

Abstract
We have previously reported development of biomimetic, asymmetric [3+2] photocycloadditions between 3-hydroxyflavones and cinnamate dipolarophiles to access (−)-rocaglamide and related natural products. Herein, we describe enantioselective syntheses of aglain cycloadducts leading to the first total syntheses and absolute configuration assignments of the aglain natural products (+)-ponapensin and (+)-elliptifoline.
Introduction
The plant genus Aglaia produces a series of secondary metabolites, including methyl rocaglate (1),1 the densely functionalized cyclopenta[b,c]benzofuran (rocaglate) anticancer agent silvestrol (2),2 and the cyclopenta[b,c]benzopyrans (aglains) (−)-ponapensin (3)3 and (−)-elliptifoline (4)4 (Figure 1). Relative stereochemical configurations of 3 and 4 were proposed in their respective isolation reports. Although aglains do not typically possess the strong biological activity of their rocaglate counterparts, (−)-ponapensin (3) was found to exhibit potent NF-κB inhibitory activity in an enzyme-based ELISA assay (IC50 = 60 nM) while rocaglamide5 and methyl rocaglate (1) exhibited IC50 values of 2.0 and 2.3 µM, respectively.3 Other aglain natural products including 4-epi-aglain A,3 aglain B,6 10-O-acetylaglain B,7 and aglain C6 were not active in the NF-κB assay (IC50 > 5 µM).3 Unlike silvestrol and related rocaglates,8,9 thus far no total syntheses of aglain natural products including 3 and 4 have been reported to date.
Figure 1.

Representative natural products from the genus Aglaia.
We have developed a biomimetic approach to the rocaglates using [3+2] photocycloaddition between 3-hydroxyflavones (3-HF’s) 5 and cinnamate derivatives 6 as dipolarophiles followed by a ketol rearrangement/reduction sequence.8a,9b,10 Herein, we present the first enantioselective total syntheses and absolute configuration assignments of the aglain natural products (+)-ponapensin (3) and (+)-elliptifoline (4), the optical antipodes of metabolites found in nature.
Results and Discussion
Cyclopenta[b,c]benzopyran Core Synthesis
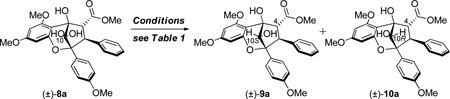
Based on previous results,10c [3+2] photocycloaddition between oxidopyrylium 7 derived from Excited State Intramolecular Proton Transfer (ESIPT) of 3-HF 5 and methyl cinnamate 6a afforded the hydrated aglain (±)-8a in 61% yield which was recrystallized from acetonitrile (Scheme 1).11 Various conditions were evaluated on substrate 8a in order to achieve stereoselective reduction of the bridgehead ketone including DIBAL-H, L-Selectride, and aluminum isopropoxide (Al(OiPr)3), all of which did not lead to reduced products. When 8a was treated with NaBH4 in MeOH, no reaction was observed (Table 1, Entry 1). NaBH4 reduction proceeded when aglain 8a was better solubilized in THF (Entry 2) and afforded reduced products 9a and 10a in a 1:5 ratio. Improved diastereoselectivity was not observed using Bu4NBH412 which afforded reduced aglain 10a as the major product (dr = 5:1) in 98% yield (Table 1, Entry 3). Fortunately, treatment of 8a with Me4NBH(OAc)313 in benzene (entry 4) afforded reduction product 9a which has the 10S configuration required for synthesis of both ponapensin (3) and elliptifoline (4). Relative configuration assignment of C10 was determined via NOE correlation observed between H10/H4 of aglain 10a and an absence of this correlation in its epimer 9a.11
Scheme 1.

Cycloaddition of 3-HF 5 and Cinnamic Esters 6
Table 1.
Diastereoselective Reduction of Aglain (±)-8a
 | |||||
|---|---|---|---|---|---|
| entry | reagenta | solvent | additive | 9a:10ab | yield (%)c |
| 1 | NaBH4 | MeOH | - | - | 0 |
| 2 | NaBH4 | THF | - | 1:5 | 93 |
| 3 | Bu4NBH4 | THF | - | 1:5 | 98 |
| 4 | Me4NBH(OAc)3 | benzene | AcOH | 3:1 | 95 |
| 5 | Me4NBH(OAc)3 | PhCF3 | AcOH | 5:1 | 95 |
| 6 | NaBH(OAc)3 | PhCF3 | AcOH | 10:1 | 95 |
See Supporting Information for detailed experimental procedures.
Determined by 1H NMR analysis of the crude reaction mixture.
Isolated after purification by silica gel chromatography.
After evaluation of solvents, we found that 2,2,2-trifluoromethylbenzene (PhCF3) was an optimal solvent for hydroxyl-directed reduction of aglain 8a to afford 9a in 10:1 dr (Entry 5). The counterion size of the reducing agent appears to be significant in enhancing diastereoselectivity of the reduction as NaBH(OAc)314 provided the 10S diastereomer 9a in 10:1 dr (Table 1, Entry 6). Treatment of hydrated ketone 8a with 4Å MS in CH2Cl2 led to the formation of the derived bridgehead ketone 11 (Figure 2).11 Reduction of 11 with NaBH(OAc)3 led to full conversion to 9a in a shorter time (2 h vs. 16 h for 8a) without loss of diastereoselectivity.
Figure 2.

Proposed assemblies for diastereoselective reduction.
A rationale for reagent-controlled, diastereoselective reduction of bridgehead ketone 11 is shown in Figure 2. When using a borohydride reducing agent (e.g. NaBH4, Bu4NBH4), the bridgehead ketone 11 may be preferentially attacked on the si face due to larger dihedral angles for O-C10-C5-C4 and O-C10-C2-C3 (134° and 138°, respectively) versus the smaller dihedral angles of O-C10-C5-C5a and O-C10-C2-O (113° and 102°, respectively), thereby allowing more optimal orbital alignment for hydride addition to the bridgehead carbonyl.15 Furthermore, use of a borohydride with a large counterion (e.g. Bu4N+) does not affect the diastereoselectivity. When a hydroxyl-directed reducing reagent13 was used (e.g. NaBH(OAc)3), diastereoselectivity did not appear to be derived from the 5-hydroxyl group in the usual fashion as this moiety is coplanar with the C10-carbonyl. We propose that borate complexation with the 5-hydroxyl takes place (cf. Figure 2, assembly 12) leading to selective hydride delivery to the re face of the activated carbonyl due to preferred orientation of the borate near the less sterically encumbered phloroglucinol ring system. Furthermore, we considered that the oxygen lone pair of the 6-methoxy group may also exchange with the borate and further direct the reduction to the re face as a secondary effect (cf. Figure 2, assembly 13). The latter issue was examined using the model aglain 1416 which lacks a methoxy at C6 (Scheme 2). Sodium triacetoxyborohydride reduction of 14 furnished reduced aglain 15 in 8:1 dr which was not significantly different from the corresponding reduction of aglain 8a (10:1 dr). Therefore, the secondary effect illustrated in assembly 13 (Figure 2) is not likely a significant factor to the diastereoselectivity for the reduction. Furthermore, reduction of ketone 14 with Bu4NBH4 yielded the reduced aglain 16 in 7:1 dr, which is comparable to the diastereoselectivity observed in the Bu4NBH4 reduction of aglain 8a (5:1 dr). Relative configurations of 15 and 16 were determined by NOESY NMR spectroscopy.11
Scheme 2.

Reductions of Demethoxy Aglain 14
Enantioselective Photocycloaddition to the Aglain Core
Having established a method to access aglain core structure 8a, we synthesized enantioenriched aglain 9a using the tetrakis-9-phenanthrenyl TADDOL 17 (Scheme 3).10a Host-guest complexation of 17 and 3-HF 5 promoted enantioselective [3+2] photocycloaddition leading to isolation of the hydrated-aglain cycloadduct (+)-8a in 69% yield and 85.5:14.5 er. We attempted recrystallization of (+)-8a to obtain enantiopure crystals. However, in this case we observed crystals of centrosymmetric racemate (±)-8a10a,11 with enantiomeric enrichment of the mother liquor. Fortunately, enantiopure crystals of the reduction product (+)-9a were obtained after two recrystallizations (>96:04 er after recrystallization from isopropanol and >98.5:1.5 er after recrystallization from acetonitrile).11 Relative and absolute stereochemistries of enantiopure aglain (+)-9a were confirmed by X-ray analysis using Cu-Kα radiation (Figure 3)17 and the absolute configuration further confirmed by conversion of (+)-8a to (−)-methyl rocaglate (1) (Scheme 4).10a, 11
Scheme 3.

Enantioselective Photocycloaddition and Subsequent Reduction
Figure 3.

Determination of the absolute stereochemistry of enantiopure aglain (+)-9a using Cu-Kα radiation X-ray analysis.
Scheme 4.

Confirmation of the Absolute Stereochemistry of Aglain (+)-8a by Synthesis of (−)-Methyl Rocaglate (1)
Further Elaboration of the Aglain Core
Before using enantiopure material, we first optimized our synthetic route to ponapensin (3) and elliptifoline (4) using chiral, racemic materials. In initial experiments, several attempts to hydrolyze methyl ester (±)-9a were made. However, in general decomposition resulted under basic or nucleophilic (e.g. LiBr) deprotection conditions (Scheme 5). We next turned our attention to hydrolysis and further advancement of the reduced aglain core structure 9b. Photocycloaddition of 3-HF 5 and benzyl cinnamate 6b led to the production of benzyl ester aglain (±)-8b in 60% yield (Scheme 1). The corresponding endo cycloadduct was then recrystallized from acetonitrile11 and was subsequently reduced to afford the 10S-aglain (±) 9b (10:1 dr). Benzyl ester 9b was hydrogenolyzed and the derived carboxylic acid subsequently coupled with 4,4-dimethoxy-1-butanamine18 18 to provide amidoacetal (±)-19 in 95% yield (two steps, Scheme 6). In an alternative pathway, transesterification of methyl ester (±)-9a was achieved using Otera’s catalyst19 in benzyl alcohol as solvent to afford benzyl ester (±)-9b in 63% yield along with recovered starting material (28%). Several reagents were evaluated in order to achieve direct ester-amide exchange of 9a including Otera’s catalyst, Sc(OTf)3, Zn4(OCOCF3)6O, Mg(OTf)2, and Zr(OtBu)4 either as catalysts or stoichiometric additives.20 Modest conversions of aglain (±)-9a to (±)-19 were observed using Otera’s catalyst and Mg(OTf)2 in 25% and 38% yields, respectively. Interestingly, when using trimethylaluminum(III)21 for ester amide exchange, we observed clean formation of amido acetal (+)-19 in 64% yield without epimerization of (+)-19 (Scheme 6).
Scheme 5.

Attempted Hydrolysis of 9a
Scheme 6.

Transesterification/ Ester-Amide Exchange toward Ponapensin and Elliptifoline
a) From 9a: Me3Al (20 equiv), amine 18 (20 equiv) toluene (0.3 M) 70 °C for 12 h (64%); from 9b: i) H2-Pd/C then ii) EDCI (1.5 equiv), HOBt (1.2 equiv), amine 18 (2.5 equiv) (95%, 2 steps); b) Otera’s catalyst (20 mol%), BnOH, 110 °C, house vacuum, 24 h (64%, recovery 28% 9b).
Final Cyclizations to Ponapensin and Elliptifoline
With enantiopure aglain amide (+)-19 in hand, we next examined cyclization to the hemiaminal of ponapensin (Scheme 7).22 Treatment of amido acetal (+)-19 with CSA (50 mol%) in EtOAc led to a mixture of products that were isolated after two purifications. The major product observed was (+)-ponapensin in 30% yield as a single hemiaminal diastereomer ([α]D25 = +170° synthetic (c 0.2, CHCl3), [α]D22 = −167° natural (c 0.4, CHCl3)). The other major product isolated was the ethyl hemiaminal derivative of ponapensin, aglain 20, isolated in 12% yield. The production of this compound was due to trace ethanol present in the ethyl acetate. Cyclic intermediate 21 was also isolated in 2% yield and its relative configuration was assigned via NOE correlation observed between H13 and H4 (Figure 4). Cyclic intermediate 21 provides important evidence for the intermediacy of acyliminium23 22 and accounts for formation of a single diastereomer of 3. However, the tertiary hydroxyl may not be the only contributing factor to the diastereoselectivity. Figure 5 shows a Spartan ’10 model of the ground state conformation (B3LYP/6-31G**) of putative acyliminium intermediate 22. Trans24 acyliminium 22 may be formed to avoid unfavorable A1,3 strain interactions25 leading to the conformer depicted in Figure 5. In this conformer, the oxygen of the 6-methoxy group is approximately 2.7 Å from C13 which bears a π* orbital.26 A n → π* interaction appears to stabilize this conformation and allows nucleophilic attack specifically from the opposite face of iminium 22. Furthermore, we also isolated trace amounts of the dihydropyrrole derived from 22 and possible dimeric products from the reaction mixture. Cyclizations were attempted in other solvents such as THF and 1,4-dioxane to avoid formation of 13-OEt derivative 20, in which case ponapensin was still not produced cleanly. After considerable optimization, we found that treatment of (+)-19 with CSA in EtOAc containing methanol (15 equiv.) led to the production of ponapensin (3) in 68% yield (Scheme 8) as a single diastereomer. When methanol was used as solvent, the reaction stopped at approximately 50% conversion to 3 suggesting that an equilibrium had been reached. In a similar manner, treatment of aglain (+)-9a with 5 equivalents of tigloyl amide and 50 mol% CSA in EtOAc led to the production of (+)-elliptifoline (4) as a single diastereomer. Elliptifoline was isolated in 81% yield ([α]D25 = +172° synthetic (c 0.2, CHCl3), [α]D22 = −88.9° natural (c 0.6, CHCl3)).
Scheme 7.

Acid-Catalyzed Cyclization without Additional Methanol
Figure 4.

B3LYP/6-31G* ground state conformer of cyclic aminal 21 and assigned relative configuration based on the observed 2D NOESY
Figure 5.

B3LYP/6-31G** ground state conformer of acyliminium 22 and stereoselective approach of the nucleophile.
Scheme 8.

Optimized Syntheses of Ponapensin (3) and Elliptifoline (4)
The relative configurations at C13 of both 3 and 4 were confirmed via X-ray crystallography using chiral, racemic natural products to be the same for ponapensin (3) and elliptifoline (4) (Figure 6). The X-ray structures show that the proline amide moiety is folded as a trans-rotamer over the neighboring aromatic ring. A C–H–π interaction may also stabilize this conformation where the phenyl ring donates electron density to the electron deficient C–H bonds of the 13–OMe in ponapensin (3).24a Kinghorn and coworkers correctly predicted this conformation for ponapensin (3) using molecular modeling and observed the 1H NMR shielding of the C13 methoxy protons.3 It should be noted that synthetic elliptifoline (4) was determined to have the opposite configuration at C13 than reported by Duh and coworkers (cf. Scheme 8).4 The crucial NOE between H4 and H13 was observed in our hands as reported by Duh and coworkers.4 The major rotamer in solution is likely the trans conformation displayed in the crystal structure (Figure 6). Although there is a large discrepancy in magnitude between the synthetic and natural samples’ specific rotations (172° vs. 88.9°), the 1H and 13C NMR data support that we have made elliptifoline (4)11 and the configuration at C13 is therefore opposite of what Duh and coworkers reported.4
Figure 6.

X-ray crystal structures of the racemic natural products. The structures shown have the absolute configuration of (+)-ponapensin (3) and (+)-elliptifoline (4), enantiomers of the natural products.
As previously described, the specific optical rotations of synthetic (3) and (4) were determined to be dextrorotatory. Accordingly, the enantiomers of both natural products 3 and 4 were synthesized. Figure 7 shows the assigned absolute configurations of the natural products which were further confirmed by the synthesis of enantioenriched (−)-methyl rocaglate (1) from enantioenriched (71% ee) (+)-9a (c.f. Scheme 4).11 We propose that a kinetic resolution may occur in nature wherein (−)-aglain ketone core structures undergo reduction and (+)-aglains do not (Figure 8).8a,27 Racemic bridgehead ketones have been utilized as substrates for kinetic resolution using microbes such as C. lunata and R. rubra and also in the presence of ketoreductase/NADP.28 (+)-Aglains may then be converted to their (−)-rocaglate counterparts via a reductoisomerase/NADPH pathway (e.g. DXR, ketol-acid reductoisomerase). Keto-acid reductoisomerase is known to catalyze the conversion of 2-acetolactate to 2,3-dihydroxyisovalerate and DXR is involved in terpenoid biosynthesis in E. coli.29 Furthermore, it is also possible that a parallel kinetic resolution (PKR)30 may occur where (+)-aglains selectively undergo ketol rearrangement/reduction and (−)-aglains are reduced via reductoisomerase/NADPH leading to enantiopure (−)-rocaglates and (−)-aglain structures, respectively.
Figure 7.

Relative and absolute stereochemical assignments of (−)-ponapensin (3) and (−)-elliptifoline (4). (−)-Elliptifoline (4) is drawn with its reassigned configuration at C13.
Figure 8.

Proposed kinetic resolution of racemic aglains.
Conclusion
In summary, we have employed enantioselective [3+2] photocycloaddition methodology to synthesize the aglain natural products (+)-ponapensin and (+)-elliptifoline. Diastereoselective, reagent-controlled reduction of the hydrated bridgehead ketone led to an intermediate with the correct S configuration at C10. Subsequent ester-amide exchange and acid-catalyzed cyclization provided the natural products. These expedient syntheses demonstrated that late stage installation of the C13 hemiaminal and aminal stereocenters may be substrate controlled by stereoelectronic stabilization of an acyliminium species. This may explain why the relative stereochemical outcome at C13 is similar for both (−)-ponapensin and (−)-elliptifoline. The unexpected finding that (−)-ponapensin and (−)-elliptifoline are enantiomeric to (−)-methyl rocaglate has led to the hypothesis for biosynthesis involving kinetic resolution of aglain ketone core structures. Further work toward development of enantioselective photocycloadditions as well as syntheses and biological evaluations of additional aglain natural products are currently in progress and will be reported in due course.
Supplementary Material
Acknowledgment
We thank the National Institutes of Health (R01 GM073855) for research support and Dr. Jeffrey Bacon (Boston University) and Dr. Emil Lobkovsky (Cornell University) for X-ray crystal structure analyses. We also thank Professor Richard Johnson (University of New-Hampshire), Professor John Snyder (Boston University), Professor Larry Overman (University of California, Irvine), Professor A. Douglas Kinghorn (The Ohio State University), Professor Chang-Yih Duh (National Sun Yatsen University) and Dr. Angela Salim (Institute for Molecular Bioscience, University of Queensland) for extremely helpful and stimulating discussions.
Footnotes
Supporting Information Available: Experimental procedures, and characterization data for all new compounds described herein, including CIF files for compounds 3, 4, and 9a. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Ko F-N, Wu T-S, Liou M-J, Huang T-F, Teng C-M. Eur. J. Pharmacol. 1992;218:129–135. doi: 10.1016/0014-2999(92)90156-x. [DOI] [PubMed] [Google Scholar]
- 2.Hwang BY, Su B, Chai H, Mi Q, Kardono LB, Afriastini JJ, Riswan S, Santarsiero BD, Mesecar AD, Wild R, Fairchild CR, Vite GD, Rose WC, Farnsworth NR, Cordell GA, Pezzuto JM, Swanson SM, Kinghorn AD. J. Org. Chem. 2004;69:3350–3358. doi: 10.1021/jo040120f. [DOI] [PubMed] [Google Scholar]
- 3.Salim AA, Pawlus AD, Chai H-B, Farnsworth NR, Kinghorn AD, Carcache-Blanco EJ. Bioorg. Med. Chem. Lett. 2007;17:109–112. doi: 10.1016/j.bmcl.2006.09.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang S-K, Cheng YJ, Duh C-Y. J. Nat. Prod. 2001;64:92–94. doi: 10.1021/np000341q. [DOI] [PubMed] [Google Scholar]
- 5.King ML, Chiang CC, Ling HC, Fujita E, Ochiai M, McPhail AT. J. Chem. Soc. Chem. Commun. 1982:1150–1151. [Google Scholar]
- 6.Dumontet V, Thoison O, Omobuwajo OR, Martin M-T, Perromat G, Chiaroni A, Riche C, Pais M, Sevenet T, Hadi AH. Tetrahedron. 1996;52:6931–6942. [Google Scholar]
- 7.Inada A, Sorano T, Murata H, Inatomi Y, Darnaedi D, Nakanishi T. Chem. Pharm. Bull. 2001;49:1226–1228. doi: 10.1248/cpb.49.1226. [DOI] [PubMed] [Google Scholar]
- 8.For select reviews on the biology and syntheses of flavaglines, see: Ebada SS, Lajkiewicz N, Porco Jr JA, Jr, Li-Weber M, Proksch P. Prog. Chem. Org. Nat. Prod. 2011;94:1–58. doi: 10.1007/978-3-7091-0748-5_1. Ribeiro N, Thuaud F, Nebigil C, Désaubry L. Bioorg. Med. Chem. 2012;20:1857–1864. doi: 10.1016/j.bmc.2011.10.048.
- 9.For recent work on rocaglates and related derivatives, see: Thuaud F, Ribeiro N, Gaiddon C, Cresteil T, Desaubry L. J. Med. Chem. 2011;54:411–415. doi: 10.1021/jm101318b. Rodrigo CM, Cencic R, Roche SP, Pelletier J, Porco JA., Jr J. Med. Chem. 2012;55:558–562. doi: 10.1021/jm201263k. Malona JA, Cariou K, Spencer WT, Frontier JA. J. Org. Chem. 2012;77:1891–1908. doi: 10.1021/jo202366c. Magnus P, Freund WA, Moorhead EJ, Rainey T. J. Am. Chem. Soc. 2012;134:6140–6142. doi: 10.1021/ja300900p.
- 10.(a) Gerard B, Sangji S, O'Leary D, Porco JA., Jr J. Am. Chem. Soc. 2006;128:7754–7756. doi: 10.1021/ja062621j. [DOI] [PubMed] [Google Scholar]; (b) Gerard B, Cencic R, Pelletier J, Porco JA., Jr Angew. Chem. Int. Ed. 2007;46:7831–7834. doi: 10.1002/anie.200702707. [DOI] [PubMed] [Google Scholar]; (c) Roche SP, Cencic R, Pelletier J, Porco JA., Jr Angew. Chem. Int. Ed. 2010;49:6533–6538. doi: 10.1002/anie.201003212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.See Supporting Information for complete experimental details.
- 12.For higher diastereoselection in reductions of hindered ketones with Bu4NBH4 vs. NaBH4 see: Tsuda Y, Sakai Y, Akiyama K, Isobe K. Chem. Pharm. Bull. 1991;39:2120–2123. Schoop A, Greiving H, Göhrt A. Tetrahedron Lett. 2000;41:1913–1916.
- 13.(a) Hoveyda AH, Evans DA, Fu GC. Chem. Rev. 1993;93:1307–1370. [Google Scholar]; (b) Gribble GW. Org. Proc. Res. Dev. 2006;10:1062–1075. [Google Scholar]
- 14.Saksena AK, Mangiaracina P. Tetrahedron Lett. 1983;24:273–276. [Google Scholar]
- 15.For trajectories of hydride/nucleophile addition to ketones, see: Bürgi HB, Dunitz JD, Shefter E. J. Am. Chem. Soc. 1973;95:5065–5067. Lodge EP, Heathcock CH. J. Am. Chem. Soc. 1987;109:2819–2820.
- 16.Gerard B, Jones G, II, Porco JA., Jr J. Am. Chem. Soc. 2004;126:13620–13621. doi: 10.1021/ja044798o. [DOI] [PubMed] [Google Scholar]
- 17.For anomalous scattering of Cu X-ray by oxygen atoms to refine absolute stereochemistry, see: Moncrief W, Sims SP. Chem. Commun. 1969:914–916. Flack HD, Bernardinelli G. Acta Cryst. A. 1999;55:908–915. doi: 10.1107/s0108767399004262. Frackenpohl J, Adelt I, Antonicek H, Arnold C, Behrmann P, Blaha N, Böhmer J, Gutbrod O, Hanke R, Hohmann S, Van Houtdreve M, Lösel P, Malsam O, Melchers M, Neufert V, Peschel E, Reckmann U, Schenke T, Thiesen H-P, Velten R, Vogelsang K, Weiss H-C. Bioorg. Med. Chem. 2009;17:4160–4184. doi: 10.1016/j.bmc.2009.01.042. Herzon SB, Calandra NA, King SM. Angew. Chem. Int. Ed. 2011;50:8863–8866. doi: 10.1002/anie.201102226.
- 18.Watanabe T, Takeuchi T, Kohzuma S, Umezawa K, Otsuka M. Chem. Commun. 1998:1097–1098. [Google Scholar]
- 19. Otera J, Dan-oh N, Nozaki N. J. Org. Chem. 1991;56:5307–5311. For reviews on transesterification, see: Otera J. Chem. Rev. 1993;93:1449–1470. Otera J. Acc. Chem. Res. 2004;37:288–296. doi: 10.1021/ar030146d.
- 20.(a) Lipton MF, Basha A, Weinreb SM. Org. Synth. Coll. 1980;59:49–53. [Google Scholar]; (b) Eldred SE, Stone DA, Gellman SH, Stahl SS. J. Am. Chem. Soc. 2003;125:3422–3423. doi: 10.1021/ja028242h. [DOI] [PubMed] [Google Scholar]; (c) Han C, Lee JP, Lobkovsky E, Porco JA., Jr J. Am. Chem. Soc. 2005;127:10039–10044. doi: 10.1021/ja0527976. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Ohshima T, Iwasaki T, Mashima K. Chem. Commun. 2006:2711–2713. doi: 10.1039/b605066b. [DOI] [PubMed] [Google Scholar]
- 21.(a) Sim TB, Yoon NM. Synlett. 1994:827–828. [Google Scholar]; (b) Pirrung MC, Chau JH-L. J. Org. Chem. 1995;60:8084–8085. [Google Scholar]
- 22.For recent examples of cyclizations of amido acetals to hemiaminals, see: Buller MJ, Gilley CB, Nguyen B, Olshansky L, Fraga B, Kobayashi Y. Synlett. 2008;15:2244–2248. King FD, Caddick S. Org. Biomol. Chem. 2011;9:4361–4366. doi: 10.1039/c0ob01149e.
- 23.Speckamp WN, Moolenaar MJ. Tetrahedron. 2000;56:3817–3856. [Google Scholar]
- 24.For references on cis/trans folding of amide bonds and electronic effects, see: Thomas KM, Naduthambi D, Zondlo NJ. J. Am. Chem. Soc. 2006;128:2216–2217. doi: 10.1021/ja057901y. Sui Q, Borchardt D, Rabenstein DL. J. Am. Chem. Soc. 2007;129:12042–12048. doi: 10.1021/ja0740925.
- 25.For A1,3 strain in acyliminiums, see: Hart DJ. J. Am. Chem. Soc. 1980;102:397–398.
- 26.For stereoelectronic and hyperconjugative stabilization of iminium ions, see: Nagase H, Yamamoto N, Nemoto T, Yoza K, Kamiya K, Hirono S, Momen S, Izumimoto N, Hasebe K, Mochizuki H, Fujii H. J. Org. Chem. 2008;73:8093–8096. doi: 10.1021/jo801276z. Mersmann S, Raabe G, Bolm C. Tetrahedron Lett. 2011;52:5424–5426.
- 27.For proposed biosyntheses of the aglains/rocaglates, see: Bacher M, Hofer O, Brader G, Vajrodaya S, Greger H. Phytochemistry. 1999;52:253–263. Proksch P, Edrada R, Ebel R, Bohnenstengel FI, Nugroho BW. Curr. Org. Chem. 2001;5:923–938.
- 28.(a) Nakazaki M, Chikamatsu H, Naemura K, Nishino M, Murakami H, Asao M. J. Org. Chem. 1979;44:4588–4593. [Google Scholar]; (b) Truppo MD, Kim J, Brower M, Madin A, Sturr MG, Moore JC. J. Mol. Catal. B: Enzym. 2006;38:158–162. [Google Scholar]
- 29.For references on reductoisomerase and biosynthesis of terpene metabolites, see: Takahashi S, Kuzuyama T, Watanabe H, Seto H. Proc. Natl. Acad. Sci. U.S.A. 1998;95:9879–9884. doi: 10.1073/pnas.95.17.9879. O’Connor SE, Maresh JJ. Nat. Prod. Rep. 2006;23:532–537. doi: 10.1039/b512615k. Munos JW, Pu X, Mansoorabadi SO, Kim HJ, Liu H-W. J. Am. Chem. Soc. 2009;131:2048–2049. doi: 10.1021/ja807987h. Fung PK, Krushkal J, Weathers PJ. Chem. Biodivers. 2010;7:1098–1110. doi: 10.1002/cbdv.200900313. Bastian S, Liu X, Meyerowitz JT, Snow CD, Chen MMY, Arnold FH. Metab. Eng. 2011;13:345–352. doi: 10.1016/j.ymben.2011.02.004.
- 30.For references on Parallel Kinetic Resolution (PKR), see: Bertozzi F, Crottie P, Macchia F, Pineschi M, Feringa BL. Angew. Chem. Int. Ed. 2001;40:930–932. Dehli JR, Gotor V. Chem. Soc. Rev. 2002;31:365–370. doi: 10.1039/b205280f. Miller LC, Sarpong R. Chem. Soc. Rev. 2011;40:4550–4562. doi: 10.1039/c1cs15069c.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.