

Abstract
Spinal muscular atrophy (SMA) is an autosomal recessive neuromuscular disease caused by mutations in the survival of motor neuron 1 (SMN1) gene and deficient expression of the ubiquitously expressed SMN protein. Pathologically, SMA is characterized by motor neuron loss and severe muscle atrophy. During muscle atrophy, the E3 ligase atrogenes, atrogin-1 and muscle ring finger 1 (MuRF1), mediate muscle protein breakdown through the ubiquitin proteasome system. Atrogene expression can be induced by various upstream regulators. During acute denervation, they are activated by myogenin, which is in turn regulated by histone deacetylases 4 and 5. Here we show that atrogenes are induced in SMA model mice and in SMA patient muscle in association with increased myogenin and histone deacetylase-4 (HDAC4) expression. This activation during both acute denervation and SMA disease progression is suppressed by treatment with a histone deacetylase inhibitor; however, this treatment has no effect when atrogene induction occurs independently of myogenin. These results indicate that myogenin-dependent atrogene induction is amenable to pharmacological intervention with histone deacetylase inhibitors and help to explain the beneficial effects of these agents on SMA and other denervating diseases.
INTRODUCTION
Spinal muscular atrophy (SMA) is a neuromuscular disorder characterized by loss of motor neurons and skeletal muscle weakness and atrophy. SMA is caused by deficient expression of the survival motor of neuron (SMN) protein due to a deletion or other mutations in the SMN1 gene (1). A nearly identical gene, SMN2, is retained in SMA patients. SMN1 primarily produces a full-length SMN transcript, while SMN2 primarily produces an alternatively spliced isoform lacking exon 7 (2,3). Nonetheless, increased SMN2 copy number ameliorates SMA disease severity in a dose-dependent fashion (4–6). SMA model mice, which are null for the endogenous mouse Smn gene, but express the human SMN2 transgene (‘delta 7’ mice) show progressive weakness, decreased motor neuron number, low body weight and an average lifespan of just 13 days (7). Treatment of SMA mice with the histone deacetylase (HDAC) inhibitor trichostatin A (TSA) improves body weight, life span, myofiber number and myofiber cross-sectional area (8). Beneficial effects of TSA on survival, muscle mass and motor function have also been shown in a mouse model of amyotrophic lateral sclerosis (9). TSA has been shown to modestly increase SMN expression (8,10,11), but as an HDAC inhibitor it may also inhibit HDAC4 activity, which has been shown to modulate the expression of genes involved in muscle atrophy. We hypothesized that TSA improves SMA muscle pathology, in part, by inhibiting this muscle atrophy pathway.
Muscle atrophy occurs in a variety of denervating diseases (12) and with disuse (13). During atrophy, muscle protein is degraded through the ubiquitin proteasome pathway (14). Expression of genes encoding the skeletal muscle-specific E3 ligases atrogin-1 and muscle ring finger 1 (MuRF1), referred to as atrogenes, is increased in atrophy models, including fasting and glucocorticoid treatment, as well as denervation (15). MuRF1 and atrogin-1 null mice are resistant to loss of muscle mass after denervation (15).
Upregulation of the atrogenes is mediated by multiple pathways, including reduction in the activity of the PI3K/Akt signaling cascade, which activates the FoxO1/3 transcription factors (16,17). MuRF1 upregulation in tumor-bearing mice is mediated by the transcription factor NF-κB, which is activated by tumor necrosis factor alpha (TNF-α) and other pro-inflammatory cytokines (18). In atrophy due to acute denervation, atrogin-1 and MuRF1 expression is regulated in part by HDACs and myogenin. Following denervation, HDAC4 is upregulated and suppresses the expression of Dach2, a transcription factor and a repressor of the myogenin promoter (19,20). Myogenin is a muscle-specific transcription factor essential for muscle development (21). Myogenin expression increases following denervation via increased HDAC4 and decreased Dach2 expression (19). Myogenin, in turn, positively regulates HDAC4 expression following denervation, creating a positive feedback loop. Atrogin-1 and MuRF1 expression and muscle atrophy following denervation are attenuated in myogenin-null mice (22,23). While these studies modeled neurogenic atrophy by severing the sciatic nerve, it remains to be determined whether (i) this pathway is conserved in a chronic motor neuron disease such as SMA and (ii) myogenin expression can be pharmacologically modulated to abrogate atrogene induction.
In this study, we demonstrate that the muscle-specific E3 ligases atrogin-1 and MuRF1 are upregulated in a cellular model of muscle atrophy and in the muscle of SMA mice and patients. We provide evidence that myogenin mediates induction of these atrogenes, and that TSA inhibits their upregulation. Thus, it is likely that this HDAC inhibitor mitigates SMA disease manifestations by inhibition of atrogene upregulation in addition to upregulation of SMN expression.
RESULTS
Atrogin-1 and MuRF1 are upregulated in SMA mouse muscle and reduced by TSA treatment
We and others have previously shown that myofibers are severely reduced in size in multiple muscles of SMA mice at the end-stage of disease, postnatal day 13 (P13), but not at P5 (8,24,25). In order to evaluate whether atrogenes are induced in SMA mice and whether this activation can be inhibited by HDAC inhibition, we treated a cohort of mice with either TSA (10 mg/kg) or vehicle (dimethyl sulfoxide, DMSO) and extracted RNA from total hindlimb muscles. The expression of atrogin-1 and MuRF1 was increased by ∼20-fold and 6-fold, respectively, in SMA mice compared with phenotypically normal heterozygous littermates at P13 (Fig. 1A). As we have also previously shown that a 16% decrease in myofiber size occurs specifically between P9 and P13 in the tibialis anterior muscle of SMA mice (24), we further characterized the timing of atrogene induction. The atrogenes were similarly expressed in SMA mice and healthy littermates at P9, however they were significantly upregulated in SMA mice at P11 (Fig. 1A). Western blot analysis confirmed that increased atrogene gene expression was accompanied by increased protein expression (Fig. 1B). Thus, atrogene induction corresponds temporally with the onset of muscle atrophy as well as with the onset of weight loss in these mice (Supplementary Material, Fig. S1) (8). Treatment with TSA significantly inhibited atrogin-1 and MuRF1 activation at both P11 and P13 (Fig. 1A and B).
Figure 1.

Atrogin-1 and MuRF1 expression are upregulated in muscle of SMA mice. (A) The mice were treated starting at P5 with either vehicle (DMSO) or TSA (10 mg/kg) daily and sacrificed at P9, P11 and P13. Muscle tissue was harvested and RNA isolated for qRT-PCR analysis. (B) Mice were treated starting at P5 with either vehicle (DMSO) or TSA (10 mg/kg) daily and sacrificed at P11. Muscle tissue was harvested and protein isolated for western blot analysis. Quantification of western data is shown in the right panel. Values represent the mean ± SEM. (*, P < 0.05; **, P< 0.01).
TSA reduces atrogin-1 and MuRF1 expression and inhibits protein breakdown in C2C12 cells
To examine the molecular mechanisms by which HDAC inhibition may suppress atrogin-1 and MuRF1 expression, we treated C2C12 myotubes with dexamethasone (10 µm), TSA (50 nm) or a combination of TSA and dexamethasone for 24 h. A synthetic glucocorticoid, dexamethasone, promotes protein breakdown and induces atrogin-1 and MuRF1 expression in myotube cultures (16) and adult mouse muscle (26). We found that atrogin-1 and MuRF1 expression in the C2C12 myotubes was increased with dexamethasone treatment and that this effect was blocked by TSA (Fig. 2A). To determine whether this was due to decreased transcription at the promoter, we transfected C2C12 myoblasts with reporter constructs containing the atrogin-1 and MuRF1 promoters upstream of luciferase and quantified luciferase expression in the presence or absence of TSA. TSA blocked atrogin-1 and MuRF1 promoter activity in a dose-dependent manner (Fig. 2B). Interestingly, TSA did not affect atrogene promoter activity when the reporters were expressed in a non-muscle cell line human embryonic kidney cell line, HEK-293T, suggesting that the effect of TSA may be mediated by muscle-specific factors (Supplementary Material, Fig. S2). The structurally unrelated HDAC inhibitor valproic acid also reduced atrogin-1 and MuRF1 promoter reporter expression in C2C12 cells, indicating that atrogene downregulation was likely not due to an off-target effect of TSA (Supplementary Material, Fig. S3).
Figure 2.
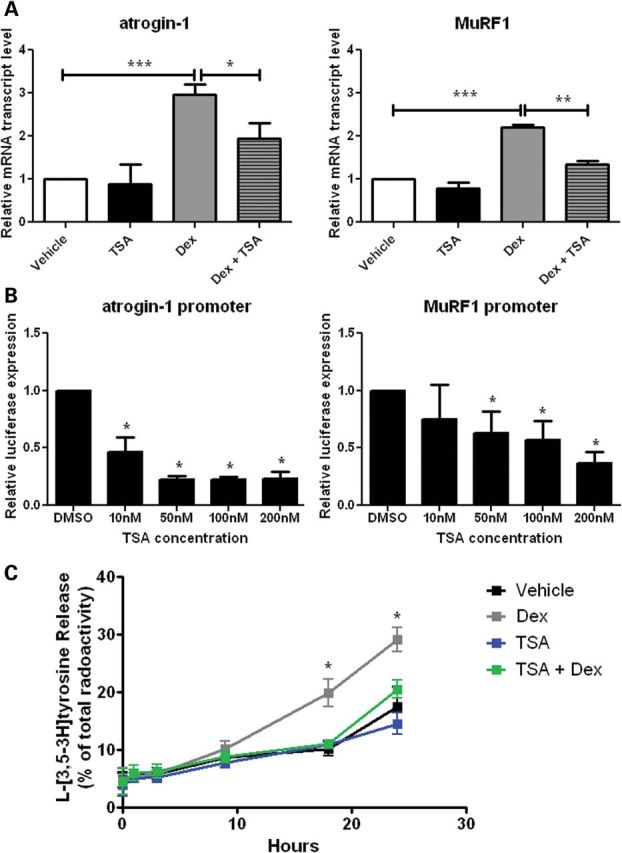
TSA inhibits atrogin-1 and MuRF1 induction and muscle protein breakdown in C2C12 myotubes. (A) qRT-PCR analysis of atrogin-1 and MuRF1 expression in C2C12 cells after 24-h treatment with dexamethasone (Dex, 10 µm), and/or TSA (50 nm). The expression level is represented relative to the vehicle control (n= 4, experiments per treatment). (B) Transfection of C2C12 myotubes with either an atrogin-1 or a MuRF1 luciferase reporter and treatment with DMSO, 10 nm, 50 nm, 100 nm or 200 nm TSA. Luciferase activity is represented relative to DMSO control (n= 4, experiments per dose). (C) The rate of protein breakdown in C2C12 myotubes labeled with l-[3,5-3H]tyrosine. Myotubes were treated with vehicle, Dex (10 µm) and/or TSA (50 nm), and release of l-[3,5-3H]tyrosine from cell proteins was used as an index of the breakdown of long-lived myofibrillar proteins. Dex treatment compared with Dex and TSA treatment (n= 3, experiments per treatment). Values represent the mean ± SEM (*, P < 0.05; **, P< 0.01;***, P< 0.001).
The maintenance of muscle mass is controlled by a balance between protein synthesis and protein degradation, which shifts toward protein degradation during atrophy. Atrogene expression stimulates proteasomal and autophagic/lysosomal proteolysis (27). We therefore tested whether TSA inhibits muscle protein degradation. After bulk labeling cellular proteins in myotubes with [3H]tyrosine for 20 h, the rate of degradation of long-lived proteins was measured in C2C12 myotubes after treatment with dexamethasone (10 µm), TSA (50 nm), both TSA and dexamethasone or vehicle alone. Treatment with dexamethasone stimulated overall proteolysis compared with vehicle-treated myotubes (Fig. 2C). However, after 18 h, TSA inhibited the increase in dexamethasone-induced proteolysis. Together, these data indicate that TSA suppresses the induction of atrogin-1 and MuRF1 and inhibits muscle protein breakdown in cultured myotubes.
TSA modulates atrogenes by reducing myogenin expression
Myogenin is upregulated in skeletal muscle following acute denervation and in turn increases the expression of MuRF1 and atrogin-1 (22,23). Reducing myogenin in mice abrogates the expression of atrogin-1 and MuRF1 following denervation and slows muscle atrophy. Thus, myogenin plays a dual role as both an inducer of neurogenic atrophy and a regulator of muscle development [reviewed by (28)]. We thus sought to determine whether the decreased expression of atrogin-1 and MuRF1 levels with TSA is associated with reduction of myogenin. We found that myogenin mRNA expression and protein levels were modestly increased in C2C12 myotubes treated with dexamethasome and this was inhibited by co-administration of TSA (Fig. 3A and B). HDAC4 is upregulated in skeletal muscle upon denervation and represses the expression of Dach2, a negative regulator of myogenin (19). We examined Dach2 levels in myotubes and found that Dach2 expression is indeed increased in the presence of TSA (Fig. 3C). Thus, in C2C12 myotubes, TSA suppresses the expression of myogenin, MuRF1 and atrogin-1, and upregulates the expression of the repressor, Dach2.
Figure 3.

TSA treatment modulates myogenin and Dach2 expression in C2C12 myotubes. C2C12 cells were treated with DMSO, dexamethasone (Dex, 10 µm) and/or TSA (50 nm). qRT-PCR analysis of myogenin (A) and Dach2 (C) expression (n= 4, experiments per treatment). Western blot analysis of myogenin (B) protein levels (n= 3, experiments per treatment). Quantification of western data is shown in the lower panel. Values represent mean ± SEM. (*, P < 0.05; **, P< 0.01).
HDAC inhibition reduces myogenin levels in SMA mice
Given that TSA repressed myogenin expression in cultured myotubes (Fig. 3A) (29), we next investigated whether the same is true in SMA mice. To examine the temporal sequence of events, we examined myogenin expression at P9, P11 and P13. While myogenin levels in untreated SMA mice were similar to heterozygous littermates at P9, TSA-treated SMA mouse muscle had significantly decreased myogenin expression (Fig. 4A). At P11, when we first detected induction of the atrogenes (Fig. 1A), we found that myogenin levels were upregulated in SMA mice, while TSA-treated SMA mice had no significant myogenin induction. Myogenin levels were similar in heterozygous littermates treated with TSA or DMSO (data not shown).
Figure 4.

Expression of myogenin is increased in SMA mice, and this effect is abrogated by TSA. (A) Mice were treated starting at P5 with either vehicle (DMSO) or TSA (10 mg/kg) daily and sacrificed at P9, P11 or P13. Muscle tissue was harvested and RNA isolated for qRT-PCR analysis of myogenin levels. (B and C) Mice were treated starting at P5 with either vehicle (DMSO) or TSA (10 mg/kg) daily and sacrificed at P11. Muscle tissue was harvested and RNA isolated for qRT-PCR analysis of Dach2 and HDAC4 levels. (D) Mice were treated starting at P5 with either vehicle (DMSO) or TSA (10 mg/kg) daily and sacrificed at P11. Muscle tissue was harvested and chromatin was isolated for ChIP analysis of myogenin association with atrogin-1 and MuRF1 promoters. Values represent the mean ± SEM. (*, P < 0.05; **, P< 0.01).
We also examined the expression of the upstream myogenin-negative regulator Dach2 at P9 and P11 in SMA muscle. Dach2 levels were unchanged at P9 (Supplementary Material, Fig. S4), but ∼150-fold lower in vehicle-treated SMA mice compared with heterozygous littermates at P11 (Fig. 4B). Furthermore, treatment with TSA resulted in ∼12-fold and 2-fold increases in Dach2 expression at P9 and P11, respectively (Supplementary Material, Fig. S4 and Fig. 4B). HDAC4 expression was increased in SMA mice compared with heterozygous littermates at P11 (Fig. 4C). TSA treatment did not decrease HDAC4 transcript levels, suggesting that TSA results in increased Dach2 expression by inhibiting HDAC4 activity rather than its expression.
We next examined whether myogenin was directly interacting with the promoters of the atrogenes to induce their expression in SMA mice. E boxes (CANNTG) that mediate myogenin binding are located in the promoters for FBXO32 (atrogin-1) (−79 bp) and TRIM63 (MuRF1) (−143 bp, −66 bp, −44 bp) (22). We examined the binding of myogenin to the endogenous MuRF1 and atrogin-1 promoters by performing chromatin immunoprecipitation assays using chromatin extracts from P11 hindlimb muscles. Chromatin was immunoprecipitated with antibodies against either myogenin or immunoglobulin G as a control. We found increased association of myogenin with the endogenous atrogin-1 and MuRF1 promoters in chromatin extracts from vehicle-treated SMA mice compared with heterozygous littermates (Fig. 4D); however, little association was observed in SMA mice treated with TSA. Together, these data suggest that myogenin directly activates atrogene expression in SMA muscle via transactivation of the atrogene promoters. Furthermore, myogenin activation in SMA possibly occurs as a consequence of increased HDAC4 and reduced Dach2 expression and this can be inhibited by HDAC inhibitors.
HDAC inhibition suppresses myogenin-dependent, but not myogenin-independent atrogene induction
Because the atrogenes can be induced by fasting, and SMA mice likely have inadequate nutrition at the end-stage of disease (11,30,31), we examined whether TSA is capable of repressing atrogene induction that occurs during fasting. Wild-type mice were treated with 10 mg/kg of TSA daily for 3 days, and then food was removed and the animals were sacrificed 18 h later. Atrogin-1 and MuRF1 expression was markedly increased in the fasting mice, but this effect was not blocked by TSA treatment (Fig. 5A). Importantly, myogenin expression was also not induced in the fasting mice, indicating that during starvation atrogene activation occurs independently of myogenin (Fig. 5B). We next examined the effects of TSA on mice with acute denervation atrophy as in this context atrogene induction is myogenin and HDAC4/5 dependent (19,20,22,23). Wild-type mice were treated with 10 mg/kg of TSA daily for 3 days before and 3 days after sciatic nerve transection. The sciatic nerve of the conralateral leg was left intact and used as a control. Increased myogenin, atrogin-1 and MuRF1 expression in the denervated muscle was almost completely inhibited by TSA treatment (Fig. 5C and D). Together these data highlight that atrogene induction can occur via distinct upstream mechanisms, and that TSA specifically inhibits the myogenin-dependent pathway.
Figure 5.

The effect of TSA on atrogene expression is myogenin-dependent. (A and B) Starved wild-type mice were treated by intraperitoneal injection with vehicle (DMSO) or 10 mg/kg of TSA daily for 3 days and sacrificed 18 h after removal of food. Muscle tissue was harvested from fed mice and DMSO and TSA-treated starved mice and RNA isolated for qRT-PCR analysis of myogenin, atrogin-1 and MuRF1 levels. (C and D) Wild-type mice were treated by intraperitoneal injection with vehicle (DMSO) or 10 mg/kg of TSA daily for 3 days before and 3 days after denervation by sciatic nerve cut. The contralateral leg remained innervated and was used as a control. Muscle tissue was harvested from both hind legs of DMSO and TSA-treated mice. RNA was isolated for qRT-PCR analysis of myogenin, atrogin-1 and MuRF1 levels. Values represent the mean ± SEM. (*, P < 0.05; **, P< 0.01;***, P< 0.001).
The atrogenes and myogenin are induced in human SMA muscle
To verify that myogenin-dependent atrogene activation is relevant to SMA in humans, we examined the expression of the atrogenes in the iliopsoas, a severely affected muscle, and the diaphragm, a relatively spared muscle, in type I SMA patients and age-matched controls (Fig. 6A). Atrogin-1, MuRF1, myogenin and HDAC4 were all significantly upregulated in SMA iliopsoas muscle, but unchanged in the diaphragm (Fig. 6B). These data are consistent with increased HDAC4 activity leading to increased expression of myogenin and upregulation of atrogenes in SMA human muscle (Fig. 7).
Figure 6.
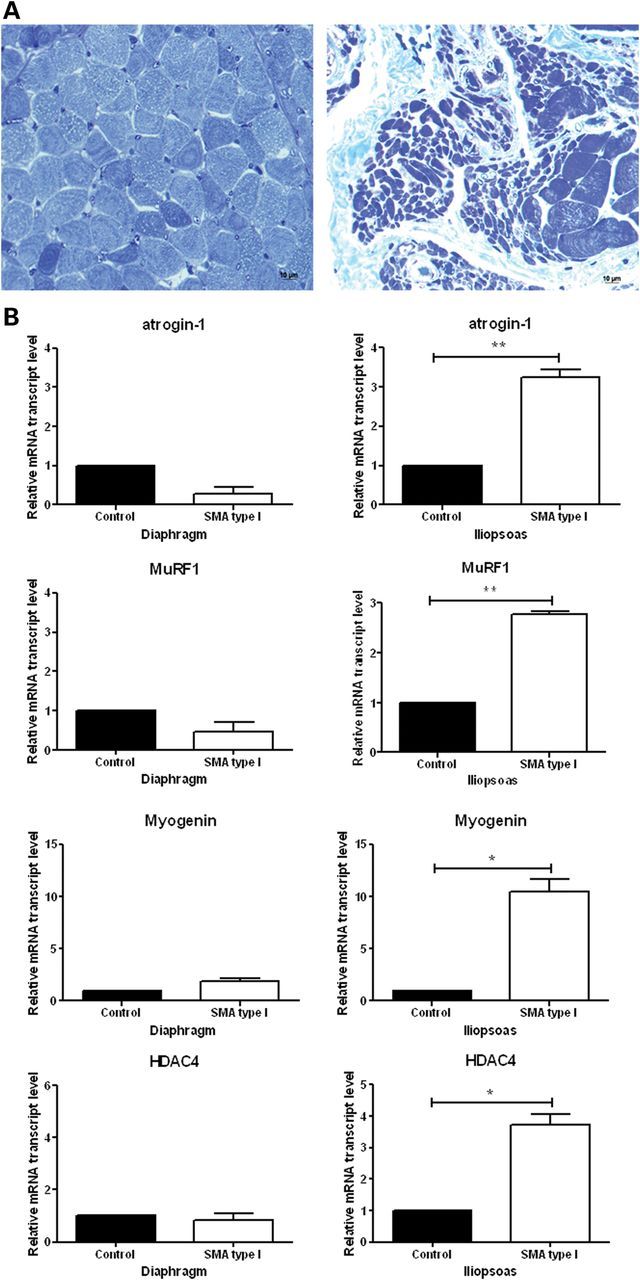
Atrophy-related genes are activated in the iliopsoas muscle, but not in the diaphragm muscle isolated from type I SMA patients. (A) Toluidine-blue stained muscle cross sections show severe muscle atrophy in the iliopsoas (right), a clinically severely affected muscle, but not in the diaphragm (left), a clinically relatively spared muscle. (B) RNA isolated from muscle tissues for qRT-PCR analysis of atrogin-1, MuRF1, myogenin and HDAC4. Values represent the mean ± SEM. (*, P < 0.05; **, P< 0.01).
Figure 7.

Proposed model of muscle atrophy in SMA. In SMA muscle, upregulation of HDAC4/5 inhibits the expression of the myogenin repressor Dach2 and results in increased myogenin expression. Myogenin then activates the atrogin-1 and MuRF1 promoters. TSA inhibits the activity of HDAC4/5, blocking the repression of Dach2. Increased Dach2 reduces myogenin expression and thus prevents the activation of atrogin-1 and MuRF1.
DISCUSSION
In this study, we show that the atrogenes atrogin-1 and MuRF1 are activated in muscle isolated from both mice and human patients with the motor neuron disease SMA. This induction is associated with increased expression of the upstream regulators, HDAC4 and myogenin, which is consistent with the pattern of gene activation previously observed during acute denervation-induced muscle atrophy (22,23). Importantly, this induction during both acute denervation and chronic SMA is markedly suppressed by the HDAC inhibitor TSA. In contrast, TSA does not suppress atrogene activation during fasting-induced muscle atrophy, which we show to occur independently of myogenin. These experiments highlight the distinct upstream pathways that regulate atrogene induction and show that myogenin-dependent atrogene activation may be suppressed pharmacologically with HDAC inhibitors. Thus, these drugs may have therapeutic relevance in multiple denervating disorders.
SMA mice show a profound reduction in the total muscle cross-sectional area and myofiber diameter at the end-stage of disease (8,24). Although specific muscle groups show structural denervation of neuromuscular junctions (NMJs) (32), the NMJ endplates of many muscles retain presynaptic terminals (24,25,33). Nonetheless, detailed structural analysis has revealed presynaptic neurofilament accumulations and reduced synaptic vesicle densities in most SMA NMJs and electrophysiological analysis has shown abnormalities of synaptic vesicle release. Furthermore, central synaptic inputs to motor neurons are also abnormal in SMA mice (32,34), and this may have downstream consequences for NMJ function. Myofiber atrophy is likely activated in SMA muscle once these functional NMJ abnormalities reach a critical threshold, without the requirement for complete structural denervation of muscle. Here we show that the atrogenes are induced at P11 in SMA mice, which corresponds well with the time period of decreasing muscle size (24) and weight loss. Importantly, this induction is near completely suppressed by treatment with the HDAC inhibitor TSA. We had previously shown that TSA improved the survival and motor function of SMA mice. Interestingly, we observed a significant improvement in muscle and myofiber size without a change in motor neuron numbers (8). Although TSA modestly increases SMN expression, we suspected that TSA may have independent beneficial effects on muscle. The current study indicates that suppression of myogenin-dependent atrogene induction with TSA is likely an important mechanism mitigating muscle atrophy in SMA mice.
Atrogin-1 and MuRF1 are induced in multiple models of skeletal muscle atrophy. While atrogin-1 targets transcription factors important for muscle growth and differentiation for degradation, MuRF1 mediates the degradation of structural proteins such as myosin heavy chain proteins (MyHC), myosin light chains 1 and 2, myosin binding protein C, muscle creatine kinase and other myofibrillar proteins (35–39). Together, these enzymes play essential roles in the loss of muscle mass.
The transcription factor myogenin induces the atrogenes by binding their promoters and activating transcription. TSA prevents induction of atrogenes by modulating the upstream regulators of myogenin expression, HDAC4 and Dach2. HDAC4 inhibits the expression of Dach2, which represses myogenin expression. We show here that HDAC4 is induced at P11 in SMA model mice, and this is associated with a marked reduction in the expression of Dach2. While TSA did not alter the expression of HDAC4 in TSA-treated mice, Dach2 expression was increased 2-fold, suggesting that TSA likely exerts its effect through HDAC4 inhibition rather than altering HDAC4 expression. Nevertheless, treatment with TSA almost completely blocked induction of myogenin in SMA mice, with only a modest increase in Dach2 levels. While it is possible that a 2-fold induction of Dach2 is sufficient to block induction of myogenin, it is likely that TSA affects other myogenin modulators. Identification of these potential modulators could provide additional therapeutic targets.
In this study, we also showed the relevance of this atrophy pathway to the human disease by examining atrogene and myogenin expression in human muscle samples isolated from type I SMA patients. We found upregulated expression of these genes in a clinically and pathologically severely affected muscle, the iliopsoas, but little induction in a relatively spared muscle, the diaphragm. Our data contrast with a previous study in which type I and type III SMA patient muscle biopsy samples were examined by microarray and quantitative reverse transcriptase-polymerase chain reaction (qRT-PCR). Downregulation of atrogin-1expression was found in SMA patients (40). Expression changes in MuRF1 were not reported in this study. The difference in atrogin-1 expression levels in the two studies could be attributed to the different muscles examined and the stage of the disease when the muscle was collected. Iliopsoas and diaphragm muscles used in this study were collected at autopsy, while the muscle samples used by Millino et al. came from biopsies of the quadriceps muscles of type I and type III patients. It is possible that the atrogenes were not yet induced at the time of biopsy, since atrogene induction occurs late in the SMA mice.
While motor neuron loss is central to SMA disease pathogenesis, therapeutic targeting of muscle may provide benefits to SMA patients. Previous studies in SMA mice investigated the effect of factors known to regulate muscle growth, such as myostatin, an inhibitor of muscle growth, and follistatin, an inhibitor of myostatin. Both genetic and therapeutic interventions to modulate these growth factors have not improved muscle mass or the overall SMA phenotype (41–43). These approaches focused on muscle growth and protein synthesis to improve the disease phenotype. However, muscle maintenance is a balance between protein synthesis induced by growth factors and protein breakdown. Targeting protein synthesis without inhibiting atrophy-related protein breakdown may be insufficient to improve the disease phenotype. Thus, modulating expression of critical genes in the atrophic pathway may represent a strategy for mitigating atrophy in SMA and perhaps other denervating diseases. Recently, a MuRF1 inhibitor (P013222) was identified with an E3 ligase screening platform (44). This small molecule selectively inhibits MuRF1 auto-ubiquitination and ubiquitination of MyHC in a cellular atrophy model. If inhibiting MuRF1 in an animal model slows muscle wasting, then this would help to validate it as a therapeutic target for denervating diseases.
Considerable research has been done with animal models to investigate the role and regulation of atrogin-1 and MuRF1 under muscle wasting conditions, yet few investigations have shown that these pathways are conserved under human conditions of muscle atrophy. The gap in knowledge between atrogin-1 and MuRF1 regulation in animal and human models of skeletal muscle atrophy makes it difficult to fully assess the validity of atrogenes as potential therapeutic targets. In this study, we approached the molecular mechanisms underlying muscle atrophy in SMA mice and patients. Expression of the muscle-specific E3 ligases is increased in SMA muscle, and this is at least partly myogenin-dependent. We show that the HDAC4-Dach2-myogenin pathway can be pharmacologically targeted to suppress atrogene induction and muscle protein breakdown. TSA likely improves the SMA phenotype not only by increasing SMN gene expression, but also by attenuating myogenin expression and inhibiting muscle atrophy.
MATERIALS AND METHODS
Mouse treatment
Experiments were approved by the National Institute for Neurological Disorders and Stroke (NINDS) Animal Care and Use Committee. An SMA mouse breeder colony was maintained as described in (8). Treated SMA mice were given intraperitoneal injections of 10 mg/kg TSA (Enzo) in DMSO beginning on postnatal day 5 (P5). Daily injections were continued until mice were sacrificed at P9, P11 or P13. Control animals received equal volumes of vehicle alone. Mice were anesthetized with isoflurane and sacrificed by cervical dislocation. Distal hindlimb muscle was isolated and flash frozen in liquid nitrogen. Tissues were stored at −80°C and thawed before RNA or protein isolation. Adult wild-type littermates of SMA mice were used for starvation and denervation studies. For starvation studies, the mice were given TSA (as described above) 2 days, 1 day and immediately before food removal. The mice were deprived of solid food for 18 h, but given free access to water. For denervation studies, the mice were given TSA (as described above) 2 days and 1 day before denervation surgery and on the day of surgery. In anesthetized mice, fur covering the lower back and proximal thigh was removed. An incision was made near the sciatic notch. The facia was cut and the hamstring and gluteal muscles separated to reveal the sciatic nerve. In one leg, a 1 cm long piece of the sciatic nerve was removed. The other leg remained innervated and served as a control. The incision was closed with sutures. Postoperative mice were given either TSA or DMSO 1, 2 and 3 days after surgery and sacrificed on the third day.
Cell culture
C2C12 myoblasts were grown to confluency in DMEM with 10% fetal bovine serum and antibiotics and differentiated for 4 days in DMEM with 10% horse serum and antibiotics. Cells were treated with 50 nm TSA in DMSO, 10 μm dexamethasone (Sigma) in ethanol or both for 24 h and then collected for RNA isolation. Control cells received vehicle only. HEK 293T cells were grown in DMEM with 10% fetal bovine serum and antibiotics.
RNA isolation
Total RNA was isolated from muscle tissue and C2C12 cells using Trizol (Invitrogen), and the RNeasy kit (Qiagen) was used to purify the RNA following the manufacturer's instructions. One microgram RNA was converted to complementary DNA (cDNA) using the High Capacity cDNA Reverse Transcription kit (Applied Biosystems), following manufacturer's instructions. Gene expression was determined by qRT-PCR using TaqMan and SYBR Green reagents (Applied Biosystems). Primers are listed in the Supplementary Table. Transcript levels were quantified by threshold cycle values using phosphoglycerate kinase 1 as a control. For each gene, the values were normalized to vehicle-treated, unaffected animals or cells. For patient samples, the values were normalized to unaffected controls.
Western blot
C2C12 cells were lysed in 1% NP-40, 50 mm Tris–HCl (pH 8), 150 mm NaCl and protease inhibitor cocktail (Roche) on ice for 10 min. The lysates were centrifuged at 4°C for 10 min and the supernatants were collected. Protein concentration was determined using the BCA Protein Assay kit (Pierce) according to the manufacturer's protocol. Protein extraction from hindlimb muscle was done as previously described (10). Protein lysates (100 μg for C2C12 cells, 50 μg for mouse muscle) were resolved by SDS PAGE (10%) and transferred to polyvinylidene difluoride membranes. The membranes were blocked in 5% milk and probed with mouse anti-myogenin (1:500; clone F5D, Developmental Studies Hybridoma Bank), rabbit anti-atrogin 1 (1:1000, ECM Biosciences), rabbit anti-MuRF1 (1:500, Santa Cruz Biotechnology) and mouse anti-alpha tubulin (1:3000, Sigma).
Luciferase assay
C2C12 cell transfections were performed using GenJet Ver. III (SignaGen) following the manufacturer's instructions. HEK 293T cell transfections were performed using FuGENE (Roche) following the manufacturer's directions. The reporter plasmid, either atrogin-1 (Dr Stewart Lecker, Beth Israel Deaconess Medical Center) or MuRF1 (Dr. Eric Olson, University of Texas Southwestern Medical Center) was used in a ratio of 5:1 with a pGL4-TK Renilla luciferase plasmid (Promega). Twenty-four hours after transfection, the cells were treated for an additional 24 h with 10 nm, 50 nm, 100 nm, 200 nm or 500 nm TSA in DMSO or 0.5 mm, 2.5 mm or 12.5 mm valproic acid (Sigma) in water. Control cells received vehicle only. Following treatment, the cells were collected and luciferase assays were performed using the Dual Luciferase Reporter Assay system (Promega) following the manufacturer's instructions.
Protein breakdown assay
C2C12 cells were grown for 3 days in differentiation media, DMEM, with 10% horse serum and antibiotics. The media was supplemented with l-[3,5-3H]tyrosine (5 μCi/ml, PerkinElmer) for an additional 22 h. The cells were then washed with phosphate-buffered saline (PBS) and returned to differentiation media with 50 nm TSA in DMSO, 10 μm dexamethasone in ethanol or both. Control cells received vehicle only. 200 μl aliquots of media were taken at 0, 1, 3, 9, 18 and 24 h for quantification of l-[3,5-3H]tyrosine release. Protein precipitation and radioactivity measurement were done as previously described (45).
Chromatin immunoprecipitation
Flash frozen hindlimb muscle was minced on ice and fixed in 1% formaldehyde/PBS for 20 min and then quenched with glycine. Samples were centrifuged and washed twice in PBS with protease inhibitors. The cells were lysed and chromatin isolated using the EZ Magna ChIP A kit (Millipore) following the manufacturer's directions. Anti-myogenin antibody (M-225x, Santa Cruz) was used to immunoprecipitate the chromatin fragments. Primers for amplifying atrogin-1 and MuRF1 promoters were described previously (22).
Patient tissue
Diaphragm and iliopsoas muscle tissues were obtained from autopsies of type I SMA patients (n= 3) or age-matched controls (n= 2) (NICHD Brain and Tissue Bank for Developmental Disorders at the University of Maryland, Baltimore, MD, USA). Cross sections of muscle were stained with toluidine-blue.
Statistical analysis
Data are presented as the mean ± the standard error of the mean. t-Tests were performed using GraphPad Prism 5 software (GraphPad Software) with a significance set at P< 0.05.
SUPPLEMENTARY MATERIAL
Supplementary material is available at HMG online.
ACKNOWLEDGEMENTS
We thank Dr Stewart Lecker (Beth Israel Deaconess Medical Center) and Dr Eric Olson (University of Texas Southwestern Medical Center) for providing the atrogin-1 and MuRF1 reporter plasmids and Dr Peter Macpherson (University of Michigan) for the Dach2 primer sequences. Human tissue was obtained from the NICHD brain and Tissue Bank for Developmental Disorders at the University of Maryland, Baltimore, MD. This work was supported by intramural NINDS research funds. C.J.S. was supported by NINDS grant R01NS062869 and funds from the Spinal Muscular Atrophy Foundation.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Lefebvre S., Burglen L., Reboullet S., Clermont O., Burlet P., Viollet L., Benichou B., Cruaud C., Millasseau P., Zeviani M., et al. Identification of spinal muscular atrophy-determining gene. Cell. 1995;80:155–165. doi: 10.1016/0092-8674(95)90460-3. [DOI] [PubMed] [Google Scholar]
- 2.Monani U.R., Lorson C.L., Parsons D.W., Prior T.W., Androphy E.J., Burghes A.H., McPherson J.D. A single nucleotide difference that alters splicing patterns distinguishes the SMA gene SMN1 from the copy gene SMN2. Hum. Mol. Genet. 1999;8:1177–1183. doi: 10.1093/hmg/8.7.1177. [DOI] [PubMed] [Google Scholar]
- 3.Lorson C.L., Hahnen E., Androphy E.J., Wirth B. A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc. Natl Acad. Sci. U S A. 1999;96:6307–6311. doi: 10.1073/pnas.96.11.6307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hsieh-Li H.M., Chang J.G., Jong Y.J., Wu M.H., Wang N.M., Tsai C.H., Li H. A mouse model for spinal muscular atrophy. Nat. Genet. 2000;24:66–70. doi: 10.1038/71709. [DOI] [PubMed] [Google Scholar]
- 5.Le T.T., Coovert D.D., Monani U.R., Morris G.E., Burghes A.H. The survival motor neuron (SMN) protein: effect of exon loss and mutation on protein localization. Neurogenetics. 2000;3:7–16. doi: 10.1007/s100480000090. [DOI] [PubMed] [Google Scholar]
- 6.Prior T.W., Swoboda K.J., Scott H.D., Hejmanowski A.Q. Homozygous SMN1 deletions in unaffected family members and modification of the phenotype by SMN2. Am. J. Med. Genet. A. 2004;130A:307–310. doi: 10.1002/ajmg.a.30251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Le T.T., Pham L.T., Butchbach M.E., Zhang H.L., Monani U.R., Coovert D.D., Gavrilina T.O., Xing L., Bassell G.J., Burghes A.H. SMNDelta7, the major product of the centromeric survival motor neuron (SMN2) gene, extends survival in mice with spinal muscular atrophy and associates with full-length SMN. Hum. Mol. Genet. 2005;14:845–857. doi: 10.1093/hmg/ddi078. [DOI] [PubMed] [Google Scholar]
- 8.Avila A.M., Burnett B.G., Taye A.A., Gabanella F., Knight M.A., Hartenstein P., Cizman Z., Di Prospero N.A., Pellizzoni L., Fischbeck K.H., et al. Trichostatin A increases SMN expression and survival in a mouse model of spinal muscular atrophy. J. Clin. Invest. 2007;117:659–671. doi: 10.1172/JCI29562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yoo Y.E., Ko C.P. Treatment with trichostatin A initiated after disease onset delays disease progression and increases survival in a mouse model of amyotrophic lateral sclerosis. Exp. Neurol. 2011;231:147–159. doi: 10.1016/j.expneurol.2011.06.003. [DOI] [PubMed] [Google Scholar]
- 10.Kwon D.Y., Motley W.W., Fischbeck K.H., Burnett B.G. Increasing expression and decreasing degradation of SMN ameliorate the spinal muscular atrophy phenotype in mice. Hum. Mol. Genet. 2011;20:3667–3677. doi: 10.1093/hmg/ddr288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Narver H.L., Kong L., Burnett B.G., Choe D.W., Bosch-Marce M., Taye A.A., Eckhaus M.A., Sumner C.J. Sustained improvement of spinal muscular atrophy mice treated with trichostatin A plus nutrition. Ann. Neurol. 2008;64:465–470. doi: 10.1002/ana.21449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lecker S.H., Solomon V., Mitch W.E., Goldberg A.L. Muscle protein breakdown and the critical role of the ubiquitin-proteasome pathway in normal and disease states. J. Nutr. 1999;129:227S–237S. doi: 10.1093/jn/129.1.227S. [DOI] [PubMed] [Google Scholar]
- 13.Tischler M.E., Rosenberg S., Satarug S., Henriksen E.J., Kirby C.R., Tome M., Chase P. Different mechanisms of increased proteolysis in atrophy induced by denervation or unweighting of rat soleus muscle. Metabolism. 1990;39:756–763. doi: 10.1016/0026-0495(90)90113-q. [DOI] [PubMed] [Google Scholar]
- 14.Solomon V., Baracos V., Sarraf P., Goldberg A.L. Rates of ubiquitin conjugation increase when muscles atrophy, largely through activation of the N-end rule pathway. Proc. Natl Acad. Sci. U S A. 1998;95:12602–12607. doi: 10.1073/pnas.95.21.12602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bodine S.C., Latres E., Baumhueter S., Lai V.K., Nunez L., Clarke B.A., Poueymirou W.T., Panaro F.J., Na E., Dharmarajan K., et al. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science. 2001;294:1704–1708. doi: 10.1126/science.1065874. [DOI] [PubMed] [Google Scholar]
- 16.Sandri M., Sandri C., Gilbert A., Skurk C., Calabria E., Picard A., Walsh K., Schiaffino S., Lecker S.H., Goldberg A.L. Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell. 2004;117:399–412. doi: 10.1016/s0092-8674(04)00400-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stitt T.N., Drujan D., Clarke B.A., Panaro F., Timofeyva Y., Kline W.O., Gonzalez M., Yancopoulos G.D., Glass D.J. The IGF-1/PI3K/Akt pathway prevents expression of muscle atrophy-induced ubiquitin ligases by inhibiting FOXO transcription factors. Mol. Cell. 2004;14:395–403. doi: 10.1016/s1097-2765(04)00211-4. [DOI] [PubMed] [Google Scholar]
- 18.Cai D., Frantz J.D., Tawa N.E., Jr., Melendez P.A., Oh B.C., Lidov H.G., Hasselgren P.O., Frontera W.R., Lee J., Glass D.J., et al. IKKbeta/NF-kappaB activation causes severe muscle wasting in mice. Cell. 2004;119:285–298. doi: 10.1016/j.cell.2004.09.027. [DOI] [PubMed] [Google Scholar]
- 19.Tang H., Macpherson P., Marvin M., Meadows E., Klein W.H., Yang X.J., Goldman D. A histone deacetylase 4/myogenin positive feedback loop coordinates denervation-dependent gene induction and suppression. Mol. Biol. Cell. 2009;20:1120–1131. doi: 10.1091/mbc.E08-07-0759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tang H., Goldman D. Activity-dependent gene regulation in skeletal muscle is mediated by a histone deacetylase (HDAC)-Dach2-myogenin signal transduction cascade. Proc. Natl Acad. Sci. U S A. 2006;103:16977–16982. doi: 10.1073/pnas.0601565103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hasty P., Bradley A., Morris J.H., Edmondson D.G., Venuti J.M., Olson E.N., Klein W.H. Muscle deficiency and neonatal death in mice with a targeted mutation in the myogenin gene. Nature. 1993;364:501–506. doi: 10.1038/364501a0. [DOI] [PubMed] [Google Scholar]
- 22.Moresi V., Williams A.H., Meadows E., Flynn J.M., Potthoff M.J., McAnally J., Shelton J.M., Backs J., Klein W.H., Richardson J.A., et al. Myogenin and class II HDACs control neurogenic muscle atrophy by inducing E3 ubiquitin ligases. Cell. 2010;143:35–45. doi: 10.1016/j.cell.2010.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Macpherson P.C., Wang X., Goldman D. Myogenin regulates denervation-dependent muscle atrophy in mouse soleus muscle. J. Cell Biochem. 2011;112:2149–2159. doi: 10.1002/jcb.23136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kong L., Wang X., Choe D.W., Polley M., Burnett B.G., Bosch-Marce M., Griffin J.W., Rich M.M., Sumner C.J. Impaired synaptic vesicle release and immaturity of neuromuscular junctions in spinal muscular atrophy mice. J. Neurosci. 2009;29:842–851. doi: 10.1523/JNEUROSCI.4434-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee Y.I., Mikesh M., Smith I., Rimer M., Thompson W. Muscles in a mouse model of spinal muscular atrophy show profound defects in neuromuscular development even in the absence of failure in neuromuscular transmission or loss of motor neurons. Dev. Biol. 2011;356:432–444. doi: 10.1016/j.ydbio.2011.05.667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Baehr L.M., Furlow J.D., Bodine S.C. Muscle sparing in muscle RING finger 1 null mice: response to synthetic glucocorticoids. J. Physiol. 2011;589:4759–4776. doi: 10.1113/jphysiol.2011.212845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mammucari C., Milan G., Romanello V., Masiero E., Rudolf R., Del Piccolo P., Burden S.J., Di Lisi R., Sandri C., Zhao J., et al. FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab. 2007;6:458–471. doi: 10.1016/j.cmet.2007.11.001. [DOI] [PubMed] [Google Scholar]
- 28.Rudnicki M.A., Le Grand F., McKinnell I., Kuang S. The molecular regulation of muscle stem cell function. Cold Spring Harb. Symp. Quant. Biol. 2008;73:323–331. doi: 10.1101/sqb.2008.73.064. [DOI] [PubMed] [Google Scholar]
- 29.Hagiwara H., Saito F., Masaki T., Ikeda M., Nakamura-Ohkuma A., Shimizu T., Matsumura K. Histone deacetylase inhibitor trichostatin A enhances myogenesis by coordinating muscle regulatory factors and myogenic repressors. Biochem. Biophys. Res. Commun. 2011;414:826–831. doi: 10.1016/j.bbrc.2011.10.036. [DOI] [PubMed] [Google Scholar]
- 30.Bowerman M., Swoboda K.J., Michalski J.-P., Wang G.-S., Reeks C., Beauvais A., Murphy K., Woulfe J., Screaton R.A., Scott F.W., et al. Glucose metabolism and pancreatic defects in spinal muscular atrophy. Ann. Neurol., 2012 doi: 10.1002/ana.23582. doi:10.1002/ana.23582. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Butchbach M.E., Rose F.F., Jr., Rhoades S., Marston J., McCrone J.T., Sinnott R., Lorson C.L. Effect of diet on the survival and phenotype of a mouse model for spinal muscular atrophy. Biochem. Biophys. Res. Commun. 2010;391:835–840. doi: 10.1016/j.bbrc.2009.11.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ling K.K., Gibbs R.M., Feng Z., Ko C.P. Severe neuromuscular denervation of clinically relevant muscles in a mouse model of spinal muscular atrophy. Hum. Mol. Genet. 2012;21:185–195. doi: 10.1093/hmg/ddr453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kariya S., Park G.H., Maeno-Hikichi Y., Leykekhman O., Lutz C., Arkovitz M.S., Landmesser L.T., Monani U.R. Reduced SMN protein impairs maturation of the neuromuscular junctions in mouse models of spinal muscular atrophy. Hum. Mol. Genet. 2008;17:2552–2569. doi: 10.1093/hmg/ddn156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mentis G.Z., Blivis D., Liu W., Drobac E., Crowder M.E., Kong L., Alvarez F.J., Sumner C.J., O'Donovan M.J. Early functional impairment of sensory-motor connectivity in a mouse model of spinal muscular atrophy. Neuron. 2011;69:453–467. doi: 10.1016/j.neuron.2010.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Clarke B.A., Drujan D., Willis M.S., Murphy L.O., Corpina R.A., Burova E., Rakhilin S.V., Stitt T.N., Patterson C., Latres E., et al. The E3 Ligase MuRF1 degrades myosin heavy chain protein in dexamethasone-treated skeletal muscle. Cell Metab. 2007;6:376–385. doi: 10.1016/j.cmet.2007.09.009. [DOI] [PubMed] [Google Scholar]
- 36.Cohen S., Brault J.J., Gygi S.P., Glass D.J., Valenzuela D.M., Gartner C., Latres E., Goldberg A.L. During muscle atrophy, thick, but not thin, filament components are degraded by MuRF1-dependent ubiquitylation. J. Cell Biol. 2009;185:1083–1095. doi: 10.1083/jcb.200901052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Koyama S., Hata S., Witt C.C., Ono Y., Lerche S., Ojima K., Chiba T., Doi N., Kitamura F., Tanaka K., et al. Muscle RING-finger protein-1 (MuRF1) as a connector of muscle energy metabolism and protein synthesis. J. Mol. Biol. 2008;376:1224–1236. doi: 10.1016/j.jmb.2007.11.049. [DOI] [PubMed] [Google Scholar]
- 38.Mearini G., Gedicke C., Schlossarek S., Witt C.C., Kramer E., Cao P., Gomes M.D., Lecker S.H., Labeit S., Willis M.S., et al. Atrogin-1 and MuRF1 regulate cardiac MyBP-C levels via different mechanisms. Cardiovasc. Res. 2010;85:357–366. doi: 10.1093/cvr/cvp348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Witt S.H., Granzier H., Witt C.C., Labeit S. MURF-1 and MURF-2 target a specific subset of myofibrillar proteins redundantly: towards understanding MURF-dependent muscle ubiquitination. J. Mol. Biol. 2005;350:713–722. doi: 10.1016/j.jmb.2005.05.021. [DOI] [PubMed] [Google Scholar]
- 40.Millino C., Fanin M., Vettori A., Laveder P., Mostacciuolo M.L., Angelini C., Lanfranchi G. Different atrophy-hypertrophy transcription pathways in muscles affected by severe and mild spinal muscular atrophy. BMC Med. 2009;7:14. doi: 10.1186/1741-7015-7-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rindt H., Buckley D.M., Vale S.M., Krogman M., Rose F.F., Jr., Garcia M.L., Lorson C.L. Transgenic inactivation of murine myostatin does not decrease the severity of disease in a model of Spinal Muscular Atrophy. Neuromuscul. Disord. 2012;22:277–285. doi: 10.1016/j.nmd.2011.10.012. [DOI] [PubMed] [Google Scholar]
- 42.Rose F.F., Jr., Mattis V.B., Rindt H., Lorson C.L. Delivery of recombinant follistatin lessens disease severity in a mouse model of spinal muscular atrophy. Hum. Mol. Genet. 2009;18:997–1005. doi: 10.1093/hmg/ddn426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sumner C.J., Wee C.D., Warsing L.C., Choe D.W., Ng A.S., Lutz C., Wagner K.R. Inhibition of myostatin does not ameliorate disease features of severe spinal muscular atrophy mice. Hum. Mol. Genet. 2009;18:3145–3152. doi: 10.1093/hmg/ddp253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Eddins M.J., Marblestone J.G., Suresh Kumar K.G., Leach C.A., Sterner D.E., Mattern M.R., Nicholson B. Targeting the ubiquitin E3 ligase MuRF1 to inhibit muscle atrophy. Cell Biochem. Biophys. 2011;60:113–118. doi: 10.1007/s12013-011-9175-7. [DOI] [PubMed] [Google Scholar]
- 45.Sacheck J.M., Ohtsuka A., McLary S.C., Goldberg A.L. IGF-I stimulates muscle growth by suppressing protein breakdown and expression of atrophy-related ubiquitin ligases, atrogin-1 and MuRF1. Am. J. Physiol. Endocrinol. Metab. 2004;287: E591–E601. doi: 10.1152/ajpendo.00073.2004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.