropathy type II. J Clin Invest. 2008;118(7):2496–505. doi: 10.1172/JCI34088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Choe KP, Strange K. Evolutionarily conserved WNK and Ste20 kinases are essential for acute volume recovery and survival after hypertonic shrinkage in Caenorhabditis elegans. Am J Physiol Cell Physiol. 2007;293(3):C915–27. doi: 10.1152/ajpcell.00126.2007. [DOI] [PubMed] [Google Scholar]
- 27.Wang Y, Liu K, Liao H, Zhuang C, Ma H, Yan X. The plant WNK gene family and regulation of flowering time in Arabidopsis. Plant Biol (Stuttg) 2008;10(5):548–62. doi: 10.1111/j.1438-8677.2008.00072.x. [DOI] [PubMed] [Google Scholar]
- 28.Welling PA, Chang YP, Delpire E, Wade JB. Multigene kinase network, kidney transport, and salt in essential hypertension. Kidney Int. 2010 Apr 14; doi: 10.1038/ki.2010.103. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Denton J, Nehrke K, Yin X, Morrison R, Strange K. GCK-3, a newly identified Ste20 kinase, binds to and regulates the activity of a cell cycle-dependent ClC anion channel. J Gen Physiol. 2005;125(2):113–25. doi: 10.1085/jgp.200409215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gamba G. Molecular physiology and pathophysiology of electroneutral cationchloride cotrans-porters. Physiol Rev. 2005;85:423–493. doi: 10.1152/physrev.00011.2004. [DOI] [PubMed] [Google Scholar]
- 31.Simon DB, Nelson-Williams C, Bia MJ, Ellison D, Karet FE, Molina AM, Vaara I, Iwata F, Cushner HM, Koolen M, Gainza FJ, Gitleman HJ, Lifton RP. Gitelman’s variant of Bartter’s syndrome, inherited hypokalaemic alkalosis, is caused by mutations in the thiazide-sensitive Na-Cl cotransporter. Nat Genet. 1996;12:24–30. doi: 10.1038/ng0196-24. [DOI] [PubMed] [Google Scholar]
- 32.Simon DB, Karet FE, Hamdan JM, DiPietro A, Sanjad SA, Lifton RP. Bartter’s syndrome, hypokalaemic alkalosis with hypercalciuria, is caused by mutations in the Na-K-2Cl cotransporter NKCC2. Nat Genet. 1996;13:183–188. doi: 10.1038/ng0696-183. [DOI] [PubMed] [Google Scholar]
- 33.Gillen CM, Brill S, Payne JA, Forbush BIII. Molecular cloning and functional expression of the K-Cl cotransporter from rabbit, rat and human. A new member of the cation-chloride cotransporter family. J Biol Chem. 1996;271:16237–16244. doi: 10.1074/jbc.271.27.16237. [DOI] [PubMed] [Google Scholar]
- 34.Payne JA, Stevenson TJ, Donaldson LF. Molecular characterization of a putative K-Cl cotransporter in rat brain. A neuronal-specific isoform. J Biol Chem. 1996;271(27):16245–52. doi: 10.1074/jbc.271.27.16245. [DOI] [PubMed] [Google Scholar]
- 35.Mercado A, Mount DB, Gamba G. Electroneutral cation-chloride cotransporters in the central nervous system. Neurochem Res. 2004;29:17–25. doi: 10.1023/b:nere.0000010432.44566.21. [DOI] [PubMed] [Google Scholar]
- 36.Howard HC, Mount DB, Rochefort D, Byun N, Dupre N, Lu J, Fan X, Song L, Riviere JB, Prevost C, Horst J, Simonati A, Lemcke B, Welch R, England R, Zhan FQ, Mercado A, Siesser WB, George AL, Jr, McDonald MP, Bouchard JP, Mathieu J, Delpire E, Rouleau GA. The K-Cl cotransporter KCC3 is mutant in a severe peripheral neuropathy associated with agenesis of the corpus callosum. Nat Genet. 2002;32:384–392. doi: 10.1038/ng1002. [DOI] [PubMed] [Google Scholar]
- 37.Boettger T, Hubner CA, Maier H, Rust MB, Beck FX, Jentsch TJ. Related articles, links deafness and renal tubular acidosis in mice lacking the K-Cl cotransporter Kcc4. Nature. 2002;416:874–878. doi: 10.1038/416874a. [DOI] [PubMed] [Google Scholar]
- 38.Lang F, Busch GL, Volkl H. The diversity of volume regulatory mechanisms. Cell Physiol Biochem. 1998;8:1–45. doi: 10.1159/000016269. [DOI] [PubMed] [Google Scholar]
- 39.Hoffmann EK, Lambert IH, Pedersen SF. Physiology of cell volume regulation in vertebrates. Physiol Rev. 2009;89(1):193–277. doi: 10.1152/physrev.00037.2007. [DOI] [PubMed] [Google Scholar]
- 40.Strange K. Cellular volume homeostasis. Adv Physiol Educ. 2004;28(1–4):155–9. doi: 10.1152/advan.00034.2004. [DOI] [PubMed] [Google Scholar]
- 41.Lytle C, McManus T. Coordinate modulation of Na-K-2Cl cotransport and KCl cotransport by cell volume and chloride. Am J Physiol Cell Physiol. 2002;283:C1422–C1431. doi: 10.1152/ajpcell.00130.2002. [DOI] [PubMed] [Google Scholar]
- 42.Russell JM. Sodium-potassium-chloride cotransport. Physiol Rev. 2000;80:211–276. doi: 10.1152/physrev.2000.80.1.211. [DOI] [PubMed] [Google Scholar]
- 43.Haas M, Forbush B. The Na-K-Cl cotransporter of secretory epithelia. Annu Rev Physiol. 2000;62:515–534. doi: 10.1146/annurev.physiol.62.1.515. [DOI] [PubMed] [Google Scholar]
- 44.Lytle C, Forbush B., 3rd Regulatory phosphorylation of the secretory Na-K-Cl cotransporter: modulation by cytoplasmic Cl. Am J Physiol. 1996;270(2 Pt 1):C437– 48. doi: 10.1152/ajpcell.1996.270.2.C437. [DOI] [PubMed] [Google Scholar]
- 45.Jennings ML, Adame MF. Direct estimate of 1:1 stoichiometry of K(+)-Cl(-) cotransport in rabbit erythrocytes. Am J Physiol Cell Physiol. 2001;281(3):C825–32. doi: 10.1152/ajpcell.2001.281.3.C825. [DOI] [PubMed] [Google Scholar]
- 46.Parker JC, Colclasure GC, McManus TJ. Coordinated regulation of shrinkageinduced Na/H exchange and swelling-induced [K-Cl] cotransport in dog red cells. Further evidence from activation kinetics and phosphatase inhibition. J Gen Physiol. 1991;98(5):869–80. doi: 10.1085/jgp.98.5.869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lytle C. Activation of the avian erythrocyte Na-K-Cl cotransport protein by cell shrinkage, cAMP, fluoride, and calyculin-A involves phosphorylation at common sites. J Biol Chem. 1997;272:15069–15077. doi: 10.1074/jbc.272.24.15069. [DOI] [PubMed] [Google Scholar]
- 48.Lytle C. A volume-sensitive protein kinase regulates the Na-K-2Cl cotransporter in duck red blood cells. Am J Physiol Cell Physiol. 1998;274:C1002–C1010. doi: 10.1152/ajpcell.1998.274.4.C1002. [DOI] [PubMed] [Google Scholar]
- 49.Rust MB, Alper SL, Rudhard Y, Shmukler BE, Vicente R, Brugnara C, Trudel M, Jentsch TJ, Hübner CA. Disruption of erythroid K-Cl cotransporters alters erythrocyte volume and partially rescues erythrocyte dehydration in SAD mice. J Clin Invest. 2007;117(6):1708–17. doi: 10.1172/JCI30630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brugnara C, De Franceschi L, Bennekou P, Alper SL, Christophersen P. Novel therapies for prevention of erythrocyte dehydration in sickle cell anemia. Drug News Perspect. 2001;(4):208–20. doi: 10.1358/dnp.2001.14.4.858404. [DOI] [PubMed] [Google Scholar]
- 51.Lew VL, Bookchin RM. Ion transport pathology in the mechanism of sickle cell dehydration. Physiol Rev. 2005;85(1):179–200. doi: 10.1152/physrev.00052.2003. [DOI] [PubMed] [Google Scholar]
- 52.Byun N, Delpire E. Axonal and periaxonal swelling precede peripheral neurodegeneration in KCC3 knockout mice. Neurobiol Dis. 2007;28(1):39–51. doi: 10.1016/j.nbd.2007.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Payne JA, Rivera C, Voipio J, Kaila K. Cation-chloride co-transporters in neuronal communication, development and trauma. Trends Neurosci. 2003;26:199–206. doi: 10.1016/S0166-2236(03)00068-7. [DOI] [PubMed] [Google Scholar]
- 54.Kahle KT, Staley KJ, Nahed BV, Gamba G, Hebert SC, Lifton RP, Mount DB. Roles of the cation-chloride cotransporters in neurological disease. Nat Clin Pract Neurol. 2008;4(9):490–503. doi: 10.1038/ncpneuro0883. [DOI] [PubMed] [Google Scholar]
- 55.Plotkin MD, Snyder EY, Hebert SC, Delpire E. Expression of the Na-K-2Cl cotransporter is developmentally regulated in postnatal rat brains: a possible mechanism underlying GABA's excitatory role in immature brain. J Neurobiol. 1997;33(6):781–95. doi: 10.1002/(sici)1097-4695(19971120)33:6<781::aid-neu6>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 56.Rivera C, Voipio J, Payne JA, Ruusuvuori E, Lahtinen H, Lamsa K, Pirvola U, Saarma M, Kaila K. The K+/Cl- co-transporter KCC2 renders GABA hyperpolarizing during neuronal maturation. Nature. 1999;397:251–255. doi: 10.1038/16697. [DOI] [PubMed] [Google Scholar]
- 57.Delpire E. Cation-chloride cotransporters in neuronal communication. News Physiol Sci. 2000;15:309–312. doi: 10.1152/physiologyonline.2000.15.6.309. [DOI] [PubMed] [Google Scholar]
- 58.Dzhala VI, Brumback AC, Staley KJ. Bumetanide enhances phenobarbital efficacy in a neonatal seizure model. Ann Neurol. 2008;63(2):222–35. doi: 10.1002/ana.21229. [DOI] [PubMed] [Google Scholar]
- 59.Ben-Ari Y. Excitatory actions of GABA during development: the nature of the nurture. Nat Rev Neurosci. 2002;(9):728–39. doi: 10.1038/nrn920. [DOI] [PubMed] [Google Scholar]
- 60.Wagner S, Castel M, Gainer H, Yarom Y. GABA in the mammalian suprachiasmatic nucleus and its role in diurnal rhythmicity. Nature. 1997;387:598–603. doi: 10.1038/42468. [DOI] [PubMed] [Google Scholar]
- 61.Choi HJ, Lee CJ, Schroeder A, Kim YS, Jung SH, Kim JS, Kim do Y, Son EJ, Han HC, Hong SK, Colwell CS, Kim YI. Excitatory actions of GABA in the suprachiasmatic nucleus. J Neurosci. 2008;28(21):5450–9. doi: 10.1523/JNEUROSCI.5750-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kahle KT, Staley KJ. The bumetanide-sensitive Na-K-2Cl cotransporter NKCC1 as a potential target of a novel mechanism-based treatment strategy for neonatal seizures. Neurosurg Focus. 2008;25(3):E22. doi: 10.3171/FOC/2008/25/9/E22. [DOI] [PubMed] [Google Scholar]
- 63.Palma E, Amici M, Sobrero F, Spinelli G, Di Angelantonio S, Ragozzino D, Mascia A, Scoppetta C, Esposito V, Miledi R, Eusebi F. Anomalous levels of Cltransporters in the hippocampal subiculum from temporal lobe epilepsy patients make GABA excitatory. Proc Natl Acad Sci U S A. 2006;103(22):8465–8. doi: 10.1073/pnas.0602979103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Huberfeld G, Wittner L, Clemenceau S, Baulac M, Kaila K, Miles R, Rivera C. Perturbed chloride homeostasis and GABAergic signaling in human temporal lobe epilepsy. J Neurosci. 2007;27(37):9866–73. doi: 10.1523/JNEUROSCI.2761-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Coull JA, Boudreau D, Bachand K, Prescott SA, Nault F, Sík A, De Koninck P, De Koninck Y. Trans-synaptic shift in anion gradient in spinal lamina I neurons as a mechanism of neuropathic pain. Nature. 2003;424(6951):938–42. doi: 10.1038/nature01868. [DOI] [PubMed] [Google Scholar]
- 66.Coull JA, Gagnon M. The manipulation of cation-chloride co-transporters as a novel means to treat persistent pain, epilepsy and other neurological disorders. Curr Opin Investig Drugs. 2009;10(1):56–61. [PubMed] [Google Scholar]
- 67.Zagórska A, Pozo-Guisado E, Boudeau J, Vitari AC, Rafiqi FH, Thastrup J, Deak M, Campbell DG, Morrice NA, Prescott AR, Alessi DR. Regulation of activity and localization of the WNK1 protein kinase by hyperosmotic stress. J Cell Biol. 2007;176(1):89–100. doi: 10.1083/jcb.200605093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Moriguchi T, Urushiyama S, Hisamoto N, Iemura S, Uchida S, Natsume T, Matsumoto K, Shibuya H. WNK1 regulates phosphorylation of cationchloride- coupled cotransporters via the STE20-related kinases, SPAK and OSR1. J Biol Chem. 2005;280:42685–42693. doi: 10.1074/jbc.M510042200. [DOI] [PubMed] [Google Scholar]
- 69.Vitari AC, Thastrup J, Rafiqi FH, Deak M, Morrice NA, Karlsson HK, Alessi DR. Functional interactions of the SPAK/OSR1 kinases with their upstream activator WNK1 and downstream substrate NKCC1. Biochem J. 2006;397(1):223–31. doi: 10.1042/BJ20060220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Anselmo AN, Earnest S, Chen W, Juang YC, Kim SC, Zhao Y, Cobb MH. WNK1 and OSR1 regulate the Na+, K+, 2Cl- cotransporter in HeLa cells. Proc Natl Acad Sci U S A. 2006;103(29):10883–8. doi: 10.1073/pnas.0604607103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tsutsumi T, Ushiro H, Kosaka T, Kayahara T, Nakano K. Proline- and alanine-rich Ste20-related kinase associates with F-actin and translocates from the cytosol to cytoskeleton upon cellular stresses. J Biol Chem. 2000;275:9157–9162. doi: 10.1074/jbc.275.13.9157. [DOI] [PubMed] [Google Scholar]
- 72.Ushiro H, Tsutsumi T, Suzuki K, Kayahara T, Nakano K. Molecular cloning and characterization of a novel Ste20-related protein kinase enriched in neurons and transporting epithelia. Arch Biochem Biophys. 1998;355:233–240. doi: 10.1006/abbi.1998.0736. [DOI] [PubMed] [Google Scholar]
- 73.Chen W, Yazicioglu M, Cobb MH. Characterization of OSR1, a member of the mammalian Ste20p/germinal center kinase subfamily. J Biol Chem. 2004;279:11129–11136. doi: 10.1074/jbc.M313562200. [DOI] [PubMed] [Google Scholar]
- 74.Gagnon KB, England R, Delpire E. Characterization of SPAK and OSR1, regulatory kinases of the Na-K-2Cl cotransporter. Mol Cell Biol. 2006;26:689–698. doi: 10.1128/MCB.26.2.689-698.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Vitari AC, Deak M, Morrice NA, Alessi DR. The WNK1 and WNK4 protein kinases that are mutated in Gordon’s hypertension syndrome phosphorylate and activate SPAK and OSR1 protein kinases. Biochem J. 2005;391:17–24. doi: 10.1042/BJ20051180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gagnon KB, England R, Delpire E. A single binding motif is required for SPAK activation of the Na-K-2Cl cotransporter. Cell Physiol Biochem. 2007;20(1–4):131–42. doi: 10.1159/000104161. [DOI] [PubMed] [Google Scholar]
- 77.Ponce-Coria J, San-Cristobal P, Kahle KT, Vazquez N, Pacheco-Alvarez D, de Los Heros P, Juárez P, Muñoz E, Michel G, Bobadilla NA, Gimenez I, Lifton RP, Hebert SC, Gamba G. Regulation of NKCC2 by a chloride-sensing mechanism involving the WNK3 and SPAK kinases. Proc Natl Acad Sci U S A. 2008;105(24):8458–63. doi: 10.1073/pnas.0802966105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Villa F, Goebel J, Rafiqi FH, Deak M, Thastrup J, Alessi DR, van Aalten DM. Structural insights into the recognition of substrates and activators by the OSR1 kinase. EMBO Rep. 2007;8(9):839–45. doi: 10.1038/sj.embor.7401048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Delpire E, Gagnon KB. Genome-wide analysis of SPAK/OSR1 binding motifs. Physiol Genomics. 2007;28(2):223–31. doi: 10.1152/physiolgenomics.00173.2006. [DOI] [PubMed] [Google Scholar]
- 80.Rafiqi FH, Zuber AM, Glover M, Richardson C, Fleming S, Jovanović S, Jovanović A, O'Shaughnessy KM, Alessi DR. Role of the WNK-activated SPAK kinase in regulating blood pressure. EMBO Mol Med. 2010;2(2):63–75. doi: 10.1002/emmm.200900058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Gagnon KB, Delpire E. Multiple pathways for PP1 regulation of NKCC1 function: the N-terminal tail of the Na-K-2Cl cotransporter serves as a regulatory scaffold for SPAK and PP1. J Biol Chem. 2010 Mar 11; doi: 10.1074/jbc.M110.112672. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.de Los Heros P, Kahle KT, Rinehart J, Bobadilla NA, Vazquez N, San Cristobal P, Mount DB, Lifton RP, Hebert SC, Gamba G. WNK3 bypasses the tonicity requirement for K-Cl cotransporter activation via a phosphatase-dependent pathway. Proc Natl Acad Sci USA. 2006;103:1976–1981. doi: 10.1073/pnas.0510947103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Garzón-Muvdi T, Pacheco-Alvarez D, Gagnon KB, Vázquez N, Ponce-Coria J, Moreno E, Delpire E, Gamba G. WNK4 kinase is a negative regulator of K+-Clcotransporters. Am J Physiol Renal Physiol. 2007;292(4):F1197–207. doi: 10.1152/ajprenal.00335.2006. [DOI] [PubMed] [Google Scholar]
- 84.Rinehart J, Maksimova YD, Tanis JE, Stone KL, Hodson CA, Zhang J, Risinger M, Pan W, Wu D, Colangelo CM, Forbush B, Joiner CH, Gulcicek EE, Gallagher PG, Lifton RP. Sites of regulated phosphorylation that control K-Cl cotransporter activity. Cell. 2009;138(3):525–36. doi: 10.1016/j.cell.2009.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Rivera C, Voipio J, Thomas-Crusells J, Li H, Emri Z, Sipilä S, Payne JA, Minichiello L, Saarma M, Kaila K. Mechanism of activity-dependent downregulation of the neuron-specific K-Cl cotransporter KCC2. J Neurosci. 2004;24(19):4683–91. doi: 10.1523/JNEUROSCI.5265-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Holden S, Cox J, Raymond FL. Cloning, genomic organization, alternative splicing and expression analysis of the human gene WNK3 (PRKWNK3) Gene. 2004;23:335, 109–19. doi: 10.1016/j.gene.2004.03.009. [DOI] [PubMed] [Google Scholar]
- 87.Belenky MA, Sollars PJ, Mount DB, Alper SL, Yarom Y, Pickard GE. Cell-type specific distribution of chloride transporters in the rat suprachiasmatic nucleus. Neuroscience. 2010;165(4):1519–37. doi: 10.1016/j.neuroscience.2009.11.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Denton J, Nehrke K, Yin X, Morrison R, Strange K. GCK-3, a newly identified Ste20 kinase, binds to and regulates the activity of a cell cycle-dependent ClC anion channel. J Gen Physiol. 2005;125(2):113–25. doi: 10.1085/jgp.200409215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kleefuss-Lie A, Friedl W, Cichon S, Haug K, Warnstedt M, Alekov A, Sander T, Ramirez A, Poser B, Maljevic S, Hebeisen S, Kubisch C, Rebstock J, Horvath S, Hallmann K, Dullinger JS, Rau B, Haverkamp F, Beyenburg S, Schulz H, Janz D, Giese B, Müller-Newen G, Propping P, Elger CE, Fahlke C, Lerche H. CLCN2 variants in idiopathic generalized epilepsy. Nat Genet. 2009;41(9):954–5. doi: 10.1038/ng0909-954. [DOI] [PubMed] [Google Scholar]
- 90.Hisamoto N, Moriguchi T, Urushiyama S, Mitani S, Shibuya H, Matsumoto K. Caenorhabditis elegans WNK-STE20 pathway regulates tube formation by modulating ClC channel activity. EMBO Rep. 2008;9(1):70–5. doi: 10.1038/sj.embor.7401128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Strange K, Denton J, Nehrke K. Ste20-type kinases: evolutionarily conserved regulators of ion transport and cell volume. Physiology Bethesda. 2006;21:61–68. doi: 10.1152/physiol.00139.2005. [DOI] [PubMed] [Google Scholar]