

Abstract
MUC4 is a type-1 transmembrane mucin differentially expressed in multiple cancers and has previously been shown to potentiate progression and metastasis of pancreatic cancer. In this study, we investigated the molecular mechanisms associated with the MUC4-induced invasion and metastasis in pancreatic cancer. Stable silencing of MUC4 in multiple pancreatic cancer cells resulted in the downregulation of N-cadherin and its interacting partner fibroblast growth factor receptor 1 (FGFR1) through downregulation of partly by pFAK, pMKK7, pJNK and pc-Jun pathway and partly through PI-3K/Akt pathway. The downregulation of FGFR1 in turn led to downregulation of pAkt, pERK1/2, pNF-κB, pIkBα, uPA, MMP-9, vimentin, N-cadherin, Twist, Slug and Zeb1 and upregulation of E-cadherin, Occludin, Cytokeratin-18 and Caspase-9 in MUC4 knockdown BXPC3 and Capan1 cells compared with scramble vector transfected cells. Further, downregulation of FGFR1 was associated with a significant change in morphology and reorganization of the actin-cytoskeleton, leading to a significant decrease in motility (P < 0.00001) and invasion (P < 0.0001) in vitro and decreased tumorigenicity and incidence of metastasis in vivo upon orthotopic implantation in the athymic mice. Taken together, the results of the present study suggest that MUC4 promotes invasion and metastasis by FGFR1 stabilization through the N-cadherin upregulation.
Introduction
Despite a welcome decline in mortality rate over the past decade, pancreatic cancer (PC) still remains the 10th most commonly diagnosed cancer and the 4th leading cause of cancer-related death in the USA (1,2). The median survival of PC patients is about 4.1 months with the overall 5-year survival rate being less than 5% (2–4). The clinical manifestations of PC usually occur at a late stage, at which time the disease has already spread to local and distant organs (in 85% of patients) (5). To acquire such invasive abilities, epithelial cancer cells undergo several phenotypic changes, similar to those seen during embryonic development. This process is termed epithelial to mesenchymal transition (EMT). Despite growing knowledge about the events underlying PC development, translation of this information into effective therapies and treatments are limited. Besides, precise molecular mechanisms by which PC cells progress from a non-invasive to a highly metastatic stage are largely unclear. Hence, in the present study, efforts are being made to identify the molecular events that underlie the metastatic ability of this lethal disease.
Previous reports have shown that around 90% of cancer-related deaths are mainly due to metastasis, not due to primary tumors (6). The process of invasion and metastasis in PC is still inadequately understood. Normally, invasion and metastasis occurs in sequential steps, which involves detachment of cancer cells from the primary tumor and invasion into the surrounding healthy tissues followed by intravasation, extravasation and finally colonization at distant sites. However, in recent years, an enormous amount of data has suggested that cancer cells utilize the same mechanisms as healthy embryonic cells (i.e. grastulation by the process of changing from an epithelial to a mesenchymal-like phenotype) called EMT. This is a phenomenon whereby malignant cells contribute to invasion, metastatic dissemination and acquisition of therapeutic resistance (7,8). The process of EMT involves the disruption of cell–cell and cell-extracellular matrix interactions, loss of cell polarity, reorganization of the actin cytoskeleton, acquisition of a mesenchymal phenotype with reduced intercellular interactions and increased migratory capacity. This is associated with a significant increase in the expression of mesenchymal markers such as vimentin and vitronectin-75 (9), downregulation of epithelial markers such as E-cadherin and cytokeratin-18 (10) and upregulation of transcription factors associated with the EMT process such as Twist, Snail and Slug (11), leading to invasion and metastasis.
MUC4 is a large membrane-anchored glycoprotein that is aberrantly expressed in many cancers (12–18). Its expression is undetectable in the normal pancreas but increases progressively in pancreatic intraepithelial neoplasia (19,20) and is strongly expressed in PC (20–23). We have previously shown that MUC4 induces cellular transformation of NIH 3T3 fibroblast cells, potentiates PC cell growth and metastasis and contributes to gemcitabine resistance (24–27). Subsequently, we have also reported that MUC4, via its interaction with the epidermal growth factor receptor family member human epidermal growth factor receptor-2, induces downstream signaling that favors proliferation, motility, invasion and promotes cell survival in PC and other malignancies (25,28). Further, human epidermal growth factor receptor-2 also activates focal adhesion kinase (FAK), a key protein involved in PC metastasis and invasion (25,28), highlighting its role as a promoter of aggressiveness in PC cells. However, its precise involvement in the metastasis and invasion of PC through a process of EMT has not been explored. In the current study, we have explored the signaling mechanism by which MUC4 potentiates invasion and metastasis, partly through regulating the EMT process and stabilizing fibroblast growth factor receptor 1 (FGFR1), which may improve our understanding of the events involved in the progression and metastasis of PC and may aid in the identification of novel targets for better management of PC.
Materials and methods
Antibodies
The anti-MUC4 mouse monoclonal antibody (8G7) used in this study was developed by our laboratory (29). The antibodies, cleaved caspase-9 (Asp330), phosphorylated/total pAkt (Serine 473)/tAkt, pMKK7 (Ser271/Thr275)/tMKK7, pJNK(Thr183/Tyr185)/tJNK, pc-JUN(Ser63)/tc-JUN and phospho-ERK1/2 / tERK1/2, phospho–FAK (pFAK-Tyr 925, Tyr 576/577)/tFAK, pHER2 (Tyrosine 1248)/tHER2, NFκB, and pIκB/IκB were obtained from cell signaling (Danvers, MA, USA). The antibodies against MMP-9 and N and E-cadherin were gifts from Dr. Keith R. Johnson (University of Nebraska Medical Center). The β-actin antibody was obtained from Sigma-Aldrich (St. Louis, MO). The secondary antibodies, anti-mouse and anti-rabbit IgGs conjugated to horseradish peroxidase, were obtained from GE Healthcare Biosciences, (Uppsala, Sweden). The fluorescein isothiocyanate–conjugated anti-mouse secondary antibody was obtained from Invitrogen (California, USA).
ShRNA-mediated MUC4 silencing in pancreatic cells Capan1 and BxPC3
Capan1 and BxPC3 PC cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum and antibiotics (100 µg/ml of penicillin and streptomycin) at 37°C with 5% CO2 in a humidified atmosphere. The shRNA-mediated silencing of MUC4 was carried out as described previously (25). In brief, two 64mer oligonucleotides (forward and reverse) containing the 19-bp target sequence in sense and antisense orientation were designed on both sides of the linker TTCAAGAGA. The Bgl II and Hind III restriction sites were introduced at 5ʹ and 3ʹ ends of 64mers, respectively, to allow direct ligation of the annealed MUC4 double-strand insert into the pSUPER-retro-puro vector. The annealed double-stranded oligonucleotides were phosphorylated using polynucleotide kinase (Roche Diagnostics, Mannheim, Germany) and ligated into the digested pSUPER-retro-puro vector. Successful cloning was ascertained by restriction digestion and sequencing. Ecotropic phoenix packaging cells were transfected with the pSUPER-retro-puro vector containing either the MUC4 shRNA insert (pSUPER-retro-puro.shMUC4) or a scrambled sequence (pSUPER-retro-puro.Scr) using Lipofectamine 2000 (Invitrogen, San Diego, CA), following the manufacturer’s protocol. Supernatant containing infection-competent retroviruses was collected 48h after transfection and used for stable transduction of Capan1 and BxPC3 cells using polybrene (4 µg/ml) to augment the infection efficiency. Stable clones were then selected in a medium containing puromycin (5 µg/ml; InvivoGen, San Diego, CA).
Real-time PCR
Total RNA was isolated and the cDNA was synthesized by reverse transcription as described previously (30). The real-time primers for MUC4, N-cadherin, FGFR1, Slug, Zeb1 and β-actin (Supplementary Table 1, available at Carcinogenesis Online) were designed by using primer 3 software. Real-time PCR was performed on a Roche 480 Real-Time PCR System, Indianapolis, USA. Real-time PCRs were performed in triplicate and non template controls (NTCs) were run for each assay under the same conditions. PCR was then performed in 10 μl reactions containing 5 μl 2x SBYR green Master Mix, 3.2 μl of autoclaved nuclease free water, 1 μl diluted RT product (1:10) and 0.4 μl each of forward and reverse primer (5 pmol). The cycling conditions were comprised of 95°C for 10min, followed by 40 cycles of 95°C for 15 s and 58°C for 1min. Gene expression levels were normalized to the level of β-actin expression and used as a control.
Immunoblot and confocal laser scanning microscopy
The MUC4-silenced Capan1/BxPC3 and scrambled control cells were processed for western blotting and confocal microscopy as described previously (26,30). In brief, a total of 30–60 µg of protein from the cell extracts were resolved by electrophoresis on either a 2.0% sodium dodecyl sulfate–agarose gel (for MUC4) or a 10% sodium dodecyl sulfate–polyacrylamide gel (for other proteins) and transferred to polyvinylidene difluoride membrane (PVDF). After blocking with 5% non-fat milk in Tris-buffered saline (0.1M, pH = 7.4), blots were incubated with specific antibodies at 40C overnight. The membrane was then incubated with horseradish peroxidase-labeled appropriate secondary immunoglobulin, and the signal was detected using an electro chemiluminescence reagent kit (Amersham Pharmacia, Piscataway, NJ). Western blots were quantified using the software ChemiImager4400 and the fold change was calculated. The numerical values specified beneath the respective bands of western blots represent the fold change in protein expression as compared to that of the control.
For confocal laser scanning microscopy, 5×103 cells were plated on cover slips and grown for 24h. Expression and localization of proteins were observed using confocal laser scanning microscopy as described by us (31). After washing with phosphate-buffered saline (pH = 7.4), the cells were fixed in ice-cold methanol at −20°C for 2min and blocked with 10% goat serum containing 0.05% Tween-20 for 1h followed by incubation with the specific antibodies overnight at 4°C. Cells were then washed and incubated with FITC-conjugated goat antimouse/rabbit secondary antibodies for 60min. Thereafter, the cover slips were mounted on glass slides with antifade Vectashield mounting medium (Vector Laboratories, Burlingame, CA, and USA). Immunostaining was observed under a Zeiss (Carl Zeiss Microimaging, Thornwood, NY) confocal laser scanning microscope, and representative photographs were captured digitally using the 510 LSM software.
Apoptosis assay
Apoptosis was measured by Annexin-V FITC staining as described by us previously (30). Briefly, 50×103 Capan1, BxPC3 and derived cells were seeded and allowed to grow for 48h and trypsinized, washed with ice-cold phosphate-buffered saline (pH = 7.4), resuspended in cold calcium-binding medium (10mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid, pH 7.4, 140mM NaCl and 2.5mM CaCl2) and stained with FITC-labeled annexin V and propidium iodide (Roche Diagnostics, Indianapolis, IN). Following incubation at room temperature for 15min in the dark, cells were analyzed using a FACS CantoTM flow cytometer.
Motility and invasion assay
Motility and invasion assays were carried out as described by us previously (30). Briefly, for motility assays, 1×106 cells suspended in serum-free medium were plated in the top chamber of polyethylene teraphthalate membranes (six-well insert, pore size 8 µm; Becton Dickinson). For invasion assay, 2×106 cells were seeded on Matrigel-coated membrane inserts (BD Biosciences, Bedford, MA). Two ml of 10% serum-containing medium was added to the lower chamber of the well and the cells were allowed to migrate for 24h under chemotactic drive. Cells that did not migrate through the pores were removed by scraping the membrane with a cotton swab. The migrated cells on the lower side of the membrane were stained with a Diff-Quick cell stain kit (Dade-Behring Inc, Newark, DE 19714, USA) and photographed in 10 random fields of view at ×10 magnification. Cell numbers were counted and expressed as the average number of cells/field of view. The experiments were repeated three times and data were represented as the average of the three independent experiments with the standard error of the mean.
Tumorigenicity assay
Subconfluent cultures of Capan1-shMUC4 and Capan1-Scr cells were trypsinized and washed with phosphate-buffered saline. Cell viability was determined by trypan blue staining and single-cell suspensions of >90% viability was used for the orthotopic injections. The cells were resuspended in a normal saline solution at a concentration of 5×105 cells/50 µl. Immunodeficient mice were purchased from the Animal Production Area of the National Cancer Institute-Frederick Cancer Research and Development Center (Frederick, MD, USA). The mice were treated in accordance with Institutional Animal Care and Use Committee guidelines. The orthotopic implantation was performed as described previously (30). All mice were killed after 21 days of implantation. The presence of metastatic lesions to different organs was determined by thorough gross inspection and histological analysis. Pancreatic tumors were excised, weighed and measured.
Co-immunoprecipitation
Co-immunoprecipitation assay was carried out as described previously by us (28). Cells were grown to 60–70% confluency, washed once with ice-cold phosphate-buffered saline and then lysed in RIPA buffer (150mM NaCl, 2mM ethylenediaminetetraacetic acid, 50mM Tris–Cl (pH 8.0), 1mM NaF, 1mM sodium orthovanadate, 1mM phenylmethylsulfonyl fluoride, 5mg of aprotinin per ml, 5mg of leupeptin per ml and containing 1% Triton X-100) for 25–35min at 4°C. The lysates were centrifuged at 16×103 g for 30min at 4°C and protein concentrations determined using a Bio-Rad D/C protein estimation kit. Equal amounts of protein were precleaned with Protein G-Sepharose beads (Oncogene Research, Boston, MA, USA) for 4h at 4°C on a rotor. Cleaned lysates were incubated overnight with anti-FGFR1/anti N-cadherin antibodies or with IgG at 4°C on a rotor. Protein G-Sepharose beads were added to the lysates and incubated for 4h at 4°C on a rotating platform. Immunocomplexes were pulled down, washed with the lysis buffer (3–4 times) and immunoblotted with anti-rabbit FGFR1 or N-cadherin.
Statistical analyses
Mean tumor weights and mean tumor volumes were compared between groups using a two-tailed independent sample student’s t-test. A P-value of <0.05 was considered statistically significant.
Results
Stable silencing of MUC4 expression by shRNA
PC cells Capan1 and BxPC3 were infected with the viral supernatant carrying either the MUC4 shRNA or the scrambled shRNA expression plasmids for effective silencing of MUC4 or to serve as a control, respectively. The pooled populations were selected in 10% DMEM medium containing puromycin (5.0 µg/ml). The stable silencing of MUC4 in Capan1-shMUC4 and BxPC3-shMUC4 cells and their scramble shRNA-transfected cells (Capan1-Scr and BxPC3-Scr) was confirmed by immunoblotting at the protein level and by real-time PCR analysis at the transcript level (Figure 1A and 1B). Western blot analysis showed 80 and 70% downregulation of MUC4 expression at the protein level in Capan1-shMUC4 and BxPC3-shMUC4 cells, respectively, when compared with scramble vector transfected cells (Figure 1A). Subsequently, our real-time PCR analysis revealed consistent results with the western blot analysis, confirming the reduced expression of MUC4. RNA from immortalized but untransformed human pancreatic ductal epithelial HPDE cells was used as a negative control for MUC4 expression (Figure 1B). In addition, to strengthen the aforementioned results, we also examined the expression of MUC4 using immunofluorescence, with results congruent with immunoblot and real-time PCR analysis (Figure 1C and 1D).
Fig. 1.

Strategy for shRNA-mediated silencing of oncogenic MUC4 in Capan1 and BxPC3 cells. (A) Analysis of MUC4 expression in BxPC3/Capan1-shMUC4, BxPC3/Capan1-Scr by immunoblotting shows significant inhibition in protein expression (70–80%), respectively. β-actin was used as a loading control. (B) Real-time PCR analysis using primers that specifically amplify the MUC4 gene. Capan1 and BxPC3 pooled population cells show a 50–60% decrease in the expression of MUC4 at the mRNA level compared with the control population. No amplification was observed in HPDE, which is negative for the MUC4 expression. (C and D). Confocal analysis showed a decreased expression of MUC4 on the cell membrane in Capan1 and BxPC3-shMUC4 cells compared with scramble cells. (E and F) A morphological comparison between the Capan1/BxPC3-shMUC4 and Capan1/BxPC3-Scr cells. The MUC4 knockdown Capan1 and BxPC3 cells grow in aggregates compared with the scramble cells. (G and H) The phalloidin-rhodamine (phalloidin-RITC) staining of actin–cytoskeleton. MUC4-expressing Capan1 and BxPC3-Scr cells revealed the presence of more microspikes, lamellopodia and filopodia-like cellular projections compared with the MUC4 knockdown Capan1/BxPC3 cells.
MUC4 knockdown leads to altered morphology in PC cells
We next examined the cellular morphology and growth patterns of Capan1-shMUC4/BxPC3-shMUC4 and scramble vector transfected cells. The Capan1-shMUC4 and BxPC3-shMUC4 cells showed a spherical (rounded) morphology and a tendency to grow in clumps compared with the Capan1-Scr and BxPC3-Scr cells which displayed a flat- and spindle-shaped morphology, with loose association (Figure 1E and 1F). Further, we also observed that MUC4 knockdown resulted in a significant reduction in lamellipodia and filopodia in BxPC3-shMUC4 and Capan1-shMUC4 cells (Figure 1G and 1H) when compared with scramble vector transfected cells.
MUC4 knockdown alters the expression of EMT markers in PC cells
To investigate the mechanism underlying the morphological changes in MUC4 knockdown Capan1 and BxPC3 cells, we examined the expression of known epithelial markers E-cadherin (CDH1), Occludin, Cytokeratin-18 (CK-18) and mesenchymal markers, such as N-cadherin (CDH2), Twist, Slug, Zeb1, uPA and vimentin, by western blotting QPCR and immunofluorescence. Both western blot and immunofluorescence analysis revealed that the expression of E-cadherin, Occludin and CK-18 significantly increased (Figure 2A and 2B), whereas expression of N-cadherin, Twist, uPA, vimentin, Slug and Zeb1 significantly decreased in both Capan1-shMUC4 and BxPC3-shMUC4 cells (Figure 2A–2C) when compared with their respective control cells. Therefore, these results demonstrate that the MUC4 mediates the EMT in PC, leading to increased invasion and metastasis.
Fig. 2.
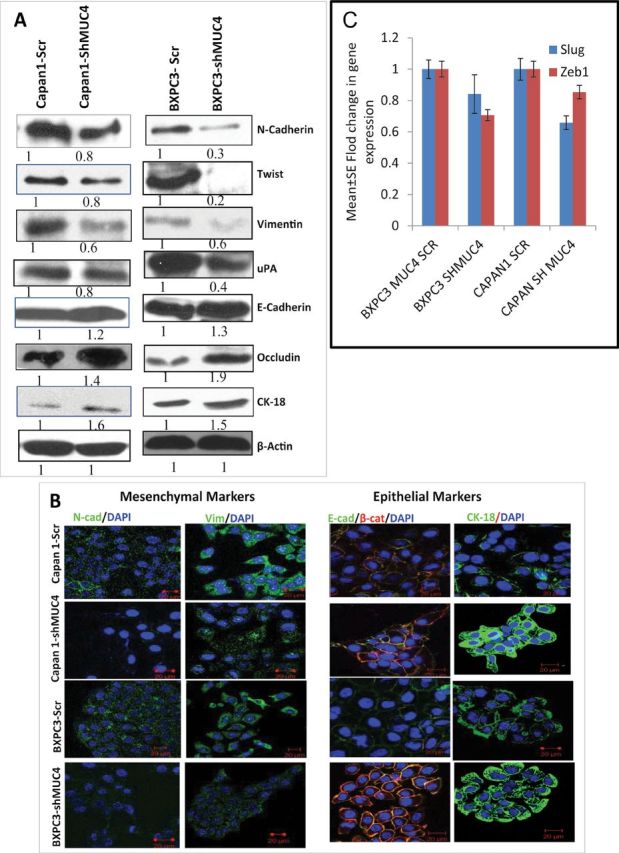
Role of MUC4 in the EMT process. (A) The immunoblot analysis showed significant upregulation of epithelial markers such as E-cadherin, Occludin and CK-18, and downregulation of mesenchymal markers, such as N-cadherin, Twist, uPA and vimentin, in Capan1/BxPC3-shMUC4 cells compared with Capan1/BxPC3-Scr vector cells. (B) Confocal microscopy showed increased staining for E-cadherin and CK-18 and a faint expression of N-cadherin and vimentin was observed in Capan1/BxPC3-shMUC4 compared with Capan1/BxPC3-Scr vector cells. FITC-conjugated goat anti-mouse IgG for secondary antibody and DAPI was used for nuclear staining. β-actin was used as a loading control in immunoblotting. (C) Real-time PCR analysis using primers that specifically amplify the Slug and Zeb1 genes showed reduced expression Slug and Zeb1 in MUC4 knockdown Capan1 and BxPC3 cells.
MUC4 upregulates N-cadherin expression through the FAK pathway in Capan1 and BxPC3 cells
N-cadherin is a key mediator of the EMT process in PC cells (32), and FAK activates N-cadherin expression in PC cells through the c-jun N-terminal kinase (JNK)-mediated pathway (33). A previous study from our laboratory has also shown that downregulation of MUC4 leads to decreased activation of FAK in pancreatic and ovarian cancer cells (22,28,31). As such, we were interested in looking at the effect of MUC4 knockdown on N-cadherin expression and the signalling pathway(s) involved. Interestingly, our results showed a significant downregulation of N-cadherin (Figures 2A, 2B and 4D), as well as pFAK (Y576/577, Y925) expression (Figure 3A and 3B), in the MUC4-silenced Capan1 and BxPC3 cells when compared with scramble vector transfected cells. Phosphorylated HER2 and pSrc are the key molecules involved in FAK activation by MUC4 (31). We checked the expression of these molecules in MUC4 knockdown Capan1/BXPC3 cells. Our western blot analysis showed a significant decrease in pHER2 and pSrc expression in Capan1 and BxPC3sh-MUC4 cells when compared with scramble vector transfected cells (Figure 3A and 3B). Difference in the levels of total FAK and HER2 was not observed (Figure 3A and 3B). We also analyzed the expression of downstream signaling molecules of FAK and observed that MUC4 knockdown leads to a decreased expression of activated pMKK7, pJNK and pc-Jun in both Capan1 and BxPC3-shMUC4 cells (Figure 3A and 3B); however, no change was observed in their total protein level (Figure 3A and 3B). Further, a recent study has shown that cadherin switch regulated at transcriptional level by Twist, Snail through PI3K/PTEN pathway (34). In the present study, we also observed the downregulation of pAkt, Twist, Slug and Zeb1 in MUC4 knockdown Capan1 and BXPC3 cells (Figures 2A, 2C, 4A and 4B). Therefore, N-cadherin expression is regulated partly through PI3K and JNK pathways.
Fig. 3.

MUC4-mediated upregulation of N-cadherin through FAK signaling. (A and B) Western blot analysis showed a significant upregulation of pHer2, pScr, pFAK, pMKK7, pJNK, pc-Jun and upregulate N-cadherin in Capan1/BxPC3-Scr vector cells as compared with Capan1/BxPC3-shMUC4. The total form of FAK, MKK7, JNK1/2, c-Jun molecules remains unchanged. β-actin was used as a loading control.
Fig. 4.
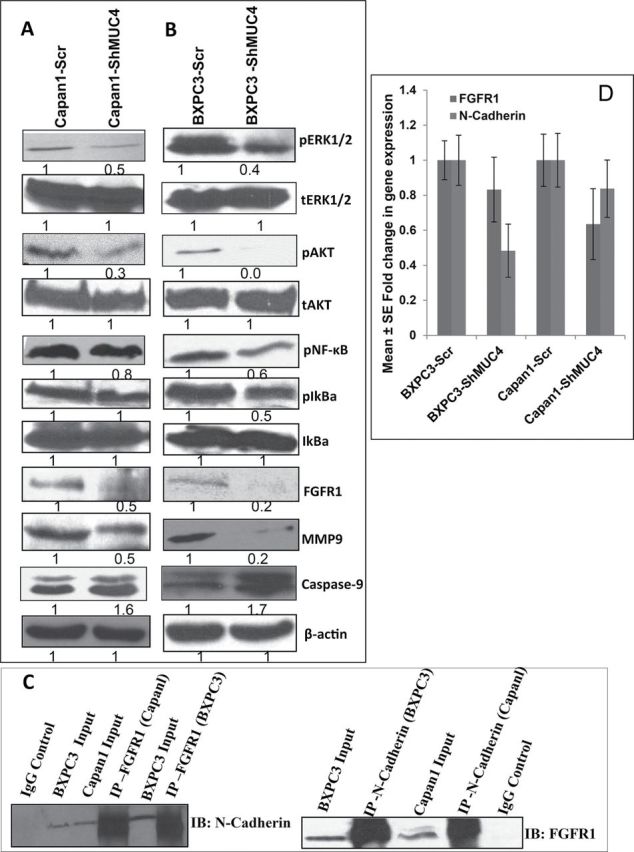
Effect of MUC4 knockdown on key signaling molecules in PC cells (A and B) involved in apoptosis (caspase 9), metastasis and invasion (pScr, phospho and total FAK, phospho and total Akt and phospho and total ERK1/2, NFkB, FGFR1, MMP-9) in Capan1/BxPC3-shMUC4 and Capan1/BxPC3-Scr vector cells. The results showed upregulation of Caspase-9 and downregulation of pSrc, pFAK (Tyr576/577, Tyr925), pERK1/2, PAkt, NFkB, FGFR1 in Capan1/BXPC3-shMUC4 compared with scramble vector transfected cells. The total form of FAK, Akt and ERK molecules remains unchanged. β-actin was used as a loading control. (C) Reciprocal co-immunoprecipitation analysis to show the interactions between N-cadherin and FGFR1. Lysates from the MUC4-expressing Capan1/BxPC3 cell lines were utilized for immunoprecipitation with N-cadherin and FGFR1 antibodies. The immunoprecipitates were electrophoretically resolved on 10% polyacrylamide gel and immunoblotted with anti-N-cadherin or anti-FGFR1 antibodies. The isotype antibodies were used as controls. (D) Real-time PCR analysis using primers that specifically amplify the FGFR1 and N-cadherin genes showed reduced expression of FGFR1 and N-cadherin in MUC4 knockdown Capan1and BxPC3 cells lines were utilized for immunoprecipitation with N-cadherin and FGFR1 antibodies.
MUC4 stabilizes FGFR1 through N-cadherin upregulation and activates downstream signaling pathways in Capan1 and BxPC3 cells
Several studies have shown that FGFR1 and FGFR2 are overexpressed in Pancreatic ductal adenocarcinoma (PDAC) and PC cell lines and it has also been shown that FGFR1 is associated with tumor growth and metastasis. Further, inhibition of FGFR1 leads to decreased invasion (35–38). Whereas N-cadherin interacts with and stabilizes FGFR1 by preventing receptor internalization and degradation, leading to MAPK/ERK1/2 activation and MMP-9 expression (39). Similarly, Peluso et al. (41) and Cavallaro et al. (40) also showed N-cadherin and FGFR1 interaction by co-immunoprecipitation (40,41). Our results showed that MUC4 knockdown resulted in the downregulation of N-cadherin (Figures 2A, 2B and 4D), as well as FGFR1 in both Capan1 and BxPC3-shMUC4 when compared with scramble vector transfected cells (Figure 4A, 4B and 4D). These observations led us to hypothesize that MUC4 upregulates N-cadherin expression, which interacts with FGFR1 and activates downstream signalling pathways resulting EMT, which facilitates invasion and metastasis of PC cells. To investigate this hypothesis, we first checked whether N-cadherin interacts with FGFR1 in PC cells. The reciprocal immunoprecipitation analysis revealed a clear interaction between N-cadherin and FGFR1, both in BxPC3 and Capan1 cells (Figure 4C). Subsequently, we investigated the effect of MUC4 knockdown on signaling pathways downstream of FGFR1. MUC4 knockdown resulted in a significant decrease in the activation of downstream signaling molecules such as pERK1/2 and pAkt (Ser473) (Figure 4A and 4B). Further, downregulation of pNFκB, pIkBα, MMP-9, uPA (Figure 4A and 4B) and upregulation of E-cadherin (Figure 2A and 2B) when compared with scramble vector transfected Capan1 and BXPC3 cells was also observed. The level of total ERK-1/2, and Akt, however, remained unchanged (Figure 2A 4A and 4B). These results support the hypothesis that silencing of MUC4 downregulates N-cadherin expression partly via Akt (34) and JNK1/2 mediated pathways, which in turn, destabilizes FGFR1, thus inhibiting its downstream signaling pathways.
MUC4 knockdown reduces motility/invasive potential and induces apoptosis in PC cells
Previous studies have demonstrated an important role of EMT in tumor progression where in tumor cells become more invasive and metastatic (42,43). We were also interested in examining the effect of MUC4 knockdown on the motility of Capan1 and BxPC3 PC cells. MUC4-knockdown (Capan1 and BxPC3-shMUC4) cells showed a significant decrease in motility (P < 0.0001) when compared with control Capan1-Scr and BxPC3-Scr cells as studied by trans-well migration (Supplementary Figure 1A and 1B, available at Carcinogenesis Online), as well as scratch assay (Supplementary Figure 1C and 1D, available at Carcinogenesis Online). MUC4 knockdown cells also demonstrated a significant decrease in their invasive potential of (Capan1 and BxPC3-shMUC4) when compared with scramble vector transfected cells (Supplementary Figure 1E and 1F, available at Carcinogenesis Online).
We have reported previously that MUC4 knockdown cells have reduced growth (25). Because the overall rate of cell growth is determined by the balance between cell proliferation and apoptosis, we were interested in looking at the effect of MUC4 knockdown on the apoptotic indices of Capan1 and BxPC3 cells. To analyze this, MUC4 knockdown and control cells were grown for 48h and trypsinized. The extent of apoptosis and necrosis was determined by Annexin-V and propidium iodide staining, followed by flow cytometry. The apoptotic, as well as necrotic, cells of both Capan1-shMUC4 and BxPC3-shMUC4 were significantly higher (P < 0.01 and 0.05, respectively) as compared with their respective scramble vector transfected cells (Supplementary Figure 1G, available at Carcinogenesis Online). Taken together, our results showed that MUC4 knockdown led to a decrease in cell survivability, as well as a decrease in the migration/invasion potential, of PC cells. Consistent with the enhanced apoptosis, we also observed an increased expression of cleaved Caspase-9 in the MUC4-silenced cells when compared with scramble vector transfected cells (Figure 4A and 4B).
In order to strengthen the aforementioned results, we knocked down N-cadherin in both Capan1 and BxPC3 cells and then checked the expression of pERK1/2, FGFR1, uPA and E-cadherin. Our western blot analysis showed that knockdown of N-cadherin resulted in the downregulation of FGFR1, N-cadherin, uPA, inactivation of pERK1/2 and upregulation of E-cadherin (Figure 5A and 5J). Our confocal analysis of BxPC3 cells also revealed that N-cadherin knockdown leads to an increased expression of E-cadherin (Figure 5B). Further, we also observed that N-cadherin knockdown also resulted in a significant reduction in the motility of BxPC3 and Capan1 cells when compared with scramble vector transfected cells (Figure 5C).
Fig. 5.


N-cadherin mediated stabilization of FGFR1. (A) Western blot analysis showed that significant upregulation of E-cadherin, downregulation of FGFR1, uPA, pERK1/2, in N-cadherin knockdown Capan1/BxPC3 cells as compared with vector controls (Capan1/BxPC3-scr/ShN-cadherin). The total form of ERK1/2 remains unchanged. β-actin was used as a loading control. (B) Confocal microscopy shows increased staining for E-cadherin in N-cadherin knockdown BxPC3-cells and diminished staining in BxPC3-Scr vector cells (scale bar 20 µm) (FITC-conjugated goat anti-mouse IgG for secondary antibody and DAPI was used for nuclear staining). (C). Capan1 and BxPC3 cells (1×106) were seeded on noncoated or matrigel-coated membranes for motility incubated for 24h. The lower chamber was filled with DMEM medium containing 10% fetal bovine serum used as a chemo attractant. The cells that migrate through trans-well membrane were fixed, stained and photographed for 10 random fields under bright-field microscopy (Magnification X10). The decreased motility and invasion were observed in N-cadherin knockdown Capan1/BxPC3 cells compared to scramble controls (***P < 0.00001 and ***P < 0.00001). (D) Effect of oncogenic MUC4 knockdown on size of orthotopically grown primary tumors. The mean weight of tumors formed by Capan1-shMUC4 cells was significantly less than that formed by the scrambled cells (P < 0.05). (E and F) are the hemotoxalin and eosin staining of primary tumors and (G–I) metastatic lesions from liver, intestine and spleen. (Δ indicates tumor, * indicates normal tissue). (J) Real-time PCR analysis using primers that specifically amplify the N-cadherin gene showed reduced expression N-cadherin in N-cadherin knockdown Capan1and BxPC3 cells.
Inhibition of MUC4 expression in PC cells results in the suppression of tumorigenicity and metastasis
To examine the effect of oncogenic MUC4 knockdown in vivo, Capan1-shMUC4 and Capan1-Scr cells (5 × 105 cell/animal) were orthotopically implanted into the pancreas of nude mice (six mice per group). The animals were killed at 21 days postimplantation, and the pancreatic tumors were removed and weighed (Figure 5D, 5E and 5F). The liver, lung, diaphragm, intestine, kidneys and mesenteric lymph nodes were carefully examined for the presence of metastatic lesions. A primary pancreatic tumor and metastatic lesions in the spleen, mesenteric lymph nodes and on the peritoneal wall were found in the majority of the mice implanted with Capan1-Scr cells. In this group, some animals also had metastasis in the liver (n = 3) and/or diaphragm (n = 1). In contrast, animals injected with Capan1-shMUC4 cells had significantly smaller tumors (P < 0.03) and had fewer or no metastatic lesions when compared with scramble vector transfected cells injected animals (Table 1, Figure 5G, 5H and 5I).
Table I.
Incidence of metastases developed by orthotopic implantation of pooled population Capan1-Scr cells and Capan1-shMUC4 in immune-deficient mice
| Cell type | Spleen | Liver | Peritoneum | Mesentric lymph nodes | Diaphram | Intestinal wall |
| Capan1 Scr | 5/6(83.33%) | 3/6(50%) | 4/6(66.66%) | 5/6(83.33%) | 1/6(16.66%) | 2/6(33.33%) |
| Capan1-shMUC4 | 0/6(0%) | 0/6(0%) | 2/6(33.33%) | 0/6(0%) | 0/6(0%) | 0/6(0%) |
| P-value (chi-square test) | 0.015 | 0.18 | 0.57 | 0.015 | 1 | 0.45 |
Discussion
MUC4 is aberrantly overexpressed in various carcinomas, including PC (12–18,20,22,23). Previous studies by our group showed a significant role of MUC4 in facilitating pancreatic and ovarian cancer cell motility by altering morphology, actin-cytoskeleton and downstream signaling events (25,28). Cancer invasion and metastasis occurs as a result of epithelial to mesenchymal transition and its underlying mechanisms that harmonize these processes in PC are not fully understood. In order to decipher the role of the glycoprotein MUC4 in regulating EMT in PC cells, we selectively silenced MUC4 expression in Capan1 and BXPC3 cells using specific shRNA. Subsequently, we studied the effect of MUC4 knockdown on EMT by performing various assays in vitro (motility, invasion, and apoptosis) and in vivo (tumorigenesis and metastasis). In addition, the downstream signaling pathways involved in EMT, which leads to invasion and metastasis of PC, were also observed. The stable expression of a shRNA against MUC4 resulted in a significant decrease in MUC4 expression at the mRNA, protein and cellular levels in Capan1-shMUC4 and BxPC3-shMUC4 compared with scramble vector transfected cells (Figure 1A, 1B, 1C and 1D).
The foremost indicator of EMT is a change in cellular morphology (44). In the present study, inhibition of MUC4 resulted in the cells becoming more circular in shape compared with the elongated, flat and spindle-shaped scramble vector transfected cells (Figure 1E and 1F). This was expected because of the established role of MUC4 in cell proliferation and survival as reorganization of the actin-cytoskeleton results in an altered morphology (25,26,28,31). The aggressive nature of cancer cells is partly due to increased cellular motility, which plays a significant role in the invasive properties of PC cells. We also observed a decreased in the cellular motility and invasion ability of MUC4 knockdown Capan1 and BxPC3 PC cells (Supplementary Figure 1A–1F, available at Carcinogenesis Online), which were further related to decreased activation of Akt, FAK and ERK pathways, downregulation of MMP9, uPA and an upregulation of E-cadherin, Occludin and CK18 (Figures 2A, 2B, 3A, 3B, 4A and 4B). These findings reinforce previous observations by our group that MUC4 plays a significant role in modulating cell motility, invasion and re-reorganizing the cytoskeleton of cancer cells (22,25–28,31).
EMT results in phenotypic changes that are accompanied by an increased cellular motility (8) and production of proteolytic enzymes resulting in the disruption of cell–cell adhesions, which are mediated by E-cadherin (45,46). In most solid tumors, the function of epithelial E-cadherin is altered due to various genetic and epigenetic mechanisms, such as activation of ERK1/2 signaling pathway. This, in turn, activates signaling molecules that promote tumor-cell migration, invasion and dissemination (8). We observed a downregulation of pERK1/2 (Figure 4A and 4B) and an increased expression of E-cadherin in the MUC4 knockdown cells (Figure 2A and 2B). These results suggest that oncogenic MUC4 inhibits E-cadherin function partly by upregulating Twist, Slug, Zeb1 via Akt pathway (Figures 2A, 2C, 4A and 4B), as well as N-cadherin and its interacting partner FGFR1 via JNK1/2 mediated pathways (Figures 2A, 4A, 4B), thus effecting downstream molecules such as ERK1/2 and MMP9. Further, this study, we also observed a decreased expression of matrix metalloproteinase-9 (MMP-9) in MUC4 knockdown Capan1 and BxPC3 cells (Figure 4A and 4B), which is a key mediator of the invasive property of tumor cells (47). Similarly, MUC4-mediated activation of the ERK1/2 pathway also promotes production of MMP-9, which in turn causes cleavage of E-cadherin, leading to the disruption of cell–cell contacts. Our results suggest that MUC4-mediated downregulation of E-cadherin may contribute to the highly metastatic property of Capan1and BxPC3 cells.
Suppression of MUC4 expression in Capan1 and BxPC3 cells led to a significant reduction in the expression of mesenchymal markers, such as Twist, Slug, Zeb1, N-cadherin, and vimentin, and an upregulation of epithelial markers, such as E-cadherin, Cytokeratin18, and Occludin (Figure 2A, 2B and 2C). During cancer metastasis, EMT is initiated and the process upregulates mesenchymal markers such as vimentin, N-cadherin and Vitronectin-75 while reducing the expression of epithelial markers such as E-cadherin and cytokeratin (9,10,48–50). Similarly, stable ectopic expression of MUC4 in SKOV3 cells led to the downregulation of E-cadherin and CK-18 and upregulation of N-cadherin and vimentin expression in cells (31).
Recent reports have shown that upregulation of N-cadherin through the activation of FAK (33) in PC cells. Subsequently, it has been shown that MUC4 upregulates N-cadherin in OC cells through FAK activation (31). In the current study, we further analyzed the mechanism(s) underlying MUC4-mediated upregulation of N-cadherin through the FAK signaling pathway in human PC cells and their further downstream signaling pathways (Figure 3A and 3B). The results of the current study provide a mechanistic basis for our previous studies on the role of MUC4 in PC (22,25) and OC (31). The results of the present study revealed that knockdown of oncogenic MUC4 resulted in decreased stabilization of pHER2, followed by decreased activation of pScr, FAK and its downstream molecules, such as MKK7, JNK1/2 and c-Jun, which further result in the downregulation of N-cadherin in PC cells (Figure 3A and 3B). Further, we also observed the downregulation of pAkt, Twist, Slug and Zeb1 in MUC4 knockdown Capan1 and BXPC3 cells (Figures 2A, 2C, 4A and 4B). Our results further corroborate the recent result showing role of PI3K/PTEN pathway role in cadherin switch at transcriptional level via regulating Twist and Snail in melanoma cells (34). Therefore, the present study indicated that MUC4 mediates N-cadherin upregulation partly through Akt pathway and partly by the JNK1/2 pathway in PC cells and EMT resulted in a switch between E-cadherin and N-cadherin expression levels (51,52). N-cadherin stabilizes FGFR1 by interacting with first FGFR1 Ig-like domain 1 (D1) and two (D2) extracellular domains (39). This interaction activates MAPK/ERK1/2, which in turn upregulates MMP-9 (39) Akt, AP1, uPA, and downregulates E-cadherin expression. We also observed an interaction between N-cadherin and FGFR1 in BxPC3 and Capan1 cells (Figure 4C) and effects downstream signalling molecules (Figure 2A, 4A and 4B).
Our data further showed that MUC4 has an anti-apoptotic effect, which is mediated by activation of the PI3-K/Akt pathway (53). In our study, inhibition of MUC4 also resulted in the suppression of ERK1/2 and pAkt (Figure 4A and 4B). Similarly, there was a significant decrease in the cellular levels of pNFkB and pIκB and an increase in the levels of caspase-9 in both Capan1-shMUC4 and BXPC3-shMUC4 cells (Figure 4A and 4B). These results suggest that MUC4 has a potential role in facilitating survival, proliferative ability, motility and invasive ability of PC cells. Thus, MUC4 emerges as a significant player in controlling important cellular processes during PC progression and metastasis, reinforcing its importance as a target for anticancer therapy.
Upon orthotopic implantation of MUC4 knockdown cells into the pancreas of nude mice, the Capan1-shMUC4 cells formed smaller tumors when compared with the Capan1-Scr control cells and did not cause morbidity (Figure 5D–5F, Table 1). However, there was no decrease in the incidence of tumors formed by Capan1-shMUC4 cells, but a significant decrease in metastasis to the vital organs was observed, suggesting that MUC4 may play a role in progression rather than the initiation of PC (Figure 5G–5I). The higher apoptotic and antitumorigenic effects of MUC4 knockdown cells were partially compensated by other unknown signaling pathways. The present study revealed that oncogenic MUC4 not only regulates tumor cell growth but also plays an important role in modulating the invasive nature of the malignant cells (Table 1).
Finally, we conclude that silencing of oncogenic MUC4 results in decreased expression of HER2, Src and FAK, which in turn leads to downregulation of the signalling pathway, resulting in reduced levels of N-cadherin partly through Akt- and JNK-mediated pathways. N-cadherin then stabilizes FGFR1 through its direct association and potentiates the downstream signaling pathways involved in promoting cell proliferation, inhibition of apoptosis, breaking cell–cell contacts and regulating expression of proteases such as MMP-9 and uPA. The observed inhibition of cell proliferation in the MUC4 knockdown cells may be mediated through the inhibition of the MAPK pathway, whereas the increase in metastatic properties of MUC4-expressing cells might be at least partly due to the activation of FAK and a decrease in the expression of E-cadherin (Figure 6). Overall, our studies demonstrate that PC cells that overexpress MUC4 promote the EMT process, leading to increased invasion and metastasis. The results of this study could be useful for targeting novel proteins downstream of MUC4 in order to disrupt signaling pathways involved in proliferation, anti-apoptosis, motility, invasion and metastasis.
Fig. 6.

Proposed model for MUC4-mediated signaling events that promote progression and metastasis of pancreatic cancer. MUC4-mediated activation of FAK induces N-cadherin expression via pMKK7, pJNK and pc-Jun activation. N-cadherin in turn interacts with FGFR1 at the plasma membrane and prevents its internalization and degradation promoting continuous activation of downstream signaling molecules Akt, ERK1/2, AP1 and NF-kB. Continuous activation of Akt, ERK1/2, AP1 and NF-kB will promote cell proliferation, survival, migration and metastasis. Moreover, MUC4-mediated upregulation of NF-kB can repress E-cadherin expression. The upregulated AP1 and NF-kB also induces expression of uPA, which promotes cancer cell invasion and metastasis.
Supplementary material
Supplementary Table 1 and Figure 1 can be found at http://carcin.oxfordjournals.org/
Funding
National Institute of Health (CA RO1 CA78590, RO1 CA133774, RO1 CA131944 and U54 CA163120).
Supplementary Material
Acknowledgements
We thank Ms. Kristi L. Berger for editing the manuscript. The authors acknowledge the invaluable technical support from Mr. Erik Moore and Ms. Kavita Mallya. We also thank Janice A. Taylor and James R. Talaska of the confocal laser scanning microscope core facility at UNMC for their support.
Glossary
Abbreviations:
- DMEM
Dulbecco's modified Eagle's medium
- EMT
epithelial to mesenchymal transition
- FAK
focal adhesion kinase
- FGFR1
fibroblast growth factor receptor 1
- MMP-9
matrix metalloproteinase-9
- PC
pancreatic cancer
Footnotes
Conflict of Interest Statement: None declared.
References
- 1. American Cancer Society (2010). Cancer Facts & Figures 2010 American Cancer Society; Atlanta: The top 5 cancer killers are (in order): lung, colon, breast, pancreatic, and prostate [Google Scholar]
- 2. Jemal A., et al. (2009). Cancer statistics, 2009 CA Cancer J.Clin. 59 225–249 [DOI] [PubMed] [Google Scholar]
- 3. Heinemann V., et al. (2008). Meta-analysis of randomized trials: evaluation of benefit from gemcitabine-based combination chemotherapy applied in advanced pancreatic cancer BMC Cancer 8:82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sultana A., et al. (2008). Meta-analyses of chemotherapy for locally advanced and metastatic pancreatic cancer: results of secondary end points analyses Br. J. Cancer 99 6–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Matsuno S., et al. (2004). Pancreatic Cancer Registry in Japan: 20 years of experience Pancreas 28 219–230 [DOI] [PubMed] [Google Scholar]
- 6. Mehlen P., et al. (2006). Metastasis: a question of life or death Nat. Rev. Cancer 6 449–458 [DOI] [PubMed] [Google Scholar]
- 7. Polyak K., et al. (2009). Transitions between epithelial and mesenchymal states: acquisition of malignant and stem cell traits Nat. Rev. Cancer 9 265–273 [DOI] [PubMed] [Google Scholar]
- 8. Thiery J.P. (2002). Epithelial-mesenchymal transitions in tumour progression Nat. Rev. Cancer 2 442–454 [DOI] [PubMed] [Google Scholar]
- 9. Ngan C.Y., et al. (2007). Quantitative evaluation of vimentin expression in tumour stroma of colorectal cancer Br. J. Cancer 96 986–992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kowalski P.J., et al. (2003). E-cadherin expression in primary carcinomas of the breast and its distant metastases Breast Cancer Res. 5 R217–R222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Peinado H., et al. (2007). Snail, Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype? Nat. Rev. Cancer 7 415–428 [DOI] [PubMed] [Google Scholar]
- 12. Carraway K.L., et al. (2001). Muc4/sialomucin complex in the mammary gland and breast cancer J. Mammary Gland Biol. Neoplasia 6 323–337 [DOI] [PubMed] [Google Scholar]
- 13. Chauhan S.C., et al. (2006). Aberrant expression of MUC4 in ovarian carcinoma: diagnostic significance alone and in combination with MUC1 and MUC16 (CA125) Mod. Pathol. 19 1386–1394 [DOI] [PubMed] [Google Scholar]
- 14. Handra-Luca A., et al. (2005). MUC1, MUC2, MUC4, and MUC5AC expression in salivary gland mucoepidermoid carcinoma: diagnostic and prognostic implications Am. J. Surg. Pathol. 29 881–889 [DOI] [PubMed] [Google Scholar]
- 15. Hollingsworth M.A., et al. (2004). Mucins in cancer: protection and control of the cell surface Nat. Rev. Cancer 4 45–60 [DOI] [PubMed] [Google Scholar]
- 16. Llinares K., et al. (2004). Diagnostic value of MUC4 immunostaining in distinguishing epithelial mesothelioma and lung adenocarcinoma Mod. Pathol. 17 150–157 [DOI] [PubMed] [Google Scholar]
- 17. Rakha E.A., et al. (2005). Expression of mucins (MUC1, MUC2, MUC3, MUC4, MUC5AC and MUC6) and their prognostic significance in human breast cancer Mod. Pathol. 18 1295–1304 [DOI] [PubMed] [Google Scholar]
- 18. Tsutsumida H., et al. (2007). MUC4 expression correlates with poor prognosis in small-sized lung adenocarcinoma Lung Cancer 55 195–203 [DOI] [PubMed] [Google Scholar]
- 19. Park H.U., et al. (2003). Aberrant expression of MUC3 and MUC4 membrane-associated mucins and sialyl Le(x) antigen in pancreatic intraepithelial neoplasia Pancreas 26 e48–e54 [DOI] [PubMed] [Google Scholar]
- 20. Swartz M.J., et al. (2002). MUC4 expression increases progressively in pancreatic intraepithelial neoplasia Am. J. Clin. Pathol. 117 791–796 [DOI] [PubMed] [Google Scholar]
- 21. Andrianifahanana M., et al. (2001). Mucin (MUC) gene expression in human pancreatic adenocarcinoma and chronic pancreatitis: a potential role of MUC4 as a tumor marker of diagnostic significance Clin. Cancer Res. 7 4033–4040 [PubMed] [Google Scholar]
- 22. Chaturvedi P., et al. (2008). MUC4 mucin interacts with and stabilizes the HER2 oncoprotein in human pancreatic cancer cells Cancer Res. 68 2065–2070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhu Y., et al. (2010). The increase in the expression and hypomethylation of MUC4 gene with the progression of pancreatic ductal adenocarcinoma Med. Oncol. 28 (suppl 1)S175–S184 [DOI] [PubMed] [Google Scholar]
- 24. Bafna S., et al. (2008). MUC4, a multifunctional transmembrane glycoprotein, induces oncogenic transformation of NIH3T3 mouse fibroblast cells Cancer Res. 68 9231–9238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chaturvedi P., et al. (2007). MUC4 mucin potentiates pancreatic tumor cell proliferation, survival, and invasive properties and interferes with its interaction to extracellular matrix proteins Mol. Cancer Res. 5 309–320 [DOI] [PubMed] [Google Scholar]
- 26. Moniaux N., et al. (2007). Human MUC4 mucin induces ultra-structural changes and tumorigenicity in pancreatic cancer cells Br. J. Cancer. 97 345–357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Singh A.P., et al. (2004). Inhibition of MUC4 expression suppresses pancreatic tumor cell growth and metastasis Cancer Res. 64 622–630 [DOI] [PubMed] [Google Scholar]
- 28. Ponnusamy M.P., et al. (2008). MUC4 activates HER2 signalling and enhances the motility of human ovarian cancer cells Br. J. Cancer 99 520–526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Moniaux N., et al. (2004). Generation and characterization of anti-MUC4 monoclonal antibodies reactive with normal and cancer cells in humans J. Histochem. Cytochem. 52 253–261 [DOI] [PubMed] [Google Scholar]
- 30. Rachagani S., et al. (2011). Activated KrasG(1)(2)D is associated with invasion and metastasis of pancreatic cancer cells through inhibition of E-cadherin Br. J. Cancer 104 1038–1048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ponnusamy M.P., et al. (2010). MUC4 mucin-induced epithelial to mesenchymal transition: a novel mechanism for metastasis of human ovarian cancer cells Oncogene 29 5741–5754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nakajima S., et al. (2004). N-cadherin expression and epithelial-mesenchymal transition in pancreatic carcinoma Clin. Cancer Res. 10 4125–4133 [DOI] [PubMed] [Google Scholar]
- 33. Shintani Y., et al. (2008). Collagen I-mediated up-regulation of N-cadherin requires cooperative signals from integrins and discoidin domain receptor 1 J. Cell Biol. 180 1277–1289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hao L., et al. (2012). Cadherin switch from E- to N-cadherin in melanoma progression is regulated by the PI3K/PTEN pathway through Twist and Snail Br. J. Dermatol. 166 1184–1197 [DOI] [PubMed] [Google Scholar]
- 35. Ishiwata T., et al. (1998). Characterization of keratinocyte growth factor and receptor expression in human pancreatic cancer Am. J. Pathol. 153 213–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kobrin M.S., et al. (1993). Aberrant expression of type I fibroblast growth factor receptor in human pancreatic adenocarcinomas Cancer Res. 53 4741–4744 [PubMed] [Google Scholar]
- 37. Liu Z., et al. (2007). Identification of a fibroblast growth factor receptor 1 splice variant that inhibits pancreatic cancer cell growth Cancer Res., 67 2712–2719 [DOI] [PubMed] [Google Scholar]
- 38. Taeger J, et al. (2011). Targeting FGFR/PDGFR/VEGFR impairs tumor growth, angiogenesis, and metastasis by effects on tumor cells, endothelial cells, and pericytes in pancreatic cancer Mol. Cancer Ther., 10 2157–2167 [DOI] [PubMed] [Google Scholar]
- 39. Suyama K, et al. (2002). A signaling pathway leading to metastasis is controlled by N-cadherin and the FGF receptor Cancer Cell 2 301–314 [DOI] [PubMed] [Google Scholar]
- 40. Cavallaro U, et al. (2001). N-CAM modulates tumour-cell adhesion to matrix by inducing FGF-receptor signalling Nat. Cell Biol., 3 650–657 [DOI] [PubMed] [Google Scholar]
- 41. Peluso J.J. (2000). N-cadherin-mediated cell contact regulates ovarian surface epithelial cell survival Biol. Signals Recept., 9 115–121 [DOI] [PubMed] [Google Scholar]
- 42. Kalluri R, et al. (2009). The basics of epithelial-mesenchymal transition J. Clin. Invest., 119 1420–1428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kalluri R. (2009). EMT: when epithelial cells decide to become mesenchymal-like cells J. Clin. Invest., 119 1417–1419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Voulgari A, et al. (2009). Epithelial-mesenchymal transition in cancer metastasis: mechanisms, markers and strategies to overcome drug resistance in the clinic Biochim. Biophys. Acta., 1796 75–90 [DOI] [PubMed] [Google Scholar]
- 45. Christofori G, et al. (1999). The role of the cell-adhesion molecule E-cadherin as a tumour-suppressor gene Trends Biochem. Sci., 24 73–76 [DOI] [PubMed] [Google Scholar]
- 46. Takeichi M. (1995). Morphogenetic roles of classic cadherins Curr. Opin. Cell Biol., 7 619–627 [DOI] [PubMed] [Google Scholar]
- 47. Fridman R, et al. (2003). Cell surface association of matrix metalloproteinase-9 (gelatinase B) Cancer Metastasis Rev., 22 153–166 [DOI] [PubMed] [Google Scholar]
- 48. Stockinger A, et al. (2001). E-cadherin regulates cell growth by modulating proliferation-dependent beta-catenin transcriptional activity J. Cell Biol., 154 1185–1196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Valdes F, et al. (2002). The epithelial mesenchymal transition confers resistance to the apoptotic effects of transforming growth factor Beta in fetal rat hepatocytes Mol. Cancer Res., 1 68–78 [PubMed] [Google Scholar]
- 50. Yang J, et al. (2004). Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis Cell 117 927–939 [DOI] [PubMed] [Google Scholar]
- 51. Gravdal K, et al. (2007). A switch from E-cadherin to N-cadherin expression indicates epithelial to mesenchymal transition and is of strong and independent importance for the progress of prostate cancer Clin. Cancer Res., 13 7003–7011 [DOI] [PubMed] [Google Scholar]
- 52. Maeda M, et al. (2005). Cadherin switching: essential for behavioral but not morphological changes during an epithelium-to-mesenchyme transition J. Cell Sci., 118 873–887 [DOI] [PubMed] [Google Scholar]
- 53. Downward J. (2004). PI 3-kinase, Akt and cell survival Semin. Cell Dev. Biol., 15 177–182 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.