

Abstract
The vitamin D receptor (VDR) has both 1,25-dihydroxyvitamin D-dependent and -independent actions in the epidermis. Ligand-dependent actions of the VDR have been shown to promote keratinocyte differentiation and to regulate formation of the epidermal barrier. In contrast, the actions of the VDR that regulate postmorphogenic hair cycling do not require 1,25-dihydroxyvitamin D. The VDR also has immunomodulatory actions that are dependent on its ligand, 1,25-dihydroxyvitamin D. To determine whether the ligand-dependent or -independent actions of the VDR regulate the inflammatory response to cutaneous injury, studies were performed in control, VDR knockout, and vitamin D-deficient mice. These investigations demonstrate that absence of receptor or ligand impairs the dermal response to cutaneous injury. Although neutrophil recruitment is not affected, the absence of VDR signaling leads to defects in macrophage recruitment and granulation tissue formation. Studies performed to identify the molecular basis for this phenotype demonstrate that absence of the VDR, or its ligand, impairs TGF-β signaling in the dermis, characterized by decreased expression of monocyte chemotactic protein-1 and reduced phosphorylation of phosphorylated Smad-3 as well as attenuated phosphorylated Smad-3 phosphorylation in response to TGF-β in primary dermal fibroblasts lacking the VDR. Thus, these data demonstrate that the liganded VDR interacts with the TGF-β signaling pathway to promote the normal inflammatory response to cutaneous injury.
Vitamin D is an important regulator of many biological processes. The active hormone, 1,25-dihydroxyvitamin D, exerts its cellular effects by binding the vitamin D receptor (VDR), a member of the nuclear receptor superfamily. The VDR has both 1,25-dihydroxyvitamin d-dependent and -independent actions in the epidermis. Ligand-dependent interactions of the VDR with distinct cofactors regulate normal keratinocyte proliferation and differentiation (1–5). Although the effects of 1,25-dihydroxyvitamin D and calcium on keratinocyte differentiation are redundant, the effects of the VDR on the epidermal barrier, including sphingolipid production by keratinocytes, require ligand-dependent VDR interactions with steroid receptor coactivator-3 in differentiated keratinocytes (6). VDR ablation in humans and mice causes rickets with alopecia (7, 8). Although the skeletal changes in the VDR knockout mice (vdr−/−) can be prevented by normalizing mineral ion homeostasis via dietary means, these mice still develop alopecia due to an inability to initiate postnatal hair cycles (7, 9, 10). The actions of the VDR required for hair follicle homeostasis are independent of 1,25-dihydroxyvitamin D (10). In the immune system, ligand-dependent actions of the VDR have been shown to be important in host defense and tissue repair (reviewed in Ref. 11). Both the innate and acquired immune responses are regulated by ligand-dependent VDR actions (11–14). Humans and animals deficient in vitamin D are more sensitive to mycobacterial infections due to impaired macrophage function (15). Furthermore, the VDR-dependent antiinflammatory actions of 1,25-dihydroxyvitamin D have been shown to play a role in the response to acute injury in both the colon and the kidney (16–20).
The mammalian cutaneous wound repair process is comprised of three overlapping phases: inflammation, new tissue formation, and tissue remodeling (21). Within 24 h of injury, inflammatory cells, including neutrophils and macrophages, are recruited to the wound. Macrophages are important regulators of all stages of wound repair (22). They have multiple functions, including the production of cytokines and growth factors important for cell recruitment to the wound and regulation of cells within the local wound microenvironment (22). Factors critical for the recruitment of macrophages to sites of injury include monocyte chemotactic protein-1 (MCP-1) and TGF-β (23). MCP-1 is derived from a number of different cell types within the wound, including endothelial cells, fibroblasts, keratinocytes, and monocytes (24, 25). Production of TGF-β is one of the earliest steps in the inflammatory response, serving to recruit macrophages to sites of injury (26). TGF-β stimulates monocyte differentiation, matrix production, and matrix remodeling (23). All cells within the wound microenvironment are capable of producing and/or responding to TGF-β.
The TGF-β signaling pathway is an important regulator of the inflammatory response to injury. TGF-β signals are mediated through phosphorylated Smads, downstream effector proteins that, upon phosphorylation, translocate to the nucleus where they activate transcription of target genes (reviewed in Ref. 27). The importance of epidermally derived TGF-β is supported by investigations demonstrating that overexpression of TGF-β in the wounded epidermis increases inflammation (28–30), whereas the inactivation of this pathway by dominant-negative expression of TGF-β in the epidermis results in an impaired inflammatory response to injury (31). Ablation of the downstream TGF-β effector Smad-3, results in decreased macrophage recruitment and a reduction in granulation tissue formation at sites of cutaneous injury (32). Because of the critical role of 1,25-dihydroxyvitamin D in epidermal barrier formation and its immunomodulatory effects, studies were performed to examine whether ligand-dependent effects of the VDR play a role in modulating the inflammatory response to cutaneous injury.
Materials and Methods
Animal maintenance and surgery
All animal studies were approved by the institutional animal care committee. Mice were maintained in a virus- and parasite-free animal facility under a 12-h light, 12-h dark cycle. Mice were on a C57BL6/J background and were weaned at 18 d on a diet that maintains normal mineral ion homeostasis in the absence of VDR signaling (2% calcium, 1.25% phosphorus, 20% lactose supplemented diet, TD96348; Harlan Teklad, Madison WI) (33). Vitamin D-deficient mice were bred and maintained in a UV-free environment and weaned onto a similar diet lacking vitamin D metabolites (TD97340; Harlan Teklad) that results in undetectable circulating 25-hydroxyvitamin D levels (9). Full-thickness punch biopsy wounds were made on mice at 8 wk of age under general anesthesia using a 3.5-mm biopsy punch (Uni-punch; Premier, Plymouth Meeting, PA). Mice were killed 2 and 5 d after biopsy to evaluate the inflammatory response.
Biochemical analyses
Serum levels of 25-hydroxyvitamin D and 1,25-dihydroxyvitamin D were evaluated using kits from Diasorin (Stillwater, MN).
Histology and immunohistochemistry
Wounds were excised and fixed in 10% formalin/PBS and embedded vertically in paraffin. The total number of dermal neutrophils was manually quantified and averaged over 3 high-power field (hpf) by a board-certified pathologist using digitally captured images of hematoxylin and eosin-stained sections. For MCP-1 and IL-1α (ab7207 and ab10955; Abcam, Cambridge, MA) immunohistochemistry (IHC), antigen retrieval was performed by boiling for 15 min in 10 mm citrate buffer. Sections were blocked and incubated with antibody in 10% BSA with 0.1% Triton X-100. For F4/80 (Abcam ab6640) IHC, after antigen retrieval, sections were permeabilized in 0.025% Triton X-100 and then blocked and incubated with antibody in 10% heat-inactivated fetal bovine serum (FBS)/PBS. Alkaline phosphatase-conjugated secondary antibodies were used (Abcam), and immunoreactivity was detected using a Vector Red alkaline phosphatase substrate kit (Vector Laboratories, Burlingame, CA) per the manufacturer's instructions. For phosphorylated Smad (pSmad)-3 (Abcam ab51177) IHC, a TSA biotin kit was used (PerkinElmer, Waltham, MA) per the manufacturer's suggestions. Sections were incubated in 10% dimethylsulfoxide in methanol for 20 min at room temperature, and endogenous peroxidase was quenched using 0.3% H2O2 in methanol for 20 min at room temperature. Antigen retrieval was performed by incubation in 10 mm citrate buffer at 97 C for 30 min. The total number of positive cells per hpf was quantified using digitally captured images of at least 3 hpf from at least three mice per genotype or condition.
Protein analysis
Wounds were excised with care, with a biopsy punch, to exclude surrounding tissue and homogenized in RIPA buffer (25 mm Tris-HCl, pH 7.4; 150 mm NaCl; 1% Nonidet P-40; 1% sodium deoxycholate; 0.1% sodium dodecyl sulfate; 10% glycerol). Cultured dermal fibroblasts were rinsed in PBS and lysed in buffer containing 85 mm KCl, 5 mm HEPES, 0.5% Nonidet P-40, 1 mm Na3VO4, 1 mm NaF, and complete miniprotease inhibitors (Roche Diagnostics, Indianapolis, IN). Total protein levels were determined using a bicinchonic protein assay (Pierce, Rockford, IL). Equal amounts of protein were resolved by SDS-PAGE and subjected to Western analyses using antibodies against phosphorylated Smad3, total Smad3 (Cell Signaling C25A9; Cell Signaling, Danvers, MA; and R&D Systems AF3797; R&D Systems, Minneapolis, MN, respectively), MCP-1 (Abcam ab7202), and actin (Santa Cruz Biotechnology, sc1616; Santa Cruz, CA). Autoradiograms of Western blots were scanned using exposures representing the linear range of the signal. Quantitation of signal intensity was performed using ImageJ 1.47b software (National Institutes of Health, Bethesda, MD). The ratio of pSmad3 to total Smad3 was determined and that of the vitamin D-deficient mice, vdr−/− mice, and vdr−/− dermal fibroblasts was compared with that of the wild-type mice and wild-type dermal fibroblasts, respectively.
Primary cell culture
Dermal fibroblasts were isolated from skin harvested from neonatal wild-type or vdr−/− mice (P0-P4) and digested overnight at 4 C in 0.25% trypsin (Invitrogen, Carlsbad, CA). The dermis was separated from the epidermis, minced, and stirred for 1 h at 37 C in 0.25% type 1 collagenase (Worthington Biochemical, Lakewood, NJ) in 1× Hanks' balanced salts solution (Mediatech, Manassas, VA). Cell suspensions were strained, pelleted, and washed twice in PBS before plating. Cells were maintained in DMEM (Invitrogen) with 10% FBS (Hyclone, Logan, UT) and penicillin/streptomycin (Invitrogen).
TGF-β treatment
Dermal fibroblasts between passages 2 and 4 were seeded overnight at a density of 1 × 105 cells/well in 12-well dishes for RNA isolation or 2 × 105 cells/well in six-well dishes for protein isolation. Media were changed to DMEM with 0.1% heat-inactivated FBS 24 h before treatment with 5 or 10 ng/ml recombinant human TGF-β1 (R&D Systems). Cells were treated for 3 h to evaluate the RNA expression or for 30 min to evaluate the Smad3 phosphorylation.
Real-time PCR
Total RNA was extracted from cultured cells using the RNeasy Plus mini kit (QIAGEN, Valencia, CA) according to the manufacturer's instructions. cDNA was synthesized using Superscript II reverse transcriptase (Invitrogen). Quantitative real-time PCR was performed using the Opticon DNA engine system (MJ Research, Waltham, MA). Primers were designed to span introns, and absence of contaminating DNA was confirmed in all samples. RNA levels encoding each gene of interest were normalized for actin RNA in the same sample using the formula of Livak and Schmittgen (34).
Results
The 1,25-dihydroxyvitamin d-dependent actions of the VDR are required for the inflammatory response to cutaneous injury
Mice lacking a functional VDR are born phenotypically normal in normal Mendelian ratios (7). Restoration of mineral ion homeostasis via dietary means prevents both the skeletal and keratinocyte differentiation defects in the vdr−/− mice, however, alopecia and abnormalities in production of sphingolipids that contribute to the epidermal barrier persist (6). To determine whether the VDR regulates the inflammatory response to injury, eight-week old wild-type control and vdr−/− mice were subjected to 3.5-mm, full-thickness punch biopsy wounds. Wounds were harvested 1, 2, and 5 d after the injury to permit evaluation of the epidermal and dermal responses to cutaneous injury. Histological analyses revealed that the number of neutrophils (polymorphonuclear cells) recruited in response to cutaneous injury 1, 2, and 5 d after the wounding was not affected by VDR ablation (Fig. 1, A and 1B); however a dramatic defect in the inflammatory response to injury was seen in the dermis of vdr−/− mice 2 and 5 d after the injury compared with controls (Fig. 1, B and C). This impaired granulation tissue formation is characterized by hypocellularity and a reduction in extracellular matrix.
Fig. 1.

Abnormalities in the inflammatory response to cutaneous wounding are apparent in vdr−/− and vitamin D-deficient mice. Hematoxylin-and-eosin-stained sections from the center of wounds isolated 1 (A), 2 (B), or 5 (C) d after the injury from control, vdr−/− (VDR-KO), and vitamin D-deficient mice (D Deficient; housed in a UV free environment and fed a high calcium, high phosphorus, lactose supplemented diet lacking vitamin D metabolites). Boxes denote area magnified in bottom panel. Original magnification (top panel), ×4, (bottom panel), ×20. Data are based on at least two sections obtained per wound from wounds isolated from at least three mice per genotype or condition. Graph in A represents the number of polymorphonuclear cells/hpf. Data were manually quantified and averaged over 3 hpf from at least three mice per genotype/condition. Care was taken to exclude fibroblasts, macrophages, and endothelial cells and to avoid performing cell counts within the scale crust. D, Representative VDR IHC analyses 2 d after the injury for the VDR in the wound granulation tissue of control (left panel), vdr−/− (VDR-KO, middle panel), and vitamin D-deficient (D Def, right panel) mice. Original magnification, ×40. Data are based on at least two sections obtained per wound from wounds isolated from at least three mice per genotype or condition.
To determine whether the actions of the VDR required for the dermal response to cutaneous injury are ligand dependent, analogous studies were performed in wild-type mice raised in a UV-free environment on a vitamin D-deficient diet. A marked reduction in granulation tissue formation was apparent in vitamin D-deficient mice compared with controls (Fig. 1, B and C), a phenotype analogous to that observed with VDR ablation. Serum 25-hydroxyvitamin D and 1,25-dihydroxyvitamin D levels were below the lower limit of detection (<5 ng/ml and < 5.8 pg/ml, respectively) in the vitamin D-deficient mice. IHC analyses demonstrated nuclear VDR immunoreactivity only in wounds isolated from control animals: nonnuclear staining was observed in wounds isolated from vitamin D-deficient mice, whereas no immunoreactivity was observed in wounds isolated from vdr−/− mice (Fig. 1d). Thus, the ligand-dependent actions of the VDR are required for the normal dermal response to cutaneous injury.
Studies undertaken to identify the basis for this finding revealed a marked reduction in the number of F4/80-positive macrophages in the wounds of vdr−/− and vitamin D-deficient mice (Fig. 2A). To determine whether this defect could be explained by altered cytokine expression, the expression of IL-1α, which plays a role in neutrophil recruitment, and MCP-1, which specifically recruits macrophages (23), was evaluated by IHC 2 d after wounding. No differences were observed in the IL-1α expression, consistent with the normal recruitment of neutrophils to the wounds of vdr−/− mice (Figs. 1A and 2B). However, a significant reduction in the MCP-1 expression was observed in the granulation tissue of the vdr−/− mice and vitamin D-deficient animals compared with the controls, suggesting that the ligand-dependent actions of the VDR are required for normal expression of MCP-1 and macrophage recruitment during the inflammatory response to cutaneous injury (Fig. 2, C and D).
Fig. 2.

Impaired macrophage recruitment and decreased MCP-1 expression are observed in the wounds of vdr−/− and vitamin D-deficient mice. A, Representative IHC analyses 2 d after the injury for the macrophage marker F4/80 (red) in the wound granulation tissue of control (left panel), vdr−/− (VDR-KO; middle panel), and vitamin D-deficient (D Def; right panel) mice. The total number of F4/80-positive cells/hpf was quantified ±sem (n ≥ 3 mice). B, Representative IHC analyses 2 d after the injury for IL-1α (red) in the wound granulation tissue of control (left panel), vdr−/− (middle panel), and vitamin D-deficient (right panel) mice. The total number of IL-1α-positive cells/hpf was quantified ±sem (n ≥ 3 mice). C, Representative IHC analyses 2 d after the injury for MCP-1 (red) in the wound granulation tissue of control (left panel), vdr−/− (middle panel), and vitamin D-deficient (right panel) mice. The total number of MCP-1-positive cells/hpf was quantified ±sem (n ≥ 3 mice). Sections were counterstained with fast green. All images were taken at ×20 magnification. Statistical significance was determined using the Student's t test. **, P ≤ 0.001. Data were manually quantified and averaged over 3 hpf from at least three mice per genotype or condition. D, Total protein (20 μg) isolated from wounds of control (Ctrl), vdr−/− (VDR-KO), and vitamin D-deficient (D Def) mice 2 d after the injury was subjected to SDS-PAGE and immunoblotted for MCP-1 (top panel) and actin (bottom panel). Data are representative of protein lysates obtained from the wounds of four animals per genotype or condition.
Activation of the TGF-β signaling pathway is dependent on intact 1,25-dihydroxyvitamin D/VDR signaling
Induction of TGF-β signaling is an early event in the inflammatory response to injury (26). Several studies demonstrate that TGF-β can induce MCP-1 expression in a variety of cell types (35–38). Consistent with this observation, macrophage recruitment in response to cutaneous injury has been shown to require the actions of Smad3, a downstream effector of the TGF-β signaling pathway (32). To determine whether the decrease in MCP-1 expression observed in the absence of the VDR could be a consequence of impaired activation of the TGF-β signaling pathway, IHC analysis for the activated form of Smad3, pSmad 3, was performed. A significant reduction in the number of pSmad3-positive nuclei was observed in the granulation tissue of vdr−/− mice compared with that of controls 2 d after the injury (Fig. 3, A and B). To determine whether the VDR actions on the TGF-β signaling pathway during the inflammatory response to cutaneous injury are ligand dependent, studies were also performed in vitamin D-deficient wild-type animals. As with the vdr−/− mice, significantly fewer pSmad3-positive nuclei were detected in the granulation tissue of vitamin D-deficient mice compared with controls (Fig. 3, A and B).
Fig. 3.
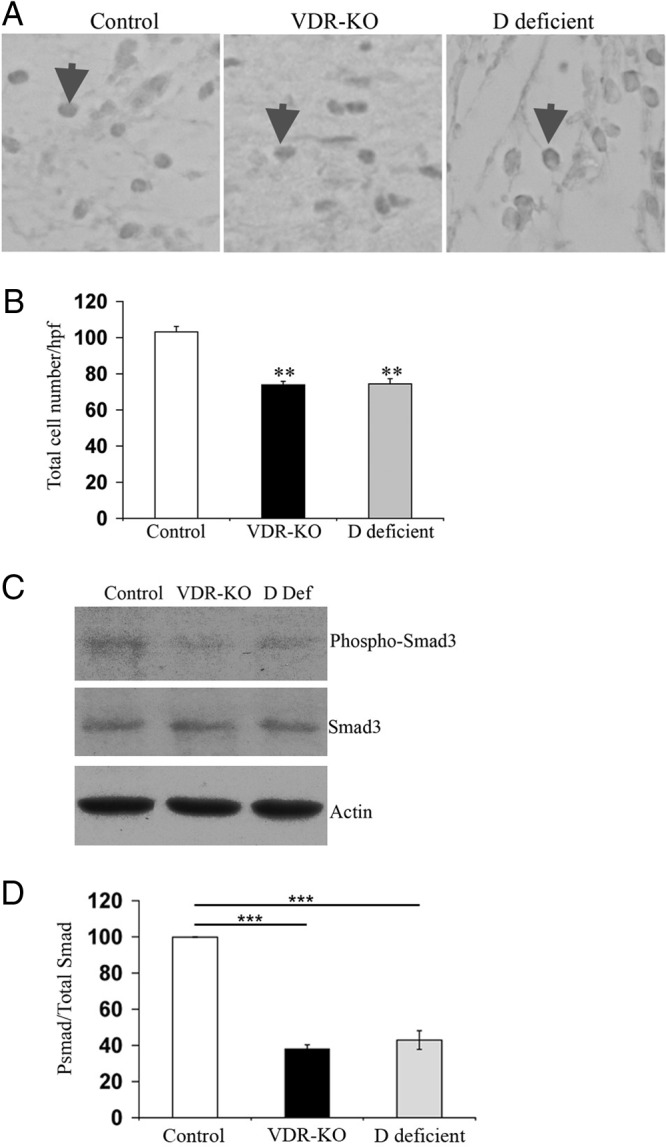
Activation of the TGF-β signaling pathway in response to cutaneous injury requires ligand-dependent actions of the VDR. A, Representative IHC analyses 2 d after the injury for nuclear pSmad3 in the wound granulation tissue of control (left panel), vdr−/− (middle panel), and vitamin D-deficient (right panel) mice. Arrows indicate examples of pSmad3 nuclear immunoreactivity. B, The total number of pSmad3-positive nuclei/hpf was quantified ±sem (n ≥ 3 mice). Statistical significance was determined using the Student's t test. **, P ≤ 0.001. Data are based on at least two sections obtained per wound from wounds isolated from at least three mice per genotype or condition. C, Total protein (20 μg) isolated from wounds of control (Ctrl), vdr−/− (VDR-KO), and vitamin D-deficient (D Def) mice 2 d after the injury was subjected to SDS-PAGE and immunoblotted for pSmad3 (top panel), total Smad3 (middle panel), and actin (bottom panel). D, The ratio of pSmad3 to total Smad3 was determined by the quantitation of signal intensity of the relevant bands. The ratio of pSmad3 to total Smad3 in the wounds of vitamin D-deficient and vdr−/− mice was normalized to that obtained from wounds of control mice. Statistical significance was determined using the student's test. ***, P ≤ 0.0005. Data represent those obtained from the wound protein lysates of four animals per genotype or condition.
Western analyses were performed to further evaluate Smad3 expression and activation. Total protein was isolated from wounds of control, vdr−/−, and vitamin D-deficient mice 2 d after the injury. The ratio of pSmad3 to total Smad3 in the wounds of vdr−/− and vitamin D-deficient mice was 38 ± 2 and 43 ± 5% that observed in the wounds of control mice, demonstrating that, in the absence of ligand-dependent actions of the VDR, there is impaired activation of the TGF-β signaling pathway during the inflammatory response to cutaneous injury (Fig. 3, C and D).
The VDR is required for activation of the TGF-β signaling pathway in dermal fibroblasts
Because activation of the TGF-β signaling pathway within the dermal component of cutaneous wounds is VDR dependent, studies were undertaken to determine whether TGF-β signaling was impaired in dermal fibroblasts isolated from vdr−/− mice. Primary dermal fibroblasts were treated with TGF-β for 3 h, and quantitative real-time PCR (qRT-PCR) analyses were performed. Although TGF-β treatment induced TGF-β, α-smooth muscle actin, and MCP-1 mRNA expression in wild-type cells, no induction of these target genes was detected in the absence of the VDR (Fig. 4, A and B), demonstrating that the VDR is required for the induction of TGF-β target genes in dermal fibroblasts. Western analyses showed a small (1.2-fold) but significant increase in total Smad3 in response to TGF-β treatment in the wild-type but not VDR knockout cells. Most notably, after 30 min of TGF-β treatment, the ratio of pSmad3 to total Smad3 in the vdr−/− dermal fibroblasts was 39 ± 6% that observed in the wild-type dermal fibroblasts (Fig. 4, C and D).
Fig. 4.
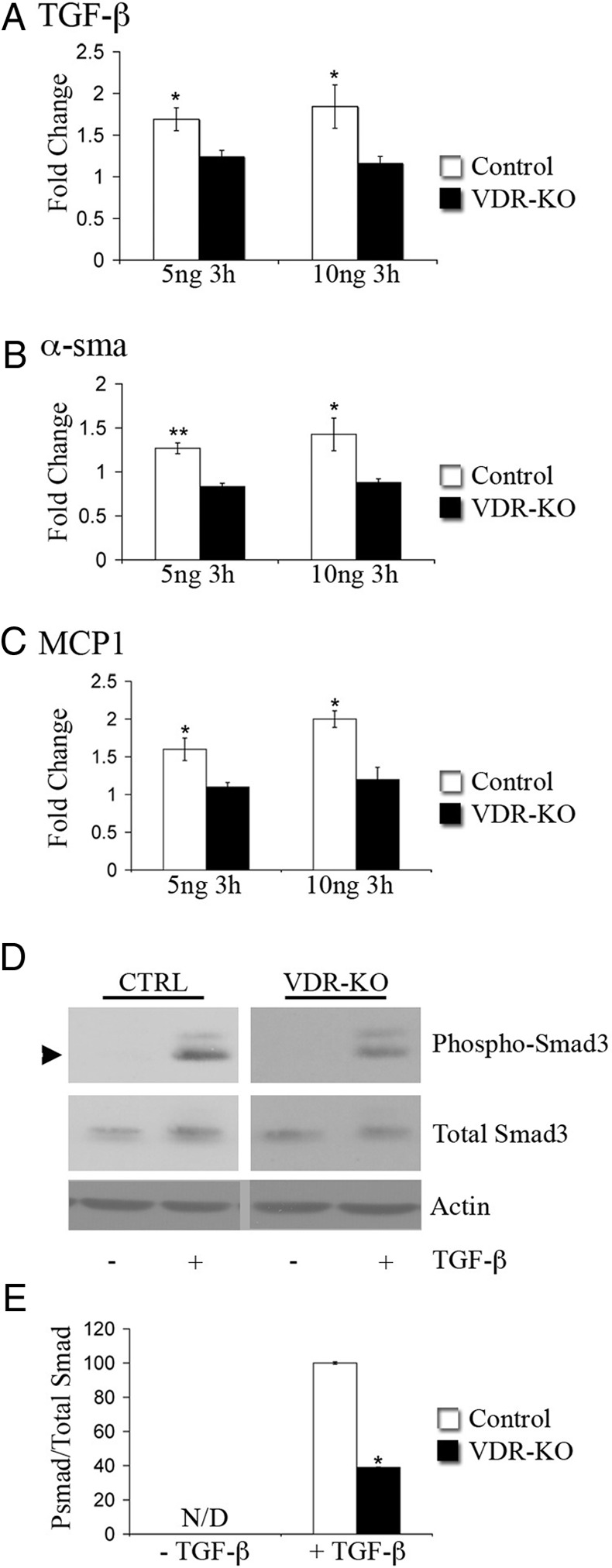
The VDR is required for the activation of TGF-β target gene expression in dermal fibroblasts. A–C, RNA isolated from primary wild-type (Control, white bars) or vdr−/− dermal fibroblasts(VDR-KO, black bars) treated for 3 h with 5 or 10 ng/ml TGF-β was subjected to qRT-PCR analysis for the genes indicated. Values are expressed as the relative fold change in expression of each transcript compared with unstimulated controls. RNA levels encoding each gene of interest were normalized for actin RNA in the same sample. Data shown are based on at least three independent RNA isolations ±sem. *, P ≤ 0.05, **, P ≤ 0.005. D, Total protein (7 μg) isolated from control (CTRL) or vdr−/− (VDR-KO) primary dermal fibroblasts treated with (+) or without (−) 10 ng TGF-β for 30 min was subjected to SDS-PAGE and immunoblotted for pSmad3, total Smad3, and actin. E, The ratio of pSmad3 to total Smad3 was determined by the quantitation of signal intensity of the relevant bands. The ratio of pSmad3 to total Smad3 of the TGF-β-treated vdr−/− dermal fibroblasts was normalized to that of the TGF-β-treated wild-type fibroblasts. Statistical significance was determined using the Student's t test, *, P ≤ 0.05. N/D, pSmad3 band was not detected in the absence of TGF-β. Data are representative of protein lysates obtained from four independent experiments.
Discussion
These studies identify novel 1,25-dihydroxyvitamin d-dependent actions of the VDR that are required for the inflammatory response to cutaneous injury. The presence of a normal neutrophil response combined with unimpaired reepithelization in the wounds of the vdr−/− and vitamin D-deficient mice (data not shown) suggest that ligand-dependent actions of the VDR are not required for these processes. In contrast, a significant decrease in MCP-1 expression was evident in the granulation tissue of vdr−/− and vitamin D-deficient wounds compared with controls, likely contributing to the impaired recruitment of macrophages to the wound.
Studies performed in mice expressing a human diphtheria toxin receptor transgene that allows inducible macrophage ablation at different stages of wound repair clearly demonstrate the importance of macrophages in the inflammatory response to injury (39, 40). Although the induction of macrophage depletion immediately before injury did not affect the recruitment of monocytes and neutrophils (40), macrophage depletion 3 d before wounding significantly decreased macrophage, but not neutrophil, recruitment (39). The impaired macrophage recruitment to the wounds of vdr−/− and vitamin D-deficient mice closely mimics this latter phenotype. The observed decrease in MCP-1 expression suggests that this defect in macrophage recruitment is attributable to impaired expression of cytokines that recruit the macrophages, rather than to an intrinsic macrophage defect. Although chemotaxis of macrophages isolated from vdr−/− mice is impaired, this defect is reversed when normocalcemia in the vdr−/− mice is restored by dietary means (41). Thus, to circumvent the effects of hypocalcemia in our investigations, all animals were maintained on a diet that prevents impaired mineral ion homeostasis in the vdr−/− and vitamin D-deficient mice (9, 33).
TGF-β signaling is required not only for initiating the inflammatory response to injury but also for the formation of granulation tissue (42). Because TGF-β signaling enhances MCP-1 expression by dermal fibroblasts (35), impaired TGF-β signaling likely contributes to the decrease in MCP-1 expression observed in the absence of the liganded VDR. Consistent with this hypothesis, deletion of Smad3, a downstream effector of TGF-β signaling, results in both impaired macrophage recruitment and reduced granulation tissue in response to cutaneous injury (32), a phenotype strikingly similar to that observed in the vdr−/− and vitamin D-deficient mice. A significant reduction in the number of pSmad3-positive cells in the granulation tissue along with decreased levels of pSmad3 in the wounds of vdr−/− and vitamin D-deficient mice demonstrate impaired activation of the TGF-β signaling pathway in the absence of the liganded VDR. This observation is supported by both the qRT-PCR studies demonstrating that, although TGF-β mRNA is not significantly altered in the wounds of mice with impaired VDR signaling, there is significant impairment in the activation of TGF-β target genes in primary dermal fibroblasts lacking the VDR. Furthermore, Western analyses demonstrate impaired induction of Smad3 protein phosphorylation in dermal fibroblasts lacking the VDR. Cross talk between the VDR and TGF-β signaling pathways has been established previously (43, 44). TGF-β not only enhances ligand-dependent VDR transactivation, but Smad3 is also a known coactivator of VDR transactivation: in the presence of 1,25-dihydroxyvitamin D, the formation of a VDR-Smad3 complex is enhanced by steroid receptor coactivator-1 (44). In a reciprocal fashion, 1,25-dihydroxyvitamin D has been shown to increase TGF-β expression and activity in cultured cells (45, 46). Disruption of VDR/Smad3 interactions likely underlies both the impaired inflammatory response and decreased granulation tissue observed in the wounds of vdr−/− and vitamin D-deficient mice. Thus, our studies demonstrate that ligand-dependent actions of the VDR are required for a normal inflammatory response to cutaneous injury.
Acknowledgments
We thank Richa Khatri for assistance with histology and Caren Gundberg for biochemical analyses.
This work was supported by Grant R01 DK-46974 (to M.B.D.) from the National Institutes of Health.
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- FBS
- Fetal bovine serum
- hpf
- high-power field
- IHC
- immunohistochemistry
- MCP-1
- monocyte chemotactic protein-1
- qRT-PCR
- quantitative real-time PCR
- pSmad
- phosphorylated Smad
- VDR
- vitamin D receptor.
References
- 1. Bikle DD, Oda Y, Xie Z. 2004. Calcium and 1,25(OH)2D: interacting drivers of epidermal differentiation. J Steroid Biochem Mol Biol 89–90:355–360 [DOI] [PubMed] [Google Scholar]
- 2. Sakai Y, Demay MB. 2000. Evaluation of keratinocyte proliferation and differentiation in vitamin D receptor knockout mice. Endocrinology 141:2043–2049 [DOI] [PubMed] [Google Scholar]
- 3. Xie Z, Komuves L, Yu QC, Elalieh H, Ng DC, Leary C, Chang S, Crumrine D, Yoshizawa T, Kato S, Bikle DD. 2002. Lack of the vitamin D receptor is associated with reduced epidermal differentiation and hair follicle growth. J Invest Dermatol 118:11–16 [DOI] [PubMed] [Google Scholar]
- 4. Oda Y, Ishikawa MH, Hawker NP, Yun QC, Bikle DD. 2007. Differential role of two VDR coactivators, DRIP205 and SRC-3, in keratinocyte proliferation and differentiation. J Steroid Biochem Mol Biol 103:776–780 [DOI] [PubMed] [Google Scholar]
- 5. Oda Y, Sihlbom C, Chalkley RJ, Huang L, Rachez C, Chang CP, Burlingame AL, Freedman LP, Bikle DD. 2003. Two distinct coactivators, DRIP/mediator and SRC/p160, are differentially involved in vitamin D receptor transactivation during keratinocyte differentiation. Mol Endocrinol (Baltimore, Md) 17:2329–2339 [DOI] [PubMed] [Google Scholar]
- 6. Oda Y, Uchida Y, Moradian S, Crumrine D, Elias PM, Bikle DD. 2009. Vitamin D receptor and coactivators SRC2 and 3 regulate epidermis-specific sphingolipid production and permeability barrier formation. J Invest Dermatol 129:1367–1378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Li YC, Pirro AE, Amling M, Delling G, Baron R, Bronson R, Demay MB. 1997. Targeted ablation of the vitamin D receptor: an animal model of vitamin D-dependent rickets type II with alopecia. Proc Natl Acad Sci USA 94:9831–9835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Malloy PJ, Pike JW, Feldman D. 1999. The vitamin D receptor and the syndrome of hereditary 1,25-dihydroxyvitamin D-resistant rickets. Endocr Rev 20:156–188 [DOI] [PubMed] [Google Scholar]
- 9. Sakai Y, Kishimoto J, Demay MB. 2001. Metabolic and cellular analysis of alopecia in vitamin D receptor knockout mice. J Clin Invest 107:961–966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Skorija K, Cox M, Sisk JM, Dowd DR, MacDonald PN, Thompson CC, Demay MB. 2005. Ligand-independent actions of the vitamin D receptor maintain hair follicle homeostasis. Mol Endocrinol (Baltimore, Md) 19:855–862 [DOI] [PubMed] [Google Scholar]
- 11. Bouillon R, Carmeliet G, Verlinden L, van Etten E, Verstuyf A, Luderer HF, Lieben L, Mathieu C, Demay M. 2008. Vitamin D and human health: lessons from vitamin D receptor null mice. Endocr Rev 29:726–776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. White JH. 2012. Vitamin D metabolism and signaling in the immune system. Rev Endocr Metab Disord 13:21–29 [DOI] [PubMed] [Google Scholar]
- 13. Baeke F, Takiishi T, Korf H, Gysemans C, Mathieu C. 2010. Vitamin D modulator of the immune system. Curr Opin Pharmacol 10:482–496 [DOI] [PubMed] [Google Scholar]
- 14. Hewison M. 2012. Vitamin D and the immune system: new perspectives on an old theme. Rheum Dis Clin North Am 38:125–139 [DOI] [PubMed] [Google Scholar]
- 15. Liu PT, Stenger S, Li H, Wenzel L, Tan BH, Krutzik SR, Ochoa MT, Schauber J, Wu K, Meinken C, Kamen DL, Wagner M, Bals R, Steinmeyer A, Zügel U, Gallo RL, Eisenberg D, Hewison M, Hollis BW, Adams JS, Bloom BR, Modlin RL. 2006. Toll-like receptor triggering of a vitamin D-mediated human antimicrobial response. Science (New York, NY) 311:1770–1773 [DOI] [PubMed] [Google Scholar]
- 16. Froicu M, Cantorna MT. 2007. Vitamin D and the vitamin D receptor are critical for control of the innate immune response to colonic injury. BMC Immunol 8:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tan X, Li Y, Liu Y. 2006. Paricalcitol attenuates renal interstitial fibrosis in obstructive nephropathy. J Am Soc Nephrol 17:3382–3393 [DOI] [PubMed] [Google Scholar]
- 18. Tan X, Wen X, Liu Y. 2008. Paricalcitol inhibits renal inflammation by promoting vitamin D receptor-mediated sequestration of NF-κB signaling. J Am Soc Nephrol 19:1741–1752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. He W, Kang YS, Dai C, Liu Y. 2011. Blockade of Wnt/β-catenin signaling by paricalcitol ameliorates proteinuria and kidney injury. J Am Soc Nephrol 22:90–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhang Y, Kong J, Deb DK, Chang A, Li YC. 2010. Vitamin D receptor attenuates renal fibrosis by suppressing the renin-angiotensin system. J Am Soc Nephrol 21:966–973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gurtner GC, Werner S, Barrandon Y, Longaker MT. 2008. Wound repair and regeneration. Nature 453:314–321 [DOI] [PubMed] [Google Scholar]
- 22. Rodero MP, Khosrotehrani K. 2010. Skin wound healing modulation by macrophages. International J Clin Exp Pathol 3:643–653 [PMC free article] [PubMed] [Google Scholar]
- 23. Barrientos S, Stojadinovic O, Golinko MS, Brem H, Tomic-Canic M. 2008. Growth factors and cytokines in wound healing. Wound Repair Regen 16:585–601 [DOI] [PubMed] [Google Scholar]
- 24. Gillitzer R, Goebeler M. 2001. Chemokines in cutaneous wound healing. J Leukocyte Biol 69:513–521 [PubMed] [Google Scholar]
- 25. Deshmane SL, Kremlev S, Amini S, Sawaya BE. 2009. Monocyte chemoattractant protein-1 (MCP-1): an overview. J Interferon Cytokine Res 29:313–326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. O'Kane S, Ferguson MW. 1997. Transforming growth factor βs and wound healing. Int J Biochem Cell Biol 29:63–78 [DOI] [PubMed] [Google Scholar]
- 27. Santibanez JF, Quintanilla M, Bernabeu C. 2011. TGF-β/TGF-β receptor system and its role in physiological and pathological conditions. Clin Sci (Lond) 121:233–251 [DOI] [PubMed] [Google Scholar]
- 28. Wang XJ, Han G, Owens P, Siddiqui Y, Li AG. 2006. Role of TGFβ-mediated inflammation in cutaneous wound healing. J Invest Dermatol Symp Proc 11:112–117 [DOI] [PubMed] [Google Scholar]
- 29. Liu X, Alexander V, Vijayachandra K, Bhogte E, Diamond I, Glick A. 2001. Conditional epidermal expression of TGFβ1 blocks neonatal lethality but causes a reversible hyperplasia and alopecia. Proc Natl Acad Sci USA 98:9139–9144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Li AG, Wang D, Feng XH, Wang XJ. 2004. Latent TGFβ1 overexpression in keratinocytes results in a severe psoriasis-like skin disorder. EMBO J 23:1770–1781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Amendt C, Mann A, Schirmacher P, Blessing M. 2002. Resistance of keratinocytes to TGFβ-mediated growth restriction and apoptosis induction accelerates re-epithelialization in skin wounds. J Cell Sci 115:2189–2198 [DOI] [PubMed] [Google Scholar]
- 32. Ashcroft GS, Yang X, Glick AB, Weinstein M, Letterio JL, Mizel DE, Anzano M, Greenwell-Wild T, Wahl SM, Deng C, Roberts AB. 1999. Mice lacking Smad3 show accelerated wound healing and an impaired local inflammatory response. Nat Cell Biol 1:260–266 [DOI] [PubMed] [Google Scholar]
- 33. Li YC, Amling M, Pirro AE, Priemel M, Meuse J, Baron R, Delling G, Demay MB. 1998. Normalization of mineral ion homeostasis by dietary means prevents hyperparathyroidism, rickets, and osteomalacia, but not alopecia in vitamin D receptor-ablated mice. Endocrinology 139:4391–4396 [DOI] [PubMed] [Google Scholar]
- 34. Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2[-ΔΔC(T)] method. Methods (San Diego, Calif) 25:402–408 [DOI] [PubMed] [Google Scholar]
- 35. Slavin J, Unemori E, Hunt TK, Amento E. 1995. Monocyte chemotactic protein-1 (MCP-1) mRNA is down-regulated in human dermal fibroblasts by dexamethasone: differential regulation by TGF-beta. Growth Factors (Chur, Switzerland) 12:151–157 [DOI] [PubMed] [Google Scholar]
- 36. Qi W, Chen X, Polhill TS, Sumual S, Twigg S, Gilbert RE, Pollock CA. 2006. TGF-βinduces IL-8 and MCP-1 through a connective tissue growth factor-independent pathway. Am J Physiol 290:F703–F709 [DOI] [PubMed] [Google Scholar]
- 37. Wu X, Ma J, Han JD, Wang N, Chen YG. 2006. Distinct regulation of gene expression in human endothelial cells by TGF-β and its receptors. Microvasc Res 71:12–19 [DOI] [PubMed] [Google Scholar]
- 38. Zhang F, Tsai S, Kato K, Yamanouchi D, Wang C, Rafii S, Liu B, Kent KC. 2009. Transforming growth factor-β promotes recruitment of bone marrow cells and bone marrow-derived mesenchymal stem cells through stimulation of MCP-1 production in vascular smooth muscle cells. J Biol Chem 284:17564–17574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Goren I, Allmann N, Yogev N, Schürmann C, Linke A, Holdener M, Waisman A, Pfeilschifter J, Frank S. 2009. A transgenic mouse model of inducible macrophage depletion: effects of diphtheria toxin-driven lysozyme M-specific cell lineage ablation on wound inflammatory, angiogenic, and contractive processes. Am J Pathol 175:132–147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mirza R, DiPietro LA, Koh TJ. 2009. Selective and specific macrophage ablation is detrimental to wound healing in mice. Am J Pathol 175:2454–2462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mathieu C, Van Etten E, Gysemans C, Decallonne B, Kato S, Laureys J, Depovere J, Valckx D, Verstuyf A, Bouillon R. 2001. In vitro and in vivo analysis of the immune system of vitamin D receptor knockout mice. J Bone Miner Res 16:2057–2065 [DOI] [PubMed] [Google Scholar]
- 42. McCartney-Francis NL, Wahl SM. 1994. Transforming growth factor β: a matter of life and death. J Leukocyte Biol 55:401–409 [DOI] [PubMed] [Google Scholar]
- 43. Subramaniam N, Leong GM, Cock TA, Flanagan JL, Fong C, Eisman JA, Kouzmenko AP. 2001. Cross-talk between 1,25-dihydroxyvitamin D3 and transforming growth factor-β signaling requires binding of VDR and Smad3 proteins to their cognate DNA recognition elements. J Biol Chem 276:15741–15746 [DOI] [PubMed] [Google Scholar]
- 44. Yanagisawa J, Yanagi Y, Masuhiro Y, Suzawa M, Watanabe M, Kashiwagi K, Toriyabe T, Kawabata M, Miyazono K, Kato S. 1999. Convergence of transforming growth factor-β and vitamin D signaling pathways on SMAD transcriptional coactivators. Science (New York, NY) 283:1317–1321 [DOI] [PubMed] [Google Scholar]
- 45. Kim HJ, Abdelkader N, Katz M, McLane JA. 1992. 1,25-Dihydroxy-vitamin-D3 enhances antiproliferative effect and transcription of TGF-β1 on human keratinocytes in culture. J Cell Physiol 151:579–587 [DOI] [PubMed] [Google Scholar]
- 46. Koli K, Keski-Oja J. 1995. 1,25-Dihydroxyvitamin D3 enhances the expression of transforming growth factor β1 and its latent form binding protein in cultured breast carcinoma cells. Cancer Res 55:1540–1546 [PubMed] [Google Scholar]