

SUMMARY
Myotonic dystrophy type 1 (DM1) is a genetic disease caused by the pathological expansion of a CTG trinucleotide repeat in the 3′ UTR of the DMPK gene. In the DMPK transcripts, the CUG expansions sequester RNA-binding proteins into nuclear foci, including transcription factors and alternative splicing regulators such as MBNL1. MBNL1 sequestration has been associated with key features of DM1. However, the basis behind a number of molecular and histological alterations in DM1 remain unclear. To help identify new pathogenic components of the disease, we carried out a genetic screen using a Drosophila model of DM1 that expresses 480 interrupted CTG repeats, i(CTG)480, and a collection of 1215 transgenic RNA interference (RNAi) fly lines. Of the 34 modifiers identified, two RNA-binding proteins, TBPH (homolog of human TAR DNA-binding protein 43 or TDP-43) and BSF (Bicoid stability factor; homolog of human LRPPRC), were of particular interest. These factors modified i(CTG)480 phenotypes in the fly eye and wing, and TBPH silencing also suppressed CTG-induced defects in the flight muscles. In Drosophila flight muscle, TBPH, BSF and the fly ortholog of MBNL1, Muscleblind (Mbl), were detected in sarcomeric bands. Expression of i(CTG)480 resulted in changes in the sarcomeric patterns of these proteins, which could be restored by coexpression with human MBNL1. Epistasis studies showed that Mbl silencing was sufficient to induce a subcellular redistribution of TBPH and BSF proteins in the muscle, which mimicked the effect of i(CTG)480 expression. These results provide the first description of TBPH and BSF as targets of Mbl-mediated CTG toxicity, and they suggest an important role of these proteins in DM1 muscle pathology.
INTRODUCTION
Myotonic dystrophy type 1 (DM1) is the most common type of muscular dystrophy in adults, with a prevalence of 1 in 8000 (OMIM #160900). DM1 is caused by a dynamic expansion of non-coding CTG repeats in the 3′ untranslated region (UTR) of the Dystrophia Myotonica Protein Kinase gene (DMPK) (Sicot et al., 2011). In the mutant transcripts, a toxic RNA gain of function of the CUG expansions has been demonstrated (Mankodi et al., 2000). CUG repeats have the ability to form RNA hairpins that are accumulated into ribonuclear foci (Taneja et al., 1995) and sequester a number of RNA-binding factors, including the alternative splicing regulators of the Muscleblind family MBNL1-MBNL3 (Miller et al., 2000; Fardaei et al., 2002). MBNL1 sequestration disrupts the normal activity of the protein, and results in mis-splicing of a growing number of target transcripts (Osborne and Thornton, 2006; Du et al., 2010). A key role of MBNL1 loss of function in DM1 was originally demonstrated by the generation of Mbnl1−/− knockout mice, which reproduced the most relevant features of the disease (Kanadia et al., 2003). Additional support came later from studies in which overexpression of Mbnl1 in mice carrying expanded CTG repeats reversed DM1-like phenotypes (Kanadia et al., 2006).
The study of Muscleblind in different organisms has shown that these proteins are predominantly expressed in skeletal muscle and nervous system, where they carry out specific functions during terminal tissue differentiation (Begemann et al., 1997; Artero et al., 1998; Lin et al., 2006; Wang, L. C. et al., 2008; Fernandez-Costa et al., 2011). In the Drosophila muscle, ultrastructural studies on muscleblind (mbl) mutants revealed a compromised organization of the muscle structural unit, the sarcomere, where thick and thin filaments were less ordered and densely packed than in wild-type individuals, and the I-bands and mesh-like matrix of Z-bands were absent, indicative of a hypercontracted state (Artero et al., 1998). In Caenorhabditis elegans, RNAi silencing of muscleblind also caused a severe disruption of the alignment pattern of the dense bodies, a nematode structure similar to the Z-bands of higher eukaryotic muscles (Wang, L. C. et al., 2008). Disorganization of the sarcomere and somatic Z-band disruption have also been reported in DM1 patients, with defects that include duplication or splitting of the Z-bands, degeneration of the thin filaments of the I-bands and sarcoplasmic swelling (Aleu and Afifi, 1964; Ludatscher et al., 1978). Muscleblind regulates the alternative splicing of transcripts that encode proteins associated with the Z-bands, including ZASP or Tnnt2. Therefore, its sequestration by the CUG hairpins could trigger the structural abnormalities observed in DM1 muscles (Lin et al., 2006; Machuca-Tzili et al., 2006; Osborne and Thornton, 2006; Garcia-Lopez et al., 2008; Wang, L. C. et al., 2008).
Characteristic mis-splicing events in DM1 are mediated mainly, but not entirely, by MBNL1. CTG expansions also trigger hyperphosphorylation, subcellular mislocalization and stabilization of a second key alternative splicing regulator, CELF1. CELF1 is not sequestered by CUG hairpins. Instead, CELF1-mediated alterations in DM1 models require the presence of DMPK (Timchenko et al., 1996; Mahadevan et al., 2006). The alternative splicing activity of CELF1 has been shown to be antagonistic to the effect of MBNL1, at least on some transcripts (Kuyumcu-Martinez et al., 2007; Kalsotra et al., 2008). Consistently, genetic studies support the idea that specific muscle, eye and cardiac defects characteristic of DM1 can be regarded as MBNL1 loss-of-function or CELF1 overexpression phenotypes (Kanadia et al., 2003; Ho et al., 2005; de Haro et al., 2006; Kalsotra et al., 2008).
TRANSLATIONAL IMPACT.
Clinical issue
Myotonic dystrophy type 1 (DM1) is a multisystemic disease that affects mainly the muscle and the central nervous system. DM1 is caused by the expansion of an unstable CTG-repeat tract in the 3′ untranslated region (UTR) of the DMPK gene. At the RNA level, expanded CUG repeats form a hairpin that sequesters muscleblind-like protein 1 (MBNL1) and other nuclear factors into ribonuclear foci in a manner proportional to the CUG expansion size. Sequestration has been proposed to cause a loss of function of these proteins that in some cases has been linked to defined symptoms. Despite advancing molecular studies on DM1, newly discovered factors continue to add complexity, and several aspects of the pathogenesis are still unclear. A better understanding of the molecular mechanisms altered by CTG repeats, and of their interactions in a living context, is crucial for deciphering the origin of some symptoms of DM1 and to generate appropriate treatments.
Results
Here, the authors use a Drosophila model of DM1 to identify new genetic components of the CTG toxicity pathway and identify RNA-binding proteins BSF and TBPH as modifiers of the DM1 phenotype. CTG-repeat expression altered the subcellular distribution of both of these proteins in the adult sarcomere. Notably, the Drosophila ortholog of MBNL1, Mbl, regulated the subcellular distribution of BSF and TBPH, but not vice versa, indicating that both proteins are downstream of Mbl. BSF and TBPH also modified CTG toxicity in other fly tissues, including the eye and wing, indicating that the effects of these proteins are not exclusive to the muscle sarcomere. Importantly, sarcomeric distribution of Drosophila Mbl is reversibly altered by expression of CTG repeats and TBPH silencing rescued the muscle phenotype characteristic of expanded CTG-repeat expression.
Implications and future directions
These results define a role for RNA-binding proteins BSF and TBPH in modifying CTG-dependent muscle defects and point out the potential to target them for therapy. Notably, this is the first report to associate BSF and TBPH with DM1 and to identify them as structural components of muscle. These results also emphasize the pathogenic role of sarcomere disorganization in DM1. The description of Mbl localization to the sarcomere of adult Drosophila muscles reveals a new function for Mbl, adding to its well-characterized activity as an alternative splicing regulator in the nucleus. Further studies will address whether these proteins are similarly involved in DM1 patients and help to better understand their involvement in the pathogenesis of DM1.
In the past few years, there has been a growing understanding of the complexity of DM1. In addition to splicing dysregulation, changes in gene expression, protein translation, and microRNA metabolism have also been suggested to contribute to DM1 (Du et al., 2010; Gambardella et al., 2010; Perbellini et al., 2011; Rau et al., 2011), indicating that multiple pathways are involved in the disease pathology (Sicot et al., 2011). To help identify new components of CTG toxicity, we previously used a fly model that expresses 480 interrupted CTG repeats in an enhancer-suppressor genetic screen of 695 lethal P-element insertion mutant lines, covering ∼5% of the Drosophila genome (Garcia-Lopez et al., 2008). This approach identified putative cellular components altered by CTG expansions, such as extracellular matrix proteins, mRNA export factors, apoptosis regulators and chromatin remodeling proteins. Some of these mechanisms were later confirmed by independent groups to be altered in DM1 myotubes and mouse models (Du et al., 2010; Loro et al., 2010). Here, we have extended this study by using a collection of RNA interference (RNAi) fly lines covering a total of 1215 genes (∼8% of the Drosophila genome). This screen allowed the identification of 34 new modifiers of CTG-mediated toxicity. Of these, RNA-binding factors BSF (Bicoid stability factor) and TBPH (TAR-binding protein homolog), which modify phenotypes in eye and wing, were also present in the muscle sarcomere, where their subcellular localization was altered by the expression of CTG expansions or Muscleblind (Mbl) depletion. Therefore, these results contribute to improve our understanding of the factors causing muscle defects in DM1.
RESULTS
A genetic screen identified bsf and TBPH as dominant modifiers of expanded-CTG toxicity
Expression of expanded CTG repeats during the development of the Drosophila eye brings about a rough eye phenotype that is sensitive to the genetic dose of a number of factors associated with DM1 (de Haro et al., 2006; Garcia-Lopez et al., 2008). To exploit this phenotype in a genetic screen of new dominant modifiers of CTG toxicity, we first generated a recombinant fly line expressing 480 interrupted CTG repeats [referred to here as i(CTG)480] in the eye, under the control of the Glass Multiple Reporter (GMR) promoter GMR-Gal4 UAS-i(CTG)480. The rough eye phenotype of these flies was enhanced by coexpression of an RNAi (IR) construct against mbl (UAS-IR-mbl) generated in our laboratory. This construct was targeted against all mbl isoforms and silenced mbl gene expression by approximately 50%, equivalent to having one copy of the loss-of-function alleles mblE27 or Df(2R)BSC154 (supplementary material Fig. S1). GMR-Gal4 UAS-i(CTG)480>UAS-IR-mbl flies showed ommatidial fusion and smaller and rougher eyes compared with control flies coexpressing i(CTG)480 and the gratuitous GFP reporter [GMR-Gal4 UAS-i(CTG)480>UAS-GFP], with occasional appearance of necrotic dots (Fig. 1A,B).
Fig. 1.

bsf and TBPH modify CTG-induced phenotypes in the Drosophila eye and wing. (A–H) SEM images of Drosophila eyes of the indicated genotypes. (A) GMR-Gal4 driven expression of i(CTG)480 [GMR-Gal4 UAS-i(CTG)480>UAS-GFP] in the eye precursors originates small and rough eyes. (B) Silencing of mbl in these flies [GMR-Gal4 UAS-i(CTG)480>UAS-IR-mbl] enhanced the phenotype (note a necrotic patch; arrowhead). (C) Silencing of bsf [GMR-Gal4 UAS-i(CTG)480>UAS-IR-bsf] and (D) TBPH [GMR-Gal4 UAS-i(CTG)480>UAS-IR-TBPH] enhanced and suppressed the phenotype, respectively. (E) External morphology of a normal eye (yw). (F) sev-Gal4 driven expression of mblC in the eye precursors (sev-Gal4 UAS-mblC>UAS-GFP) originates mild rough eyes. (G) This phenotype was not modified by silencing of bsf (sev-Gal4 UAS-mblC>UAS-IR-bsf), whereas reduction of TBPH (sev-Gal4 UAS-mblC>UAS-IR-TBPH) enhanced roughness and originated smaller eyes (H). (I,J) Representative bright field microscopy images of wing morphologies of flies expressing i(CTG)480 [vg-Gal4 UAS-i(CTG)480>UAS-GFP], showing the phenotypic variability. (K) mbl silencing increased the percentage of flies with altered wing morphology compared with vg-Gal4 UAS-i(CTG)480 flies, whereas silencing of bsf or TBPH significantly reduced the number of flies with wing defects. ***P<0.001; *P<0.05.
GMR-Gal4 UAS-i(CTG)480 recombinant flies were then crossed with 1215 RNAi-knockdown fly lines randomly chosen from the NIG-Fly collection. Using the phenotype of GMR-Gal4 UAS-i(CTG)480>UAS-GFP flies as a reference, we identified 202 RNAi lines (16.6% of total tested) that significantly modified the eye phenotype in at least two independent crosses. Of these, 62 suppressed the rough eye phenotype, 127 enhanced it and 13 caused lethality (Table 1; supplementary material Table S1).
Table 1.
Genetic modifiers of expanded CTG repeat toxicity

To rule out suppressors of CTG toxicity that were eye-specific, the 202 modifiers identified were re-tested using a second phenotype caused by expression of i(CTG)480 in the wing, under the control of the vestigial (vg) promoter [vg-Gal4 UAS-i(CTG)480>UAS-GFP]. The expression of expanded CTG repeats in the wing originated a variable phenotype, which was sensitive to temperature changes. At 25°C, ∼100% of the flies had altered wing morphology, ranging from small size and notches to puckered wings (Fig. 1I,J). At 22°C only 75% of the flies showed wing morphology alterations, and the remaining 25% were completely normal. We crossed our 202 candidate RNAi lines with vg-Gal4 UAS-i(CTG)480 at 22°C, taking the above percentages for this temperature as our reference phenotype. Of the 202 lines tested, ten significantly increased the percentage of flies with altered wing morphology, whereas 24 reduced it in at least two independent crosses (Table 1; supplementary material Table S1). Among these 34 modifiers, we found transcriptional regulators, including purine-rich binding protein α (Pur-α), Lilliputian (lilli), hyrax (hyx) and enhancer of yellow 3 [e(y)3]; structural and cytoskeletal components, such as Sarcoglycan δ (Scgδ), capulet (capt), capping protein α (cpa), zormin and bent (bt); and genes coding for RNA-binding factors bsf (Fig. 1C,K), TBPH (Fig. 1D,K) and CG10341. In some cases, the directions of these genetic interactions differed between wing and eye, suggesting the participation of tissue-specific pathways mediating the link of these genes with CTG-dependent toxicity.
mbl overexpression phenotype is modified by silencing of RNA-binding proteins TBPH and BSF
Because of the demonstrated relevance of RNA-binding proteins in DM1, we decided to focus on this group of modifiers, which was formed by bsf, TBPH and CG10341. CG10341 is the Drosophila ortholog of human NAF1, which is involved in ribosome biogenesis. Such genes tend to be recovered as nonspecific modifiers in genetic screens, and therefore CG10341 was not prioritized for further analysis. For bsf and TBPH, two independently generated RNAi lines were used to confirm their genetic interaction with i(CTG)480. In addition, silencing of both genes in the absence of i(CTG)480 did not cause any apparent phenotype, further demonstrating the specificity of their interaction with i(CTG)480 (supplementary material Fig. S2).
We next studied whether a connection between bsf or TBPH and mbl function in the eye existed. mbl silencing alleles could not be used, given the lack of an externally visible eye phenotype produced by mbl loss-of-function alleles (not shown). However, we previously showed that overexpression of the isoform C of mbl (mblC) driven by the sevenless (sev) promoter (sev-Gal4 UAS-mblC) resulted in a rough eye phenotype (Vicente-Crespo et al., 2008). Therefore, we tested the ability of bsf or TBPH to modify the eye defects of the sev-Gal4 UAS-mblC flies. Although bsf silencing did not modify the mblC-induced eye phenotype (Fig. 1F,G), a reduction of TBPH enhanced it, giving a rougher and narrower eye (Fig. 1F,H). Thus, these results pointed out a link between bsf or TBPH and mbl, which could in turn explain their interaction with i(CTG)480.
TBPH silencing suppressed CTG-induced muscle defects
DM1 is primarily a neuromuscular disease. We therefore studied whether candidate modifiers of i(CTG)480 eye and wing phenotypes could also modify CTG toxicity in the muscle. Previously, our laboratory and others have demonstrated that expression of i(CTG)480 in the Drosophila indirect flight muscles (IFMs) under the control of the Myosin heavy chain (Mhc) promoter, Mhc-Gal4 UAS-i(CTG)480, causes muscle defects, including fiber loss and disorganization, that worsened over the course of time and led to flightless flies (de Haro et al., 2006; Garcia-Lopez et al., 2008). To study the role of TBPH and bsf in CTG-induced muscle pathology, we first analyzed changes in the IFM morphology of flies with reduced levels of BSF or TBPH proteins. Quantification of the total muscle area of heterozygous TBPHΔ23 flies (TBPHΔ23/+), as well as bsf- and TBPH-silenced flies (Mhc-Gal4>UAS-IR-bsf and Mhc-Gal4>UAS-IR-TBPH, respectively) did not show significant changes in the IFM total area compared with control flies (yw or Mhc-Gal4>UAS-GFP; Fig. 2A–E). However, silencing of TBPH in CTG-expressing flies [Mhc-Gal4 UAS-i(CTG)480>UAS-IR-TBPH] partially rescued CTG-induced muscle fiber loss, with an increase in total muscle area of ∼15% compared with control flies coexpressing i(CTG)480 and GFP [Mhc-Gal4 UAS-i(CTG)480>UAS-GFP; Fig. 2F–J]. bsf silencing caused a variable phenotype with a tendency to enhance the CTG-induced IFM size reduction, although this difference was not significant. These results suggest a role for TBPH in the muscle pathology induced by CTG repeat expression; and indicate that, although bsf interacts genetically with i(CTG)480 in other tissues, its silencing is not enough to prevent muscle fiber loss.
Fig. 2.

Silencing of TBPH suppressed CTG-induced muscle degeneration. Dorsoventral sections of resin-embedded adult thoraces showing their IFMs. (AD) The IFM area in control fly thoraces (Mhc-Gal4>UAS-GFP; A) was not significantly altered by silencing of bsf (Mhc-Gal4>UAS-IR-bsf; B,E), TBPH (Mhc-Gal4>UAS-IR-TBPH; C,E) or by introducing a copy of the mutant TBPH allele TBPHΔ23 (TBPHΔ23/+; D,E). (F–I) Expression of i(CTG)480 under the control of the Mhc-Gal4 driver [Mhc-Gal4 UAS-i(CTG)480>UAS-GFP] caused a phenotype characterized by lower density of myofibrils that resulted in a reduced muscle area (F–J). Silencing of bsf [Mhc-Gal4 UAS-i(CTG)480>UAS-IR-bsf] weakly enhanced the CTG-induced reduction in total muscle area, although this change was not statistically significant (H,J). TBPH silencing, however, rescued the CTG-dependent muscle defects, inducing a significant increase in total muscle area (I,). All graphs show means ± s.e.m. Quantifications shown in E are relative to the control genotype shown in A. Quantifications in J are relative to the wild-type control represented in F. **P<0.01.
Expanded CTG repeats disrupted TBPH subcellular distribution in the adult muscle
TBPH, and its human ortholog TDP-43, are mainly expressed in the central nervous system, where they have been widely studied due to the implication of this protein in neurodegenerative disorders (Feiguin et al., 2009; Armstrong and Cairns, 2011a). However, no detailed information about expression of this protein in muscle has been reported to date. To help understand the role of TBPH in CTG-induced muscle pathology, we first studied its expression pattern in the adult IFMs of control flies (Mhc>yw). Immunostaining of rostrocaudal cryosections of fly thoraces using an anti-TBPH antibody showed a disperse and weak signal for TBPH in the nucleus, and a stronger presence in cytoplasmic bands transversal to the muscle fibers (Fig. 3A,E–H). Double staining using phalloidin, which detects actin filaments, revealed that TBPH was localized in the sarcomeric H-bands (Fig. 3A,D). Abolishment of TBPH detection upon TBPH silencing (Mhc-Gal4>UAS-IR-TBPH) confirmed the specificity of the antibody signal (see later). Immunodetection of TBPH in recombinant Mhc-Gal4 UAS-i(CTG)480 flies showed a stronger distribution of TBPH in the nucleus upon CTG expression (Fig. 3I–L) and a change in the sarcomeric distribution of the protein, which now included the Z-bands (Fig. 3B,D). This is consistent with current data on the implication of TBPH in other degenerative conditions, in which the distribution of this protein is altered. Importantly, coexpression of i(CTG)480 with human MBNL1 [Mhc-Gal4 UAS-i(CTG)480>UAS-MBNL1] rescued the original pattern of the protein, although TBPH nuclear signal was still higher than in the control flies (Fig. 3C). Moreover, fluorescent in situ hybridization to detect CUG-RNA foci ruled out the possibility that nuclear TBPH colocalizes with RNA aggregates (Fig. 3I–L).
Fig. 3.

Expression of expanded CTG repeats modifies TBPH localization in the Drosophila muscle. (A–C) Fluorescent confocal images of rostrocaudal cryosections from adult Drosophila thoraces stained with an anti-TBPH antibody (green), and counterstained with phalloidin (red). In control flies (Mhc>yw; A) TBPH was detected preferentially in the cytoplasm, as a part of the sarcomeric H-bands. (B) Expression of i(CTG)480 in the muscle [Mhc-Gal4 UAS-i(CTG)480] enhanced the TBPH signal in the sarcomeric Z-bands. (C) Coexpression of i(CTG)480 with human MBNL1 [Mhc-Gal4 UAS-i(CTG)480>UAS-MBNL1] partially rescued the sarcomeric localization of TBPH. (D) Representation of the basic organization of a sarcomere. (E–L) Fluorescent confocal images comparing the subcellular distribution of i(CUG)480 RNA (fluorescent in situ hybridization using a CAG red-labeled probe; E,I) with TBPH protein (green; G,K; see merge in H,L), in control (E–H) and CTG-expressing flies (I–L). Expression of CTG repeats in the muscle caused a marked increase in nuclear TBPH, which did not seem to colocalize with nuclear CUG-RNA foci. Nuclei were counterstained with DAPI (blue; F,J).
To investigate whether TBPH transcript levels were increased by expression of i(CTG)480, we performed a quantitative RT-PCR (qRT-PCR) analysis from Mhc-Gal4 i(CTG)480 and Mhc-Gal4>UAS-GFP (control) adult flies. No significant changes were detected compared with controls. Moreover, TBPH levels were not modified when i(CTG)480 was coexpressed with human MBNL1 (Fig. 4A). To assess whether TBPH alternative splicing was altered, we measured the levels of TBPH isoforms by qRT-PCR. The TBPH gene encodes six transcripts that produce two different TBPH proteins of different sizes (isoform A and isoforms B–F; Fig. 4B,C). When we measured the levels of different combinations of TBPH isoforms, no significant differences were detected when i(CTG)480 was expressed alone or coexpressed with MBNL1 (Fig. 4B). Thus, the levels of TBPH transcripts remain unchanged in the presence of CTG repeats, and the increased nuclear TBPH pattern suggested by our immunodetection experiments might originate from increased protein synthesis or stability, or from a change in the subcellular localization of the protein. To test this, we measured the total levels of TBPH protein by western blot, using two different i(CTG)480 lines. No differences were observed in the amount of TBPH protein compared with controls (Fig. 4D), ruling out an effect on protein translation or stability. Therefore, our results indicate that TBPH subcellular distribution is altered in the muscle of CTG-expressing flies, and that this mislocalization can be partially rescued by MBNL1.
Fig. 4.

CTG repeats do not affect the levels of TBPH transcripts nor TBPH alternative splicing. (A) qRT-PCR analysis of the levels of TBPH transcripts showed no differences between control (Mhc-Gal4 UAS-GFP>UAS-GFP) and CTG-expressing flies or flies coexpressing i(CTG)480 and human MBNL1. Expression of a TBPH RNAi line (Mhc-Gal4>UAS-IR-TBPH; IR-TBPH column) and heterozygous TBPHΔ23 flies was reduced. Measurements were normalized using the housekeeping gene Rp49 and are shown relative to the control genotype. (B) qRT-PCR analysis of the alternative splicing of TBPH transcripts showed no significant differences between control (Mhc-Gal4 UAS-GFP>UAS-GFP), CTG-expressing [Mhc-Gal4 UAS-i(CTG)480>UAS-GFP] and rescued [Mhc-Gal4 UAS-i(CTG)480>UAS-MBNL1 flies. Measurements are shown relative to the levels of TBPH constitutive exon 5 and to the housekeeping gene Rp49. All graphs show the means + s.e.m. of three biological replicates (n=50 flies per replicate) and three technical replicates per biological sample. (C) Representation of the six transcript variants of TBPH (TBPH-A to TBPH-F) taken from Flybase database, indicating the regions that were amplified by the primer pairs used in A and B, in order to measure TBPH transcript levels (which comprises a region overlapping constitutive exons 2 and 3) or different TBPH isoform combinations. The later included a tract from the 5′ UTR region of TBPH transcripts that detected isoforms A and C–E (labeled 5′ UTR 1); a tract from the 5′ UTR region of TBPH transcripts that detected isoform C (labeled 5′ UTR 2); and a fragment of the alternative exon 4, which is excluded in the TBPH isoform A (labeled alternative exon). (D) Western blot analysis of TBPH protein levels in control flies (Mhc-Gal4/+ and Mhc-Gal4>UAS-LacZ) and CTG-expressing flies [Mhc-Gal4 UAS-i(CTG)480 line A and Mhc-Gal4 UAS-i(CTG)480 line B] revealed no significant differences between genotypes. Tubulin was used as the loading control. Graphs show means + s.e.m. from three experimental replicates.
Expanded CTG repeats disrupted BSF localization in the adult sarcomere
Previous works have characterized BSF as a cytoplasmic protein during early embryonic development, whereas a ubiquitous mitochondrial pattern has been described in adult flies (Chintapalli et al., 2007; Bratic et al., 2011). Here, an immunohistochemical analysis was also performed for BSF that showed a strong signal in the Z- and H-bands of the IFM sarcomeres, but did not reveal a mitochondrial pattern (Fig. 5A,D–G). Abolishment of BSF detection upon bsf silencing (Mhc-Gal4>UAS-IR-bsf) confirmed the specificity of the signal (see later). These results contrast with the notion that BSF is present exclusively in the mitochondria, as previously described by authors using cell fractioning from whole flies and the same antibody as that used here (Mancebo et al., 2001; Bratic et al., 2011) (supplementary material Fig. S3). As similarly described for TBPH, the BSF distribution in sarcomeric bands was abolished upon CTG expression, whereas the BSF signal in the nucleus was strongly enhanced (Fig. 5B,D–K), again suggesting a CTG-induced relocalization of BSF from cytoplasm to nucleus. In situ hybridization also confirmed that nuclear BSF did not accumulate into CUG-RNA foci. Finally, coexpression of i(CTG)480 with human MBNL1 [Mhc-Gal4 UAS-i(CTG)480>UAS-MBNL1] also rescued the normal distribution of BSF in sarcomeric bands, although the nuclear signal was again more intense than in the controls (Fig. 5C).
Fig. 5.
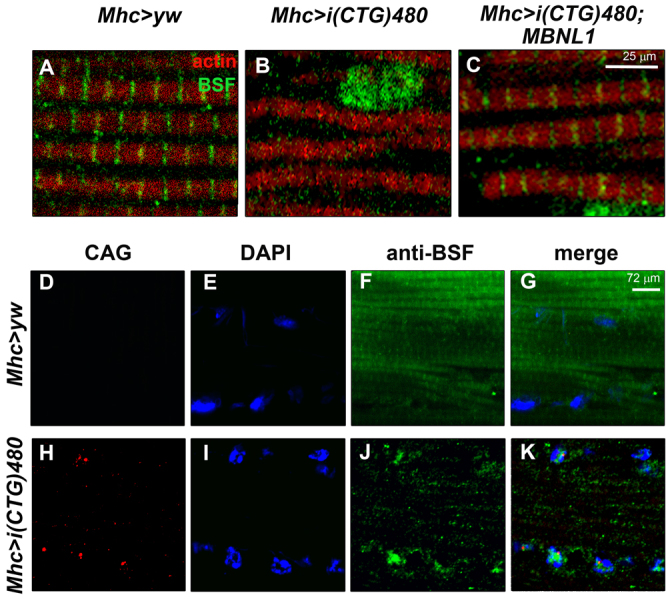
Expression of expanded CTG repeats impaired BSF distribution in the Drosophila muscle sarcomere. (A–C) Fluorescent confocal images of rostrocaudal cryosections from adult Drosophila thoraces stained with an anti-BSF antibody (green), and counterstained with phalloidin (red). In control flies (Mhc>yw; A) BSF was detected preferentially in the cytoplasm as a constituent part of the sarcomeric bands (see also Fig. 3C). (B) Expression of i(CTG)480 in the muscle [Mhc-Gal4 UAS-i(CTG)480] disrupted cytoplasmic BSF signal. (C) Coexpression of i(CTG)480 with human MBNL1 [Mhc-Gal4 UAS-i(CTG)480>UAS-MBNL1] partially rescued the sarcomeric localization of BSF. (D–K) Fluorescent confocal images comparing the subcellular distribution of i(CUG)480 RNA (fluorescent in situ hybridization using a CAG red-labeled probe; D,H) with BSF protein (green; F,J; see merge in G,K) in control (D–G) and CTG-expressing flies (H–K). Expression of CTG repeats in the muscle not only abolished cytoplasmic BSF signal, but also enhanced its detection in the nuclei. Nuclear BSF signal did not seem to colocalize with CUG-RNA foci. Nuclei were counterstained with DAPI (blue; E,I).
Drosophila Mbl is present in the sarcomeric bands of adult fly muscles
Given the effect of human MBNL1 on BSF and TBPH localization in adult Drosophila muscle, we decided to study the subcellular distribution of the endogenous Drosophila Mbl protein throughout muscle development. At the time of this study, the Mbl expression pattern had only been described for embryonic stages (Artero et al., 1998). We confirmed the nuclear pattern of Mbl in the embryonic central nervous system, as well as the somatic and visceral musculatures (Fig. 6A and not shown). In the somatic muscle of third instar larvae, we observed that Mbl was no longer only present in the nucleus, but was also present in the cytoplasm, where it showed a transversal banding distribution (Fig. 6B). In rostrocaudal sections of adult thoraces, a neat signal with a regular transversal banding pattern spanning throughout the IFM fiber width was observed (Fig. 6C). Counterstaining of these sections with phalloidin showed that Mbl colocalized with sarcomeric Z-and H-bands (Fig. 6D). Staining with the pre-immune serum did not reveal any of these patterns (not shown), confirming specificity of the antibody signal. Moreover, we also studied the sarcomeric bands of the IFMs in adult thorax sections from flies overexpressing MblC (Mhc-Gal4>UAS-mblC) or silencing mbl (Mhc-Gal4>UAS-IR-mbl) (Fig. 6D). When we overexpressed MblC, we detected an increase in the signal intensity, both in the bands (mainly Z-bands) and in the nucleus (not shown), whereas mbl silencing abolished all detection of cytoplasmic bands. Finally, simultaneous detection of endogenous Mbl and the GFP signal of a GFP-tagged MblC protein (Mhc-Gal4>UAS-MblC:GFP) confirmed that the MblC isoform is present in sarcomeric Z-bands, but not in the H-bands (Fig. 6E). Thus, these results demonstrate that Mbl localizes in the sarcomeric bands and also indicate that there is a differential sarcomeric distribution between Mbl protein isoforms. As expected, in situ hybridization confirmed that nuclear Mbl colocalizes with CUG-RNA foci in CTG-expressing flies (Fig. 6F–M).
Fig. 6.

Sarcomeric distribution of Drosophila Mbl is reversibly altered by expression of CTG repeats. (A–M) Fluorescence images showing staining with anti-Mbl antibody. (A–C) Analysis of the endogenous Mbl expression (green) in the muscle throughout the different stages of development in wild-type individuals. In the embryonic somatic musculature, Mbl expression was restricted to the nucleus (A; compare with cytoplasmic signal of myosin heavy chain protein, MHC, shown in red below). (B) In the larval body wall muscles, Mbl was detected both in the nucleus (counterstained with DAPI in blue) and in cytoplasmic transversal bands. (C) In adults, the nuclear localization of Mbl was almost undetectable and the protein was preferentially detected in cytoplasmic transversal bands. (D) Double staining with anti-Mbl and phalloidin revealed that Mbl was localized in the sarcomeric H- and Z-bands. mbl silencing (Mhc-Gal4>UAS-IR-mbl), abolished Mbl signal in the bands, whereas overexpression of mblC (Mhc-Gal4>UAS-mblC), increased it mainly in the Z-bands. (E) GFP detection of a MblC:GFP fusion protein (green) coupled with an anti-Mbl antibody (red) confirmed the specificity of the signal in the Z-bands. However, MblC was not detected in the H-bands, suggesting a differential distribution of Mbl protein isoforms within the sarcomere. (F–M) Fluorescent confocal images comparing the subcellular distribution of i(CUG)480 RNA (fluorescent in situ hybridization using a CAG red-labeled probe; F,J) with Mbl protein (green; G,K; see merge in I,M) in control (J–M) and CTG-expressing flies (F–I). Nuclear signal of endogenous Mbl colocalized with CUG-RNA foci in CTG-expressing muscles. Nuclei were counterstained with DAPI (blue; H,L). (D–M) are confocal images using the 100× objective.
Depletion of Drosophila Mbl mimics the effect of CTG-repeat expression on BSF and TBPH subcellular localization
To assess the potential contribution of endogenous Mbl to the alterations of BSF and TBPH originated by i(CTG)480 expression, we studied the distribution of these proteins in thorax sections of Mhc-Gal4 UAS-IR-mbl flies after mbl silencing. We observed that Mbl reduction in these flies induced a subcellular redistribution of BSF and TBPH similar to that caused by i(CTG)480. Namely, BSF accumulated in the nucleus and disappeared from the cytoplasm, whereas TBPH signal redistributed to both the nucleus and the Z-bands (Fig. 7A–C). Neither BSF nor TBPH silencing altered Mbl subcellular distribution in the adult IFMs, indicating that the localization of these proteins is a process that occurs downstream of Mbl (Fig. 7D,G). Moreover, silencing of either bsf or TBPH did not affect the protein patterns of the other (Fig. 7F,H).
Fig. 7.
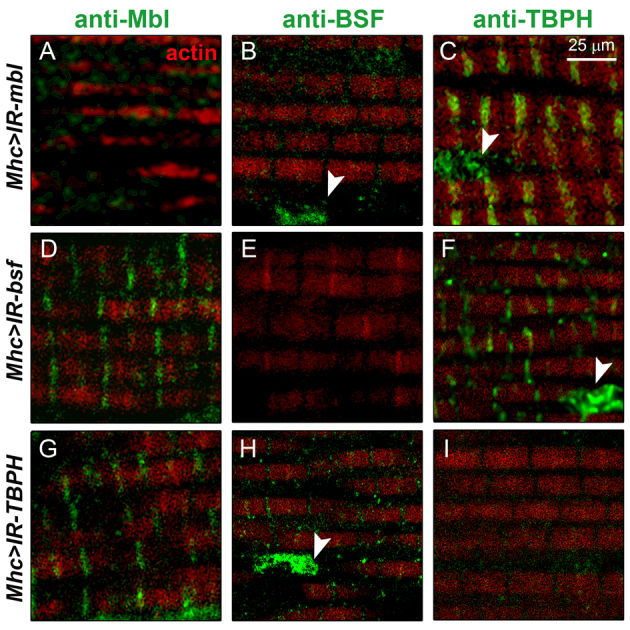
Mbl is necessary for the localization of BSF and TBPH in sarcomeric bands. Fluorescent confocal images showing immunodetection of Mbl (A,D,G), BSF (B,E,H) or TBPH (C,F,I) in green, counterstained with phalloidin (red), in adult thoraces upon silencing of mbl (Mhc-Gal4>-UAS-IR-mbl; A–C), bsf (Mhc-Gal4>-UAS-IR-bsf; D–F) or TBPH (Mhc-Gal4>-UAS-IR-TBPH; G–I). Silencing of mbl depleted Mbl (A) and BSF (B) from sarcomeric bands as well as increased TBPH in the Z-bands and in the nucleus (C). Neither bsf (D–F) nor TBPH silencing (G–I) affected the expression of Mbl in the muscle, nor did TBPH or bsf modify each other’s protein localization patterns. Arrowheads point at nuclei.
Taken together, our results demonstrate that Drosophila Mbl, BSF and TBPH localize in the sarcomere in the adult muscle, and that their distribution patterns are altered by expression of expanded CTG repeats and Mbl depletion.
DISCUSSION
RNAi has proven to be a powerful tool for conducting genetic screens in Drosophila. In this study we have used a large collection of RNAi fly lines to identify new modifiers of CTG toxicity. Our findings suggest the existence of previously unidentified components of the pathogenic mechanisms behind DM1. We isolated a number of cytoskeletal components, transcription factors and RNA-binding proteins as modifiers of CTG-induced phenotypes in the eye and wing. Some of these modifiers were the Amyloid precursor protein (APP)-like protein interacting protein 1 and zormin, the Drosophila ortholog of Titin, which have been previously associated with DM1 (Lin et al., 2006; Dickson and Wilusz, 2010); and the transcription factor Pur-α, which has been found to bind rCGG repeats in a FXTAS model (Jin et al., 2007). The implication of RNA-binding proteins, including the splicing regulator MBNL1, in DM1 has been demonstrated in animal models and human cells (Ebralidze et al., 2004; Lin et al., 2006; Osborne et al., 2009; Du et al., 2010). For this reason, we focused on two RNA-binding proteins identified in our screen, BSF and TBPH, which have not yet been associated with DM1. In our screening, silencing of bsf in GMR-Gal4 UAS-i(CTG)480 flies enhanced the eye phenotype, whereas TBPH suppressed it. However, toxicity of expanded CTG repeats in the wing was rescued by both bsf and TBPH. The tissue-specific direction of a genetic interaction has been widely reported for other genes (Port et al., 2011; Gregory et al., 2007) and supports the finding that factors involved in CTG toxicity can vary between different contexts. Additionally, the two driver lines used for expression in the eye (GMR-Gal4) and wing (vg-Gal4) produce different transcript levels, thus originating non-comparable stoichiometric proportions between bsf or TBPH and the CUG transcripts, which could further contribute to the difference observed between the tissues.
BSF is the homolog of human LRPPRC (Sterky et al., 2010; Bratic et al., 2011). This protein was originally identified as a post-transcriptional regulator that mediated the stability of the Drosophila bicoid (bcd) mRNA during oogenesis by binding to RNA structures within the 3′ UTR of the transcripts that resemble the CUG hairpins (Mancebo et al., 2001). More recently, BSF has been found to have a function in the regulation of mitochondrial gene expression, where its loss of function leads to a reduced activity of enzymes from the respiratory chain (Bratic et al., 2011). Interestingly, Cyp6a17, a gene also identified as a modifier of CTG toxicity in our screen, has been predicted to have electron carrier activity (McQuilton et al., 2012). However, in our immunohistochemistry experiments we could not detect BSF in the mitochondria of the adult IFMs, although the protein was localized in the cytoplasm and nucleus (supplementary material Fig. S3). Given that the cell fractionation studies by Bratic and coworkers were carried out using whole fly body extracts, we do not rule out a minor mitochondrial presence of BSF in the IFMs, or a more prevalent mitochondrial role in other tissues.
TDP-43, the human homolog of TBPH, has been widely studied due to its implication in a number of sporadic and inherited neurodegenerative diseases, including amyotrophic lateral sclerosis (ALS) and frontotemporal dementias (Moisse et al., 2009; Armstrong and Cairns, 2011b; Neumann et al., 2007; Geser et al., 2009; Salajegheh et al., 2009). TDP-43 is a heterogeneous nuclear ribonucleoprotein (hnRNP) that undergoes nucleocytoplasmic shuttling (Ayala et al., 2008; Wang, I. F. et al., 2008; Ritson et al., 2010). In the nervous system, upregulation of TDP-43 coupled with protein redistribution to the cytoplasm is recognized as a pathological feature (Colombrita et al., 2009; Moisse et al., 2009). Interestingly, MBNL1 also associates with components of the stress granules (Onishi et al., 2008). In our Drosophila model, TBPH subcellular localization in the muscle was also altered. However, the change occurred in the opposite direction, with TBPH protein being increased in the nucleus and partially decreased in the cytoplasm. This mislocalization, which suggests a toxic effect of nuclear TBPH accumulation, was not accompanied by changes in the levels of TBPH transcripts, mis-splicing events or increased protein levels. Therefore, it is likely that the CTG-induced effect on TBPH occurs at a more downstream level, for example by affecting the transport of TBPH mRNAs and/or TBPH protein, or by triggering post-translational modifications that affect protein localization. In our experiments, the subcellular distribution of BSF in the muscle was also altered from exclusively cytoplasmic to mainly nuclear. Both TBPH and bsf genes interacted with mblC overexpression in the eye, and their localization to the muscle sarcomere seemed to be regulated by Mbl, but not vice versa. Human MBNL2 was previously described to mediate integrin alpha3 transcript localization to the adhesion complexes (Adereth et al., 2005). Therefore, it is possible that Mbl could be involved in the subcellular localization of these proteins or their transcripts.
No function in the muscle has been previously described for BSF or TBPH. In this study, TBPH modified CTG-induced muscle fiber loss. Restoration of muscle mass in the adult IFMs by reducing TBPH expression suggests a role for this protein in the molecular mechanisms responsible for the muscle pathology characteristic of DM1. Although this rescue was only of ∼15%, a recent work demonstrated that small changes in the number of muscle fibers can trigger big differences in overall muscle strength (Moyer et al., 2011). In addition, our laboratory has found that expression of human MBNL1, a key factor in DM1, in CTG-expressing flies can rescue fiber loss in the IFMs by a ∼20% (unpublished results), which closely resembles the rescue achieved by TBPH silencing. The subcellular distribution patterns of TBPH and BSF in the muscle indicate that these proteins play a role associated with the muscle sarcomere, which was altered by expanded CTG-repeat expression. We also found a sarcomeric localization for Mbl and a differential isoform distribution, the MblC isoform being exclusive to the Z-bands. BSF not only colocalized with endogenous Mbl in the Z-and H-bands of wild-type fly muscles (not shown), but both proteins also revealed a striking parallelism in their response to CTG-repeat expression, disappearing from the sarcomeric bands and redistributing to the nucleus. MBNL1 overexpression rescued the patterns of both BSF and TBPH proteins in CTG-expressing muscles. Moreover, endogenous mbl silencing caused alterations in these proteins that resembled the alterations caused by CTG repeats. These observations support the notion that the CTG-induced effects on BSF and TBPH occur downstream of Mbl sequestration by the CUG hairpins (supplementary material Fig. S4).
Three more sarcomeric proteins were identified in our genetic screen: zormin, which encodes the fly ortholog of Titin, the splicing of which is altered in DM1 patients (Lin et al., 2006); bent (bt), which encodes Proyectin, an invertebrate protein with a structure similar to Titin; and p21-activated kinase 3 (Pak-3), whose vertebrate ortholog is localized in the Z-bands. Intriguingly, all these proteins were identified as modifiers of the eye and wing phenotypes, in which the sarcomeric structure does not exist. We previously reported that overexpression of mblC, which we identified here to be localized in the sarcomere, caused phenotypes in the eye and wing (Vicente-Crespo et al., 2008). Like Mbl, BSF and TBPH could also have additional roles in these tissues, possibly mediated by their RNA-binding ability. Consistently, TBPH silencing enhanced a rough eye phenotype caused by mblC overexpression. If the CTG-induced effect on TBPH expression observed in the muscle was mediated by Mbl, it would not be surprising that a similar interaction occurs in the developing eye, where Mbl is also present.
The localization of Drosophila Mbl in the sarcomeric bands suggests a new role for this protein in the cytoplasm, which would differ from its demonstrated activity as a splicing regulator (Machuca-Tzili et al., 2006; Vicente-Crespo et al., 2008). Given that Mbl, BSF and TBPH undergo important redistribution from the sarcomeric bands in the presence of CTG repeats, these proteins could contribute to the changes in the sarcomere structure described for DM1. However, further studies will be necessary to decipher the role of Mbl in the regulation of BSF and TBPH, as well as the position of BSF and TBPH in the CTG toxicity pathway. Traditionally, the Z-bands have been viewed as a passive constituent of the sarcomere, being important only for the cross-linking of thin filaments and for the transmission of force generated by myofilaments. However, recent studies have confirmed that various signaling molecules interact with sarcomeric Z-band proteins, several of which shuttle between the Z-bands and other cellular compartments, including the nucleus (Frank et al., 2006). Interestingly, mutations in a number of Z-band proteins have been shown to cause cardiomyopathies and/or muscular dystrophies (Hauser et al., 2000; Gerull et al., 2002; Mavroidis et al., 2008). Our study supports the implication of the sarcomere in DM1 muscle pathology and identifies two new components of the sarcomeric structure that are affected by expanded CTG repeats.
METHODS
Drosophila genetics
All RNAi fly lines used during the screen were provided by Fly Stocks of National Institute of Genetics (NIG, Mishima, Japan), the Vienna Drosophila RNAi Center (VDRC, Vienna, Austria) and the Transgenic RNAi project (TRiP, Harvard, MA). The act5C-Gal4, vg-Gal4, UAS-GFP, UAS-mitoGFP and y1w1118 flies were from the Bloomington Drosophila Stock Center (Bloomington, IN). Mhc-Gal4 and GMR-Gal4 are described in the literature (Garcia-Lopez et al., 2008). Mhc-Gal4 UAS-GFP was obtained from Eric Olson (UT Southwestern Medical Center, Dallas, TX), and Df(2R)BSC154 and TBPHΔ23 from Francisco Baralle (International Centre for Genetic Engineering and Biotechnology, Trieste). The following transgenic lines were generated in our laboratory: UAS-MBNL1, UAS-mblC and UAS-mblC-GFP (García-Casado et al., 2002; Pascual et al., 2010), UAS-i(CTG)480 (Garcia-Lopez et al., 2008), and UAS-IR-mbl. For the generation of the UAS-IR-mbl line, RNAi fragments against all mbl isoforms were designed using the web tool E-RNAi (Arziman et al., 2005). The RNAi cassette, consisting of two inverted repeats of the RNAi fragment and a DNA spacer from the GFP gene, was generated by overlapping nested PCRs (Pwo polymerase, Roche; see supplementary material Table S2). The RNAi cassette was cloned into the Bglll site of the pUAST vector (Brand and Perrimon, 1993) and microinjected into w1118 embryos (VANEDIS Drosophila injection service). The efficiency of mbl silencing was analyzed by qRT-PCR in flies ubiquitously expressing the UAS-IR-mbl transgene under the control of the Actin5C (Act5C)-Gal4 driver. The following recombinant lines were generated during this study: GMR-Gal4 UAS-i(CTG)480, vg-Gal4 UAS-i(CTG)480 and Mhc-Gal UAS-i(CTG)480, for expression of i(CTG)480 in the eye, wing and muscle, respectively. All flies were maintained at 25°C with standard food except for the crosses with vg-Ga4 UAS-i(CTG)480, which were performed at 22°C.
Reverse transcription and polymerase chain reaction
Total RNA from 50 females was extracted using Trizol (Sigma). Then, 2 μg of RNA was digested with DNaseI (Invitrogen) and retrotranscribed with SuperScript II (Invitrogen) using random hexanucleotides (García-López et al., 2008; García-López et al., 2011). qPCR was carried out from 10 ng (mbl and tubulin 84B), 4 ng (TBPH) or 0.4 ng (Rp49) of cDNA template with SYBR Green PCR Master Mix (Applied Biosystems), using tubulin 84B (mbl) or Rp49 (TBPH) as the reference gene (supplementary material Table S2). Thermal cycling was performed in an ABi 7000 sequence detection system (Applied Biosystems). Three biological replicates and three technical replicates per biological sample were carried out. Expression levels were normalized relative to the reference gene using the 2−ΔΔCt method.
Western blot
Thirty flies per genotype were processed. Thoraxes were homogenized in lysis buffer (10 mM Tris, 150 mM NaCl, 5 mM EDTA, 5 mM EGTA, 10% glycerol, 50 mM NaF, 5 mM DTT and 4 M urea, pH 7.4) plus Protease Inhibitors (Roche). After 5 minutes of centrifugation at 400 g, lysates were quantified with Quant-iT Protein Assay Kit (#Q33211, Invitrogen) and separated on 8% SDS-PAGE. After transferring to a nitrocellulose membrane (Protran #NBA083C, Whatman), blots were blocked with 5% non-fat milk and incubated with primary anti-TBPH antibody (produced in house, amino acids 1–268; 1:3000 dilution) (Feiguin et al., 2009), followed by anti-rabbit horseradish peroxidase (HRP)-conjugated secondary antibody (1:30,000; #31460, Pierce). Loading control was anti-tubulin (1:3000; #CP06, Calbiochem) followed by incubation with anti-mouse HRP-conjugated secondary antibody (1:30,000; #31430, Pierce). Bands were detected using SuperSignal West Femto Maximum Sensitivity Substrate Kit (#PR34095, Pierce).
Fluorescent immunohistochemical analysis
Fly thoraces from 3-day-old females were dissected and fixed in 4% paraformaldehyde (PFA) overnight at 4°C, followed by cryoprotection with 30% sucrose for 48 hours at 4°C. Thoraces were then embedded in OCT, and transversal sections (10 μm) obtained with a Leica CM 1510S cryostate. Cryosections were washed in PBS containing Triton 0.3% (PBT), blocked (PBT containing 5% donkey serum and 0.5% BSA) for 30 minutes at room temperature and incubated with the corresponding primary antibody (1:500) overnight at 4°C. Primary antibodies were sheep anti-Mbl (Houseley et al., 2005), rat anti-BSF (Mancebo et al., 2001) and rabbit anti-TBPH (Feiguin et al., 2009). After washes with PBT, the tissue was incubated for 45 minutes with biotin-conjugated secondary antibodies (Sigma) at 1:200 dilution. Cryosections were then incubated with ABC solution (ABC kit, VECTASTAIN) for 30 minutes at room temperature, followed by washes and incubation with streptavidin-FITC (1:1000) (Vector) for 45 minutes. Phalloidin (Sigma) was then added at 1:1000 for 20 minutes and samples mounted in Vectashield (Vector) with 2 μg/ml DAPI. To improve detection of endogenous Mbl in the muscle, the primary antibody was preincubated with 1- to 6-hour-old embryos, which do not express the protein. For double detection of CUG-RNA foci and endogenous Mbl, sections were processed for in situ hybridization with a Cy3-double-labeled (Cy3-CAG10) probe prior to immunostaining with the anti-Mbl antibody as described (Houseley et al., 2005). All confocal images were taken on a LEICA SP1.
Non-fluorescent histological analysis
For analysis of the IFMs, Drosophila thoraces (n=6 flies) were embedded in Epon for transversal, semithin sectioning (1.5 μm) following standard procedures. Images were taken at 10× and muscle area quantified by binarizing a fixed section of all images containing the IFMs (five per fly; NIH ImageJ software), considering the percentage area within this section that corresponded to muscle tissue (García-López et al., 2011). P-values were obtained using a two-tailed, non-paired t-test (α=0.05). Welch’s correction was applied when variances were significantly different. For scanning electron microscopy (SEM), adult fly eyes were processed as described (Garcia-Lopez et al., 2008).
Supplementary Material
Acknowledgments
We thank Patrick Morcillo for his guidance in the design of the genetic screen and Maria Lloret for technical support. Most of the RNAi fly lines used in this study were obtained from Fly Stocks of National Institute of Genetics (Japan). Emissions caused by this work, estimated at some 7 tonnes of CO2, have been compensated through a reforestation project.
Footnotes
COMPETING INTERESTS
The authors declare that they do not have any competing or financial interests.
AUTHOR CONTRIBUTIONS
R.A., B.L. and J.M.F. conceived and designed the experiments. B.L., J.M.F., A.B. and A.G.L. performed the experiments. All authors analyzed data and contributed to the final paper. A.G.L., B.L., J.M.F. and A.B. prepared the manuscript with input by R.A.
FUNDING
This work was supported by research grants from Genoma España Foundation, Generalitat Valenciana (Prometeo/2010/081) and Ministerio de Ciencia e Innovacion (BFU2009-10940) to R.A. A.B. was supported by Generalitat Valenciana (Prometeo/2010/081). J.M.F.C. was supported by a predoctoral fellowship from Generalitat Valenciana and grants from Fundacion Ramon Areces and Caja Navarra. A.G.L. was supported by a FPU fellowship from Ministerio de Educacion y Ciencia.
SUPPLEMENTARY MATERIAL
Supplementary material for this article is available at http://dmm.biologists.org/lookup/suppl/doi:10.1242/dmm.009563/-/DC1
REFERENCES
- Adereth Y., Dammai V., Kose N., Li R., Hsu T. (2005). RNA-dependent integrin alpha3 protein localization regulated by the Muscleblind-like protein MLP1. Nat. Cell Biol. 7, 1140–1147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aleu F. P., Afifi A. K. (1964). Ultrastructure of muscle in myotonic dystrophy: preliminary observations. Am. J. Pathol. 45, 221–231 [PMC free article] [PubMed] [Google Scholar]
- Armstrong R. A., Cairns N. J. (2011a). Spatial patterns of TDP-43 neuronal cytoplasmic inclusions (NCI) in fifteen cases of frontotemporal lobar degeneration with TDP-43 proteinopathy (FTLD-TDP). Neurol. Sci. 32, 653–659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong R. A., Cairns N. J. (2011b). A morphometric study of the spatial patterns of TDP-43 immunoreactive neuronal inclusions in frontotemporal lobar degeneration (FTLD) with progranulin (GRN) mutation. Histol. Histopathol. 26, 185–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artero R., Prokop A., Paricio N., Begemann G., Pueyo I., Mlodzik M., Perez-Alonso M., Baylies M. K. (1998). The muscleblind gene participates in the organization of Z-bands and epidermal attachments of Drosophila muscles and is regulated by Dmef2. Dev. Biol. 195, 131–143 [DOI] [PubMed] [Google Scholar]
- Arziman Z., Horn T., Boutros M. (2005). E-RNAi: a web application to design optimized RNAi constructs. Nucleic Acids Res. 33, W582–W588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayala Y. M., Zago P., D’Ambrogio A., Xu Y. F., Petrucelli L., Buratti E., Baralle F. E. (2008). Structural determinants of the cellular localization and shuttling of TDP-43. J. Cell Sci. 121, 3778–3785 [DOI] [PubMed] [Google Scholar]
- Begemann G., Paricio N., Artero R., Kiss I., Pérez-Alonso M., Mlodzik M. (1997). muscleblind, a gene required for photoreceptor differentiation in Drosophila, encodes novel nuclear Cys3His-type zinc-finger-containing proteins. Development 124, 4321–4331 [DOI] [PubMed] [Google Scholar]
- Brand A. H., Perrimon N. (1993). Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development 118, 401–415 [DOI] [PubMed] [Google Scholar]
- Bratic A., Wredenberg A., Grönke S., Stewart J. B., Mourier A., Ruzzenente B., Kukat C., Wibom R., Habermann B., Partridge L., et al. (2011). The bicoid stability factor controls polyadenylation and expression of specific mitochondrial mRNAs in Drosophila melanogaster. PLoS Genet. 7, e1002324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chintapalli V. R., Wang J., Dow J. A. T. (2007). Using FlyAtlas to identify better Drosophila models of human disease. Nat. Genet. 39, 715–720 [DOI] [PubMed] [Google Scholar]
- Colombrita C., Zennaro E., Fallini C., Weber M., Sommacal A., Buratti E., Silani V., Ratti A. (2009). TDP-43 is recruited to stress granules in conditions of oxidative insult. J. Neurochem. 111, 1051–1061 [DOI] [PubMed] [Google Scholar]
- de Haro M., Al-Ramahi I., De Gouyon B., Ukani L., Rosa A., Faustino N. A., Ashizawa T., Cooper T. A., Botas J. (2006). MBNL1 and CUGBP1 modify expanded CUG-induced toxicity in a Drosophila model of myotonic dystrophy type 1. Hum. Mol. Genet. 15, 2138–2145 [DOI] [PubMed] [Google Scholar]
- Dickson A. M., Wilusz C. J. (2010). Repeat expansion diseases: when a good RNA turns bad. Wiley Interdiscip. Rev. RNA 1, 173–192 [DOI] [PubMed] [Google Scholar]
- Du H., Cline M. S., Osborne R. J., Tuttle D. L., Clark T. A., Donohue J. P., Hall M. P., Shiue L., Swanson M. S., Thornton C. A., et al. (2010). Aberrant alternative splicing and extracellular matrix gene expression in mouse models of myotonic dystrophy. Nat. Struct. Mol. Biol. 17, 187–193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebralidze A., Wang Y., Petkova V., Ebralidse K., Junghans R. P. (2004). RNA leaching of transcription factors disrupts transcription in myotonic dystrophy. Science 303, 383–387 [DOI] [PubMed] [Google Scholar]
- Fardaei M., Rogers M. T., Thorpe H. M., Larkin K., Hamshere M. G., Harper P. S., Brook J. D. (2002). Three proteins, MBNL, MBLL and MBXL, co-localize in vivo with nuclear foci of expanded-repeat transcripts in DM1 and DM2 cells. Hum. Mol. Genet. 11, 805–814 [DOI] [PubMed] [Google Scholar]
- Feiguin F., Godena V. K., Romano G., D’Ambrogio A., Klima R., Baralle F. E. (2009). Depletion of TDP-43 affects Drosophila motoneurons terminal synapsis and locomotive behavior. FEBS Lett. 583, 1586–1592 [DOI] [PubMed] [Google Scholar]
- Fernandez-Costa J. M., Llamusi M. B., Garcia-Lopez A., Artero R. (2011). Alternative splicing regulation by Muscleblind proteins: from development to disease. Biol. Rev. Camb. Philos. Soc. 86, 947–958 [DOI] [PubMed] [Google Scholar]
- Frank D., Kuhn C., Katus H. A., Frey N. (2006). The sarcomeric Z-disc: a nodal point in signalling and disease. J. Mol. Med. (Berl.) 84, 446–468 [DOI] [PubMed] [Google Scholar]
- Gambardella S., Rinaldi F., Lepore S. M., Viola A., Loro E., Angelini C., Vergani L., Novelli G., Botta A. (2010). Overexpression of microRNA-206 in the skeletal muscle from myotonic dystrophy type 1 patients. J. Transl. Med. 8, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Casado M. Z., Artero R. D., Paricio N., Terol J., Pérez-Alonso M. (2002). Generation of GAL4-responsive muscleblind constructs. Genesis 34, 111–114 [DOI] [PubMed] [Google Scholar]
- Garcia-Lopez A., Monferrer L., Garcia-Alcover I., Vicente-Crespo M., Alvarez-Abril M. C., Artero R. D. (2008). Genetic and chemical modifiers of a CUG toxicity model in Drosophila. PLoS ONE 3, e1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-López A., Llamusí B., Orzáez M., Pérez-Payá E., Artero R. D. (2011). In vivo discovery of a peptide that prevents CUG-RNA hairpin formation and reverses RNA toxicity in myotonic dystrophy models. Proc. Natl. Acad. Sci. USA 108, 11866–11871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerull B., Gramlich M., Atherton J., McNabb M., Trombitás K., Sasse-Klaassen S., Seidman J. G., Seidman C., Granzier H., Labeit S., et al. (2002). Mutations of TTN, encoding the giant muscle filament titin, cause familial dilated cardiomyopathy. Nat. Genet. 30, 201–204 [DOI] [PubMed] [Google Scholar]
- Geser F., Martinez-Lage M., Kwong L. K., Lee V. M., Trojanowski J. Q. (2009). Amyotrophic lateral sclerosis, frontotemporal dementia and beyond: the TDP-43 diseases. J. Neurol. 256, 1205–1214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory S. L., Shandala T., O’Keefe L., Jones L., Murray M. J., Saint R. B. (2007). A Drosophila overexpression screen for modifiers of Rho signalling in cytokinesis. Fly (Austin) 1, 13–22 [DOI] [PubMed] [Google Scholar]
- Hauser M. A., Horrigan S. K., Salmikangas P., Torian U. M., Viles K. D., Dancel R., Tim R. W., Taivainen A., Bartoloni L., Gilchrist J. M., et al. (2000). Myotilin is mutated in limb girdle muscular dystrophy 1A. Hum. Mol. Genet. 9, 2141–2147 [DOI] [PubMed] [Google Scholar]
- Ho T. H., Bundman D., Armstrong D. L., Cooper T. A. (2005). Transgenic mice expressing CUG-BP1 reproduce splicing mis-regulation observed in myotonic dystrophy. Hum. Mol. Genet. 14, 1539–1547 [DOI] [PubMed] [Google Scholar]
- Houseley J. M., Wang Z., Brock G. J., Soloway J., Artero R., Perez-Alonso M., O’Dell K. M., Monckton D. G. (2005). Myotonic dystrophy associated expanded CUG repeat muscleblind positive ribonuclear foci are not toxic to Drosophila. Hum. Mol. Genet. 14, 873–883 [DOI] [PubMed] [Google Scholar]
- Jin P., Duan R., Qurashi A., Qin Y., Tian D., Rosser T. C., Liu H., Feng Y., Warren S. T. (2007). Pur alpha binds to rCGG repeats and modulates repeat-mediated neurodegeneration in a Drosophila model of fragile X tremor/ataxia syndrome. Neuron 55, 556–564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalsotra A., Xiao X., Ward A. J., Castle J. C., Johnson J. M., Burge C. B., Cooper T. A. (2008). A postnatal switch of CELF and MBNL proteins reprograms alternative splicing in the developing heart. Proc. Natl. Acad. Sci. USA 105, 20333–20338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanadia R. N., Johnstone K. A., Mankodi A., Lungu C., Thornton C. A., Esson D., Timmers A. M., Hauswirth W. W., Swanson M. S. (2003). A muscleblind knockout model for myotonic dystrophy. Science 302, 1978–1980 [DOI] [PubMed] [Google Scholar]
- Kanadia R. N., Shin J., Yuan Y., Beattie S. G., Wheeler T. M., Thornton C. A., Swanson M. S. (2006). Reversal of RNA missplicing and myotonia after muscleblind overexpression in a mouse poly(CUG) model for myotonic dystrophy. Proc. Natl. Acad. Sci. USA 103, 11748–11753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuyumcu-Martinez N. M., Wang G. S., Cooper T. A. (2007). Increased steady-state levels of CUGBP1 in myotonic dystrophy 1 are due to PKC-mediated hyperphosphorylation. Mol. Cell 28, 68–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin X., Miller J. W., Mankodi A., Kanadia R. N., Yuan Y., Moxley R. T., Swanson M. S., Thornton C. A. (2006). Failure of MBNL1-dependent post-natal splicing transitions in myotonic dystrophy. Hum. Mol. Genet. 15, 2087–2097 [DOI] [PubMed] [Google Scholar]
- Loro E., Rinaldi F., Malena A., Masiero E., Novelli G., Angelini C., Romeo V., Sandri M., Botta A., Vergani L. (2010). Normal myogenesis and increased apoptosis in myotonic dystrophy type-1 muscle cells. Cell Death Differ. 17, 1315–1324 [DOI] [PubMed] [Google Scholar]
- Ludatscher R. M., Kerner H., Amikam S., Gellei B. (1978). Myotonia dystrophica with heart involvement: an electron microscopic study of skeletal, cardiac, and smooth muscle. J. Clin. Pathol. 31, 1057–1064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machuca-Tzili L., Thorpe H., Robinson T. E., Sewry C., Brook J. D. (2006). Flies deficient in Muscleblind protein model features of myotonic dystrophy with altered splice forms of Z-band associated transcripts. Hum. Genet. 120, 487–499 [DOI] [PubMed] [Google Scholar]
- Mahadevan M. S., Yadava R. S., Yu Q., Balijepalli S., Frenzel-McCardell C. D., Bourne T. D., Phillips L. H. (2006). Reversible model of RNA toxicity and cardiac conduction defects in myotonic dystrophy. Nat. Genet. 38, 1066–1070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancebo R., Zhou X., Shillinglaw W., Henzel W., Macdonald P. M. (2001). BSF binds specifically to the bicoid mRNA 3′ untranslated region and contributes to stabilization of bicoid mRNA. Mol. Cell. Biol. 21, 3462–3471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mankodi A., Logigian E., Callahan L., McClain C., White R., Henderson D., Krym M., Thornton C. A. (2000). Myotonic dystrophy in transgenic mice expressing an expanded CUG repeat. Science 289, 1769–1772 [DOI] [PubMed] [Google Scholar]
- Mavroidis M., Panagopoulou P., Kostavasili I., Weisleder N., Capetanaki Y. (2008). A missense mutation in desmin tail domain linked to human dilated cardiomyopathy promotes cleavage of the head domain and abolishes its Z-disc localization. FASEB J. 22, 3318–3327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McQuilton P., St Pierre S. E., Thurmond J., FlyBase Consortium (2012). FlyBase 101 – the basics of navigating FlyBase. Nucleic Acids Res. 40, D706–D714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller J. W., Urbinati C. R., Teng-Umnuay P., Stenberg M. G., Byrne B. J., Thornton C. A., Swanson M. S. (2000). Recruitment of human muscleblind proteins to (CUG)(n) expansions associated with myotonic dystrophy. EMBO J. 19, 4439–4448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moisse K., Volkening K., Leystra-Lantz C., Welch I., Hill T., Strong M. J. (2009). Divergent patterns of cytosolic TDP-43 and neuronal progranulin expression following axotomy: implications for TDP-43 in the physiological response to neuronal injury. Brain Res. 1249, 202–211 [DOI] [PubMed] [Google Scholar]
- Moyer M., Berger D. S., Ladd A. N., Van Lunteren E. (2011). Differential susceptibility of muscles to myotonia and force impairment in a mouse model of myotonic dystrophy. Muscle Nerve 43, 818–827 [DOI] [PubMed] [Google Scholar]
- Neumann M., Kwong L. K., Sampathu D. M., Trojanowski J. Q., Lee V. M. (2007). TDP-43 proteinopathy in frontotemporal lobar degeneration and amyotrophic lateral sclerosis: protein misfolding diseases without amyloidosis. Arch. Neurol. 64, 1388–1394 [DOI] [PubMed] [Google Scholar]
- Onishi H., Kino Y., Morita T., Futai E., Sasagawa N., Ishiura S. (2008). MBNL1 associates with YB-1 in cytoplasmic stress granules. J. Neurosci. Res. 86, 1994–2002 [DOI] [PubMed] [Google Scholar]
- Osborne R. J., Thornton C. A. (2006). RNA-dominant diseases. Hum. Mol. Genet. 15, R162–R169 [DOI] [PubMed] [Google Scholar]
- Osborne R. J., Lin X., Welle S., Sobczak K., O’Rourke J. R., Swanson M. S., Thornton C. A. (2009). Transcriptional and post-transcriptional impact of toxic RNA in myotonic dystrophy. Hum. Mol. Genet. 18, 1471–1481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascual M., Monferrer L., Fernandez-Costa J. M., Bargiela A., Artero R., Llamusi B. (2010). A GFP-tagged muscleblind C protein isoform reporter construct. Fly (Austin) 4, 333–337 [DOI] [PubMed] [Google Scholar]
- Perbellini R., Greco S., Sarra-Ferraris G., Cardani R., Capogrossi M. C., Meola G., Martelli F. (2011). Dysregulation and cellular mislocalization of specific miRNAs in myotonic dystrophy type 1. Neuromuscul. Disord. 21, 81–88 [DOI] [PubMed] [Google Scholar]
- Port F., Hausmann G., Basler K. (2011). A genome-wide RNA interference screen uncovers two p24 proteins as regulators of Wingless secretion. EMBO Rep. 12, 1144–1152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rau F., Freyermuth F., Fugier C., Villemin J. P., Fischer M. C., Jost B., Dembele D., Gourdon G., Nicole A., Duboc D., et al. (2011). Misregulation of miR-1 processing is associated with heart defects in myotonic dystrophy. Nat. Struct. Mol. Biol. 18, 840–845 [DOI] [PubMed] [Google Scholar]
- Ritson G. P., Custer S. K., Freibaum B. D., Guinto J. B., Geffel D., Moore J., Tang W., Winton M. J., Neumann M., Trojanowski J. Q., et al. (2010). TDP-43 mediates degeneration in a novel Drosophila model of disease caused by mutations in VCP/p97. J. Neurosci. 30, 7729–7739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salajegheh M., Pinkus J. L., Taylor J. P., Amato A. A., Nazareno R., Baloh R. H., Greenberg S. A. (2009). Sarcoplasmic redistribution of nuclear TDP-43 in inclusion body myositis. Muscle Nerve 40, 19–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sicot G., Gourdon G., Gomes-Pereira M. (2011). Myotonic dystrophy, when simple repeats reveal complex pathogenic entities: new findings and future challenges. Hum. Mol. Genet. 20 R2, R116–R123 [DOI] [PubMed] [Google Scholar]
- Sterky F. H., Ruzzenente B., Gustafsson C. M., Samuelsson T., Larsson N. G. (2010). LRPPRC is a mitochondrial matrix protein that is conserved in metazoans. Biochem. Biophys. Res. Commun. 398, 759–764 [DOI] [PubMed] [Google Scholar]
- Taneja K. L., McCurrach M., Schalling M., Housman D., Singer R. H. (1995). Foci of trinucleotide repeat transcripts in nuclei of myotonic dystrophy cells and tissues. J. Cell Biol. 128, 995–1002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timchenko L. T., Miller J. W., Timchenko N. A., DeVore D. R., Datar K. V., Lin L., Roberts R., Caskey C. T., Swanson M. S. (1996). Identification of a (CUG)n triplet repeat RNA-binding protein and its expression in myotonic dystrophy. Nucleic Acids Res. 24, 4407–4414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicente-Crespo M., Pascual M., Fernandez-Costa J. M., Garcia-Lopez A., Monferrer L., Miranda M. E., Zhou L., Artero R. D. (2008). Drosophila muscleblind is involved in troponin T alternative splicing and apoptosis. PLoS ONE 3, e1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang I. F., Wu L. S., Shen C. K. (2008). TDP-43: an emerging new player in neurodegenerative diseases. Trends Mol. Med. 14, 479–485 [DOI] [PubMed] [Google Scholar]
- Wang L. C., Hung W. T., Pan H., Chen K. Y., Wu Y. C., Liu Y. F., Hsiao K. M. (2008). Growth-dependent effect of muscleblind knockdown on Caenorhabditis elegans. Biochem. Biophys. Res. Commun. 366, 705–709 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.