

Abstract
1-(1-Acetyl-piperidin-4-yl)-3-adamantan-1-yl-urea 14a (AR9281), a potent and selective soluble epoxide hydrolase inhibitor, was recently tested in a phase 2a clinical setting for its effectiveness in reducing blood pressure and improving insulin-resistance in pre-diabetic patients. In a mouse model of diet induced obesity, AR9281 attenuated the enhanced glucose excursion following an intraperitoneal glucose tolerance test. AR9281 also attenuated the increase in blood pressure in angiotensin-II-induced hypertension in rats. These effects were dose-dependent and well correlated with inhibition of the sEH activity in whole blood, consistent with a role of sEH in the observed pharmacology in rodents.
Soluble epoxide hydrolase (sEH, Epxh2) has attracted considerable interest over the last decade as a novel pharmaceutical target primarily for cardiovascular and inflammatory disease as well as a number of other indications. Considerable effort has been expended both in academia and the pharmaceutical industry to develop novel, drug-like inhibitors.1 We wish to report the chemical details and rationale for the selection of the sEH inhibitor, 1-(1-acetyl-piperidin-4-yl)-3-adamantan-1-yl-urea2 14a (AR9281), as a candidate for human clinical trials in hypertension and metabolic syndrome.3
sEH is responsible for the conversion of C18, C20 and C22 epoxy lipids, including epoxyeicosatrienoic acids (EETs), which are formed by the monooxygenase epoxidation of arachidonic acid by CYP2J and CYP2C epoxygenases, to the corresponding dihydroxyeicosatrienoic acids (DHETs). EETs are recognized modulators of biological function eliciting a range of pharmacological effects, while the corresponding DHETs are less active.4 Inhibition of sEH is proposed to stabilize EETs, and effectively increase EET concentrations in relevant compartments to produce biological effects. sEH attracted attention as a potential pharmaceutical target based on reports of lipid modulatory, anti-hypertensive, cardioprotective, organ protective, and anti-inflammatory effects in a variety of animal models.5–17 The observed profile of effects suggest that sEH inhibition should have a beneficial impact on the component processes and symptoms of metabolic syndrome.5
Prototypical sEH inhibitors, dicyclohexyl urea (DCU) 1 and adamantyl ureido dodecanoic acid (AUDA) 2, are potent enzyme inhibitors (DCU IC50 = ca. 52 nM, AUDA IC50 = ca. 3 nM),18 but suffer from less than optimal physical properties and lack appropriate pharmacokinetic behavior for drug development.19 Subsequent efforts have involved modification of the lipophilic aliphatic chain of AUDA on the right hand side (RHS) of the urea primary pharmacophore and introduction of rigid cyclic moieties on both the right and left hand side (LHS) resulting in compounds with improved potency and drug-like properties.1 Most of the potent sEH inhibitors published to date contain urea as the primary pharmacophore. However, Boehringer Ingelheim and Merck have recently reported potent sEH inhibitors with improved pharmacokinetic behavior with amide as the primary pharmacophore utilizing rigid, biaryl and/or spirocyclic moieties as the LHS/RHS portions. Boehringer reports optimization of its amide-based diphenylmethane series for metabolic stability and reports compound 3 as an optimal compound showing excellent potency and in vitro microsomal stability resulting in good plasma exposure in rat.20 A second, amide-based series, exemplified by compound 4, is also reported.21 No animal pharmacology studies are reported for either series. Merck has disclosed three structural classes of potent, urea-based inhibitors exemplified by compounds 5, 6, and 7, as well as a series of non-urea, non-amide based inhibitors.22, 23 However, despite desirable PK and demonstrated target engagement, compounds 5, 6, and 7 failed to show efficacy in an SHR hypertensive model.22 No results for AngII-hypertensive rat models were reported by Merck in spite of this being one of the standard paradigms used in previous sEH hypertension studies.6, 7, 9
One of the many structural variations on sEH inhibitors published by the Hammock laboratory involved incorporation of a solubilizing group into one of the cyclohexane rings of DCU 1. The resulting 4-piperidinyl ureas were reported to be potent sEH inhibitors.2 We focused on expanding the SAR around the piperidinyl urea based sEH inhibitors 14 and wish to report the details of the chemistry SAR and the rationale for the selection of one member of this chemical series, 1-(1-acetyl-piperidin-4-yl)-3-adamantan-1-yl-urea2 14a (AR9281), as a clinical candidate in hypertension and metabolic syndrome.3
The synthesis of 14a and its analogs via key intermediates 10 and 11 is shown in Scheme 1. This library approach allows facile preparation of various LHS and RHS combinations around the piperidinyl urea nucleus. An additional advantage of this approach is that it allows urea formation using a nucleophilic amine via intermediate 11 rather than via an isocyanate.24 In some cases, especially with UV transparent amines such as adamantylamine and cyclohexylamine, the corresponding symmetrical ureas, e.g., dicyclohexyl- and bisadamantyl urea, which are unavoidable impurities in the isocyanates, can be almost impossible to remove efficiently from a reaction product and are potent inhibitors of sEH in their own right. Urea formation with aryl isocyanates is less problematic.
Scheme 1.

Preparation of piperidinyl ureas 14: a) NH2OH·HCl, EtOH, Pyridine, reflux, 1h, 82% b) RaNi, MeOH, rt, 4 h, 83% c) p-nitrophenyl chloroformate, DCM, 2 h, 87% d) DCM, rt, 12 h, 82% e) THF, reflux, 12 h, 85% f) TFA, DCM, rt, 12 h, 88% g) Ac2O or MeSO2Cl, DCM, rt, 18 h, 70–90%
The readily available N-Boc protected piperidone 8 is reacted with hydroxyl amine to obtain the corresponding oxime 9 which is subsequently reduced with Raney nickel to afford the N-Boc aminopiperidine 10. Treatment of N-Boc-4-piperidineamine 10 with p-nitrobenzene chloroformate affords key intermediate 11. At this point amine 10 can be treated with an isocyanate to provide the Boc protected urea 12. This sequence was typically used with aromatic isocyanates. An alternate procedure was employed for certain more nucleophilic, UV transparent amines. Reaction of intermediate 11 with nucleophilic amines at room temperature affords LHS substituted products 12. Less nucleophilic amines, e.g., aromatic amines can also be reacted with 11 but require elevated reaction temperatures. The Boc group on 12 can then be removed with TFA to give the corresponding amine intermediate 13. Amine 13 can be treated with various reagents, e.g., activated carboxylic acids or sulfonyl chlorides, to afford the desired substituted piperidinyl urea 14. Representative compounds are shown in Table 1. An alternate preparation of 14a useful for large scale preparations has been disclosed in the patent literature.25
Table 1.
Enzyme and cell IC50 values, hERG inhibition and oral exposure for selected sEH inhibitors
| Cmpd | R1 | R | hsEHa IC50 (nM) | Cellb IC50 (nM) | hERGc % Inhibition at 30 μM | Rat AUC24h (ng h/mL)e |
|---|---|---|---|---|---|---|
| 14a | Adamantyl | COCH3 | 8 | 57 | < 8d | 720 |
| 14b | 3-Fluoroadamantyl | COCH3 | 58 | - | - | - |
| 14c | 3,5,7-Trifluoroadamantyl | COCH3 | 120 | - | - | - |
| 14d | 4-Methyl[2,2,2]bicyclooctyl | COCH3 | 25 | 300 | - | 1480 |
| 14e | [2,2,1]Cycloheptyl | COCH3 | 14 | 120 | - | - |
| 14f | Cyclohexyl | COCH3 | 29 | - | - | - |
| 14g | Cycloheptyl | COCH3 | 0.8 | 27 | 9 | 7840 |
| 14h | 4-t-Butylcyclohexyl | COCH3 | 13 | 270 | 7 | 1780 |
| 14i | 4,4-Dimethylcyclohexyl | COCH3 | 6.0 | 38 | 0.8 | f |
| 14j | 4,4-Difluorocyclohexyl | COCH3 | 185 | - | - | - |
| 14k | 4-CF3Phenyl | COCH3 | 11 | 98 | 35 | 13100 |
| 14l | 3-CF3Phenyl | COCH3 | 10 | 226 | 10 | 19600 |
| 14m | 4-CF3OPhenyl | COCH3 | 12 | 59 | - | - |
| 14n | Adamantyl | SO2CH3 | 1.9 | 3.9 | −2.8 | 640 |
| 14o | 4-CF3Phenyl | SO2CH3 | 3.7 | 26 | 34 | 6850 |
| 14p | 4-CFO3Phenyl | CO(3-pyridyl) | 1.6 | 6.7 | 57 | 11300 |
| 14q | Cyclohexyl | CO(2-pyridyl) | 6.4 | 67 | 3.4 | 8320 |
| 14r | 4-CF3Phenyl | SO2CH(Me)2 | 2.9 | 13 | 13 | 5890 |
| 14s | 4-CF3OPhenyl | SO2CH(Me)2 | 1.6 | 64 | 11 | 7300 |
Enzyme activity was determined in duplicate.18
Conversion of 14,15-EET to 14,15-DHET was measured in HUVEC cell.
hERG activity inhibition was determined by a high throughput patch clamp method.
8% at 100 μM
Data for PO dosing normalized to 10 mg/Kg.
PO data not available; IV showed a very short half life.
Piperidinyl urea 14a exhibited excellent enzyme and cell-based assay IC50 values, but the pharmacokinetic profile in rat was less than ideal. The poor pharmacokinetic behaviour was identified as due to rapid CYP mediated metabolic oxidation at the adamantyl group in vivo.26 Introduction of electronegative fluorine(s) on the LHS to circumvent CYP oxidation was investigated, but analogues with mono fluoro or trifluoro substitution on the adamantyl ring27 (Table 1: 14b, 14c) afforded reduced enzyme potency. A similar drop in potency with fluorine introduction was also observed with a cyclohexyl LHS, e.g., 14f and 4, 4-difluorocyclohexyl analogue 14j. Replacement of the adamantyl group with a 4-trifluoromethylphenyl, e.g., 14k, or 4-trifluoromethoxyphenyl, e.g., 14m, group gave comparable enzyme and cell-based assay potency and afforded a dramatic improvement in the pharmacokinetic profile. Unfortunately, replacing the adamantyl group on the LHS with substituted phenyl had a decidedly negative effect on hERG channel liability. While 14a showed only 8% hERG channel block at 100 μM, both 14k and 14o showed ca. 35% inhibition at 30 μM, which was judged to be unacceptable. A large number of analogues with various electron withdrawing groups, e.g., F, Cl, Br, CF3, CF3O, CHF2O, and CF3CH2, substituted independently at one or two of the 3, 4, 5 positions on the aromatic ring to alter the electronic nature and/or steric requirements of the LHS aryl system afforded no significant improvement in hERG liability with the exception of the 3-trifluoromethylphenyl analogue 14l which shows a significant loss in cell potency. Use of one or more electron donating groups on the LHS aryl ring or use of a LHS heteroaryl ring also gave no improvement in hERG liability and typically resulted in less effective sEH enzyme inhibition (data not shown). While most of the examples given in the table contain N-acetyl as the RHS functionality, additional analogues with an N-sulfonyl RHS were also prepared. These analogues were, in general, more potent than the N-acetyl analogues (e.g., 14n, o vs. 14a, k) but the increase in potency was compromised by decreased oral exposure. While the methyl sulfonamide analogue 14o had very similar hERG liability compared to the acetamide analogue 14k, the isopropyl sulphonamide 14r was found to block the hERG channel by three-fold less. Introduction of aryl or pyridyl rings in the RHS along with aryl LHS substitution, e.g., 14p, gave increased hERG liability. The hERG liability with an aryl RHS series could be ameliorated by substituting cycloalkyl for aryl on the LHS as shown in example 14q. While cycloalkyl replacements for adamantyl LHS (14g–14i) afforded reduced hERG liability, no significant improvement in pharmacokinetic profile was observed.
Piperidinyl urea compounds with a cycloalkyl LHS including adamantyl, e.g., 14a, typically showed an oral pharmacokinetic profile that indicated a rapid absorption with a high Cmax, but rapid metabolism manifested by a short half life and relatively high oral clearance. Reduction of metabolic liability in non-adamantyl LHS cycloalkyl groups was attempted by introducing steric hindrance or electron withdrawing fluorine groups. None of these compounds afforded the desired improvement in pharmacokinetic profile, although all gave significant reduction in hERG inhibition. Replacement of the adamantyl group with aryl did result in potent sEH inhibitors with the desired pharmacokinetic profile but they all suffered from increased inhibition of hERG activity. Since the targeted disease states, i.e., hypertension and metabolic syndrome are chronic indications, it was decided to advance compounds having reasonable PK and extended inhibition of sEH with minimal risk of cardiovascular toxicity. Compound 14a was the candidate of choice based on scalable PK, target engagement, and its consistent efficacy in animal models of hypertension and diet induced obesity.
Compound 14a is a potent inhibitor with a human sEH enzyme IC50 value of 8 nM and mouse sEH enzyme IC50 value of 3 nM. The cell-based human sEH IC50 value was found to be 57 nM consistent with a ca.10 fold decrease in potency seen for most of the sEH inhibitors between isolated enzyme and cell-based assays. In murine PK/PD experiments, the plasma concentrations of 14a were directly correlated with the inhibition of sEH activity in the whole blood, as measured by the hydrolysis rate of 14,15-EET. Plasma exposure was also directly associated with increases in ratios of endogenous epoxy-to-dihydroxy lipids. Total EET/DHET and EpOME/DiHOME (9,10- and 12,13-octadecenoic acid) ratios in 14a treated wild type mice, where blood sEH activity was inhibited 90%, were similar to those in Ephx deficient mice. An oral dosing schedule of 100 mg/kg BID in mice resulted in an extended period of 90% or greater inhibition of blood sEH activity. Compound 14a was found to be highly selective with no inhibitory activity against microsomal epoxide hydrolase or an extended panel of ca.150 other enzyme and receptor targets when tested at 10 μM. No significant inhibition of drug metabolizing CYP enzymes such as CYP1A2, CYP2B6, CYP2C9, CYP2C19 and CYP3A4, as well as CYP2J and CYP2C, which are essential for the production of EETs was noted with 14a. The plasma protein binding of the compound was modest, ranging from 58–61% across multiple species. The compound was highly permeable across a Caco-2 monolayer. When administered orally, 14a was well absorbed in various species with oral bioavailability ranging from 25% in cynomolgus monkey to 100% in rat. The oral exposure, as measured by the area under the plasma concentration-time curve, increased in approximate proportion to dose up to 2000 mg/kg in rats, 500 mg/kg in dogs, and 180 mg/kg in cynomolgus monkeys. The compound had a relatively moderate to high clearance and short half life, and is eliminated primarily by the oxidative modifications on the adamantyl ring, in the form of hydroxylated adamantyl metabolites.
Following the establishment of potency and pharmacokinetic properties, several compounds from different series as well as 14a were evaluated in an AngII-infused rat model of hypertension. This model was selected for early pharmacological evaluation due to the published efficacy of AUDA.7 Compounds were tested at 50 mg/kg, dosed orally twice daily in a suspension in 0.5% carboxymethylcellulose, 0.1% Tween 80 in Sprague-Dawley rats that were induced to be hypertensive by a continuous infusion of 65 ng/min of AngII. The blood pressure was assessed continuously by telemetric recording. Of the compounds tested, 14a showed superior efficacy in reducing systolic and diastolic blood pressure. Relative to vehicle control treatment, systolic blood pressure was decreased by 8–10% (or 14–16 mm Hg) with a more modest effect on diastolic pressure. This result was confirmed independently in a second laboratory.28
Compound 14a was also tested in an SHR rat model of hypertension8 and was found to be moderately efficacious at a dose of 300 mg/kg with a 14% decrease in mean blood pressure, but no effect was noted at 100 mg/kg. These data are consistent with the recent report22 showing a lack of correlation between potency and exposure with blood pressure lowering in the SHR model. sEH inhibitors exert antihypertensive effects in some but not all animal models of hypertension.28 Collectively, the lack of a robust relationship between target engagement, exposure, and the pharmacological endpoint was insufficient to support solely a human hypertension indication for clinical evaluation of sEH inhibitors.
The hypertension models utilized were based on the literature precedence for a blood pressure lowering effect of sEH inhibitors both in AngII-induced hypertension,7 and in SHR.8 Compound 14a was subsequently evaluated in a diet induced-obesity (DIO) mouse model of aberrant glucose regulation.29 The DIO mouse model tested the pharmacology of these inhibitors in a model of insulin resistance with anticipated activity due in part to the vasodilator, lipid modulatory and anti-inflammatory effects of EETs.14 EETs have been shown, to play a role in endothelial cell function and vascular inflammatory processes, involving modulation of NF-κB-mediated transcription.30 Vascular inflammation has been implicated as an important factor in insulin sensitivity.31 EETs are regulated by sEH,32 and EETs have been shown to modulate PPARγ33 and eNOS.34 The enzymes, PPARγ and eNOS, are regulators in insulin resistance.35 Thus, sEH inhibition was proposed as a mechanism by which insulin resistance could be improved in a model of diet-induced obesity.
The efficacy of 14a was evaluated in DIO male C57Bl/6 mice with emphasis on the impact of administration of 14a on glucose excursions following a bolus administration of glucose. C57BL/6 mice were fed a high fat, high fructose diet using Research Diets 45% fat diet, D12451, with administration of high fructose corn syrup in the liquid portion of the diet at 0.42 kcal/mL for 6 weeks prior to initiation of AR9281 administration and throughout AR9281 administration.29 Accordingly, 14a or vehicle was administered via oral gavage at a dose of 100 mg/kg twice a day for an additional 12 weeks. After 4 weeks treatment, a glucose tolerance test was conducted by intraperitoneal injection of glucose (2 g/kg) at 4 hours after the dose of 14a. Blood glucose measurements were taken with a glucometer at time intervals up to 2 hours following glucose administration. The vehicle-treated mice had an impaired glucose tolerance evidenced by the blood glucose excursions being in excess of mice fed normal chow and that the blood glucose concentration was still not restored to the baseline at 2 hour after glucose load (Figure 1, left). However, in comparison to vehicle-treated animals, the 14a-treated mice had a lower glucose AUC as well as lower maximal glucose excursion. At the end of the study, blood samples were taken at specific times after the last dose and processed to plasma for 14a concentration measurements using LC/MS/MS and blood sEH activity. Blood sEH activity was defined as the rate of 14, 15 EET hydrolysis, corrected for non-specific hydrolysis in the presence of 5 mM AUDA.29 sEH activity was undetectable in whole blood from animals receiving 14a for up to 7 hours post administration. The sEH-catalyzed EET hydrolysis activity continued to be inhibited by ~70% for 12 hours after the last dose, suggesting that at trough there continued to be significant target inhibition (Figure 1, right).29 These results indicate that 14a inhibited sEH activity and significantly improved glucose tolerance in a DIO mouse model.
Figure 1.
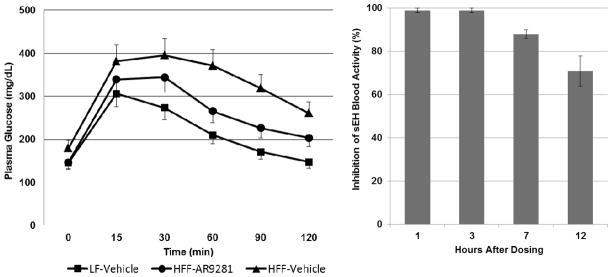
Evaluation of 14a (AR9281) in DIO mouse model. Left is the blood glucose levels over time following an intraperitoneal administration in a typical experiment. Right is the whole blood sEH activity following the last dose of the study.
In conclusion we have shown that piperidinyl urea 14a (AR9281) is a potent sEH inhibitor with excellent selectivity against other targets. AR9281 was well tolerated in early animal and human dose range finding toxicology studies.29 AR9281 affords an acceptable half-life in mouse for pharmacology testing with whole blood sEH activity essentially completely inhibited for up to 7 hours post administration and still inhibited by ~70% for twelve hours after the last 100 mg/Kg dose. In humans it was shown that greater than 90% inhibition of blood sEH was possible with once a day and twice a day dosing.36 Based on the blood pressure lowering activity in the AngII-induced hypertension and SHR models and the more robust activity in the DIO mouse, 14a (AR9281) was selected for proof of concept studies in a human clinical trial involving obese patients with stage 1 hypertension and impaired glucose tolerance.
Figure 3.

Structures 1–7
Figure 4.
Structure 14a
Acknowledgments
Partial support for P.D.J. and B.D.H. came from NIEHS R01 ES002710. B.D.H. is a George and Judy Marcus Senior Fellow of the American Asthma Foundation. P.D.J. was supported in part by an NIH/NHLBI Ruth L. Kirchstein NRSA Grant (F32 HL078096).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.(a) Marino JP., Jr Curr Top Med Chem. 2009;9:452. doi: 10.2174/156802609788340805. [DOI] [PubMed] [Google Scholar]; (b) Shen HC. Expert Opin Ther Patents. 2010;20:1. doi: 10.1517/13543776.2010.484804. [DOI] [PubMed] [Google Scholar]
- 2.(a) Jones PD, Tsai HJ, Do ZN, Morisseau C, Hammock BD. Biorg Med Chem Lett. 2006;16:5212. doi: 10.1016/j.bmcl.2006.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Rose TE, Morisseau C, Liu JY, Inceoglu B, Jones PD, Sanborn JR, Hammock BD. J Med Chem. 2010;53:7067. doi: 10.1021/jm100691c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.A Randomized, Double-Blind, Placebo-Controlled, Dose-Ranging, Exploratory, 28-Day Study to Examine the Effects of AR9281 on Blood Pressure and Glucose Tolerance in Patients With Mild to Moderate Hypertension and Impaired Glucose Tolerance. ClinicalTrials.gov. ID NCT00847899. [Google Scholar]
- 4.(a) Spector AA, Norris AW. Am J Physiol Cell Physiol. 2007;292:C996. doi: 10.1152/ajpcell.00402.2006. [DOI] [PubMed] [Google Scholar]; (b) Lason BT, Gutterman DD, Hatoum OA. Eur J Clin Invest. 2006;36:293. doi: 10.1111/j.1365-2362.2006.01634.x. [DOI] [PubMed] [Google Scholar]; (c) Spector AA, Fang X, Snyder GD, Weintraub NL. Prog Lipid Res. 2004;434:55. doi: 10.1016/s0163-7827(03)00049-3. [DOI] [PubMed] [Google Scholar]
- 5.(a) Luo P, Chang H-H, Zhou Y, Zhang S, Hwang SH, Morisseau C, Wang C-Y, Inscho EW, Hammock BD, Wang M-H. J Pharmacol Exp Ther. 2010;334:430. doi: 10.1124/jpet.110.167544. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) De Taeye BM, Morisseau C, Coyle J, Covington JW, Luria A, Yang J, Murphy SB, Friedman DB, Hammock BD, Vaughan DE. Obesity. 2010;18:489. doi: 10.1038/oby.2009.227. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Lyer A, Fairlie DP, Prins JB, Hammock BD, Brown L. Nat Rev Endocrinol. 2010;6:71. doi: 10.1038/nrendo.2009.264. [DOI] [PubMed] [Google Scholar]; (d) Sodhi K, Inoue K, Gotlinger KH, Canestraro M, Vanella L, Kim DH, Manthati VL, Koduru SR, Falck JR, Schwartzman ML, Abraham NG. J Pharmacol Exp Ther. 2009;331:906. doi: 10.1124/jpet.109.157545. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Mustafa S, Sharma V, McNeill JH. Exp Clin Cardiol. 2009;14:e41. [PMC free article] [PubMed] [Google Scholar]; (f) Burdon KP, Lehtinen AB, Langefeld CD, Carr JJ, Rich SS, Freedman BI, Herrington D, Bowden DW. Diab Vasc Dis Res. 2008;5:128. doi: 10.3132/dvdr.2008.021. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Ohtoshi K, Kaneto H, Node K, Nakamura Y, Shiraiwa T, Matsuhisa M, Yamasaki Y. Biochem and Biophysical Res Comm. 2005;331:347. doi: 10.1016/j.bbrc.2005.03.171. [DOI] [PubMed] [Google Scholar]; (h) Webb H. WO 08058033 A2. 2008; (i) Larsen BT, Campbell WB, David D, Gutterman DD. Trends Pharmacol Sci. 2007;28:32. doi: 10.1016/j.tips.2006.11.002. [DOI] [PubMed] [Google Scholar]
- 6.Jung O, Brandes RP, Kim I, Schweda F, Schmidt R, Hammock BD, Busse R, Fleming I. Hypertension. 2005;45:759. doi: 10.1161/01.HYP.0000153792.29478.1d. [DOI] [PubMed] [Google Scholar]
- 7.Imig JD, Zhao X, Zaharis CZ, Olearczyk JJ, Pollock DM, Newman JW, Kim IH, Watanabe T, Hammock BD. Hypertension. 2005;46:975. doi: 10.1161/01.HYP.0000176237.74820.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yu Z, Xu F, Huse LM, Morisseau C, Draper AJ, Newman JW, Parker C, Graham L, Engler MM, Hammock BD, Zeldin DC, Kroetz DL. Circ Res. 2000;87:992. doi: 10.1161/01.res.87.11.992. [DOI] [PubMed] [Google Scholar]
- 9.(a) Zhao X, Yamamoto T, Newman JW, Kim I-H, Watanabe T, Hammock BD, Stewart J, Pollock JS, Pollock DM, Imig JD. J Am Soc Nephrol. 2004;15:1244. [PubMed] [Google Scholar]; (b) Imig JD. Am J Physiol Renal Physiol. 2005;289:F496. doi: 10.1152/ajprenal.00350.2004. [DOI] [PubMed] [Google Scholar]
- 10.Dorrance AM, Rupp N, Pollock DM, Newman JW, Hammock BD, Imig JD. J Cardiovasc Pharm. 2005;46:842. doi: 10.1097/01.fjc.0000189600.74157.6d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xu D, Li N, He Y, Timofeyev V, Lu L, Tsai HJ, Kim IH, Tuteja D, Mateo RKP, Singapuri A, Davis BB, Low R, Hammock BD, Chiamvimonvat N. Proc Nat Acad Sci. 2006;103:18733. doi: 10.1073/pnas.0609158103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.(a) Liu Y, Webb H, Kroetz DL. FASEB J. 2008;22:479.19. [Google Scholar]; (b) Parrish AR, Chen G, Burghardt RC, Watanabe T, Morisseau C, Hammock BD. Cell Biol Toxicol. 2009;25:217. doi: 10.1007/s10565-008-9071-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.(a) Zhang L-N, Vincelette J, Cheng Y, Mehra U, Chen D, Anandan S-K, Gless R, Webb HK, Wang Y-X. Arterioscler Thromb Vasc Biol. 2009;29:1265. doi: 10.1161/ATVBAHA.109.186064. [DOI] [PubMed] [Google Scholar]; (b) Ulu A, Davis BB, Tsai HJ, Kim IH, Morisseau C, Inceoglu B, Fiehn O, Hammock BD, Weiss RH. J Cardiovasc Pharm. 2008;52:314. doi: 10.1097/FJC.0b013e318185fa3c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Node K, Huo Y, Ruan X, Yang B, Spiecker M, Ley k, Zeldin DC, Liao JK. Science. 1999;285:1276. doi: 10.1126/science.285.5431.1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schmelzer KR, Kubala L, Newman JW, Kim IH, Eiserich JP, Hammock BD. Proc Nat Acad Sci. 2005;102:9772. doi: 10.1073/pnas.0503279102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Smith KR, Pinkerton KE, Watanabe T, Pedersen TL, Ma SJ, Hammock BD. Proc Nat Acad Sci. 2005;102:2186. doi: 10.1073/pnas.0409591102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.(a) Inceoglu B, Jinks SL, Schmelzer KR, Waite T, Kim IH, Hammock BD. Life Sci. 2006;79:2311. doi: 10.1016/j.lfs.2006.07.031. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Inceoglu B, Schmelzer KR, Morisseau C, Jinks SL, Hammock BD. Prostag Oth Lipid M. 2007;82:42. doi: 10.1016/j.prostaglandins.2006.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Inceoglu B, Jinks SL, Ulu A, Hegedus CM, Georgi K, Schmelzer KR, Wagner K, Jones PD, Morisseau C, Hammock BD. Proc Natl Acad Sci USA. 2008;105:18901. doi: 10.1073/pnas.0809765105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jones PD, Wolf NM, Morisseau C, Whetstone P, Hock B, Hammock BD. Anal Biochem. 2005;343:66. doi: 10.1016/j.ab.2005.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim IH, Nishi K, Tsai HJ, Bradford T, Koda Y, Watanabe T, Morisseau C, Blanchfield J, Toth I, Hammock BD. Biorg Med Chem Lett. 2007;15:312. doi: 10.1016/j.bmc.2006.09.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Eldrup AB, Solewymanzadeh F, Taylor SJ, Muegge I, Farrow NA, Joseph D, McKellop K, Man CC, Kulkulka A, De Lombaert S. J Med Chem. 2009;52:5880. doi: 10.1021/jm9005302. [DOI] [PubMed] [Google Scholar]
- 21.Taylor SJ, Solewymanzadeh F, Eldrup AB, Muegge I, Farrow NA, Kulkulka A, Kabcenell AK, De Lombaert S. Bioorg Med Chem Lett. 2009;19:5864. doi: 10.1016/j.bmcl.2009.08.074. [DOI] [PubMed] [Google Scholar]
- 22.(a) Shen HC, Ding FX, Wang S, Xu S, Chen HS, Tong X, Tong V, Mitra K, Kumar S, Zhang X, Chen Y, Zhou G, Pai LY, Alonso-Galicia M, Chen X, Zhang B, Tata JR, Berger JP, Colletti SL. Bioorg Med Chem. 2009;19:3398. doi: 10.1016/j.bmcl.2009.05.036. [DOI] [PubMed] [Google Scholar]; (b) Shen HC, Ding FX, Wang S, Deng Q, Zhang X, Chen Y, Zhou G, Xu S, Chen HS, Tong X, Tong V, Mitra K, Kumar S, Tsai C, Stevenson AS, Pai LY, Alonso-Galicia M, Chen X, Soisson SM, Roy S, Zhang B, Tata JR, Berger JP, Colletti SL. J Med Chem. 2009;52:5009. doi: 10.1021/jm900725r. [DOI] [PubMed] [Google Scholar]; (c) Shen HC, Ding FX, Deng Q, Xu S, Chen HS, Tong X, Tong V, Zhang X, Chen Y, Zhou G, Pai LY, Alonso-Galicia M, Zhang B, Roy S, Tata JR, Berger JP, Colletti SL. Bioorg Med Chem Lett. 2009;19:5314. doi: 10.1016/j.bmcl.2009.07.138. [DOI] [PubMed] [Google Scholar]
- 23.Shen HC, Ding FX, Deng Q, Xu S, Tong X, Zhang X, Chen Y, Zhou G, Pai LY, Alonso-Galicia M, Roy S, Zhang B, Tata JR, Berger JP, Colletti SL. Bioorg Med Chem Lett. 2009;9:5716. doi: 10.1016/j.bmcl.2009.08.006. [DOI] [PubMed] [Google Scholar]
- 24.Kowalski JA, Swinamer AD, Muegge I, Eldrup AB, Kukulka A, Cywin CL, De Lombaert S. Bioorg Med Chem Lett. 2010;20:3703. doi: 10.1016/j.bmcl.2010.04.078. [DOI] [PubMed] [Google Scholar]
- 25.Gless R. WO2008094862 A1. 2008 Aug 7;
- 26.(a) Jia L, Noker PE, Coward L, Gorman GS, Protopopova M, Tomaszewski JE. Brit J Pharm. 2006;147:476. doi: 10.1038/sj.bjp.0706650. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Rohde JL, Pliushchev MA, Sorensen BK, Wodka D, Shuai Q, Wang J, Fung S, Monzon KM, Chiou WJ, Pan L, Deng X, Chovan LE, Ramaiya A, Mullally M, Henry RF, Stolarik DF, Imade HM, Marsh KC, Beno DWA, Fey TA, Droz BA, Brune ME, Camp HS, Sham HL, Frevert EU, Jacobson PB, Link JT. J Med Chem. 2007;50:149. doi: 10.1021/jm0609364. [DOI] [PubMed] [Google Scholar]; (c) Cohen ML, Bloomquist W, Calligaro D, Swanson S. Drug Dev Res. 1998;43:193. [Google Scholar]; (d) Kajbaf M, Rossato P, Barnaby JR, Pellegatti M. Xenobiotica. 1998;28:167. doi: 10.1080/004982598239669. [DOI] [PubMed] [Google Scholar]; (d) Hoffman HE, Gaylord JC, Blaseki JW, Shalaby LM, Whitney GC. Antimicrob Agents and Chemother. 1988;32:1699. doi: 10.1128/aac.32.11.1699. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Fukuda EK. Drug Metab Disp. 1988;16:773. [PubMed] [Google Scholar]
- 27.Vytautas JJ, Volkmann RA. U.S. Patent 6,057,364. 2000 May 2;
- 28.Imig JD, Carpenter MA, Shaw S. Pharmaceuticals. 2009;2:217. doi: 10.3390/ph2030217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Whitcomb R, Chen D, Wang Y-X, Anandan S-K, Gless R, Webb HK. Diabetes. 2009;58:A165, 612. [Google Scholar]; Wong K, Zhang L-N, Vincelette J, Chen D, Tran V, Mehra U, Gless R, Anandan S-K, Webb HK, MacIntyre U, Wang Y-X. Diabetes. 2009;58:A436, 1698. [Google Scholar]
- 30.Fang X, Kaduce TL, Weintraub NL, VanRollins M, Spector AA. Circ Res. 1996;79:784. doi: 10.1161/01.res.79.4.784. [DOI] [PubMed] [Google Scholar]
- 31.Kim F, Pham M, Luttrel I, Bannerman DD, Tupper J, Thaler J, Hawn TR, Rainse EW, Schwartz MW. Circ Res. 2007;100:1589. doi: 10.1161/CIRCRESAHA.106.142851. [DOI] [PubMed] [Google Scholar]
- 32.(a) Zeldin DC, Kobayashi J, Falck JR, Winder BS, Hammock BD, Snapper JR, Capdevila JH. J Biol Chem. 1993;268:6402. [PubMed] [Google Scholar]; (b) Yu Z, Xu F, Huse LM, Morisseau C, Draper AJ, Newman JW, Parker C, Graham L, Engler MM, Hammock BD, Zeldin DC, Kroetz DL. Circ Res. 2000;87:992. doi: 10.1161/01.res.87.11.992. [DOI] [PubMed] [Google Scholar]
- 33.Wray J, Bishop-Bailey D. Exp Physiol. 2008;93:148. doi: 10.1113/expphysiol.2007.038612. [DOI] [PubMed] [Google Scholar]
- 34.(a) Wang D, Hirase T, Nitto T, Soma M, Node K. J Cardiol. 2009;54:368. doi: 10.1016/j.jjcc.2009.06.004. [DOI] [PubMed] [Google Scholar]; (b) Chen R, Jiang J, Xiao X, Wang D. Sci China C Life Sci. 2005;48:495. doi: 10.1360/062004-36. [DOI] [PubMed] [Google Scholar]
- 35.Rizzo NO, Maloney E, Pham M, Luttrell I, Wessells H, Tateya S, Daum G, Handa P, Schwartz MW, Kim F. Arterioscler Thromb Vasc Biol. 2010;30:758. doi: 10.1161/ATVBAHA.109.199893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen D, Whitcomb R, MacIntyre E, Tran V, Do ZN, Sabry J, Patel DV, Anadan SK, Gless R, Webb HK. J Clin Pharma. doi: 10.1177/0091270010397049. in press. [DOI] [PubMed] [Google Scholar]