

SUMMARY
Mutations in SEC63 cause polycystic liver disease in humans. Sec63 is a member of the endoplasmic reticulum (ER) translocon machinery, although it is unclear how mutations in SEC63 lead to liver cyst formation in humans. Here, we report the identification and characterization of a zebrafish sec63 mutant, which was discovered in a screen for mutations that affect the development of myelinated axons. Accordingly, we show that disruption of sec63 in zebrafish leads to abnormalities in myelinating glia in both the central and peripheral nervous systems. In the vertebrate nervous system, segments of myelin are separated by the nodes of Ranvier, which are unmyelinated regions of axonal membrane containing a high density of voltage-gated sodium channels. We show that sec63 mutants have morphologically abnormal and reduced numbers of clusters of voltage-gated sodium channels in the spinal cord and along peripheral nerves. Additionally, we observed reduced myelination in both the central and peripheral nervous systems, as well as swollen ER in myelinating glia. Markers of ER stress are upregulated in sec63 mutants. Finally, we show that sec63 mutants develop liver pathology. As in glia, the primary defect, detectable at 5 dpf, is fragmentation and swelling of the ER, indicative of accumulation of proteins in the lumen. At 8 dpf, ER swelling is severe; other pathological features include disrupted bile canaliculi, altered cytoplasmic matrix and accumulation of large lysosomes. Together, our analyses of sec63 mutant zebrafish highlight the possible role of ER stress in polycystic liver disease and suggest that these mutants will serve as a model for understanding the pathophysiology of this disease and other abnormalities involving ER stress.
INTRODUCTION
In eukaryotic cells, most proteins destined for membrane insertion or secretion are first processed in the endoplasmic reticulum (ER). Nascent polypeptide chains, synthesized by cytoplasmic ribosomes, enter the ER lumen at specialized sites in the ER membrane called translocons, which are complexes of several ER membrane proteins that associate to form a pore (Schnell and Herbert, 2003). Sec61α, Sec61β and Sec61γ form the pore, and this trimeric complex is associated with other proteins including ERj1, Sec62 and Sec63 in mammals (Meyer et al., 2000; Zimmermann et al., 2006). Mutations in SEC63 cause polycystic liver disease (PCLD) in humans, a progressive disorder characterized by the presence of many (>20) cysts throughout the liver (Davila et al., 2004; Everson et al., 2004). PCLD often co-occurs in patients with autosomal dominant polycystic kidney disease (PCKD), but can also exist as a separate disease without kidney cysts (Torres et al., 2007). Polycystic livers can grow up to ten times their normal size, resulting in significant patient morbidity. Although a few therapeutic interventions are available to slow cyst growth, only liver transplantation can change the course of the disease (Drenth et al., 2010). It remains unclear how mutations in SEC63 cause liver cysts, but possibilities include disrupted trafficking of vital proteins such as polycystin-1, an integral cilia membrane protein mutated in PCKD (Fedeles et al., 2011) and disrupted tethering of proteins to the cytosolic face of the ER (Müller et al., 2010). Another possibility is that disruption of SEC63 triggers ER stress that contributes to the pathophysiology of PCLD.
Nascent polypeptides are transported across the ER translocon for processing, folding and maturation (Rapoport, 2007). An imbalance between the load of unfolded preproteins that enter the ER and the capacity of this organelle to properly process the load results in ER ‘stress’: in this case an accumulation of misfolded proteins in the ER lumen (Ron and Walter, 2007). This activates the unfolded protein response (UPR), a conserved cellular homeostatic mechanism, in an attempt to reconcile the imbalance. If the imbalance persists, the UPR can ultimately lead to cell death (Ron and Walter, 2007). Not surprisingly, elevation of ER stress and activation of the UPR are implicated in the pathology of many diseases, including myelin disorders such as multiple sclerosis and Charcot-Marie-Tooth disease (D’Antonio et al., 2009; Lin and Popko, 2009).
Myelin is a multilayered membrane formed by the wrapping of glial cells around axons that allows for efficient conduction of action potentials in the vertebrate nervous system (Nave and Trapp, 2008). Specialized glial cells generate the myelin sheath: oligodendrocytes in the central nervous system (CNS) and Schwann cells in the peripheral nervous system (PNS). Myelin is formed as an elaboration of the plasma membrane of the glial cells, which must generate tremendous amounts of membrane proteins and lipids (Anitei and Pfeiffer, 2006). Segments of myelin are separated by the nodes of Ranvier, which are unmyelinated regions of axonal membrane containing a high density of voltage-gated sodium channels (NaV) (Salzer, 2003; Salzer et al., 2008). These channels propagate the action potential by generating current in response to membrane depolarization (Ritchie, 1995).
TRANSLATIONAL IMPACT.
Clinical issue
Mutations in human SEC63 cause polycystic liver disease (PCLD). There are few treatment options for PCLD; only invasive surgery or liver transplantation can change the course of the disease. Although it is known that SEC63 is part of the endoplasmic reticulum (ER) translocon complex, which transports nascent polypeptides across membranes for folding and maturation in the ER, how mutations in SEC63 cause PCLD has been unclear. Among many hypotheses, it has been proposed that SEC63 mutations might trigger ER stress, which occurs when the capacity of the ER to process nascent or damaged proteins is overloaded. Many studies have linked ER stress with myelin disorders, including multiple sclerosis and peripheral neuropathy.
Results
In a forward genetic screen for zebrafish mutants with abnormal sodium channel clustering in myelinated axons, the authors identified sec63st67, a missense mutation in sec63. Mutant larvae showed multiple defects in myelinated axons in the peripheral nervous system (PNS) and central nervous system (CNS), including disruptions in myelination. Additionally, liver development was abnormal in the sec63st67 mutants; however, cysts were not observed in the liver or kidneys. Swelling and fragmentation of the ER was observed in cells of the PNS, CNS and liver, and multiple molecular markers of ER stress were activated in sec63st67 mutants.
Implications and future directions
These findings introduce the sec63st67 mutant as a new model for studying the function of a gene implicated in PCLD, as well as the role of ER stress in disorders of disrupted myelination. Given that myelinating glia and hepatocytes synthesize large amounts of membrane and secreted proteins during development, they are highly susceptible to disruptions in the translocon machinery. This report advances our understanding of how ER stress contributes to disease processes and provides a new model for investigating the underlying mechanisms.
To discover genes required for the development and organization of myelinated axons, we performed a genetic screen in zebrafish to identify mutants with disruptions in the node of Ranvier (Voas et al., 2007; Voas et al., 2009) (M.G.V. and W.S.T., unpublished data). One mutation identified in this screen disrupts the zebrafish ortholog of the translocon protein Sec63. We show that the UPR is active in zebrafish sec63 mutants and that sec63 mutant axons in the CNS and the PNS are hypomyelinated, with reduced and abnormal NaV clusters. Given the role of Sec63 in human PCLD, we also examined the livers of zebrafish sec63 mutants. We show that pathology develops in this organ, with unusual accumulations of enlarged ER cisternae, disrupted bile canaliculi and accumulation of large, debris-laden lysosomes. These results raise the possibility that ER stress contributes to PCLD caused by SEC63 mutations and offer a new model for diseases involving protein trafficking and ER stress.
RESULTS
Identification and analysis of st67 mutants
In order to understand the genetic mechanisms underlying the organization of myelinated axons, we performed a genetic screen in zebrafish to identify mutations that affect the development of the nodes of Ranvier (Voas et al., 2007; Voas et al., 2009) (M.G.V. and W.S.T., unpublished data). In this screen, we examined expression of NaV along zebrafish axons in whole-mounts of immunostained larvae. A mutation, st67, was identified with a strong defect in NaV clustering along axons. st67 mutants have morphologically abnormal and reduced numbers of NaV channel clusters along the posterior lateral line nerve (PLLn) at 5 days post-fertilization (dpf) (Fig. 1D,E). Time-course analysis determined that the number of nodal NaV clusters in the mutant PLLn was normal at 3 dpf (Fig. 1A,B,E) but was significantly reduced in mutant larvae at 4 dpf and later (Fig. 1E). In st67 mutants, axonal acetylated tubulin expression in the PLLn appeared to be normal (Fig. 1B,D) and the number of Schwann cells along the PLLN at 40 hours post-fertilization (hpf) was also similar to that in the wild type, as determined by in situ hybridization for sox10 (data not shown). The sec63st67 mutation is lethal; mutant larvae fail to inflate their swim bladders (supplementary material Fig. S1) and most do not survive past 14 dpf.
Fig. 1.
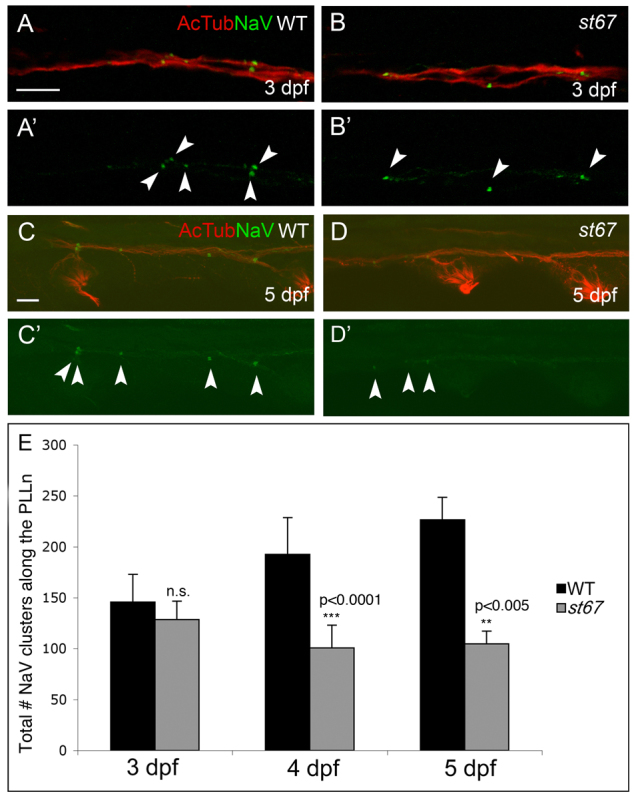
Nodes of Ranvier are abnormal in st67 mutant zebrafish at 5 dpf. (A–D) Images of axons from the posterior lateral line nerve (PLLn) in larvae of the indicated genotypes at 3 (A,B) and 5 dpf (C,D). Axons were double-labeled with antibodies against acetylated tubulin (AcTub, red) and NaV (green). NaV labeling is shown alone in A′–D′. In wild-type and st67 mutant larvae, NaV clusters (arrowheads) appear as discrete labeled puncta. At 3 dpf, no differences are observed in either frequency or morphology of NaV clusters in st67 mutants compared with wild type (A,B). At 5 dpf, st67 mutants have fewer NaV clusters and many NaV clusters are more diffuse than in wild-type PLLn (C,D). (E) Quantification of the total number of NaV clusters along the entire length of the PLLn in wild-type and st67/+ larvae (WT, black bars) compared with homozygous st67 mutants (st67, gray bars) at the indicated developmental stages. P values for unpaired t-test comparisons (two-tailed) are shown; error bars indicate s.d. Sample sizes: at 3 dpf, 23 siblings (WT and st67/+) and 9 mutants; at 4 dpf, 18 siblings and 8 mutants; at 5 dpf, 7 siblings and 9 mutants. Genotypes were assessed by PCR after photography. Scale bars: 10 μm (A–D).
To better understand myelination defects in the st67 mutants, we examined nerve ultrastructure by transmission electron microscopy (TEM). In the developing PNS of mammals and zebrafish, Schwann cells sort axonal segments away from other axons and subsequently myelinate them (Webster et al., 1973; Raphael et al., 2011). At 3 dpf, an early stage of myelination, the extent of axon sorting and myelination by Schwann cells was similar in st67 mutant PLLn and in wild-type PLLn (Fig. 2F,G). By contrast, at 5 dpf, fewer axons were sorted and myelinated in the st67 mutant PLLn compared with wild type (Fig. 2A–D,F,G), and those axons that were myelinated in st67 mutants were surrounded by fewer myelin wraps compared with wild type (wild type, 6.17±0.98 wraps, n=6 axons from four siblings; st67 mutant, 4.5±0.55 wraps, n=6 axons from three mutants; P=0.001). In st67 mutants, Schwann cells had abnormally swollen ER (Fig. 2D,E), which is indicative of ER stress and ER lumenal protein accumulation (Tsang et al., 2007; Sharma et al., 2011). Thus, the initial stages of myelination and node of Ranvier formation appear normal in st67 mutants at 3 dpf, but myelination and nodal NaV clusters are disrupted by 5 dpf.
Fig. 2.
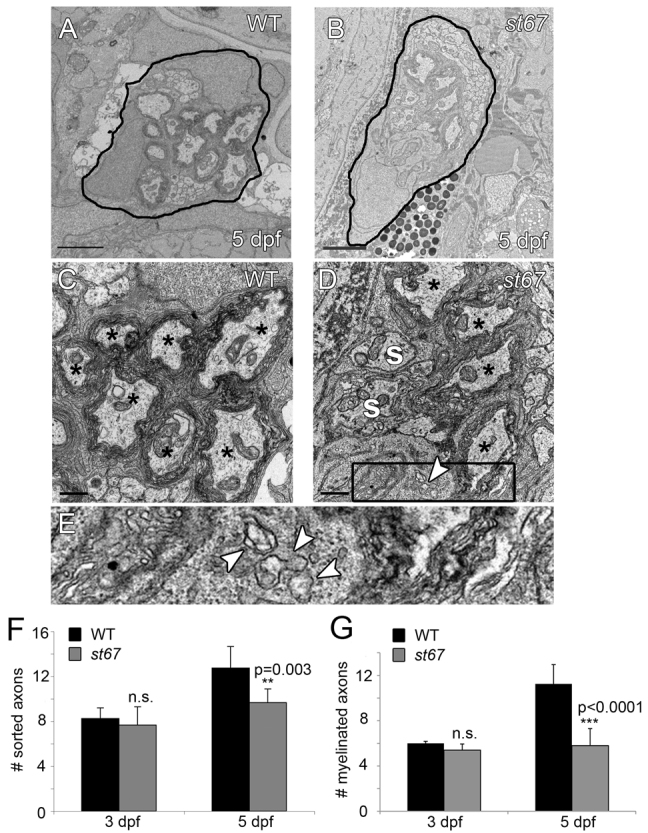
PLLn axons are hypomyelinated in st67 mutants. (A–E) TEM images showing cross-section through the PLLn at 5 dpf. Sorted axons(s) are completely surrounded by Schwann cells and some sorted axons are myelinated (*). In the wild type (A,C), many axons are surrounded by several wraps of myelin at 5 dpf. In st67 mutants (B,D), fewer axons are myelinated at 5 dpf, and irregular Schwann cell cytoplasm is observed with visibly swollen endoplasmic reticulum (D). (E) Blow-up of boxed region in D showing swollen ER (arrows). (F,G) A significant decrease in both sorted (F) and myelinated (G) axons was detected in st67 mutant larvae at 5 dpf, but not at 3 dpf. The P values for unpaired t-test comparisons (two-tailed) are shown; error bars indicate s.d. Sample sizes: at 3 dpf, 4 nerves from 4 siblings and 6 nerves from 5 mutants; at 5 dpf, 8 nerves from 5 siblings and 6 nerves from 3 mutants. All larvae were imaged at and quantifications made from approximately the same location along the anterioposterior axis, at the level of the 7th hemisegment. Scale bars: 2 μm (A,B); 0.5 μm (C,D).
Ultrastructural analysis revealed that the nodes of Ranvier are similar in wild-type and st67 mutants at 5 dpf (supplementary material Fig. S2). One notable difference, however, between mutant and wild-type siblings was that Schwann cell nuclei were often directly over or in close proximity to nodes of Ranvier in st67 mutants (supplementary material Fig. S2; 4 of 8 mutant nodes examined and 0 of 12 wild-type nodes). Schwann cell nuclei are normally located roughly in the middle of a myelin segment (Lubinska, 1959), but have been observed over nodes in mammalian models of peripheral neuropathy (Low, 1976; Robertson et al., 1997).
Examination of NaV expression and myelin in the CNS of st67 mutants revealed that the spinal cord was more severely affected than the PLLn. In contrast to the PLLn, which is indistinguishable from the wild type at 3 dpf, there were fewer NaV clusters in the ventral spinal cord of the mutant at 3 dpf (Fig. 3A,B; Fig. 4B–F). We did not observe a reduction in oligodendrocyte number in the spinal cord at 40 hpf using in situ hybridization for sox10 (data not shown). TEM analysis at 5 dpf showed that axons in the spinal cord were more thinly myelinated in the st67 mutant than in the wild type (Fig. 3C,D; wild type, 3.16±0.85 wraps, n=25 axons from four siblings; st67 mutant, 2.53±0.64 wraps, n=15 axons from three mutants; P=0.01). Similarly to Schwann cells, we also noted swollen and elaborate endoplasmic reticulum in the cytoplasm of oligodendrocytes (Fig. 3D,E).
Fig. 3.

Nodes of Ranvier are disrupted in the spinal cord of st67 mutants and the spinal cord axons are hypomyelinated. (A,B) Myelin basic protein (Mbp, red) and NaV (green) antibody staining in the ventral spinal cord at 3 dpf. No differences can be seen in the intensity of Mbp stain in st67 mutants (B) compared with siblings (A), but NaV puncta (arrowheads) are greatly reduced in st67 mutants (see also Fig. 4). (C–E), TEM images showing cross-sections through the ventral spinal cord at 5 dpf. (C) At 5 dpf, many axons in sibling spinal cord are surrounded by several wraps of myelin (*). (D) Fewer axons are myelinated (*) in st67 mutant spinal cord, and irregular oligodendrocyte cytoplasm was observed with visibly swollen ER (arrowheads) in all mutants examined. (E) Enlarged view of the boxed region in D. In D and E, arrowheads denote swollen ER. Sample sizes: 8 wild-type and heterozygous larvae and 3 mutant larvae. Scale bars: 20 μm (A,B); 1 μm (C,D).
Fig. 4.

st67 disrupts zebrafish sec63. (A) Representation of Sec63 showing functional domains and the location of the lesion in the st67 mutation. TM, transmembrane domain; DnaJ, DnaJ domain; Sec63, Sec63 domain; CC, coiled-coil region. Also shown are sequence traces from homozygous wild-type and st67 mutant larvae. The st67 mutation changes a conserved tyrosine to an aspartic acid in the Sec63 domain. A comparison of zebrafish and human Sec63 amino acid sequence in the vicinity of the st67 mutation is also shown. The light blue box indicates the location of the lesion in st67 zebrafish mutants. The asterisk marks the position of a SEC63 mutation identified in human patients with PCLD (W651G) (Waanders et al., 2010). (B–E) Representative images of antibody-stained preparations of the spinal cord of larvae of the indicated genotypes and injection treatments at 72 hpf. (B) Siblings injected with control solution show normal NaV clustering (arrowheads). (C) st67 mutants injected with control solution show aberrant NaV clustering. (D,E) Siblings and st67 mutants injected with 150 pg of synthetic sec63 mRNA show normal NaV clustering. (F) Quantification of the total number of NaV puncta in two hemisegments (∼200 μm) of ventral spinal cord of sibling (black bars) and st67 mutant larvae (gray bars) at 72 hpf following the indicated injection regimes. The P values for unpaired t-test comparisons (two-tailed) are shown; error bars indicate s.d. Sample sizes: 9 control-injected siblings. 6 control-injected mutants, 12 sec63-injected siblings and 11 sec63-injected mutants. Scale bar: 20 μm (B–E).
st67 disrupts zebrafish sec63
By high-resolution genetic mapping, we determined that st67 disrupts the zebrafish homolog of sec63. In st67, a T-to-G transversion is predicted to change a highly conserved tyrosine to an aspartic acid at amino acid position 647 (Fig. 4A). All mutants tested were homozygous for the T-to-G mutation (n>360), demonstrating that sec63 is tightly linked to the st67 mutation. To obtain additional evidence that sec63 is disrupted by the st67 mutation, we rescued the mutants by injecting synthetic mRNA encoding wild-type sec63. Injection of 150 pg of sec63 mRNA into wild-type and heterozygous embryos did not affect the number or morphology of NaV clusters in the ventral spinal cord at 3 dpf (Fig. 4D,F). st67 mutants injected with control solution showed a significant reduction in the number of NaV clusters in the ventral spinal cord at 3 dpf compared with wild type (Fig. 4C,F), whereas NaV clustering was rescued in mutants injected with 150 pg of wild-type sec63 mRNA (Fig. 4E,F). Together, these data indicate that sec63 is disrupted by the st67 mutation.
Sec63 is a member of the ER translocon machinery and is well conserved from yeast to human (Deshaies et al., 1991; Skowronek et al., 1999; Schnell and Hebert, 2003). Zebrafish Sec63 is 71% identical and 84% similar to human Sec63. Human Sec63 is predicted to span the ER membrane three times and contain a luminal N-terminus, a cytoplasmic C-terminus, with a coiled-coil region and a luminal DnaJ domain between the second and third transmembrane pass (Fig. 4A) (Davila et al., 2004; Müller et al., 2010; Waanders et al., 2010). RT-PCR showed that sec63 is expressed at all stages examined in wild-type and st67 mutants. (supplementary material Fig. S3, data not shown), and whole-mount in situ hybridization showed that sec63 is broadly expressed (supplementary material Fig. S3B,C) (Thisse and Thisse, 2004), with strong staining in the liver and pancreas (supplementary material Fig. S3C).
ER stress response is upregulated in sec63st67 mutants
To counter the accumulation of unfolded proteins in the ER lumen, genes encoding chaperone proteins are transcriptionally activated during the unfolded protein response (UPR), increasing the protein folding capacity of the ER. One of the best-characterized ER chaperones upregulated by the UPR is BiP, which encodes the immunoglobulin heavy-chain-binding protein (Lee, 2005), a chaperone that belongs to the highly conserved hsp70 protein family (Munro and Pelham, 1986; Nicholson et al., 1990). If ER stress cannot be resolved, the UPR leads to transcriptional upregulation of pro-apoptotic genes, such as CHOP (C/EBP-homologous protein), which is downstream of the PERK-eIF2α pathway (Harding et al., 2000). ER transmembrane proteins including inositol-requiring protein 1 (IRE1), a kinase that possesses site-specific endoribonuclease (RNase) activity, also control UPR signaling. The only known target of this RNase activity is X-box binding protein 1 (XBP1). Upon accumulation of misfolded proteins in the ER lumen, IRE1 splices the mRNA of XBP1, excising a 26-nucleotide fragment. The generation of this noncanonically spliced mRNA is specific to UPR activation, and the translated protein is a potent activator of UPR target genes (Yoshida et al., 2001; Calfon et al., 2002).
To test the hypothesis that the sec63st67 mutation activates the UPR, we examined xbp-1 splicing as well as bip and chop expression levels in sec63st67 mutants. The UPR-specific spliced form of xbp-1 was upregulated in sec63st67 embryos and larvae at all time points examined (Fig. 5A). Using quantitative real-time PCR (qRT-PCR), we also found that bip was upregulated in sec63st67 mutants at 3 dpf and 5 dpf, and that chop is upregulated in sec63st67 mutants at 5 dpf (Fig. 5B,C). Together, these data show an upregulation of multiple UPR markers in sec63st67 mutants, suggesting that ER stress levels are elevated, as one might expect in mutants with disruptions in sec63.
Fig. 5.
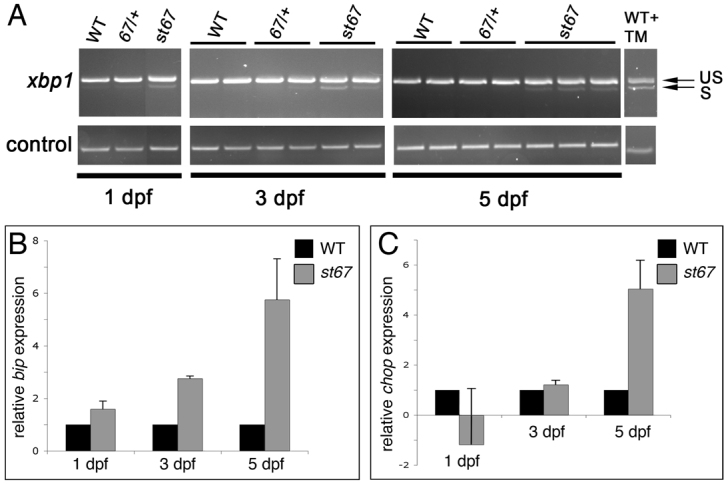
Markers of ER stress are elevated in sec63st67 mutants. (A) RT-PCR showing expression of spliced and unspliced xbp-1 in individual larvae of the indicated genotypes and developmental stages. During the UPR, a 26-nucleotide fragment of xbp-1 mRNA is spliced. RT-PCR shows that the spliced form of xbp-1 (S) is enriched in sec63st67 mutants compared with wild-type (WT) and st67/+ larvae. Unspliced xbp-1 (US) is present in all samples. As a positive control for the assay, wild-type larvae treated with the ER stressor tunicamycin show upregulation of the spliced form of xbp-1. (B,C) qRT-PCR showing relative expression of UPR markers bip (B) and chop (C) in wild-type and heterozygous siblings (WT) vs sec63st67 mutant larvae (st67) at the indicated stages. Error bars indicate s.d.
sec63st67 mutant livers develop abnormally
Mutations in SEC63 cause PCLD in humans, an inherited, progressive disorder characterized by the presence of numerous cysts throughout the liver (Everson et al., 2004). Although disease symptoms appear in adulthood, the cysts are thought to arise during embryonic development from intralobular bile ducts (Desmet, 1992, Qian et al., 2003). Interestingly, one of the disease-causing mutations in patients with PCLD affects a tryptophan residue near the st67 lesion (Fig. 4A) (Waanders et al., 2010). To determine whether the sec63st67 mutation causes liver pathology in zebrafish larvae, we examined the ultrastructure of this organ at 5 dpf and at 8 dpf. As in glia, at 5 dpf, we observed swelling and fragmenting of the ER, again indicative of ER stress (supplementary material Fig. S4). At 8 dpf, the changes were even more obvious: fragmentation and swelling of the ER with accumulation of a dense matrix in the ER lumen (Fig. 6A,B) were accompanied by regions of empty cytoplasm (Fig. 6C,D), smaller mitochondria with a dense matrix and wider cristae (Fig. 6; supplementary material Fig. S2), as well as disrupted and disorganized bile canaliculi (Fig. 6D). Finally, we observed multiple regions of sec63st67 mutant livers laden with large lysosomes filled with debris and these were more frequent and prominent at 8 dpf than at 5 dpf (Fig. 6F). This phenotype was also observed in the intestine of the mutants (data not shown), but never in wild-type siblings.
Fig. 6.

sec63st67 mutants develop numerous liver pathologies. (A–F) TEM images showing liver ultrastructure at 8 dpf in wild-type and sec63st67 mutant zebrafish. (A) Wild-type liver; arrow points to ER with normal morphology. (B) sec63st67 mutant liver; ER is swollen (arrows) and cytoplasm is disrupted. (C,D) Ultrastructure of bile canaliculi (*) in wild-type (C) and sec63st67 mutant (D) livers. sec63st67 bile canaliculi appear disorganized compared with the wild type. Lysosomes are filled with debris in sec63st67 mutants (F; arrows) but not in the wild type (E). Scale bars: 500 nm (A–D); 2 μm (E,F).
Additionally, ER stress has been linked to fatty liver disease in humans (Asselah et al., 2010; Hotamisligil, 2010) and to steatosis liver pathologies in zebrafish (Cinaroglu et al., 2011). Therefore, to assess steatosis in sec63st67 mutant livers, we performed Oil Red O staining at 3, 5 and 8 dpf. No changes in stain intensity were observed in sec63st67 mutants at 3 dpf (data not shown; sample sizes were 11 wild type, 22 heterozygotes and 8 sec63st67 mutants) or at 5 dpf (Fig. 7; sample sizes were 6 wild type, 12 heterozygotes and 7 sec63st67 mutants). However, at 8 dpf, all sec63st67 mutant livers examined showed strong Oil Red O stain compared with wild-type and heterozygous siblings (Fig. 7; sample sizes were 14 wild type, 34 heterozygous and 8 sec63st67 mutants), which is indicative of liver steatosis. Together, our analyses show that the sec63st67 mutation disrupts liver development in addition to myelinating glia in the PNS and CNS.
Fig. 7.

sec63st67 mutants develop liver steatosis. (A–F′) Lateral views of larvae stained with Oil Red O of the indicated genotypes and at the indicated developmental stages. (A–C) Arrows indicate the location of the liver in 5 dpf larvae. sec63st67 mutant livers (C, n=7) are indistinguishable from wild-type (A, n=6) or heterozygous (B, n=12) livers. (D–F) Boxed regions denote the areas enlarged in D′–F′. (D′–F′) Outlines denote the liver in 8 dpf larvae. sec63st67 mutant livers (F,F′, n=8) show stronger Oil Red O stain than wild-type (D,D′, n=14) or heterozygous (E,E′, n=34) livers. Scale bars: 200 μm.
DISCUSSION
Starting with a genetic screen for mutants with abnormal nodes of Ranvier, we identified a mutation in sec63 that disrupts nodal NaV clusters in the PNS and the CNS of zebrafish larvae. The specific disruption of myelination and liver development in sec63st67 mutants is consistent with the requirements of myelinating glia and hepatocytes to synthesize very large amounts of membrane and secreted proteins during development, making these cells especially sensitive to perturbations of the secretory pathway (Wrabetz et al., 2004; Saher et al., 2005; Suter and Scherer, 2003; Anitei and Pfeiffer, 2006; Oratz et al., 1975; Crane and Miller, 1977). We show that multiple markers of the unfolded protein response are upregulated in sec63st67 mutants and hypothesize that the pathologies we observe in myelinating glia (fragmented and swollen ER, disrupted NaV clustering and hypomyelination) are a general consequence of ER stress and disruption of the secretory pathway and not a specific function of Sec63 in node of Ranvier formation or myelination. Similarly, the liver steatosis and the pathologies we observed in hepatocytes (fragmented and swollen ER, smaller mitochondria with denser matrices and wider cristae, disrupted bile canaliculi and debris-laden lysosome accumulation) could also be caused by general ER stress in addition to disrupted protein trafficking. In this model, other cells with very active secretory pathways should be preferentially affected by the sec63st67 mutation. Indeed, we also observed debris-laden lysosome accumulation in the mutant intestine (data not shown).
Comparison with previous analyses of Sec63 in yeast and mammals suggests a number of ways in which the missense mutation in the cytosolic region of Sec63 observed in sec63st67 mutants could disrupt translocon function and protein folding. In yeast and mammals, the cytosolic region of Sec63 interacts with Sec62 (Panzner et al., 1995; Tyedmers et al., 2000; Wittke et al., 2000; Willer et al., 2003; Wang and Johnsson, 2005); in yeast, this interaction is essential for protein transport into the ER and is thought to play the same role in higher vertebrates (Müller et al., 2010). In this way, the sec63st67 mutation could lead to decreased preprotein translocation into the ER. The lumenal J-domain of Sec63 is required for interactions with the chaperone BiP (Brodsky and Schekman, 1993; Scidmore et al., 1993; Lyman and Schekman, 1995; Corsi and Schekman, 1997; Tyedmers et al., 2000). Although the sec63st67 mutation is located in the cytosolic region, the lesion might also lead to a change in Sec63 structure or function that disrupts BiP interactions and therefore protein folding in the lumen of the ER. Finally, in yeast, the Sec61p-Sec63p-BiP translocation complex can also retrogradely affect transport of misfolded proteins out of the ER for cytosolic proteasome degradation (Plemper et al., 1997). This function of the complex remains to be described in higher eukaryotes but if it is conserved, the sec63st67 mutation might also inhibit transport and degradation of misfolded proteins. In each of these scenarios, a combination of UPR activation and reduced secretion of essential proteins might underlie the pathologies in sec63st67 mutants.
Given the essential function of Sec63 in the secretory pathway, it is perhaps surprising that the sec63st67 mutants survive for approximately 14 days. Indeed, germline deletion of Sec63 in mammals results in fully penetrant early embryonic lethality (Fedeles et al., 2011). In zebrafish, maternally supplied transcripts or compensation by another translocon protein, ERj1, might account for the absence of the early, general defects observed in the sec63st67 mutants. ERj1 is an Hsp40 family member related to Sec63 that also provides a luminal J-domain for BiP interactions. Importantly, expression of human ERj1 in yeast can complement mutations in sec63p (Kroczynska et al., 2004). Another, not mutually exclusive, possibility is that the mutant protein encoded by the sec63st67 allele retains enough activity to support some essential functions of the Sec63 protein.
The unfolded protein response in myelinating disorders
After UPR was first described in Saccharomyces cerevisiae (Kozutsumi et al., 1988; Patil and Walter, 2001), a growing body of evidence has implicated this evolutionarily conserved pathway as a causative factor in many human diseases including liver disease, renal disease, diabetes, cancer, neurodegenerative disease, heart disease and myelin disorders (Kaufman, 2002; Ron and Walter, 2007; Austin, 2009; D’Antonio et al., 2009; Lin and Popko, 2009). The biogenesis of myelin requires the synthesis of extremely large amounts of myelin lipids and proteins (Wrabetz et al., 2004; Saher et al., 2005; Suter and Scherer, 2003; Anitei and Pfeiffer, 2006). Accordingly, the UPR has been implicated in the pathogenesis of myelin disorders, including Pelizaeus-Merzbacher disease (PMD), vanishing white matter disease, multiple sclerosis and peripheral neuropathies (van der Voorn et al., 2005; Lin et al., 2005; Lin et al., 2006; Mháille et al., 2008; Wrabetz et al., 2006; Pennuto et al., 2008).
In PMD, for example, several studies have shown that the mutant forms of the structural myelin protein proteolipid protein (PLP) and its alternatively spliced isoform DM20 accumulate in the ER of oligodendrocytes. This activates the UPR and leads to oligodendrocyte death (Gow et al., 1994; Gow and Lazzarini, 1996; Southwood et al., 2002; Swanton et al., 2005; Dhaunchak and Nave, 2007; Dhaunchak et al., 2011). Although, a priori, one might postulate that the pathology in PMD results from reduced levels of PLP in the myelin, a phenotypic comparison of different Plp mutant alleles provides strong evidence against this possibility. Point mutations that cause accumulation of mutant PLP in the ER cause more severe phenotypes than null mutants that lack PLP entirely (Hodes et al., 1993; Klugmann et al., 1997; Swanton et al., 2005). Interestingly, although upregulation of CHOP during the UPR leads to apoptosis in most cell types (Ron and Walter, 2007), it appears to be protective in oligodendrocytes. Analysis of double mutant CHOP null;PLP mutant mice showed that the absence of CHOP exacerbated the clinical phenotype of PLP mutant mice (Southwood et al., 2002). The mechanisms of the protective effect of CHOP on oligodendrocytes are not known, but these genetic studies emphasize the connection between UPR and diseases of the myelin. Furthermore, there is substantial evidence from numerous studies that the UPR plays a role in other myelin disorders affecting oligodendrocytes and Schwann cells (reviewed by D’Antonio et al., 2009; Lin and Popko, 2009).
Sec63 in polycystic liver disease
A number of mutations in SEC63 have been identified in patients with autosomal dominant PCLD (Waanders et al., 2010). PCLD is a progressive disorder characterized by many (>20/liver) fluid-filled liver cysts that might or might not co-occur with autosomal dominant PCKD (Torres et al., 2007). PCKD is caused by mutations in PKD1 and PKD2, which respectively encode polycystin-1 and polycystin-2 (The European Polycystic Kidney Disease Consortium, 1994; Mochizuki et al., 1996). PCLD without PCKD is caused by mutations in SEC63 (Davila et al., 2004) or PRKCSH (Drenth et al., 2003; Li et al., 2003). Although polycystic livers retain normal function, cysts lead to increased liver volume, which causes significant morbidity as the enlarged liver impinges upon other organs and impairs their functions. Currently, there are no therapeutic interventions for PCLD and invasive surgery to remove or aspirate cysts or liver transplant represent the only treatment options (Drenth et al., 2010; Janssen et al., 2010). SEC63 mutations in PCLD patients include a T-to-G missense mutation that changes a conserved tryptophan to a glycine at amino acid position 651 (Waanders et al., 2010). This mutation is located in the same cytosolic region of Sec63 as the st67 mutation (Fig. 4A). This region of Sec63 is located between two β-sheets; in humans, the W651G mutation is predicted to profoundly alter the structure and therefore function of Sec63 (Waanders et al., 2010).
PCLD can also be caused by mutations in PRKCSH (Drenth et al., 2003; Li et al., 2003), which encodes the β-subunit of glucosidase II (protein kinase C substrate 80K-H, also called hepatocystin), an enzyme involved in the oligosaccharide processing of newly synthesized glycoproteins (Drenth et al., 2004). PCLD patients are heterozygous for mutations in PRKCSH or SEC63, and loss of PRKSCH heterozygosity has been observed in cells obtained from liver cyst biopsies (Janssen et al., 2011). Loss of heterozygosity (LOH) for SEC63 mutations remains to be described, but given the observed LOH in PRKSCH mutations, a similar model has been proposed for SEC63 (Zimmermann et al., 2006). In line with this model, a recent report in mouse shows that tissue-specific homozygous loss-of-function mutations in Sec63 result in kidney and liver cyst formation (Fedeles et al., 2011). In addition, our analysis shows that homozygous sec63st67 mutants develop liver pathology. We noted multiple abnormalities in hepatocytes of sec63st67 mutants, although we did not observe cysts in mutant livers or kidneys. Although human PCLD cysts are thought to arise in bile ducts, our observations of pathology in hepatocytes are consistent with a previous report that shows robust expression of Sec63 in human fetal hepatocytes (Waanders et al., 2008). It is possible that the pathologies that we observed in hepatocytes precede cyst formation in bile ducts, and that cysts might become evident at later stages than we were able to analyze.
It is unclear how mutations in SEC63 lead to PCLD in humans, but there are at least three possible, non-mutually exclusive models. One possibility is that reduced expression of a protein(s) trafficked through the ER causes overgrowth and cyst formation in the liver. In accordance with this model, a recent study provided evidence that levels of polycystin-1 protein are reduced in Sec63 mutant mouse cells and suggested that this is a key contributing factor in cyst formation (Fedeles et al., 2011). A second possibility is that Sec63 binds to and retains specific proteins at the cytosolic face of the ER, so that Sec63 mutations mislocalize these proteins. This model is supported by the finding that Sec63 physically interacts with the cytosolic protein nucleoredoxin (Müller et al., 2010), which interacts with Dishevelled to negatively regulate the Wnt–β-cateninin and Wnt–planar cell polarity signaling pathways (Funato et al., 2006; Funato et al., 2010). Despite this possible connection to Wnt signaling, we did not observe defects in the expression of Wnt targets axin2 and naked1 in sec63st67 mutants at 24 hpf (data not shown). Our results highlight a third possible contributing factor: ER stress.
It is likely that a combination of these factors contributes to the final disease outcome. For example, Fedeles et al. reported that treatment with a proteasome inhibitor ameliorated kidney cyst pathology in mouse models of PCKD (Fedeles et al., 2011). The authors proposed a model in which this treatment raised the levels of polycystin-1 and also killed cells under severe ER stress. Future work will define the relationship between UPR, the trafficking of specific proteins such as polycystin-1, and cyst formation. We expect that the sec63st67 mutant zebrafish described in this study will prove useful in future efforts to dissect the pathogenesis of PCLD as well as the consequences of UPR induction in myelinating glia. Future chemical modifier screens might uncover small molecules that alter liver and myelin phenotypes in sec63st67 mutants and perturbation of ER stress or unbiased screens in heterozygous larvae and adults could shed light on the pathways that drive pathologic alterations in liver, myelinated axons and other organs.
METHODS
Fish strains
Zebrafish embryos were raised at 28.5°C and staged according to published methods (Kimmel et al., 1995). N-ethyl-N-nitrosourea (ENU) mutagenesis and rearing of fish for screening were performed as described (Pogoda et al., 2006).
Genetic mapping and positional cloning of sec63st67
Wild-type and st67 mutants at 5 dpf were sorted by their NaV-clustering phenotype. The st67 mutation was genetically localized via bulked segregant analysis with PCR-based simple sequence length polymorphisms (SSLPs) by standard methods (Talbot and Schier, 1999). Fully sequenced BACs within the st67 interval were identified from the zebrafish fpc database (http://www.sanger.ac.uk/Projects/D_rerio/WebFPC/zebrafish) and the Ensembl genome browser (http://useast.ensembl.org/Danio_rerio/Info/Index). Predicted coding regions in these BAC sequences were used in additional mapping experiments to further refine the st67 interval. These experiments placed the st67 mutation in a region of LG20 between the clones CH211-278N15 (GenBank accession #BX927123.8) and CH211-260K9 (GenBank accession #BX649264.4). We sequenced the coding regions of the genes in the interval to find the lesion in sec63.
Genotyping
To genotype individual larvae in the phenotypic analyses, the st67 mutation was scored in genomic DNA samples via a TaqαI restriction enzyme recognition site introduced by the lesion. We used the following primers to amplify fragments from genomic DNA templates: 5′-GGTCACTCTGTCATCGGTTCT-3′ and 5′-TGTGTGATGCTCATGTTTTGC-3′. To genotype individual larvae in the qRT-PCR analysis (see below), the st67 mutation was again scored via TaqαI cleavage. We used the following primers to amplify cDNA generated from single larvae: 5′-ACAAAGGCAGCGAATCAGAC-3′ and 5′-TGGAGCAGGGAATTTCAGTT-3′. The PCR products were digested with TaqαI to generate a smaller fragment from the mutant allele.
In situ hybridization and fluorescent antibody labeling
In situ hybridization was performed with standard protocols (Thisse and Thisse, 2008). To synthesize the sec63 riboprobe, we used the following primers to amplify an 886 bp fragment from a full-length zebrafish sec63 cDNA (GenBank accession #BC076198) obtained from Open Biosystems: 5′-TAAAGGCGGAGATGAGGCTA-3′, 5′-CTTCTCCTCCCCCAGAAATC-3′. The PCR product cloned into pCR II-TOPO vector (Invitrogen) and sequenced. This construct was linearized with NotI and transcribed with SP6 for antisense and linearized with BamHI and transcribed with T7 for sense. Antibody labeling and image acquisition was performed as described for anti-panNaV, anti-acetylated tubulin, and anti-Mbp (Voas et al., 2007; Voas et al., 2009; Monk et al., 2009). For NaV cluster quantification in the PNS, we counted the total number of NaV clusters labeled by the anti-panNaV antibody along the entire length of the posterior lateral line nerve. For NaV cluster quantification in the CNS, we counted the number of NaV clusters labeled by the anti-panNaV antibody in two hemisegments (∼200 μm) of ventral spinal cord. Genotypes were determined by PCR assay after image acquisition (immunohistochemistry) or from larval tail clips after in situ hybridization.
Microinjections
Full-length zebrafish sec63 cDNA (GenBank accession #BC076198) was obtained from Open Biosystems and subcloned into the pCS2+ expression vector. The sequence was confirmed, and synthetic mRNA encoding sec63 was generated with the SP6 mMessage mMachine kit (Ambion) after linearization with ApaI. For the rescue experiments, embryos were injected at the 1–2 cell stage with 150 pg of sec63 mRNA in 1.5 nl of 1× Danieau buffer with 5 mg/ml Phenol Red. Control embryos were injected with 1.5 nl of 1× Danieau buffer with 5 mg/ml Phenol Red.
Oil Red O stain
Oil Red O staining was performed as described (Passeri et al., 2009). Genotypes were determined by PCR from larval tail clips following Oil Red O staining.
RT-PCR
For traditional RT-PCR, total RNA was isolated from pooled wild-type embryos and larvae at the stages indicated in supplementary material Fig. S3 using TRIZOL according to standard protocols. cDNA was reverse transcribed using SuperScript II reverse transcriptase and random hexamers according to the manufacturer’s instructions. To control for genomic DNA contamination, reverse transcriptase was omitted using the same RNA samples. For RT-PCR, we used the following primers: sec63, 5′-GATTTCTGGGGGAGGAGAAG-3′ and 5′-TCCCTCTGCTTCGTCTGATT-3′ (457 bp); xbp-1, 5′-CTGTTGCGAGACAAGACGAG-3′ and 5′-GAAGAGCTCGGAGTCAAGGA-3′ (310 bp unspliced, 275 bp spliced). Either ef1a (Monk et al., 2009) or gapdh: 5′-TGACCCATTCATTGACCTTG-3′ and 5′-GCATGACCATCAATGACCAG-3′ (117 bp) were used as normalization standards. As an additional control for genomic DNA contamination, all primer pairs spanned introns. RT-PCR was performed according to standard protocols and cycling conditions.
qRT-PCR
For qRT-PCR, total RNA was isolated from individual larvae obtained from a sec63st67/+ intercross at 1, 3 and 5 dpf using the RNeasy Micro Kit (Qiagen) according to the manufacturer’s instructions. cDNA was reverse transcribed using SuperScript II reverse transcriptase and oligo(dT) primers according to the manufacturer’s instructions. To control for genomic DNA contamination, reverse transcriptase was omitted using the same RNA samples. For qRT-PCR, we used the following primers: bip, 5′-TCAGCGTCAGGCCACTAA-3′ and 5′-GTCAGCAGAGACACGTCAAA-3′ (171 bp); chop, 5′-CGGTTCCCGACACATCA-3′ and 5′-CACTTTCCTTTCATTCTCCTGTT-3′ (179 bp). gapdh was used as a normalization standard with the primers listed in RT-PCR. As an additional control for genomic DNA contamination, all primer pairs spanned introns. qRT-PCR was performed using SYBR Green Master Mix (Applied Biosystems) and a Roche LightCycler 2.0 according to the manufacturers’ instructions. Fold change was calculated using the 2−ΔΔCt method (Livak and Schmittgen, 2001).
Tunicamycin administration
Tunicamycin (2.5 μg/ml; Sigma) or an equal volume of DMSO was added to the embryo medium of manually dechorionated embryos from 3 dpf to 5 dpf. These embryos were used as a positive control for the ER stress assays (Fig. 5 and data not shown).
Transmission electron microscopy
Larval fixation and embedding for electron microscopy was performed as described (Czopka and Lyons, 2011). The number of larvae examined in each experiment are given in the figure legends. Sections were acquired and stained as described (Czopka and Lyons, 2011), and imaged on a Jeol 1230 or a Philips 410 electron microscope.
Acknowledgments
We thank members of the Talbot laboratory for helpful discussion and Dave Lyons, Florence Marlow, Julie Perlin and Alya Raphael for critical evaluation of the manuscript; Alison Walker for excellent technical support in the initial mapping of the st67 mutation; Tuky Reyes and Chenelle Hill for outstanding fish care; and Roel Nusse and Angela Bowman for sharing the Roche LightCycler.
Footnotes
COMPETING INTERESTS
The authors declare that they do not have any competing or financial interests.
AUTHOR CONTRIBUTIONS
M.G.V. and W.S.T. designed and performed the genetic screen, and M.G.V. identified the st67 mutation in the screen. K.R.M. and W.S.T. designed the experiments, and K.R.M. and I.S.H. performed the experiments. C.F.-A. obtained liver electron micrographs and analyzed the mutant liver pathology. K.R.M. and W.S.T. wrote the manuscript, and all authors read, discussed and edited the manuscript.
FUNDING
This work was supported by grants to W.S.T. from the National Multiple Sclerosis Society [grant number RG3943-A-2] and the National Institutes of Health (NIH) [grant number NS050223] and to C.F.A from the NIH [grant number AR055104 to K. G. Beam]. K.R.M. was supported by a National Multiple Sclerosis Society postdoctoral award [grant number FG1719-A-1] and by the Stanford Genome Training Program (NIH/National Human Genome Research Institute). M.G.V. was supported by a fellowship from the NIH.
SUPPLEMENTARY MATERIAL
Supplementary material for this article is available at http://dmm.biologists.org/lookup/suppl/doi:10.1242/dmm.009217/-/DC1
REFERENCES
- Anitei M., Pfeiffer S. E. (2006). Myelin biogenesis: sorting out protein trafficking. Curr. Biol. 16, R418–R421 [DOI] [PubMed] [Google Scholar]
- Asselah T., Bièche I., Mansouri A., Laurendeau I., Cazals-Hatem D., Feldmann G., Bedossa P., Paradis V., Martinot-Peignoux M., Lebrec D., et al. (2010). In vivo hepatic endoplasmic reticulum stress in patients with chronic hepatitis C. J. Pathol. 221, 264–274 [DOI] [PubMed] [Google Scholar]
- Austin R. C. (2009). The unfolded protein response in health and disease. Antioxid. Redox Signal. 11, 2279–2287 [DOI] [PubMed] [Google Scholar]
- Brodsky J. L., Schekman R. (1993). A Sec63p-BiP complex from yeast is required for protein translocation in a reconstituted proteoliposome. J. Cell Biol. 123, 1355–1363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calfon M., Zeng H., Urano F., Till J. H., Hubbard S. R., Harding H. P., Clark S. G., Ron D. (2002). IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 415, 92–96 [DOI] [PubMed] [Google Scholar]
- Cinaroglu A., Gao C., Imrie D., Sadler K. C. (2011). Activating transcription factor 6 plays protective and pathological roles in steatosis due to endoplasmic reticulum stress in zebrafish. Hepatology 54, 495–508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corsi A. K., Schekman R. (1997). The lumenal domain of Sec63p stimulates the ATPase activity of BiP and mediates BiP recruitment to the translocon in Saccharomyces cerevisiae. J. Cell Biol. 137, 1483–1493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crane L. J., Miller D. L. (1977). Plasma protein synthesis by isolated rat hepatocytes. J. Cell Biol. 72, 11–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czopka T., Lyons D. A. (2011). Dissecting mechanisms of myelinated axon formation using zebrafish. Methods Cell Biol. 105, 25–62 [DOI] [PubMed] [Google Scholar]
- D’Antonio M., Feltri M. L., Wrabetz L. (2009). Myelin under stress. J. Neurosci. Res. 87, 3241–3249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davila S., Furu L., Gharavi A. G., Tian X., Onoe T., Qian Q., Li A., Cai Y., Kamath P. S., King B. F., et al. (2004). Mutations in SEC63 cause autosomal dominant polycystic liver disease. Nat. Genet. 36, 575–577 [DOI] [PubMed] [Google Scholar]
- Deshaies R. J., Sanders S. L., Feldheim D. A., Schekman R. (1991). Assembly of yeast Sec proteins involved in translocation into the endoplasmic reticulum into a membrane-bound multisubunit complex. Nature 349, 806–808 [DOI] [PubMed] [Google Scholar]
- Desmet V. J. (1992). Congenital diseases of intrahepatic bile ducts: variations on the theme “ductal plate malformation”. Hepatology 16, 1069–1083 [DOI] [PubMed] [Google Scholar]
- Dhaunchak A. S., Nave K. A. (2007). A common mechanism of PLP/DM20 misfolding causes cysteine-mediated endoplasmic reticulum retention in oligodendrocytes and Pelizaeus-Merzbacher disease. Proc. Natl. Acad. Sci. USA 104, 17813–17818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhaunchak A. S., Colman D. R., Nave K. A. (2011). Misalignment of PLP/DM20 transmembrane domains determines protein misfolding in Pelizaeus-Merzbacher disease. J. Neurosci. 31, 14961–14971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drenth J. P., te Morsche R. H., Smink R., Bonifacino J. S., Jansen J. B. (2003). Germline mutations in PRKCSH are associated with autosomal dominant polycystic liver disease. Nat. Genet. 33, 345–347 [DOI] [PubMed] [Google Scholar]
- Drenth J. P., Martina J. A., Te Morsche R. H., Jansen J. B., Bonifacino J. S. (2004). Molecular characterization of hepatocystin, the protein that is defective in autosomal dominant polycystic liver disease. Gastroenterology 126, 1819–1827 [DOI] [PubMed] [Google Scholar]
- Drenth J. P. H., Chrispijn M., Nagorney D. M., Kamath P. S., Torres V. E. (2010). Medical and surgical treatment options for polycystic liver disease. Hepatology 52, 2223–2230 [DOI] [PubMed] [Google Scholar]
- Everson G. T., Taylor M. R., Doctor R. B. (2004). Polycystic disease of the liver. Hepatology 40, 774–782 [DOI] [PubMed] [Google Scholar]
- Fedeles S. V., Tian X., Gallagher A. R., Mitobe M., Nishio S., Lee S. H., Cai Y., Geng L., Crews C. M., Somlo S. (2011). A genetic interaction network of five genes for human polycystic kidney and liver diseases defines polycystin-1 as the central determinant of cyst formation. Nat. Genet. 43, 639–647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funato Y., Michiue T., Asashima M., Miki H. (2006). The thioredoxin-related redox-regulating protein nucleoredoxin inhibits Wnt-β-catenin signalling through dishevelled. Nat. Cell Biol. 8, 501–508 [DOI] [PubMed] [Google Scholar]
- Funato Y., Terabayashi T., Sakamoto R., Okuzaki D., Ichise H., Nojima H., Yoshida N., Miki H. (2010). Nucleoredoxin sustains Wnt/β-catenin signaling by retaining a pool of inactive dishevelled protein. Curr. Biol. 20, 1945–1952 [DOI] [PubMed] [Google Scholar]
- Gow A., Lazzarini R. A. (1996). A cellular mechanism governing the severity of Pelizaeus-Merzbacher disease. Nat. Genet. 13, 422–428 [DOI] [PubMed] [Google Scholar]
- Gow A., Friedrich V. L., Jr, Lazzarini R. A. (1994). Many naturally occurring mutations of myelin proteolipid protein impair its intracellular transport. J. Neurosci. Res. 37, 574–583 [DOI] [PubMed] [Google Scholar]
- Harding H. P., Novoa I., Zhang Y., Zeng H., Wek R., Schapira M., Ron D. (2000). Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol. Cell 6, 1099–1108 [DOI] [PubMed] [Google Scholar]
- Hodes M. E., Pratt V. M., Dlouhy S. R. (1993). Genetics of Pelizaeus-Merzbacher disease. Dev. Neurosci. 15, 383–394 [DOI] [PubMed] [Google Scholar]
- Hotamisligil G. S. (2010). Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell 140, 900–917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssen M. J., Waanders E., Woudenberg J., Lefeber D. J., Drenth J. P. H. (2010). Congenital disorders of glycosylation in hepatology: the example of polycystic liver disease. J. Hepatol. 52, 432–440 [DOI] [PubMed] [Google Scholar]
- Janssen M. J., Waanders E., Te Morsche R. H., Xing R., Dijkman H. B., Woudenberg J., Drenth J. P. (2011). Secondary, somatic mutations might promote cyst formation in patients with autosomal dominant polycystic liver disease. Gastroenterology 141, 2056–2063.e2 [DOI] [PubMed] [Google Scholar]
- Kaufman R. J. (2002). Orchestrating the unfolded protein response in health and disease. J. Clin. Invest. 110, 1389–1398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimmel C. B., Ballard W. W., Kimmel S. R., Ullmann B., Schilling T. F. (1995). Stages of embryonic development of the zebrafish. Dev. Dyn. 203, 253–310 [DOI] [PubMed] [Google Scholar]
- Klugmann M., Schwab M. H., Pühlhofer A., Schneider A., Zimmermann F., Griffiths I. R., Nave K. A. (1997). Assembly of CNS myelin in the absence of proteolipid protein. Neuron 18, 59–70 [DOI] [PubMed] [Google Scholar]
- Kozutsumi Y., Segal M., Normington K., Gething M. J., Sambrook J. (1988). The presence of malfolded proteins in the endoplasmic reticulum signals the induction of glucose-regulated proteins. Nature 332, 462–464 [DOI] [PubMed] [Google Scholar]
- Kroczynska B., Evangelista C. M., Samant S. S., Elguindi E. C., Blond S. Y. (2004). The SANT2 domain of the murine tumor cell DnaJ-like protein 1 human homologue interacts with alpha1-antichymotrypsin and kinetically interferes with its serpin inhibitory activity. J. Biol. Chem. 279, 11432–11443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee A. S. (2005). The ER chaperone and signaling regulator GRP78/BiP as a monitor of endoplasmic reticulum stress. Methods 35, 373–381 [DOI] [PubMed] [Google Scholar]
- Li A., Davila S., Furu L., Qian Q., Tian X., Kamath P. S., King B. F., Torres V. E., Somlo S. (2003). Mutations in PRKCSH cause isolated autosomal dominant polycystic liver disease. Am. J. Hum. Genet. 72, 691–703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin W., Popko B. (2009). Endoplasmic reticulum stress in disorders of myelinating cells. Nat. Neurosci. 12, 379–385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin W., Harding H. P., Ron D., Popko B. (2005). Endoplasmic reticulum stress modulates the response of myelinating oligodendrocytes to the immune cytokine interferon-gamma. J. Cell Biol. 169, 603–612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin W., Kemper A., Dupree J. L., Harding H. P., Ron D., Popko B. (2006). Interferon-gamma inhibits central nervous system remyelination through a process modulated by endoplasmic reticulum stress. Brain 129, 1306–1318 [DOI] [PubMed] [Google Scholar]
- Livak K. J., Schmittgen T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2(–ΔΔC(T)) method. Methods 25, 402–408 [DOI] [PubMed] [Google Scholar]
- Low P. A. (1976). Hereditary hypertrophic neuropathy in the trembler mouse. Part 2. Histopathological studies: electron microscopy. J. Neurol. Sci. 30, 343–368 [DOI] [PubMed] [Google Scholar]
- Lubinska L. (1959). Region of transition between preserved and regenerating parts of myelinated nerve fibers. J. Comp. Neurol. 113, 315–335 [DOI] [PubMed] [Google Scholar]
- Lyman S. K., Schekman R. (1995). Interaction between BiP and Sec63p is required for the completion of protein translocation into the ER of Saccharomyces cerevisiae. J. Cell Biol. 131, 1163–1171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer H. A., Grau H., Kraft R., Kostka S., Prehn S., Kalies K. U., Hartmann E. (2000). Mammalian Sec61 is associated with Sec62 and Sec63. J. Biol. Chem. 275, 14550–14557 [DOI] [PubMed] [Google Scholar]
- Mháille A. N., McQuaid S., Windebank A., Cunnea P., McMahon J., Samali A., FitzGerald U. (2008). Increased expression of endoplasmic reticulum stress-related signaling pathway molecules in multiple sclerosis lesions. J. Neuropathol. Exp. Neurol. 67, 200–211 [DOI] [PubMed] [Google Scholar]
- Mochizuki T., Wu G., Hayashi T., Xenophontos S. L., Veldhuisen B., Saris J. J., Reynolds D. M., Cai Y., Gabow P. A., Pierides A., et al. (1996). PKD2, a gene for polycystic kidney disease that encodes an integral membrane protein. Science 272, 1339–1342 [DOI] [PubMed] [Google Scholar]
- Monk K. R., Naylor S. G., Glenn T. D., Mercurio S., Perlin J. R., Dominguez C., Moens C. B., Talbot W. S. (2009). A G protein-coupled receptor is essential for Schwann cells to initiate myelination. Science 325, 1402–1405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller L., de Escauriaza M. D., Lajoie P., Theis M., Jung M., Müller A., Burgard C., Greiner M., Snapp E. L., Dudek J., et al. (2010). Evolutionary gain of function for the ER membrane protein Sec62 from yeast to humans. Mol. Biol. Cell 21, 691–703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munro S., Pelham H. R. (1986). An Hsp70-like protein in the ER: identity with the 78 kd glucose-regulated protein and immunoglobulin heavy chain binding protein. Cell 46, 291–300 [DOI] [PubMed] [Google Scholar]
- Nave K. A., Trapp B. D. (2008). Axon-glial signaling and the glial support of axon function. Annu. Rev. Neurosci. 31, 535–561 [DOI] [PubMed] [Google Scholar]
- Nicholson R. C., Williams D. B., Moran L. A. (1990). An essential member of the HSP70 gene family of Saccharomyces cerevisiae is homologous to immunoglobulin heavy chain binding protein. Proc. Natl. Acad. Sci. USA 87, 1159–1163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oratz M., Rothschild M. A., Schreiber S. S. (1975). Protein synthesis in the hepatocyte. Ann. N. Y. Acad. Sci. 252, 51–62 [DOI] [PubMed] [Google Scholar]
- Panzner S., Dreier L., Hartmann E., Kostka S., Rapoport T. A. (1995). Posttranslational protein transport in yeast reconstituted with a purified complex of Sec proteins and Kar2p. Cell 81, 561–570 [DOI] [PubMed] [Google Scholar]
- Passeri M. J., Cinaroglu A., Gao C., Sadler K. C. (2009). Hepatic steatosis in response to acute alcohol exposure in zebrafish requires sterol regulatory element binding protein activation. Hepatology 49, 443–452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patil C., Walter P. (2001). Intracellular signaling from the endoplasmic reticulum to the nucleus: the unfolded protein response in yeast and mammals. Curr. Opin. Cell Biol. 13, 349–355 [DOI] [PubMed] [Google Scholar]
- Pennuto M., Tinelli E., Malaguti M., Del Carro U., D’Antonio M., Ron D., Quattrini A., Feltri M. L., Wrabetz L. (2008). Ablation of the UPR-mediator CHOP restores motor function and reduces demyelination in Charcot-Marie-Tooth 1B mice. Neuron 57, 393–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plemper R. K., Böhmler S., Bordallo J., Sommer T., Wolf D. H. (1997). Mutant analysis links the translocon and BiP to retrograde protein transport for ER degradation. Nature 388, 891–895 [DOI] [PubMed] [Google Scholar]
- Pogoda H. M., Sternheim N., Lyons D. A., Diamond B., Hawkins T. A., Woods I. G., Bhatt D. H., Franzini-Armstrong C., Dominguez C., Arana N., et al. (2006). A genetic screen identifies genes essential for development of myelinated axons in zebrafish. Dev. Biol. 298, 118–131 [DOI] [PubMed] [Google Scholar]
- Qian Q., Li A., King B. F., Kamath P. S., Lager D. J., Huston J., 3rd, Shub C., Davila S., Somlo S., Torres V. E. (2003). Clinical profile of autosomal dominant polycystic liver disease. Hepatology 37, 164–171 [DOI] [PubMed] [Google Scholar]
- Raphael A. R., Lyons D. A., Talbot W. S. (2011). ErbB signaling has a role in radial sorting independent of Schwann cell number. Glia 59, 1047–1055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapoport T. A. (2007). Protein translocation across the eukaryotic endoplasmic reticulum and bacterial plasma membranes. Nature 450, 663–669 [DOI] [PubMed] [Google Scholar]
- Ritchie J. (1995). The axon structure, function and pathophysiology. In Physiology of Axons (ed. Waxman S. G., Kocsis J. D., Stys P. K.), pp. 68–96 New York: Oxford University Press [Google Scholar]
- Robertson A. M., King R. H. M., Muddle J. R., Thomas P. K. (1997). Abnormal Schwann cell/axon interactions in the Trembler-J mouse. J. Anat. 190, 423–432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ron D., Walter P. (2007). Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 8, 519–529 [DOI] [PubMed] [Google Scholar]
- Saher G., Brügger B., Lappe-Siefke C., Möbius W., Tozawa R., Wehr M. C., Wieland F., Ishibashi S., Nave K. A. (2005). High cholesterol level is essential for myelin membrane growth. Nat. Neurosci. 8, 468–475 [DOI] [PubMed] [Google Scholar]
- Salzer J. L. (2003). Polarized domains of myelinated axons. Neuron 40, 297–318 [DOI] [PubMed] [Google Scholar]
- Salzer J. L., Brophy P. J., Peles E. (2008). Molecular domains of myelinated axons in the peripheral nervous system. Glia 56, 1532–1540 [DOI] [PubMed] [Google Scholar]
- Schnell D. J., Hebert D. N. (2003). Protein translocons: multifunctional mediators of protein translocation across membranes. Cell 112, 491–505 [DOI] [PubMed] [Google Scholar]
- Scidmore M. A., Okamura H. H., Rose M. D. (1993). Genetic interactions between KAR2 and SEC63, encoding eukaryotic homologues of DnaK and DnaJ in the endoplasmic reticulum. Mol. Biol. Cell 4, 1145–1159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma R., Tsuchiya M., Tannous B. A., Bartlett J. D. (2011). Measurement of fluoride-induced endoplasmic reticulum stress using Gaussia luciferase. In The Unfolded Protein Response and Cellular Stress: Part C (Methods in Enzymology), Vol. 491 (ed. Conn P. M.), pp. 111–125 San Diego: Elsevier Academic Press; [DOI] [PubMed] [Google Scholar]
- Skowronek M. H., Rotter M., Haas I. G. (1999). Molecular characterization of a novel mammalian DnaJ-like Sec63p homolog. Biol. Chem. 380, 1133–1138 [DOI] [PubMed] [Google Scholar]
- Southwood C. M., Garbern J., Jiang W., Gow A. (2002). The unfolded protein response modulates disease severity in Pelizaeus-Merzbacher disease. Neuron 36, 585–596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suter U., Scherer S. S. (2003). Disease mechanisms in inherited neuropathies. Nat. Rev. Neurosci. 4, 714–726 [DOI] [PubMed] [Google Scholar]
- Swanton E., Holland A., High S., Woodman P. (2005). Disease-associated mutations cause premature oligomerization of myelin proteolipid protein in the endoplasmic reticulum. Proc. Natl. Acad. Sci. USA 102, 4342–4347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talbot W. S., Schier A. F. (1999). Positional cloning of mutated zebrafish genes. Methods Cell Biol. 60, 259–286 [DOI] [PubMed] [Google Scholar]
- The European Polycystic Kidney Disease Consortium (1994). The polycystic kidney disease 1 gene encodes a 14 kb transcript and lies within a duplicated region on chromosome 16. Cell 77, 881–894 [DOI] [PubMed] [Google Scholar]
- Thisse B., Thisse C. (2004). Fast release clones: a high throughput expression analysis. ZFIN direct data submission (http://zfin.org).
- Thisse C., Thisse B. (2008). High-resolution in situ hybridization to whole-mount zebrafish embryos. Nat. Protoc. 3, 59–69 [DOI] [PubMed] [Google Scholar]
- Torres V. E., Harris P. C., Pirson Y. (2007). Autosomal dominant polycystic kidney disease. Lancet 369, 1287–1301 [DOI] [PubMed] [Google Scholar]
- Tsang K. Y., Chan D., Cheslett D., Chan W. C., So C. L., Melhado I. G., Chan T. W., Kwan K. M., Hunziker E. B., Yamada Y., et al. (2007). Surviving endoplasmic reticulum stress is coupled to altered chondrocyte differentiation and function. PLoS Biol. 5, e44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyedmers J., Lerner M., Bies C., Dudek J., Skowronek M. H., Haas I. G., Heim N., Nastainczyk W., Volkmer J., Zimmermann R. (2000). Homologs of the yeast Sec complex subunits Sec62p and Sec63p are abundant proteins in dog pancreas microsomes. Proc. Natl. Acad. Sci. USA 97, 7214–7219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Voorn J. P., van Kollenburg B., Bertrand G., Van Haren K., Scheper G. C., Powers J. M., van der Knaap M. S. (2005). The unfolded protein response in vanishing white matter disease. J. Neuropathol. Exp. Neurol. 64, 770–775 [DOI] [PubMed] [Google Scholar]
- Voas M. G., Lyons D. A., Naylor S. G., Arana N., Rasband M. N., Talbot W. S. (2007). alphaII-spectrin is essential for assembly of the nodes of Ranvier in myelinated axons. Curr. Biol. 17, 562–568 [DOI] [PubMed] [Google Scholar]
- Voas M. G., Glenn T. D., Raphael A. R., Talbot W. S. (2009). Schwann cells inhibit ectopic clustering of axonal sodium channels. J. Neurosci. 29, 14408–14414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waanders E., Croes H. J. E., Maass C. N., te Morsche R. H. M., van Geffen H. J. A. A., van Krieken J. H. J. M., Fransen J. A. M., Drenth J. P. H. (2008). Cysts of PRKCSH mutated polycystic liver disease patients lack hepatocystin but express Sec63p. Histochem. Cell Biol. 129, 301–310 [DOI] [PubMed] [Google Scholar]
- Waanders E., Venselaar H., te Morsche R. H., de Koning D. B., Kamath P. S., Torres V. E., Somlo S., Drenth J. P. H. (2010). Secondary and tertiary structure modeling reveals effects of novel mutations in polycystic liver disease genes PRKCSH and SEC63. Clin. Genet. 78, 47–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X., Johnsson N. (2005). Protein kinase CK2 phosphorylates Sec63p to stimulate the assembly of the endoplasmic reticulum protein translocation apparatus. J. Cell Sci. 118, 723–732 [DOI] [PubMed] [Google Scholar]
- Webster H. D., Martin R., O’Connell M. F. (1973). The relationships between interphase Schwann cells and axons before myelination: a quantitative electron microscopic study. Dev. Biol. 32, 401–416 [DOI] [PubMed] [Google Scholar]
- Willer M., Jermy A. J., Young B. P., Stirling C. J. (2003). Identification of novel protein-protein interactions at the cytosolic surface of the Sec63 complex in the yeast ER membrane. Yeast 20, 133–148 [DOI] [PubMed] [Google Scholar]
- Wittke S., Dünnwald M., Johnsson N. (2000). Sec62p, a component of the endoplasmic reticulum protein translocation machinery, contains multiple binding sites for the Sec-complex. Mol. Biol. Cell 11, 3859–3871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrabetz L., Feltri M. L., Kleopa K. A., Scherer S. S. (2004). Inherited neuropathies: clinical, genetic and biological features. In Myelin Biology and Disorders (ed. Lazzarini R. A.), pp. 905–951 San Diego: Elsevier Academic Press [Google Scholar]
- Wrabetz L., D’Antonio M., Pennuto M., Dati G., Tinelli E., Fratta P., Previtali S., Imperiale D., Zielasek J., Toyka K., et al. (2006). Different intracellular pathomechanisms produce diverse Myelin Protein Zero neuropathies in transgenic mice. J. Neurosci. 26, 2358–2368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida H., Matsui T., Yamamoto A., Okada T., Mori K. (2001). XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 107, 881–891 [DOI] [PubMed] [Google Scholar]
- Zimmermann R., Müller L., Wullich B. (2006). Protein transport into the endoplasmic reticulum: mechanisms and pathologies. Trends Mol. Med. 12, 567–573 [DOI] [PubMed] [Google Scholar]