

SUMMARY
A subset of patients with Parkinson’s disease acquires a debilitating dementia characterized by severe cognitive impairments (i.e. Parkinson’s disease dementia; PDD). Brains from PDD patients show extensive cholinergic loss as well as dopamine (DA) depletion. We used a mutant mouse model to directly test whether combined cholinergic and DA depletion leads to a cognitive profile resembling PDD. Mice carrying heterozygous deletion of the high-affinity, hemicholinium-3-sensitive choline transporter (CHTHET) show reduced levels of acetylcholine throughout the brain. We achieved bilateral DA depletion in CHTHET and wild-type (WT) littermates via intra-striatal infusion of 6-hydroxydopamine (6-OHDA), or used vehicle as control. Executive function and memory were evaluated using rodent versions of cognitive tasks commonly used with human subjects: the set-shifting task and spatial and novel-object recognition paradigms. Our studies revealed impaired acquisition of attentional set in the set-shifting paradigm in WT-6OHDA and CHTHET-vehicle mice that was exacerbated in the CHTHET-6OHDA mice. The object recognition test following a 24-hour delay was also impaired in CHTHET-6OHDA mice compared with all other groups. Treatment with acetylcholinesterase (AChE) inhibitors physostigmine (0.05 or 0.1 mg/kg) and donepezil (0.1 and 0.3 mg/kg) reversed the impaired object recognition of the CHTHET-6OHDA mice. Our data demonstrate an exacerbated cognitive phenotype with dual ACh and DA depletion as compared with either insult alone, with traits analogous to those observed in PDD patients. The results suggest that combined loss of DA and ACh could be sufficient for pathogenesis of specific cognitive deficits in PDD.
INTRODUCTION
Parkinson’s disease (PD) has traditionally been viewed as a motor disorder caused by degeneration of dopaminergic neurons supplying dopamine (DA) to the striatum. From yearly stages, many PD patients show cognitive deficits known as mild cognitive impairment (Aarsland et al., 2011). Although its etiology is still unclear, the loss of DA in the basal ganglia circuitry appears important (Aarsland et al., 2011; Kehagia et al., 2010; Sawamoto et al., 2008). In rodents (Braga et al., 2005; Da Cunha et al., 2006; De Leonibus et al., 2007; Moriguchi et al., 2012) and primates (Decamp et al., 2004; Lipina and Colombo, 2007), DA depletion impairs cognition. Most PD patients eventually develop dementia (Aarsland et al., 2003). Patients with PD and dementia (PDD) are impaired on tasks of executive function, defined as the ability to self-generate and alter plans and rules that guide behavior (Janvin et al., 2005; Noe et al., 2004; Perretta et al., 2005). Additionally, verbal and visual memory and visual-spatial abilities are impaired (Higginson et al., 2005; Kuzis et al., 1999; Mosimann et al., 2004; Muslimovi3 et al., 2007; Noe et al., 2004) (see also Kehagia et al., 2010).
The pathogenesis of dementia in PD remains poorly understood. Because the loss of DA is comparable in non-demented PD and patients with PDD (Bychkov et al., 2008; Colloby et al., 2005; Hilker et al., 2005; Ito et al., 2002; Joyce et al., 2002), additional neural insults must contribute to dementia in PDD. Severe alterations in cholinergic pathways are found in PDD. PDD subjects show neuronal loss in the nucleus basalis (Nakano and Hirano, 1984; Whitehouse et al., 1983), the extent of which correlates with the severity of dementia (Perry et al., 1987). Choline acetyltransferase is reduced in the neocortex in PDD (Lange et al., 1993; Ruberg et al., 1982), and the reduction correlates with cognitive impairment (Dunois et al., 1983; Perry et al., 1985). The density of vesicular acetylcholine (ACh) transporters is reduced throughout the neocortex in PDD but only in the parietal and occipital cortices in PD (Kuhl et al., 1996). Positron-emission tomography (PET) markers for ACh pathways are reduced in the neocortex in PDD as compared with non-demented PD patients (Bohnen et al., 2003; Hilker et al., 2005; Klein et al., 2010). ACh esterase (AChE) inhibitors improve cognition in PDD patients (Aarsland et al., 2003; Giladi et al., 2003; Hutchinson and Fazzini, 1996). Benefits are modest, however, possibly due to dose-limiting side effects (reviewed by Kurtz and Kaufer, 2011). Thus, both cholinergic deficit (reviewed by Campbell et al., 2009; Hasselmo and Sarter, 2011; Klinkenberg and Blokland, 2010) and loss of DA (Aarsland et al., 2011; Johnson and Galvin, 2011) impair cognition in humans and animals. We hypothesize that dementia in PD results from pronounced ACh deficit on the background of extensive DA depletion.
Here we describe use of a novel mouse model to test the hypothesis that combined depletion of DA and ACh, but not the loss of ACh or DA alone, induces cognitive deficits characteristic for PDD, specifically, visual-spatial recognition and executive function.
TRANSLATIONAL IMPACT.
Clinical issue
Although Parkinson’s disease (PD) is traditionally viewed as a motor disorder, cognitive deficits and dementia are common in patients. Dementia has a major impact on the quality of life and life expectancy of PD patients, and is thus recognized as one of the most pressing and challenging problems in the clinical management of the disease. However, our understanding of the pathophysiology of PD dementia, and the ability to treat the condition in patients, lags behind what we know about motor symptoms. Previous work suggests that a loss of subcortical dopamine (DA) and/or acetylcholine (ACh) contribute to the pathophysiology of dementia in PD. To model PD dementia, this study investigated the combined effect of DA and Ach loss on cognitive function in mice.
Results
The authors report that mice with combined depletion of DA and ACh demonstrate cognitive abnormalities that resemble PD dementia, such as diminished executive functions and impaired visual-spatial memory. Importantly, mice with a reduction in only one of these factors (either DA or Ach) did not show these defects. Visual-spatial memory deficit in these mice was reversed by the acetylcholinesterase inhibitors physostigmine and dopenesil, which are the only drugs currently available to manage dementia in PD.
Implications and future directions
This study provides the first mechanistic support that combined reduction of DA and ACh can cause PD dementia. The findings provide a foundation for further mechanistic studies of PD dementia and for the search for agents to treat it using this double-depleted mouse model.
To model widespread depletion of ACh, mutant mice heterozygous for a deletion of the high affinity, hemicholinium-3-sensitive choline transporter (CHTHET) were used. CHT protein transports choline as a rate-limiting step in the synthesis of ACh (Ferguson et al., 2003). CHTHET mice show roughly half the levels of ACh as compared with wild-type (WT) littermates (Bazalakova et al., 2007) but retain intact motor functions and basic cognitive abilities. Therefore, they appear ideal for assessing possible interactions between ACh depletion and DA depletion on cognitive impairment.
RESULTS
Characterization of dopaminergic and cholinergic depletion
The parameters of the 6-hydroxydopamine (6-OHDA) lesion were optimized to achieve partial DA depletion centered on the motor region of the caudate-putamen. This lesion paradigm was chosen because deep bilateral DA depletion compromises mouse viability and might result in unwanted motor defects. Intra-striatal infusions of 6-OHDA led to a partial loss of dopaminergic fibers in the striatum, with apparent sparing of dopaminergic neurons in the substantia nigra (Fig. 1A). The lesion was similar in WT and CHTHET mice and remained stable for 4 weeks following surgery, as evidenced by the level of tyrosine hydroxylase (TH; an enzyme involved in the formation of DA) (Fig. 1B,D). The amount of TH was reduced by 6-OHDA treatment [F(1,27)=157.2, P<0.0001]. Similar declines were found in WT (∼58%) and CHTHET (∼54%) mice [no genotype effect; F(1,27)=0.03, P=0.87] and remained constant across 7, 14, 21 and 28 days following surgery [F(3,27)=1.14, P=0.35].
Fig. 1.

Partial dopaminergic lesion in the striatum was stable and independent of genotype. Mice received a unilateral partial 6-OHDA lesion (control hemisphere was injected with vehicle) as described in Methods. (A) Low power photomicrograph of representative striatal (upper panel) and nigral (lower panel) sections immunostained for TH. Photographs illustrate partial depletion of TH-positive fibers in the dorso-lateral caudato-putamen (white arrow) in the hemispheres injected intra-striatally with 6-OHDA as compared with vehicle. Note minimal loss of dopaminergic cells in the substantia nigra. (B) Representative western blot for TH showing reduced amount of TH in the hemisphere lesioned with 6-OHDA. Standards correspond to the amount of TH found in indicated amount of the total striatal proteins from the rat brain. (C) DA measured by HPLC and (D) TH measured by western blot showed significant declines in 6-OHDA groups. Data are shown as ng/mg protein for DA and arbitrary units for TH. (E) The DA metabolites DOPAC (left), HVA (middle) and 3-MT (right) (expressed as the percentage of vehicle-treated hemispheres) were also reduced following 6-OHDA treatment. (F) Norepinephrine (left) and serotonin (right) in the 6-OHDA-lesioned hemispheres expressed as percentage of values in the vehicle-treated hemispheres. *P<0.05, **P<0.01 compared with corresponding vehicle group according to one-way repeated measure ANOVA with treatment as a repeated measure factor performed separately for each genotype and each post-lesion day. All values indicate mean + s.e.m.
DA was ∼33% lower in 6-OHDA-treated than in vehicle-treated hemispheres [Fig. 1C; F(1,40)=55.9, P<0.0001], with no effect of genotype or number of recovery days. The catecholamine metabolites DOPAC, HVA and 3-MT were also significantly lower in the 6-OHDA-treated hemispheres (P<0.001), with no effect of genotype or duration of recovery (Fig. 1E). Norepinephrine (NE) content was also slightly, albeit significantly (P<0.01) decreased across genotypes and recovery days, although no significant differences were seen in either genotype on individual days (Fig. 1F). Serotonin levels (Fig. 1F) remained unaffected by the 6-OHDA lesion.
TH significantly declined in mice treated with 6-OHDA compared with vehicle (Fig. 1B,D,C). Similar declines were found in WT (∼58%) and CHTHET (∼54%) mice that remained constant across 4 weeks following surgery. The degree of TH loss was less than that seen in the brains of human PD and PDD patients at post-mortem (Bychkov et al., 2008), although post-mortem material represents end-stage disease with probably more extensive loss of TH than in living PD patients. Taken together, the data show that the partial lesion was stable and reproduced the pattern of TH and DA loss seen in PD patients. However, the magnitude of the DA depletion in the mice underrepresented that seen in PDD patients.
As found previously (Bazalakova et al., 2007), ACh levels were 52% lower in striatal and cortical tissue in CHTHET mice than in WT littermates (Fig. 2A,B; P<0.001). Intra-striatal infusions of 6-OHDA did not affect ACh levels regardless of genotype (Fig. 2A,B). Striatal ACh levels in the CHTHET-vehicle and CHTHET-6OHDA groups were similar and significantly lower than in both vehicle-and 6-OHDA-treated WT groups. The ratio of ACh to choline displayed the same pattern of effects as seen for ACh alone (Fig. 2C,D). The ratio of ACh to choline in the frontal cortex and striatum was significantly lower in CHTHET mice than in WT, regardless of 6-OHDA treatment (P=0.001 for the effect of genotype in both regions). No genotype or treatment differences were found for the ACh precursor, choline, in either brain region (Fig. 2E,F).
Fig. 2.
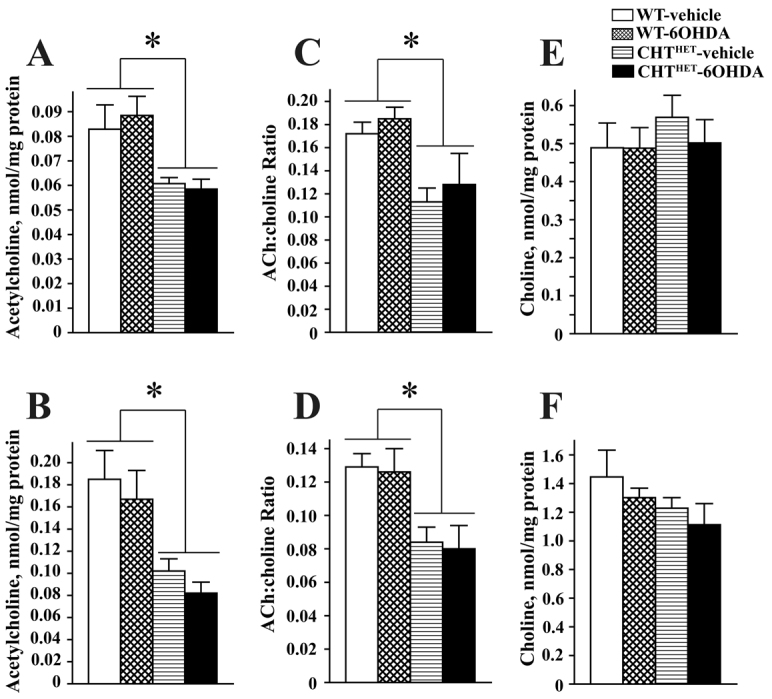
Level of ACh was not altered by dopaminergic lesion in either genotype. (A,B) ACh, measured by HPCL, in sham-operated or 6-OHDA-treated WT or CHTHET mice in the prefrontal cortex (A) or striatum (B). Tissue was collected 7 days following surgery. (C,D) The ratio of ACh to choline in the frontal cortex (C) and striatum (D). (E,F) The cortical (E) and striatal (F) levels of choline were not affected by 6-OHDA or genotype. *P<0.001 compared with corresponding vehicle group across genotypes. All values indicate mean + s.e.m.
Behavioral evaluation
Rotarod
To ascertain whether CHTHET mice with loss of DA show motor deficits, we compared the performance of all groups on rotarod (WT-vehicle n=7, WT-6OHDA n=6, CHTHET-vehicle n=8, CHTHET-6OHDA n=8) (Fig. 3). As previously shown (Bazalakova et al., 2007), we observed no effects of genotype or 6-OHDA treatment. There was a significant improvement in performance across the 3 days of testing [F(2,50)=29.26, P<0.0001], with mice remaining on the rotarod for a shorter period of time on day one compared with days two (P<0.01) and three (P<0.01). Latencies to fall from the rotarod were higher than previously published (Bazalakova et al., 2007), potentially due to the extensive handling received by mice during preceding behavioral testing. To verify proper rotarod procedures, naïve WT and CHTHET mice were tested with no prior experimental procedures (e.g. surgery or handling). Latency to fall in naïve mice was similar to previously published reports (n=6; mean ± s.e.m.: day one, 123.5±14.39 seconds; day two, 211.17±15.45 seconds; day three, 263.5±29.07 seconds). The results demonstrate intact motor performance in mice with loss of ACh, DA or combined depletion of ACh and DA, which is the prerequisite for cognitive assessments of these animals.
Fig. 3.
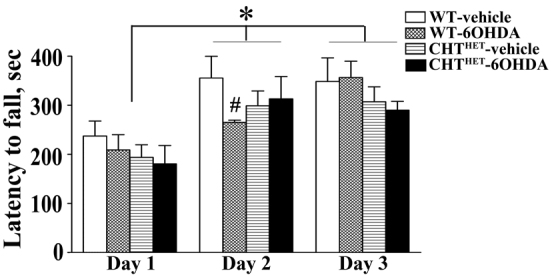
Dopaminergic lesion or genotype did not compromise rotarod behavior. Rotarod performance was measured by latency to fall in seconds. * indicates significant difference (P<0.01) in performance between day 1 and day 2 and between day 1 and day 3 across genotypes and treatments. # indicates a significant difference (P<0.01) between WT-vehicle and WT-6OHDA on day 2. All values indicate mean + s.e.m.
Set-shifting task
The impairment of executive functions is believed to be the core component of cognitive impairment in PDD (Emre, 2004; Robottom and Weiner, 2009). We evaluated the ability of DA-and ACh-depleted mice to form and shift attentional sets. WT-6OHDA mice showed 49±10.8% of TH relative to vehicle control and CHTHET-6OHDA mice showed 56.6±9.3% relative to the CHTHET-vehicle group (mean ± s.e.m.). Fig. 4A schematically shows the experimental chamber and setup, and Table 1 lists the stimuli used. Two-way ANOVA analysis of simple discrimination (SD), compound discrimination (CD) and intra-dimensional shift (IDS) I, IDS II and IDS III (see Methods for details) individually showed no significant effects of genotype (Fig. 4B), treatment or genotype × treatment interaction. During the reversal session, all groups showed markedly worsened performance as compared with IDS III [F(1,28)=50.9, P<0.0001, across genotypes and treatments by three-way repeated measure ANOVA] with no group differences, demonstrating that all groups were capable of forming affective sets. In IDS IV that followed the reversal session, both 6-OHDA-treated groups performed significantly, albeit slightly, worse than vehicle-treated groups [F(1,28)=5.1, P=0.032 for treatment] (Fig. 4B).
Fig. 4.

Double depletion of ACh and DA impaired attentional set formation. (A) Representation of the training apparatus. Food cups filled with medium (light or dark fill) scented with two different odors (black or white circles) were placed in choice compartments, one hiding a food reward (star). Trials were initiated by removing a PVC door, symbolized by the dashed line. (B) Set-shifting training procedure by sessions shown as the number of trials to reach learning criterion (eight consecutively correct responses) for each session. ^P<0.05, ^^P<0.01 according to repeated measure ANOVA with session as a repeated measure factor applied separately for each genotype-treatment combination across sessions indicated by the brackets. a, P<0.05; b, P<0.01; c, P<0.001 by repeated measure ANOVA with session as factor separate for each group. *P<0.05, **P<0.01 as compared with WT-vehicle according to Gamed-Howell post hoc test following one-way ANOVA with group (combinations of genotype-treatment) as the main factor. (C) The number of errors committed during the EDS trial. (D) The ratio of trials to criterion against the immediately preceding IDS session. *P<0.05, **P<0.01 as compared with WT-vehicle according to Gamed-Howell post hoc test following one-way ANOVA with group (combinations of genotype-treatment) as the main factor. All values indicate mean + s.e.m.
Table 1.
Summary of the set-shifting experiment, with specific odors and media used

The experimental groups demonstrated different dynamics of performance across sessions. The analysis of trials to criterion across all IDS for each experimental group by repeated-measure ANOVA with session as within-group factor yielded significant effects of session for all groups (P=0.019, 0.03 and 0.017 for WT-vehicle, WT-6OHDA and CHTHET-vehicle, respectively) except CHTHET-6OHDA (P=0.25). These data suggest that there was a distinct behavioral pattern from session to session in all groups except CHTHET-6OHDA. A more detailed analysis of the session effects revealed that prior to the reversal, WT-vehicle mice, but no other group, needed progressively fewer trials in IDS I, IDS II and IDS III to reach criterion (P=0.01) indicating consistently improved performance across sessions. After the reversal, in IDS IV, both vehicle-treated groups performed significantly worse than in IDS III preceding the reversal (P<0.05). WT-vehicle mice significantly improved performance in subsequent IDS as compared with the reversal (P<0.05). WT-vehicle was the only group to show signs of improvements immediately after the reversal in IDS IV (P=0.09, reversal-IDS IV). Other experimental groups in IDS IV performed similarly to that in the reversal (P >0.5). WT-6OHDA and CHTHET-vehicle groups (P<0.05) improved from the reversal through IDS VI as well as from IDS IV through IDS VI (P<0.01 and 0.05, respectively). CHTHET-6OHDA did not differ in performance across IDS IV to IDS VI or in comparison with the reversal (P>0.5). Thus, CHTHET-6OHDA was the only experimental group that failed to improve across a series of training sessions.
In EDS, there were several effects. Genotype [Fig. 4B; F(1,26)=17.1, P<0.0001] and treatment [F(1,26)=7.44, P=0.01] significantly affected performance. The WT-vehicle group required a greater number of trials to reach the criterion as compared with the CHTHET-vehicle and CHTHET-6OHDA (P<0.01) groups. Furthermore, CHTHET-6OHDA was the only group that committed significantly fewer errors in EDS (P<0.05) and showed significantly lower EDS to IDS VI trial-to-criterion ratio than WT control (Fig. 4C). These data are consistent with the failure of CHTHET-6OHDA mice to improve performance across training sessions and together suggest a failure to form an attentional set.
The WT-vehicle group required approximately twice as many trials to reach criterion in the EDS compared with the preceding IDS VI session (P<0.001 according to repeated measure ANOVA). Both WT-6OHDA and CHTHET-vehicle mice demonstrated impaired performance in EDS compared with IDS VI, requiring roughly 50% more trials to criterion (P<0.05). CHTHET-6OHDA showed no relative increase in trials to criterion between IDS VI and EDS, and post hoc analysis showed CHTHET-6OHDA performance to be significantly different from all other groups (Fig. 4D). The ratio of trials to criterion during the EDS versus IDS VI was not affected by the direction of shift (i.e. odor to medium or medium to odor). The data suggest that mice with combined depletion of DA and ACh were unable to form an attentional set.
Spatial and object recognition
Memory is another cognitive domain affected in PDD. To assess this domain, we used object and spatial recognition tests (Fig. 5A). Following a 5-minute retention delay, recognition scores in spatial or object test were not affected by genotype or treatment. These data indicate that dual DA and ACh depletion did not impair performance in relatively simple short-term visual-spatial memory tasks.
Fig. 5.
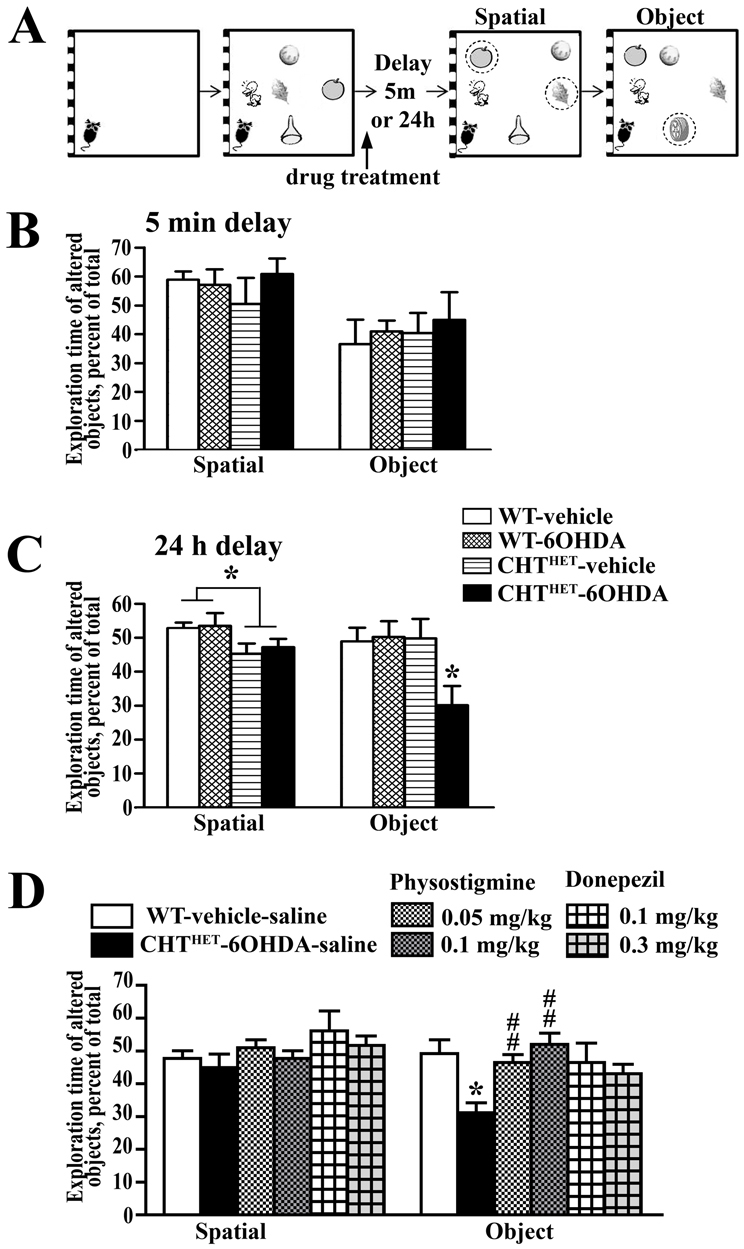
Mice with double depletion of ACh and DA demonstrate a deficit in delayed object recognition. (A) Representation of the spatial and object recognition task. An acclimation session with no objects was followed by three sessions with five objects in a specific configuration. Memory was challenged by a 5-minute or 24-hour retention delay. Two sessions with an altered spatial configuration (two object misplaces; circled) were given, the first being quantified for spatial recognition memory. One session with a novel object (circled) was scored for object recognition memory. The arrow points to the time of administration of physostigmine for the experiment depicted in D. (B) Spatial and object recognition performance with a retention delay of 5 minutes. (C) Spatial and object recognition performance with a retention delay of 24 hours. A modest impairment in performance in the spatial memory test was seen in CHTHET mice regardless of the treatment (P<0.05). *P<0.05 for all other groups according to Gamed-Howell post hoc test following one-way ANOVA with group (combinations of genotype-treatment) as main factor. (D) Physostigmine and donepezil had no effect on the spatial recognition test. Both doses of physostigmine reversed impaired object recognition memory in CHTHET-6OHDA mice. Similarly, both dozes of donepezil reversed the object recognition deficit in CHTHET-6OHDA mice. *P<0.01 compared with WT-vehicle-saline group; ##P<0.01 compared with CHTHET-6OHDA-saline group. All values indicate mean + s.e.m.
A more difficult version of the same tasks, with a 24-hour delay, revealed memory impairments in both the spatial and object tests. On the spatial memory test, a main effect of genotype (Fig. 5C; P=0.02) indicated impaired performance in CHTHET mice, either vehicle- or 6-OHDA-treated, as compared with WT groups, but post hoc comparison revealed no significant group differences. The performance in the object memory test was significantly affected by genotype [F(1,36)=4.25, P=0.046], with the effect of treatment and genotype × treatment interaction approaching significance (P=0.055 and 0.067, respectively). The CHTHET-6OHDA group was the only one that performed significantly worse than the WT-vehicle group (Fig. 5C). Both 6OHDA-treated groups had similar levels of TH depletion: WT-6OHDA mice showed 45.8±5.2% of TH relative to the vehicle control and CHTHET-6OHDA groups and 53.2±6.2% relative to the CHTHET-vehicle group (mean ± s.e.m.).
A number of confounding factors might have affected the mouse performance in the recognitions tasks. One obvious possibility is an overall lower activity or reduced motivation for novelty of CHTHET-6OHDA mice, resulting in shorter exploration time. This explanation is refuted by the fact that there were no group differences in total distance traveled or total exploration time of all objects during any of the session of the recognition protocol (Fig. 6A,B). Our finding that a deficit is revealed following a 24-hour but not 5-minute delay suggests that lower exploration of the novel object in CHTHET-6OHDA results from a memory problem rather than a defect in motivation.
Fig. 6.

No differences were detected in total exploration and distance traveled during the delayed visual-spatial recognition task. (A) Total distance traveled during visual-spatial recognition task with 24-hour delay in all sessions including acclimation, in which no objects were present, is expressed in centimeters. (B) Total exploration time of the five objects available during each session is expressed as total time in seconds. There was an overall reduction across groups in the total exploration time across the acclimation through Acquisition III session (P=0.0002), but individually only the CHTHET-vehicle group showed a significant decrease (P<0.05). Similarly, the total exploration time significantly decreased across the spatial recognition through the object recognition session (P=0.0004), with the WT-6OHDA group showing a significant effect (P<0.001). Significance levels are given for the effect of session across all sessions based on one-way repeated measure ANOVA with session as repeated measure factor and apply to all experimental groups (brackets). All values indicate mean + s.e.m.
A deficit in 24-hour delayed object recognition unique to CHTHET-6OHDA mice presented an opportunity to test whether increasing ACh via inhibition of AChE in the brain would rescue the phenotype. The previously found impaired spatial memory impairment was not replicated (Fig. 5D; P>0.7). However, significant group differences were found, again, on the object memory test [Fig. 5D; F(3,36)=4.99, P<0.01]. The performance was impaired in the CHTHET-6OHDA mice compared with WT-vehicle (P<0.01), both given peripheral saline control. The object recognition deficit in CHTHET-6OHDA mice was reversed by systemic administration of physostigmine at 0.05 mg/kg (P<0.02) and 0.1 mg/kg (P<0.01). Physostigmine-treated CHTHET-6OHDA mice demonstrated the same level of novel object exploration as WT-vehicle mice (Fig. 5D). CHTHET-6OHDA mice treated with another AChE inhibitor, donepezil, were not significantly different in the object recognition performance to WT-vehicle mice (Fig. 5D). Physostigmine or donepezil treatment did not affect locomotion or exploration behavior, as indicated by the lack of group differences in distance traveled (Fig. 7A) or total exploration time (Fig. 7B). CHTHET-6OHDA mice treated with vehicle demonstrated a TH level of 49.4±6.0% (mean ± s.e.m.) of the respective vehicle control. Drug-treated groups were also similar: mice treated with physostigmine at 0.5 mg/kg showed 35.6±5.6% TH relative to the CHTHET-vehicle group and treatment with 1 mg/kg showed 36.5±4.3%; mice treated with dopenesil at 0.1 mg/kg showed 34.3±3.1% of the TH level of the CHTHET-vehicle control and at 0.3 mg/kg showed 45.4±6.8%.
Fig. 7.

Treatment with AChE inhibitors did not affect total exploration and distance traveled during the visual-spatial recognition task. (A) Total distance traveled is expressed in centimeters during each session of training, including acclimation, in which no objects were present (B) Total time, in seconds, spent in exploration of all five objects present during a given session. Significance levels are given for the effect of session across all sessions based on one-way repeated measure ANOVA with session as the repeated measure factor and apply to experimental groups under brackets. All values indicate mean + s.e.m.
DISCUSSION
To define the role of dopaminergic and cholinergic deficits in the pathogenesis of dementia in PDD, we studied the effect of DA and ACh depletion on cognition in a mouse model. To that end, we used the CHTHET mouse, with a global ∼50% loss of ACh, that underwent bilateral 6-OHDA lesion into the motor caudate-putamen to achieve partial, bilateral striatal DA depletion. The procedure resulted in a stable depletion of both neurotransmitters (ACh and DA) in the mouse brain without causing gross motor deficits. Therefore, the double-depleted mouse is a suitable model for investigating the impact of the combined loss of DA and ACh on specific cognitive functions affected in PDD.
Executive functions are one of the cognitive domains most impaired in PDD. To evaluate executive functions, we tested mice on the set-shifting task. The test is highly relevant because it shares important features with a test commonly used in human studies of executive functioning, the Wisconsin Card Sorting Test. Non-demented PD patients display selective deficits in set-shifting performance (Cools et al., 2001; Monchi et al., 2004), possibly due to a combination of nigrostriatal and mesocortical DA deficiency. Patients with PDD demonstrate perseveration and difficulty acquiring the task (Beatty and Monson, 1990; Beatty et al., 1989; Litvan et al., 1991; Tröster et al., 1998). The set-shifting task depends on the functioning of the prefrontal cortex (Birrell and Brown, 2000; Bissonette et al., 2008; McAlonan and Brown, 2003). Dysfunction of the cholinergic system might also play a role in the pathogenesis of executive disturbances (Chen et al., 2004; Tzavos et al., 2004).
Set formation is evidenced by poorer performance during EDS trial as compared with preceding IDS in normal humans, primates and rodents (Birrell and Brown, 2000; Bissonette et al., 2008; Dias et al., 1996; Owen et al., 1993a). Because each IDS used novel odors and medias with the rewarded dimension remaining constant, mice had the opportunity to learn that one dimension consistently led to reward whereas the other dimension did not (i.e. to form an attention set). By assigning the previously correct dimension as incorrect, EDS requires an extra effort, in a process known as set-shifting, to abandon an attentional set learned in preceding IDS sessions and to learn the new rules. EDS performance is affected in the opposite directions by both set formation during IDS and set-shifting during EDS. Mice, in comparison with rats, have been reported to have difficulty in forming attentional sets (Bissonette et al., 2008; Garner et al., 2006). In our behavioral paradigm, WT-vehicle mice displayed a strong impairment in EDS, indicative of a successful formation of attentive set. Mice with DA or ACh depletion demonstrated significant, albeit less pronounced, deficit in EDS performance. Mice with double depletion showed no deficit in the EDS performance, thus presenting no evidence of attention set formation. An alternative interpretation is that better EDS learning reflects greater cognitive flexibility allowing for faster set-shifting, but this explanation is unlikely. Animals that learn an attentional set would be expected to improve performance across IDS sessions and show poor performance on the EDS, as was seen in WT-vehicle and, to some extent, in WT-6OHDA and CHTHET-vehicle mice. By contrast, CHTHET-6OHDA mice did not benefit from the repetition of IDS sessions, learning at the same rate in each subsequent IDS session. This further argues that DA-depleted CHTHET animals approached each IDS trial as a separate event to which no previous experience applied. Our data supports the idea that double-depletion of DA and ACh is detrimental for the ability to form an attentional set. Previous data indicated that the loss of NE affected set-shifting performance in rats (McGaughy et al., 2008; Tait et al., 2007). However, the degree of NE depletion (70%) far exceeded the minimal loss detected in this study. Furthermore, the NE depletion impaired set-shifting, but not set formation. Therefore, it is unlikely that NE contributed to the deficit in set-shifting performance in CHTHET-6OHDA mice. Loss of ACh alone, induced by the lesion of the basal forebrain, did not alter the set-shifting performance in rats (McGaughy et al., 2008; Tait and Brown, 2008), which underscores a unique phenotype in the set-shifting paradigm detected in mice depleted in both DA and ACh. Thus, the evidence suggests that loss of DA in the cortico-striatal circuit associated with PD and further loss of ACh in a proportion of PD patients might play an essential role in the pathogenesis of executive deficits in PDD.
Studies have demonstrated memory deficits in PDD (Higginson et al., 2005; Kuzis et al., 1999; Noe et al., 2004; Owen et al., 1993b; Varanese et al., 2010). The impairment is most evident for visual-spatial memory tasks (Noe et al., 2004; Owen et al., 1993b). A rapid decline in visual-spatial performance is characteristic for PDD patients compared with non-demented PD patients (Johnson and Galvin, 2011; Mosimann et al., 2004) and might be a contributing factor in memory deficits. Spatial and object recognition is a classic test used to evaluate visuo-spatial memory in animals. We have employed the test to examine the effect of double DA and ACh depletion in mice on their ability to recognize a new object or a new position of familiar objects. In object recognition with 24-hour delay, CHTHET-6OHDA was the only group that showed a deficit in performance. This result differs from data reported with mice hemizygous for vesicular acetylcholine transporter that were impaired on object recognition, with both short and long delays (Prado et al., 2006). The reason for this discrepancy is unclear, although differences in the method might have played a role. Importantly, our approach allowed for the selective detection of a deficit due to combined DA and ACh loss. In our model, deficits in object recognition were unique to the double-depleted group, whereas both single DA- and ACh-depleted groups showed intact performance, which suggests that combined DA and ACh depletion is required to induce cognitive deficits. It is possible that more profound ACh or DA depletion alone would lead to similar cognitive impairment. For example, Wisman et al. showed that double DA and ACh depletion with 6-OHDA and 192 IgG-saporin led to severe impairments in working and reference memory in the Morris water maze, with loss of DA alone causing similar cognitive impairments only mildly exacerbated with additional ACh lesioning (Wisman et al., 2008). However, at the level of depletion in our model, which importantly does not cause gross motor or cognitive deficits, only double DA and ACh loss resulted in impairment in cognitive domains affected in PDD. Interestingly, the deficit in object recognition seen in DA-depleted CHTHET mice was completely reversible by inhibition of ACh breakdown, suggesting that an increase in available ACh is beneficial for cognition even when the deficit is not caused exclusively by the loss of ACh.
Although PD has traditionally been viewed as a motor disorder, there is evidence that PD patients from early stages show cognitive and memory deficits collectively referred to as mild cognitive impairment (Aarsland et al., 2011; Johnson and Galvin, 2011; Sawamoto et al., 2008). Cognitive problems associated with mild cognitive impairment, although quite heterogeneous, encompass deficits in executive functions, flexibility, planning and working memory and are believed to be driven by subcortical DA deficiency (Aarsland et al., 2011; Kehagia et al., 2010). In nonhuman primates (Decamp et al., 2004; Lipina and Colombo, 2007), bilateral DA depletion impaired performance on a variety of cognitive tasks. Mice with bilateral striatal 6-OHDA lesions displayed deficiency in spatial recognition with short, but not long, delay (De Leonibus et al., 2007). Bilateral lesioning of the substantia nigra caused defects in working, cued spatial and long-term memory in rodents (Ardayfio et al., 2008; Braga et al., 2005; Da Cunha et al., 2006; Ferro et al., 2005; Hefco et al., 2003; Lindner et al., 1999; Miyoshi et al., 2002; Tadaiesky et al., 2008) (see also Dunnett and Lelos, 2010). In some cases, motor alterations (Ardayfio et al., 2008; Ferro et al., 2005; Lindner et al., 1999) have also been described. Some studies found no cognitive detriments in specific memory tasks such as novel object recognition or passive avoidance in bilaterally DA-depleted rodents (Branchi et al., 2008; De Leonibus et al., 2007). Therefore, loss of DA seems to be an important contributing factor to cognitive deficits in PDD but by itself might not result in cognitive impairment. The loss of ACh alone might be sufficient to impair cognition, as evidenced by detrimental effects of anticholinergic drugs on cognitive performance both in humans and animals (Campbell et al., 2009; Klinkenberg and Blokland, 2010). Obviously, the degree of neurotransmitter loss is an important factor. However, our data suggest that a combination of a moderate ACh deficit on the background of modest DA depletion might produce a specific cognitive state not seen in either condition alone.
In spite of a high prevalence of dementia in PD (Aarsland and Kurz, 2010), its pathophysiology remains obscure. In treatment, the PDD clinic appropriates strategies from the field of Alzheimer’s disease, capitalizing on drugs that enhance ACh signaling, i.e. AChE inhibitors. AChE agents provide only modest improvement in cognitive performance (reviewed by Aarsland et al., 2011; Kehagia et al., 2010; Maidment et al., 2006; Williams-Gray et al., 2006). The mechanistic and translational studies of dementia in PDD lag far behind those of motor deficits in PD or dementia in Alzheimer’s disease. So far, the understanding of PDD pathophysiology has relied on human post-mortem studies and imaging studies of PDD patients. Both of these approaches, although providing invaluable information, are not amenable to experimental manipulation and thus afford limited opportunities to probe for causal links or discover new therapeutic targets. Our studies with the DA-depleted CHTHET mouse demonstrate that combined modest loss of DA and ACh, but not the depletion of each neurotransmitter alone, causes deficits in the cognitive domains specifically affected in PDD, executive functions and visual-spatial memory. Because this behavioral phenotype reminiscent of PDD has been reproduced in the DA-depleted CHTHET mouse based on known neuropathological features of PDD (i.e. loss of DA in the basal ganglia combined with loss of ACh), our data suggest that both loss of DA and ACh contribute to dementia in PDD. The ability of AChE inhibitors, known to improve cognition in PDD, in reversing visual-spatial memory deficits in DA-depleted CHTHET mice serves as further confirmation of essential similarities between the neuropathology in the DA-depleted CHTHET mouse and PDD.
It is clear that the neuropathology of the DA-depleted CHTHET mouse does not cover the whole spectrum of PDD neuropathology. Post-mortem analysis of brain tissue has revealed several neuropathological features of PDD brains. The cortical Lewy bodies (Halliday et al., 2008; Mattila et al., 2000) and Alzheimer’s-related pathologies (Aho et al., 2008; Jellinger, 2009; Jellinger et al., 2002) seen post-mortem frequently correlate to ante-mortem cognition, although recent imaging studies failed to detect high amyloid load in PDD patients (Edison et al., 2008). The role of Lewy bodies and Alzheimer’s-type pathologies in PDD is a hotly debated area, with some authors favoring the accumulation of Lewy bodies and others favoring Alzheimer’s-type pathologies as the prime contributor to PDD pathogenesis (Aarsland et al., 2005; Hurtig et al., 2000; Jellinger, 2009). Importantly, ACh depletion is the most consistently found neural difference between non-demented PD and PDD and consistently correlates with cognitive score (Bohnen et al., 2006; Choi et al., 2012; Kuhl et al., 1996; Perry et al., 1985; Perry et al., 1987). Our studies in the CHTHET-6OHDA mouse suggest that downstream consequences of combined depletion of DA and ACh might be sufficient to bring about essential features of dementia in PDD, which is important for the evaluation of treatments and interventions for therapeutic potential in patients.
METHODS
Animals and stereotaxic surgery
All animal procedures were approved by the Vanderbilt University Institutional Animal Care and Use Committee. Male mice with heterozygous deletion of the CHT protein (CHTHET) have been previously described (Bazalakova et al., 2007; Ferguson et al., 2004; Ferguson et al., 2003). CHTHET are congenic on a C57BL/6 background and were housed up to five per cage on a 12-hour light-dark cycle with food and water ad libitum, except when food restriction was required for training. All experimental procedures were performed during the light phase of the diurnal cycle.
At 11–13 weeks of age, mice received bilateral intra-striatal injections of 6-OHDA (2 μg in 1 μl of 0.05% ascorbic acid in phosphate-buffered saline) or vehicle at stereotaxic coordinates (from bregma) anterior-posterior (AP) 0.65, medial-lateral (ML) ± 2.5 and dorsal-ventral (DV; skull) 3.2. Mice were anesthetized by injection (i.p.) with a mixture of ketamine (100 mg/kg) and xylazine (10 mg/kg). Desipramine (20 mg/kg, i.p.; Sigma, St Louis, MO) was given 30 minutes prior to 6-OHDA injection to protect noradrenergic terminals.
Neurotransmitter measurements
Monoamines
To assess the time course and extent of DA decline following intra-striatal 6-OHDA, unilaterally injected mice were decapitated under isoflurane anesthesia at 7 (WT n=5; CHTHETn=7), 14 (WT n=6; CHTHETn=7), 21 (WT n=4; CHTHETn=7) or 28 (WT n=6; CHTHETn=6) days following surgery. The unilateral 6-OHDA lesion was employed in this particular case in order to determine the degree of DA loss at each post-surgerical interval while conserving mice. Striatum was dissected on ice and rapidly frozen on dry ice prior to storage at −80°C until ready for monoamine analyses. HPLC determination of monoamine neurotransmitter and metabolites was performed by the Vanderbilt Center for Molecular Neuroscience Neurochemistry Core utilizing an Antec Decade II (oxidation: 0.4) electrochemical detector operated at 33°C and Phenomenex Kintex (2.6 u, 100A) C18 HPLC column (100×4.60 mm). Biogenic amines were eluted with a mobile phase consisting of 89.5% 0.1 M TCA, 10−2 M sodium acetate, 10−4 M EDTA and 10.5% methanol (pH 3.8) in the following order: noradrenaline, MHPG, adrenaline, DOPAC, dopamine, 5-HIAA, HVA, 5-HT and 3-MT (Lindley et al., 1998). Solvent was delivered at 0.6 ml/minute using a Waters 515 HPLC pump. HPLC control and data acquisition were managed by Empower software. Data from these analyses are expressed as ng/mg protein.
Acetylcholine and choline
To determine whether intra-striatal 6-OHDA altered ACh levels in the striatum or overlying cortex, bilaterally injected mice were rapidly decapitated 7 days following surgery (WT-vehicle n=6; WT-6OHDA n=6; CHTHET-vehicle n=6; CHTHET-6OHDA n=5). Striatum and frontal cortex were dissected on ice following microwaving to inactivate AChE (Bertrand et al., 1994). Dissected tissue was rapidly frozen on dry ice and stored at −80°C until ready for use. ACh and choline levels were quantified using HPLC techniques by the Vanderbilt Center for Molecular Neuroscience Neurochemistry Core. The HPLC system was composed of a Waters 717+ autosampler, Water model 515 pump and Antec Decade electrochemical detector. The column employed was from Bioanalytical Systems (acetylcholine column) coupled with post-column immobilized enzyme reactor (IMER) containing bound AChE and choline oxidase (ChO) that converted ACh to choline and further oxidized it to hydrogen peroxide. The final reaction product was detected amperometrically and quantified on a platinum working electrode (+400 mv) (Damsma et al., 1985). Levels of ACh and choline from these analyses are expressed as nmol/mg protein.
Quantitative western blot
The expression of TH was measured in all animals tested for behavior. For the TH analysis, mice were decapitated under isoflurane anesthesia following completion of behavioral measures or, to create a timeline of TH decline, following 6-OHDA lesion, at 7 (WT n=5; CHTHETn=3), 14 (WT n=3; CHTHETn=6), 21 (WT n=3; CHTHETn=5) or 28 (WT n=5; CHTHETn=5) days post-surgery. Brains were collected and rapidly frozen on dry ice. Both striata were outlined on precut 150-μm thick coronal sections and scraped into 150 μl of lysis solution (Ambion, Austin, TX). Protein concentration in the samples was determined with the Bradford reagent (Bio-Rad, Hercules, CA). Samples were stored at −80°C until needed.
Protein sample preparation and western blots were performed as previously described (Ahmed et al., 2008; Ahmed et al., 2010). Briefly, proteins were precipitated from lysis buffer with nine volumes of methanol, pelleted by centrifugation, washed with 1 ml of 90% methanol, dried and dissolved in sodium dodecyl sulfate sample buffer at the final concentration of 0.1 μg protein/μl. For electrophoresis, 0.5 μg protein from each sample was loaded on the gel. TH was detected with rabbit polyclonal primary antibody (Millipore, Temecula, CA) at 1:24,000 dilution. The secondary antibody was horseradish peroxidase-conjugated goat anti-rabbit at 1:25,000 (Jackson ImmunoResearch Laboratories, West Grove, PA). Blots were developed using SuperSignal enhanced chemiluminescence reagent WestPico (Pierce, Rockford, IL) and exposed to X-ray film for appropriate periods of time. Optical density of the bands was measured using Versadoc software (Bio-Rad). Standard curves were fitted to linear equations using Prism 4.0 (GraphPad Software, San Diego, CA).
Immunohistochemistry
TH immunostaining was performed as previously described (Ahmed et al., 2008; Ahmed et al., 2010). Briefly, mice were injected with ketamine and xylazine anesthetic at 28 days following unilateral infusions of 6-OHDA and perfused intracardially with 4% paraformaldehyde. Following cryoprotection in 30% sucrose, brains were stored at −80°C until needed. Sections (30 μM) were obtained on a cryostat and incubated with rabbit polyclonal primary antibody for TH (1:500; Millipore, Temecula, CA) for 48 hours at 4°C. TH antibody was detected using fluorescently labeled goat anti-rabbit antisera (1:200; Vector Laboratories, Burlingame, CA) and streptavidine tagged with Alexa Fluor 488 (1:200; Invitrogen, Eugene, OR), each incubated with TH antibody-labeled sections for 60 minutes at room temperature.
Behavioral tasks
Rotarod
Mice were tested for motor co-ordination and balance on the accelerating Rotarod apparatus (Model 7650, Ugo Basile, Napoli, Italy) following training in spatial and object recognition 19–21 days after surgery. Mice were placed on a cylinder, rotating at 5 r.p.m. Rotation speed increased to a maximum of 40 r.p.m. gradually over 5 minutes. Latency (in seconds) to fall from the cylinder was recorded as the main dependent variable, with a maximum testing period of 500 seconds. Mice performed three trials per day for 3 consecutive days. The mean latency was calculated per day and used in statistical analyses.
Set-shifting task (attentional set-shifting task)
A separate set of mice without previous training experience was used for these experiments. Procedures for the set-shifting task were adapted from previously used protocols for rats (Birrell and Brown, 2000; Wood et al., 1999) and mice (Bissonette et al., 2008; Colacicco et al., 2002). Procedures began following a one-week recovery period from intra-striatal vehicle or 6-OHDA infusion (WT-vehicle n=7; WT-6OHDA n=8; CHTHET-vehicle n=7; CHTHET-6OHDA n=8). Data from two additional CHTHET-vehicle mice were included in the reversal and striatal TH measures only, because these mice were trained in preliminary studies using a shorter protocol (i.e. reversal CHTHET-vehicle n=9). Mice were food-restricted to 85% of their free-feeding weight and acclimated with the testing room and clear, polycarbonate chamber (30×30×30 cm with two 15×15 cm choice compartments at one end; Fig. 4A). Acclimation consisted of, on consecutive days: (1) exposure to the empty chamber for 10 minutes, (2) four trials with food cups holding uncovered food reward (one half piece of Honey-Nut Cheerios) and (3) four trials with food reward hidden under unscented medium. Media used during acclimation were not used again during the protocol. Testing continued for the next 5 days, with two sessions per day separated by 2 hours to prevent satiation during afternoon testing. Mice were trained to find food hidden under medium scented by dried spices (i.e. odors). The location of the food reward (i.e. a correct response) was determined either by one of the odors (in which case the medium was irrelevant) or by one of the media (in which case the odor was irrelevant). Mice were randomly assigned to approach an odor or medium. Table 1 lists the odor-medium combinations used per session. At each trial, mice were allowed to explore both bowls before making a choice by digging. During the first four trials of each session, mice were allowed to retrieve the food reward from the correct bowl after initially making an incorrect response. Following the fourth trial, no digging could be done following an incorrect choice.
Mice were initially trained on a simple discrimination (SD) where two odors were each paired with only one type of medium for odor-medium set-shifting, or vice versa for medium-odor set-shifting. Referred to as direction of shift, mice in each group were counterbalanced to ensure no bias towards a particular dimension. Following the example in Table 1, mice learned that clove-scented dishes held food whereas basil-scented dishes did not, with both clove and basil mixed into the same medium. Session 2, a compound discrimination (CD), added a second medium, for which clove and basil were each mixed with the previously used medium plus an additional, novel medium. In the CD, mice learned that clove-scented dishes held food, regardless of the medium, and basil-scented dishes never held food, regardless of the medium.
An intra-dimensional shift (IDS) modeled the compound discrimination paradigm with, importantly, novel odors and media for each IDS. Reversal of the third intra-dimensional shift (session six) rewarded an odor that was incorrect in the immediately preceding session. An extra-dimensional shift (EDS) during the last session rewarded a medium instead of an odor or the reverse. Poor performance during the EDS indicates successful formation of the attentional set. Mice were trained in each session until achieving the learning criterion of eight consecutively correct trials or for a maximum of 32 trials. The session maximum of 32 trials was chosen to avoid lack of performance due to satiation. A trial maximum of 2 minutes was imposed. For each session, the correct dish was counterbalanced to the right or left chamber compartment. Also, the direction of EDS (i.e. odor-medium or medium-odor) was counterbalanced for each treatment group. For each session, trials to criterion (i.e. eight consecutively correct trials) and quantity of errors were recorded. Performance on the EDS was further analyzed as the ratio of trials to criterion for the EDS versus the immediately preceding IDS (i.e. IDS VI).
Spatial and object recognition
A separate set of mice without previous training experience was used for these experiments. Following a one-week post-lesion recovery period, mice were transferred to the Center for Molecular Neuroscience’s Laboratory for Neurobehavior Core holding facilities and given one week to acclimate. On training day, mice were placed in a clean holding cage and placed in the training anteroom for a minimum of 30 minutes prior to initiation of the task. Training occurred across four sessions, initially in an empty chamber (clear polycarbonate, 50×50×40 cm) followed by three sessions with five objects (wiffle ball, plastic orange, clay leaf, plastic funnel and rubber duck) in a specific configuration (Fig. 5A). A retention delay of either 5 minutes (WT-vehicle n=10; WT-6OHDA n=7; CHTHET-vehicle n=7; CHTHET-6OHDA n=7) or 24 hours (WT-vehicle n=11; WT-6OHDA n=11; CHTHET-vehicle n=9; CHTHET-6OHDA n=10) was imposed, followed by a spatial recognition test. Two previously encountered objects were moved to novel locations, resulting in a novel configuration of the same five objects. Mice were exposed to the novel configuration a second time, followed by an object recognition test in the final session. The object recognition test consisted of introducing a novel object (wheel caster) in place of a familiar, non-displaced object.
Mouse behavior was captured using an overhead camera and analyzed by AnyMaze tracking software (Stoelting, Wood Dale, IL) in real-time. Exploration was quantified as the amount of time spent within 1 cm of each object. Tracking was centered on the nose of the mouse, and time spent on top of an object was subtracted. For the spatial recognition test, data were expressed as the percent of total exploration time spent with the two displaced objects. Similarly, for the object recognition test, data were expressed as the percent of total exploration time spent with the one novel object. Animals were eliminated if exploration at any of the three acquisition sessions totaled less than 5 seconds. Sessions lasted 6 minutes with 5-minute inter-session intervals. Preliminary studies were performed to verify a lack of preference amongst the six objects used.
The effect of physostigmine (eserine hemisulphate salt; Sigma, St Louis, MO) and donepezil (donepezil hydrochloride; Sigma) on visual-spatial recognition was determined in a separate group of WT-vehicle and CHTHET-6OHDA mice. AChE inhibitors were chosen because they are predominantly used in the treatment and shown to effectively ameliorate cognitive impairments in PDD (Aarsland et al., 2002; Fuchs et al., 2004; Leroi et al., 2004). Immediately post-training (i.e. following the final acquisition session), mice were injected (i.p.) with 0.05 (n=10) or 0.1 (n=9) mg physostigmine/kg body weight, 0.1 (n=7) or 0.3 (n=9) mg donepezil/kg or saline vehicle (n=9). As a control, vehicle-treated WT mice (n=12) were also tested. To minimize stress from the injection, mice were handled for 5 out of 7 days prior to training. Handling occurred for 5 minutes per day outside the vivarium, in a room distinctly different from the training room.
Data analysis
All animals used in behavioral testing were analyzed for the striatal expression of TH upon completion of behavioral testing. Mice with 25% or smaller loss of TH-positive innervation in the striatum (as compared with the mean value of the respective vehicle group, WT-vehicle or CHT-vehicle) were eliminated from final analysis. StatView software (SAS Institute, Cary, NC) was used for statistical analysis. Most statistical analyses were performed using two-way ANOVA with treatment (vehicle, 6-OHDA) and genotype (WT, CHTHET) as main factors. Timelines for changes to TH, DA, DOPAC, 3-MT, HVA, norepinephrine and serotonin were analyzed using a three-way ANOVA with the additional independent variable of duration of recovery (7, 14, 21 and 28 days). Two behavioral tests were analyzed using repeated measure two-way ANOVA with treatment and genotype as between-group factors. The repeated measure was day of testing for rotarod analysis and session number for analysis of the set-shifting task. Effects of physostigmine on performance of the recognition task were analyzed using a one-way ANOVA with full treatment (WT-vehicle-saline, CHTHET-6OHDA-saline, CHTHET-6OHDA-0.05, CHTHET-6OHDA-0.1) as the independent variable. When relevant, post-hoc analysis was performed using the Games-Howell test (Games and Howell, 1976). For all tests, P<0.05 was considered significant.
Footnotes
COMPETING INTERESTS
The authors declare that they do not have any competing or financial interests.
AUTHOR CONTRIBUTIONS
E.V.G. and L.Z. conceived and designed the experiments. L.Z., E.B., E.L.T. and C.S. performed the experiments. L.Z., C.S. and E.V.G. analyzed the data. R.D.B. contributed CHT mice. L.Z., E.V.G. and R.D.B. wrote the paper.
FUNDING
This work was supported by the National Institutes of Health [grant numbers NS065868 to E.V.G. and MH073159 to R.D.B.] and by Cognitive Deficits and Mood Disorders in PD Award from the Michael J. Fox Foundation to E.V.G.
REFERENCES
- Aarsland D., Kurz M. W. (2010). The epidemiology of dementia associated with Parkinson disease. J. Neurol. Sci. 289, 18–22 [DOI] [PubMed] [Google Scholar]
- Aarsland D., Laake K., Larsen J. P., Janvin C. C. (2002). Donepezil for cognitive impairment in Parkinson’s disease: a randomised controlled study. J. Neurol. Neurosurg. Psychiatry 72, 708–712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aarsland D., Andersen K., Larsen J. P., Lolk A., Kragh-Sørensen P. (2003). Prevalence and characteristics of dementia in Parkinson disease: an 8-year prospective study. Arch. Neurol. 60, 387–392 [DOI] [PubMed] [Google Scholar]
- Aarsland D., Perry R., Brown A., Larsen J. P., Ballard C. (2005). Neuropathology of dementia in Parkinson’s disease: a prospective, community-based study. Ann. Neurol. 58, 773–776 [DOI] [PubMed] [Google Scholar]
- Aarsland D., Brønnick K., Fladby T. (2011). Mild cognitive impairment in Parkinson’s disease. Curr. Neurol. Neurosci. Rep. 11, 371–378 [DOI] [PubMed] [Google Scholar]
- Ahmed M. R., Bychkov E., Gurevich V. V., Benovic J. L., Gurevich E. V. (2008). Altered expression and subcellular distribution of GRK subtypes in the dopamine-depleted rat basal ganglia is not normalized by l-DOPA treatment. J. Neurochem. 104, 1622–1636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed M. R., Berthet A., Bychkov E., Porras G., Li Q., Bioulac B. H., Carl Y. T., Bloch B., Kook S., Aubert I., et al. (2010). Lentiviral overexpression of GRK6 alleviates L-dopa-induced dyskinesia in experimental Parkinson’s disease. Sci. Transl. Med. 2, ra28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aho L., Parkkinen L., Pirttilä T., Alafuzoff I. (2008). Systematic appraisal using immunohistochemistry of brain pathology in aged and demented subjects. Dement. Geriatr. Cogn. Disord. 25, 423–432 [DOI] [PubMed] [Google Scholar]
- Ardayfio P., Moon J., Leung K. K. A., Youn-Hwang D., Kim K.-S. (2008). Impaired learning and memory in Pitx3 deficient aphakia mice: a genetic model for striatum-dependent cognitive symptoms in Parkinson’s disease. Neurobiol. Dis. 31, 406–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazalakova M. H., Wright J., Schneble E. J., McDonald M. P., Heilman C. J., Levey A. I., Blakely R. D. (2007). Deficits in acetylcholine homeostasis, receptors and behaviors in choline transporter heterozygous mice. Genes Brain Behav. 6, 411–424 [DOI] [PubMed] [Google Scholar]
- Beatty W. W., Monson N. (1990). Problem solving in Parkinson’s disease: comparison of performance on the Wisconsin and California Card Sorting Tests. J. Geriatr. Psychiatry Neurol. 3, 163–171 [DOI] [PubMed] [Google Scholar]
- Beatty W. W., Staton R. D., Weir W. S., Monson N., Whitaker H. A. (1989). Cognitive disturbances in Parkinson’s disease. J. Geriatr. Psychiatry Neurol. 2, 22–33 [DOI] [PubMed] [Google Scholar]
- Bertrand N., Beley P., Beley A. (1994). Brain fixation for acetylcholine measurements. J. Neurosci. Methods 53, 81–85 [DOI] [PubMed] [Google Scholar]
- Birrell J. M., Brown V. J. (2000). Medial frontal cortex mediates perceptual attentional set shifting in the rat. J. Neurosci. 20, 4320–4324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bissonette G. B., Martins G. J., Franz T. M., Harper E. S., Schoenbaum G., Powell E. M. (2008). Double dissociation of the effects of medial and orbital prefrontal cortical lesions on attentional and affective shifts in mice. J. Neurosci. 28, 11124–11130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohnen N. I., Kaufer D. I., Ivanco L. S., Lopresti B., Koeppe R. A., Davis J. G., Mathis C. A., Moore R. Y., DeKosky S. T. (2003). Cortical cholinergic function is more severely affected in parkinsonian dementia than in Alzheimer disease: an in vivo positron emission tomographic study. Arch. Neurol. 60, 1745–1748 [DOI] [PubMed] [Google Scholar]
- Bohnen N. I., Kaufer D. I., Hendrickson R., Ivanco L. S., Lopresti B. J., Constantine G. M., Mathis C. A., Davis J. G., Moore R. Y., Dekosky S. T. (2006). Cognitive correlates of cortical cholinergic denervation in Parkinson’s disease and parkinsonian dementia. J. Neurol. 253, 242–247 [DOI] [PubMed] [Google Scholar]
- Braga R., Kouzmine I., Canteras N. S., Da Cunha C. (2005). Lesion of the substantia nigra, pars compacta impairs delayed alternation in a Y-maze in rats. Exp. Neurol. 192, 134–141 [DOI] [PubMed] [Google Scholar]
- Branchi I., D’Andrea I., Armida M., Cassano T., Pèzzola A., Potenza R. L., Morgese M. G., Popoli P., Alleva E. (2008). Nonmotor symptoms in Parkinson’s disease: investigating early-phase onset of behavioral dysfunction in the 6-hydroxydopamine-lesioned rat model. J. Neurosci. Res. 86, 2050–2061 [DOI] [PubMed] [Google Scholar]
- Bychkov E. R., Gurevich V. V., Joyce J. N., Benovic J. L., Gurevich E. V. (2008). Arrestins and two receptor kinases are upregulated in Parkinson’s disease with dementia. Neurobiol. Aging 29, 379–396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell N., Boustani M., Limbil T., Ott C., Fox C., Maidment I., Schubert C. C., Munger S., Fick D., Miller D., et al. (2009). The cognitive impact of anticholinergics: a clinical review. Clin. Interv. Aging 4, 225–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K. C., Baxter M. G., Rodefer J. S. (2004). Central blockade of muscarinic cholinergic receptors disrupts affective and attentional set-shifting. Eur. J. Neurosci. 20, 1081–1088 [DOI] [PubMed] [Google Scholar]
- Choi S. H., Jung T. M., Lee J. E., Lee S.-K., Sohn Y. H., Lee P. H. (2012). Volumetric analysis of the substantia innominata in patients with Parkinson’s disease according to cognitive status. Neurobiol. Aging 33, 1265–1272 [DOI] [PubMed] [Google Scholar]
- Colacicco G., Welzl H., Lipp H.-P., Würbel H. (2002). Attentional set-shifting in mice: modification of a rat paradigm, and evidence for strain-dependent variation. Behav. Brain Res. 132, 95–102 [DOI] [PubMed] [Google Scholar]
- Colloby S. J., Williams E. D., Burn D. J., Lloyd J. J., McKeith I. G., O’Brien J. T. (2005). Progression of dopaminergic degeneration in dementia with Lewy bodies and Parkinson’s disease with and without dementia assessed using 123I-FP-CIT SPECT. Eur. J. Nucl. Med. Mol. Imaging 32, 1176–1185 [DOI] [PubMed] [Google Scholar]
- Cools R., Barker R. A., Sahakian B. J., Robbins T. W. (2001). Mechanisms of cognitive set flexibility in Parkinson’s disease. Brain 124, 2503–2512 [DOI] [PubMed] [Google Scholar]
- Da Cunha C., Silva M. H. C., Wietzikoski S., Wietzikoski E. C., Ferro M. M., Kouzmine I., Canteras N. S. (2006). Place learning strategy of substantia nigra pars compacta-lesioned rats. Behav. Neurosci. 120, 1279–1284 [DOI] [PubMed] [Google Scholar]
- Damsma G., Westerink B. H. C., Horn A. S. (1985). A simple, sensitive, and economic assay for choline and acetylcholine using HPLC, an enzyme reactor, and an electrochemical detector. J. Neurochem. 45, 1649–1652 [DOI] [PubMed] [Google Scholar]
- De Leonibus E., Pascucci T., Lopez S., Oliverio A., Amalric M., Mele A. (2007). Spatial deficits in a mouse model of Parkinson disease. Psychopharmacology (Berl.) 194, 517–525 [DOI] [PubMed] [Google Scholar]
- Decamp E., Tinker J. P., Schneider J. S. (2004). Attentional cueing reverses deficits in spatial working memory task performance in chronic low dose MPTP-treated monkeys. Behav. Brain Res. 152, 259–262 [DOI] [PubMed] [Google Scholar]
- Dias R., Robbins T. W., Roberts A. C. (1996). Dissociation in prefrontal cortex of affective and attentional shifts. Nature 380, 69–72 [DOI] [PubMed] [Google Scholar]
- Dunnett S. B., Lelos M. (2010). Behavioral analysis of motor and non-motor symptoms in rodent models of Parkinson’s disease. Prog. Brain Res. 184, 35–51 [DOI] [PubMed] [Google Scholar]
- Dunois B., Ruberg M., Javoy-Agid F., Ploska A., Agid Y. (1983). A subcortico-cortical cholinergic system is affected in Parkinson’s disease. Brain Res. 288, 213–218 [DOI] [PubMed] [Google Scholar]
- Edison P., Rowe C. C., Rinne J. O., Ng S., Ahmed I., Kemppainen N., Villemagne V. L., O’Keefe G., Någren K., Chaudhury K. R., et al. (2008). Amyloid load in Parkinson’s disease dementia and Lewy body dementia measured with [11C]PIB positron emission tomography. J. Neurol. Neurosurg. Psychiatry 79, 1331–1338 [DOI] [PubMed] [Google Scholar]
- Emre M. (2004). Dementia in Parkinson’s disease: cause and treatment. Curr. Opin. Neurol. 17, 399–404 [DOI] [PubMed] [Google Scholar]
- Ferguson S. M., Savchenko V., Apparsundaram S., Zwick M., Wright J., Heilman C. J., Yi H., Levey A. I., Blakely R. D. (2003). Vesicular localization and activity-dependent trafficking of presynaptic choline transporters. J. Neurosci. 23, 9697–9709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson S. M., Bazalakova M., Savchenko V., Tapia J. C., Wright J., Blakely R. D. (2004). Lethal impairment of cholinergic neurotransmission in hemicholinium-3-sensitive choline transporter knockout mice. Proc. Natl. Acad. Sci. USA 101, 8762–8767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferro M. M., Bellissimo M. I., Anselmo-Franci J. A., Angellucci M. E., Canteras N. S., Da Cunha C. (2005). Comparison of bilaterally 6-OHDA- and MPTP-lesioned rats as models of the early phase of Parkinson’s disease: histological, neurochemical, motor and memory alterations. J. Neurosci. Methods 148, 78–87 [DOI] [PubMed] [Google Scholar]
- Fuchs G. A., Gemende I., Herting B., Lemke M. R., Oehlwein C., Reichmann H., Rieke J., Emmans D., Volkmann J., (2004) Dementia in idiopathic Parkinson’s syndrome. J. Neurol. 251 Suppl. 6, 28–32 [DOI] [PubMed] [Google Scholar]
- Games P. A., Howell J. F. (1976). Pairwase multiple comparison procedures with unequal n’s and/or variances: A Monte Carlo study. J. Educ. Stat. 1, 113–125 [Google Scholar]
- Garner J. P., Thogerson C. M., Würbel H., Murray J. D., Mench J. A. (2006). Animal neuropsychology: validation of the Intra-Dimensional Extra-Dimensional set shifting task for mice. Behav. Brain Res. 173, 53–61 [DOI] [PubMed] [Google Scholar]
- Giladi N., Shabtai H., Gurevich T., Benbunan B., Anca M., Korczyn A. D. (2003). Rivastigmine (Exelon) for dementia in patients with Parkinson’s disease. Acta Neurol. Scand. 108, 368–373 [DOI] [PubMed] [Google Scholar]
- Halliday G., Hely M., Reid W., Morris J. (2008). The progression of pathology in longitudinally followed patients with Parkinson’s disease. Acta Neuropathol. 115, 409–415 [DOI] [PubMed] [Google Scholar]
- Hasselmo M. E., Sarter M. (2011). Modes and models of forebrain cholinergic neuromodulation of cognition. Neuropsychopharmacology 36, 52–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hefco V., Yamada K., Hefco A., Hritcu L., Tiron A., Nabeshima T. (2003). Role of the mesotelencephalic dopamine system in learning and memory processes in the rat. Eur. J. Pharmacol. 475, 55–60 [DOI] [PubMed] [Google Scholar]
- Higginson C. I., Wheelock V. L., Carroll K. E., Sigvardt K. A. (2005). Recognition memory in Parkinson’s disease with and without dementia: evidence inconsistent with the retrieval deficit hypothesis. J. Clin. Exp. Neuropsychol. 27, 516–528 [DOI] [PubMed] [Google Scholar]
- Hilker R., Thomas A. V., Klein J. C., Weisenbach S., Kalbe E., Burghaus L., Jacobs A. H., Herholz K., Heiss W. D. (2005). Dementia in Parkinson disease: functional imaging of cholinergic and dopaminergic pathways. Neurology 65, 1716–1722 [DOI] [PubMed] [Google Scholar]
- Hurtig H. I., Trojanowski J. Q., Galvin J., Ewbank D., Schmidt M. L., Lee V. M., Clark C. M., Glosser G., Stern M. B., Gollomp S. M., et al. (2000). Alpha-synuclein cortical Lewy bodies correlate with dementia in Parkinson’s disease. Neurology 54, 1916–1921 [DOI] [PubMed] [Google Scholar]
- Hutchinson M., Fazzini E. (1996). Cholinesterase inhibition in Parkinson’s disease. J. Neurol. Neurosurg. Psychiatry 61, 324–325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K., Nagano-Saito A., Kato T., Arahata Y., Nakamura A., Kawasumi Y., Hatano K., Abe Y., Yamada T., Kachi T., et al. (2002). Striatal and extrastriatal dysfunction in Parkinson’s disease with dementia: a 6-[18F]fluoro-L-dopa PET study. Brain 125, 1358–1365 [DOI] [PubMed] [Google Scholar]
- Janvin C. C., Aarsland D., Larsen J. P. (2005). Cognitive predictors of dementia in Parkinson’s disease: a community-based, 4-year longitudinal study. J. Geriatr. Psychiatry Neurol. 18, 149–154 [DOI] [PubMed] [Google Scholar]
- Jellinger K. A. (2009). A critical evaluation of current staging of alpha-synuclein pathology in Lewy body disorders. Biochim. Biophys. Acta 1792, 730–740 [DOI] [PubMed] [Google Scholar]
- Jellinger K. A., Seppi K., Wenning G. K., Poewe W. (2002). Impact of coexistent Alzheimer pathology on the natural history of Parkinson’s disease. J. Neural Transm. 109, 329–339 [DOI] [PubMed] [Google Scholar]
- Johnson D. K., Galvin J. E. (2011). Longitudinal changes in cognition in Parkinson’s disease with and without dementia. Dement. Geriatr. Cogn. Disord. 31, 98–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joyce J. N., Ryoo H. L., Beach T. B., Caviness J. N., Stacy M., Gurevich E. V., Reiser M., Adler C. H. (2002). Loss of response to levodopa in Parkinson’s disease and co-occurrence with dementia: role of D3 and not D2 receptors. Brain Res. 955, 138–152 [DOI] [PubMed] [Google Scholar]
- Kehagia A. A., Barker R. A., Robbins T. W. (2010). Neuropsychological and clinical heterogeneity of cognitive impairment and dementia in patients with Parkinson’s disease. Lancet Neurol. 9, 1200–1213 [DOI] [PubMed] [Google Scholar]
- Klein J. C., Eggers C., Kalbe E., Weisenbach S., Hohmann C., Vollmar S., Baudrexel S., Diederich N. J., Heiss W. D., Hilker R. (2010). Neurotransmitter changes in dementia with Lewy bodies and Parkinson disease dementia in vivo. Neurology 74, 885–892 [DOI] [PubMed] [Google Scholar]
- Klinkenberg I., Blokland A. (2010). The validity of scopolamine as a pharmacological model for cognitive impairment: a review of animal behavioral studies. Neurosci. Biobehav. Rev. 34, 1307–1350 [DOI] [PubMed] [Google Scholar]
- Kuhl D. E., Minoshima S., Fessler J. A., Frey K. A., Foster N. L., Ficaro E. P., Wieland D. M., Koeppe R. A. (1996). In vivo mapping of cholinergic terminals in normal aging, Alzheimer’s disease, and Parkinson’s disease. Ann. Neurol. 40, 399–410 [DOI] [PubMed] [Google Scholar]
- Kurtz A., Kaufer D. (2011). Dementia in Parkinson’s disease. Curr. Treat. Options Neurol. 13, 242–254 [DOI] [PubMed] [Google Scholar]
- Kuzis G., Sabe L., Tiberti C., Merello M., Leiguarda R., Starkstein S. E. (1999). Explicit and implicit learning in patients with Alzheimer disease and Parkinson disease with dementia. Neuropsychiatry Neuropsychol. Behav. Neurol. 12, 265–269 [PubMed] [Google Scholar]
- Lange K. W., Wells F. R., Jenner P., Marsden C. D. (1993). Altered muscarinic and nicotinic receptor densities in cortical and subcortical brain regions in Parkinson’s disease. J. Neurochem. 60, 197–203 [DOI] [PubMed] [Google Scholar]
- Leroi I., Brandt J., Reich S. G., Lyketsos C. G., Grill S., Thompson R., Marsh L. (2004). Randomized placebo-controlled trial of donepezil in cognitive impairment in Parkinson’s disease. Int. J. Geriatr. Psychiatry 19, 1–8 [DOI] [PubMed] [Google Scholar]
- Lindner M. D., Cain C. K., Plone M. A., Frydel B. R., Blaney T. J., Emerich D. F., Hoane M. R. (1999). Incomplete nigrostriatal dopaminergic cell loss and partial reductions in striatal dopamine produce akinesia, rigidity, tremor and cognitive deficits in middle-aged rats. Behav. Brain Res. 102, 1–16 [DOI] [PubMed] [Google Scholar]
- Lindsey J. W., Jung A. E., Narayanan T. K., Ritchie G. D. (1998). Acute effects of a bicyclophosphate neuroconvulsant on monoamine neurotransmitter and metabolite levels in the rat brain. J. Toxicol. Environ. Health 54, 421–429 [DOI] [PubMed] [Google Scholar]
- Lipina S. J., Colombo J. A. (2007). Premorbid exercising in specific cognitive tasks prevents impairment of performance in parkinsonian monkeys. Brain Res. 1134, 180–186 [DOI] [PubMed] [Google Scholar]
- Litvan I., Mohr E., Williams J., Gomez C., Chase T. N. (1991). Differential memory and executive functions in demented patients with Parkinson’s and Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatry 54, 25–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maidment I., Fox C., Boustani M. (2006). Cholinesterase inhibitors for Parkinson’s disease dementia. Cochrane Database Syst. Rev. 1, CD004747. [DOI] [PubMed] [Google Scholar]
- Mattila P. M., Rinne J. O., Helenius H., Dickson D. W., Röyttä M. (2000). Alpha-synuclein-immunoreactive cortical Lewy bodies are associated with cognitive impairment in Parkinson’s disease. Acta Neuropathol. 100, 285–290 [DOI] [PubMed] [Google Scholar]
- McAlonan K., Brown V. J. (2003). Orbital prefrontal cortex mediates reversal learning and not attentional set shifting in the rat. Behav. Brain Res. 146, 97–103 [DOI] [PubMed] [Google Scholar]
- McGaughy J., Ross R. S., Eichenbaum H. (2008). Noradrenergic, but not cholinergic, deafferentation of prefrontal cortex impairs attentional set-shifting. Neuroscience 153, 63–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyoshi E., Wietzikoski S., Camplessei M., Silveira R., Takahashi R. N., Da Cunha C. (2002). Impaired learning in a spatial working memory version and in a cued version of the water maze in rats with MPTP-induced mesencephalic dopaminergic lesions. Brain Res. Bull. 58, 41–47 [DOI] [PubMed] [Google Scholar]
- Monchi O., Petrides M., Doyon J., Postuma R. B., Worsley K., Dagher A. (2004). Neural bases of set-shifting deficits in Parkinson’s disease. J. Neurosci. 24, 702–710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriguchi S., Yabuki Y., Fukunaga K. (2012). Reduced calcium/calmodulin-dependent protein kinase II activity in the hippocampus is associated with impaired cognitive function in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-treated mice. J. Neurochem. 120, 541–551 [DOI] [PubMed] [Google Scholar]
- Mosimann U. P., Mather G., Wesnes K. A., O’Brien J. T., Burn D. J., McKeith I. G. (2004). Visual perception in Parkinson disease dementia and dementia with Lewy bodies. Neurology 63, 2091–2096 [DOI] [PubMed] [Google Scholar]
- Muslimović D., Schmand B., Speelman J. D., de Haan R. J. (2007). Course of cognitive decline in Parkinson’s disease: a meta-analysis. J. Int. Neuropsychol. Soc. 13, 920–932 [DOI] [PubMed] [Google Scholar]
- Nakano I., Hirano A. (1984). Parkinson’s disease: neuron loss in the nucleus basalis without concomitant Alzheimer’s disease. Ann. Neurol. 15, 415–418 [DOI] [PubMed] [Google Scholar]
- Noe E., Marder K., Bell K. L., Jacobs D. M., Manly J. J., Stern Y. (2004). Comparison of dementia with Lewy bodies to Alzheimer’s disease and Parkinson’s disease with dementia. Mov. Disord. 19, 60–67 [DOI] [PubMed] [Google Scholar]
- Owen A. M., Roberts A. C., Hodges J. R., Summers B. A., Polkey C. E., Robbins T. W. (1993a). Contrasting mechanisms of impaired attentional set-shifting in patients with frontal lobe damage or Parkinson’s disease. Brain 116, 1159–1175 [DOI] [PubMed] [Google Scholar]
- Owen A. M., Beksinska M., James M., Leigh P. N., Summers B. A., Marsden C. D., Quinn N. P., Sahakian B. J., Robbins T. W. (1993b). Visuospatial memory deficits at different stages of Parkinson’s disease. Neuropsychologia 31, 627–644 [DOI] [PubMed] [Google Scholar]
- Perretta J. G., Pari G., Beninger R. J. (2005). Effects of Parkinson disease on two putative nondeclarative learning tasks: probabilistic classification and gambling. Cogn. Behav. Neurol. 18, 185–192 [DOI] [PubMed] [Google Scholar]
- Perry E. K., Curtis M., Dick D. J., Candy J. M., Atack J. R., Bloxham C. A., Blessed G., Fairbairn A., Tomlinson B. E., Perry R. H. (1985). Cholinergic correlates of cognitive impairment in Parkinson’s disease: comparisons with Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatry 48, 413–421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry R. H., Perry E. K., Smith C. J., Xuereb J. H., Irving D., Whitford C. A., Candy J. M., Cross A. J. (1987). Cortical neuropathological and neurochemical substrates of Alzheimer’s and Parkinson’s diseases. J. Neural Transm. Suppl. 24, 131–136 [PubMed] [Google Scholar]
- Prado V. F., Martins-Silva C., de Castro B. M., Lima R. F., Barros D. M., Amaral E., Ramsey A. J., Sotnikova T. D., Ramirez M. R., Kim H. G., et al. (2006). Mice deficient for the vesicular acetylcholine transporter are myasthenic and have deficits in object and social recognition. Neuron 51, 601–612 [DOI] [PubMed] [Google Scholar]
- Robottom B. J., Weiner W. J. (2009). Dementia in Parkinson’s disease. Int. Rev. Neurobiol. 84, 229–244 [DOI] [PubMed] [Google Scholar]
- Ruberg M., Ploska A., Javoy-Agid F., Agid Y. (1982). Muscarinic binding and choline acetyltransferase activity in Parkinsonian subjects with reference to dementia. Brain Res. 232, 129–139 [DOI] [PubMed] [Google Scholar]
- Sawamoto N., Piccini P., Hotton G., Pavese N., Thielemans K., Brooks D. J. (2008). Cognitive deficits and striato-frontal dopamine release in Parkinson’s disease. Brain 131, 1294–1302 [DOI] [PubMed] [Google Scholar]
- Tadaiesky M. T., Dombrowski P. A., Figueiredo C. P., Cargnin-Ferreira E., Da Cunha C., Takahashi R. N. (2008). Emotional, cognitive and neurochemical alterations in a premotor stage model of Parkinson’s disease. Neuroscience 156, 830–840 [DOI] [PubMed] [Google Scholar]
- Tait D. S., Brown V. J. (2008). Lesions of the basal forebrain impair reversal learning but not shifting of attentional set in rats. Behav. Brain Res. 187, 100–108 [DOI] [PubMed] [Google Scholar]
- Tait D. S., Brown V. J., Farovik A., Theobald D. E., Dalley J. W., Robbins T. W. (2007). Lesions of the dorsal noradrenergic bundle impair attentional set-shifting in the rat. Eur. J. Neurosci. 25, 3719–3724 [DOI] [PubMed] [Google Scholar]
- Tröster A. I., Fields J. A., Testa J. A., Paul R. H., Blanco C. R., Hames K. A., Salmon D. P., Beatty W. W. (1998). Cortical and subcortical influences on clustering and switching in the performance of verbal fluency tasks. Neuropsychologia 36, 295–304 [DOI] [PubMed] [Google Scholar]
- Tzavos A., Jih J., Ragozzino M. E. (2004). Differential effects of M1 muscarinic receptor blockade and nicotinic receptor blockade in the dorsomedial striatum on response reversal learning. Behav. Brain Res. 154, 245–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varanese S., Perfetti B., Monaco D., Thomas A., Bonanni L., Tiraboschi P., Onofrj M. (2010). Fluctuating cognition and different cognitive and behavioural profiles in Parkinson’s disease with dementia: comparison of dementia with Lewy bodies and Alzheimer’s disease. J. Neurol. 257, 1004–1011 [DOI] [PubMed] [Google Scholar]
- Whitehouse P. J., Hedreen J. C., White C. L., 3rd, Price D. L. (1983). Basal forebrain neurons in the dementia of Parkinson disease. Ann. Neurol. 13, 243–248 [DOI] [PubMed] [Google Scholar]
- Williams-Gray C. H., Foltynie T., Lewis S. J., Barker R. A. (2006). Cognitive deficits and psychosis in Parkinson’s disease: a review of pathophysiology and therapeutic options. CNS Drugs 20, 477–505 [DOI] [PubMed] [Google Scholar]
- Wisman L. A. B., Sahin G., Maingay M., Leanza G., Kirik D. (2008). Functional convergence of dopaminergic and cholinergic input is critical for hippocampus-dependent working memory. J. Neurosci. 28, 7797–7807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood E. R., Dudchenko P. A., Eichenbaum H. (1999). The global record of memory in hippocampal neuronal activity. Nature 397, 613–616 [DOI] [PubMed] [Google Scholar]