

The anions derived from metalation of allylic nitriles represent a promising class of nucleophiles for carbon–carbon bond formation. The reactions of these species with electrophiles can proceed through either α or γ addition and provide access to synthetically useful building blocks containing nitrile and alkene functional groups (Scheme 1). Achieving high site selectivity in these reactions presents a significant challenge for reaction development, especially when considering the number of metalated intermediates that can form upon activation of the allylic nitrile. Notable progress has recently been made in site-selective additions of allyl and crotyl nitriles to carbonyl compounds using pro-azaphosphatranes[1] and palladium/pincer[2] catalysts. Despite these advances, and the utility that nucleophilic allylic nitriles offer in carbon–carbon bond-forming reactions, enantioselective processes involving these species are rare.
Scheme 1.

Activation and electrophilic trapping of allylic nitriles. DMSO=dimethylsulfoxide.
To date, only the catalytic, enantioselective addition of allyl nitrile has been realized.[3] The method, reported by Shibasaki and co-workers, involves the cooperative catalytic action of a soft CuI Lewis acid, a hard lithium alkoxide brønsted base, and a chiral phosphine ligand to achieve activation/deprotonation of allyl nitrile. The enantioselective addition of the resulting metalated nitrile to ketoimines[3a] and aromatic ketones[3b,c] occurs with good yields, high site selectivities, and moderate to high enantioselectivities. Although this example represents an important advance for asymmetric additions of this nucleophile class, the scope has been limited to the reaction of allyl nitrile with carbonyl compounds that do not readily undergo base-mediated self-condensation reactions.[4] The reactions of allylic nitrile nucleophiles with aldehydes remain an unsolved problem.
An alternative strategy for creating nucleophilic allylic nitriles that avoids the use of anionic intermediates is to employ N-silyl vinylketene imines (2; see Scheme 2). These compounds could be prepared by selective N silylation of allylic nitrile anions. Utilization of these nucleophiles in catalytic, enantioselective, vinylogous aldol reactions would generate δ-hydroxy α,β-unsaturated nitriles (Scheme 2). The synthetic benefit of unsaturated nitriles has been highlighted by their ability to undergo new carbon–carbon bond-forming reactions with organometallic compounds[5] as well as allowing access to α,β-unsaturated aldehydes,[6] carboxylic acid derivatives, and allylic amines through manipulation of the nitrile.[3c] Interestingly, the vinylogous Mukaiyama aldol reaction, which is a well-established method for controlling site and stereoselectivity in the addition of ketone, ester, and amide dienolates, has not previously been reported for nitriles.[7] Herein, we describe a new approach for generating nucleophilic allylic nitriles, through the intermediacy of a silyl vinylketene imine, and the subsequent use of these reagents in enantioselective, vinylogous aldol reactions.
Scheme 2.
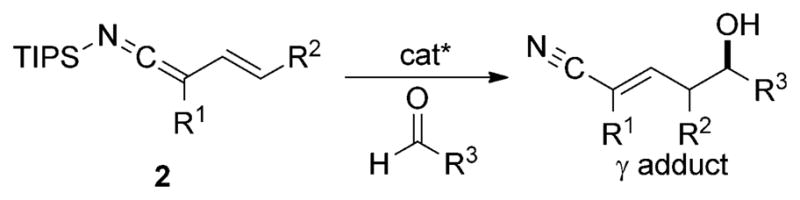
Vinylogous aldol reaction of N-silyl vinylketene imines.
The synthesis of silyl ketene imines derived from α,α-disubstituted nitriles is well documented,[8] and previous studies from these laboratories,[9] as well as others, have demonstrated their efficacy in the enantioselective synthesis of quaternary stereogenic centers.[10] However, only a single report describes the synthesis and use of N-silyl vinylketene imines. Ghosez and co-workers reported a method for converting allyl nitrile into a bis-silyl vinylketene imine by double deprotonation with lithium diisopropylamide (LDA) and silylation with excess triisopropylsilyl chloride (TIPSCl).[11] The resulting vinylketene imine is employed as a diene in cycloaddition reactions with acetylenic esters, thus affording anilines after desilylation and aromatization.
The challenge inherent to the synthesis of N-silyl vinyl-ketene imines is achieving selective N silylation instead of Cα or Cγ silylation of the metalated allylic nitrile intermediate. Foregoing studies have documented the tendency of metalated nitriles to undergo competitive C silylation; however, the use of bulky silylating agents typically results in kinetically controlled silylation at the less hindered nitrogen atom.[8b,12] The preliminary work of Ghosez and co-workers on the synthesis of N-silyl vinylketene imines also supports this trend.
Cognizant of the aforementioned precedents, studies aimed at the preparation of N-silyl vinylketene imines from allylic nitrile derivatives were initiated. 2-Methyl-3-butene-nitrile (3; Scheme 3)[13] was chosen as the pronucleophile because it is a readily available, inexpensive starting material, and the methyl substituent at C2 should inhibit competitive Cα silylation. Through optimization studies, it was found that the N-silyl vinylketene imine 4 could easily be prepared in good yield and selectivity by addition of nitrile 3 to a pre-cooled solution of LDA and TIPSCl (Scheme 3). The resulting product was isolated as a thermally stable liquid that could be purified by distillation and handled in air without significant hydrolysis. When stored at −10°C, 4 showed no signs of decomposition even after storage for eight months.
Scheme 3.

Synthesis of N-silyl vinylketene imine 4.
Earlier reports from these laboratories have shown that silyl ketene imines are susceptible to Lewis base catalyzed,[14] SiCl4-mediated carbonyl addition reactions. In general, the reactions are characterized by high yields and excellent stereoselectivities and have provided novel methods for the preparation of quaternary stereogenic carbon centers,[9a] tertiary alcohols,[15] and cross-benzoin products.[15] One major limitation for silyl ketene imine additions with this catalyst system has been the difficulty of engaging aliphatic aldehydes. The reaction of disubstituted silyl ketene imines with aliphatic aldehydes has been thwarted by the added strain energy that accrues from the formation of a quaternary carbon center as well as the reduced reactivity observed for aliphatic aldehydes in Lewis base catalyzed, SiCl4-mediated aldol reactions.[16] We postulated that N-silyl vinylketene imines would be able to react with aliphatic aldehydes through the γ-carbon atom of the ketene imine because this site is relatively free of steric encumbrance. To test this hypothesis, 4 was combined with hydrocinnamaldehyde using reaction conditions similar to those reported previously for Lewis base catalyzed, vinylogous aldol additions of enoxysilanes.[17] Gratifyingly, the reaction afforded selectively the E-γ-addition product 7a in moderate yield and with excellent enantioselectivity (Table 1, entry 1). Further analysis revealed that the moderate yield observed in the addition resulted from incomplete consumption of the aldehyde. This problem could be eliminated at slightly elevated reaction temperatures (−55°C) and nitrile 7a was isolated in a synthetically useful yield and comparable enantioselectivity. Other aliphatic aldehydes were also tested to explore the substrate generality with regard to substitution pattern. Overall, the addition was highly selective for formation of E-α,β-unsaturated nitriles in moderate to good yields and with good to excellent enantioselectivities. Aldehydes containing β branching gave nitrile products 7a–c with the highest levels of enantiomeric purity whilst aldehydes containing a linear aliphatic chain, reacted with slightly reduced enantioselectivity (Table 1, entries 5–8). Surprisingly, an even more hindered aliphatic aldehyde containing α branching underwent addition to give the E-α,β-unsaturated nitrile 7d in moderate yield and excellent enantioselectivity (Table 1, entry 4). The addition of 4 to aliphatic aldehydes containing either isolated olefins or benzyloxy ethers were also tested. The resulting γ-nitrile products 7 f–g were obtained with excellent site selectivity and good yields and stereoselectivities. However, for aldehydes containing ethereal linkages, the enantioselectivity was sensitive to the position of the oxygen atom in the alkyl side chain (Table 1, compare entries 7 and 8).
Table 1.
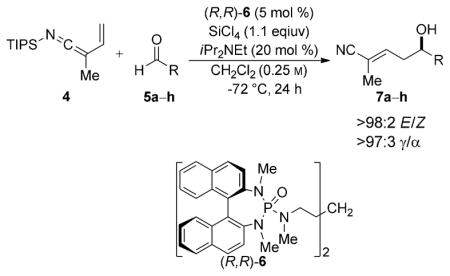
| |||
|---|---|---|---|
| Entry | Product | Yield [%][b] | e.r.[d] |
| 1 |
 7a
7a
|
65 (82)[e] | 95:5 (95:5)[e] |
| 2 |
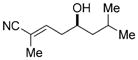 7b
7b
|
57 (86)[e] | 98.5:1.5 (98:2)[e] |
| 3 |
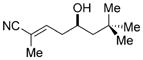 7c
7c
|
82 | 96.5:3.5 |
| 4 |
 7d
7d
|
63 | 98.5:1.5 |
| 5 |
 7e
7e
|
75 (81)[e] | 92:8 (90:10)[e] |
| 6 |
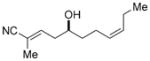 7f
7f
|
55 (86)[e] | 88:12 (87:13)[e] |
| 7 |
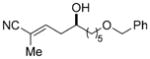 7g
7g
|
74 | 90:10 |
| 8 |
 7h
7h
|
54 (78)[e] | 77.5:22.5 (76:24)[e] |
1.0 mmol scale reaction.
Yields are for constitutionally and analytically pure material.
γ/α and E/Z ratios determined by 1H NMR analysis of crude reaction mixtures.
Enantiomeric ratios determined by CSP-SFC analysis.
Reaction performed at −55 °C.
To further elaborate the scope of the electrophiles in this reaction, a survey of α,β-unsaturated aldehydes was conducted. These reaction partners present an additional challenge because the catalyst must control the site selectivity for addition of both the nucleophile (γ versus α) and the electrophile (1,4 versus 1,2). Initial rate studies using in situ IR monitoring showed a rapid consumption of the starting material (see the Supporting Information), thus allowing a dramatic reduction in the overall reaction time. Under these modified reaction conditions, the addition of 4 to cinnamaldehyde was studied (Table 2, entry 1). Analysis of the crude reaction mixture by 1H NMR spectroscopy showed excellent constitutional site selectivity for γ attack of nucleophile 4 to the carbonyl group of the enal (1,2-addition), thus producing diene 9a in good yield and enantioselectivity. In view of this promising result, other commercially available aromatic enals were evaluated in the reaction and the diene products were isolated in good yield. Electron-rich and electron-poor aromatic enals underwent addition with similar rates yielding diene products in excellent E/Z selectivity and good site and enantioselectivity (Table 2, entries 2–4).
Table 2.
Addition of N-silyl vinylketene imine 4 to aromatic enals.[a]
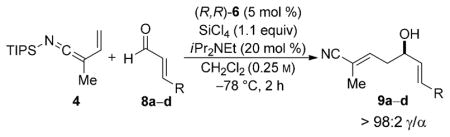
| |||||
|---|---|---|---|---|---|
| Entry | Product | Yield [%][b] | 1,2/1,4[c] | E/Z[c] | e.r.[d] |
| 1 |
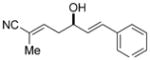 9a
9a
|
84 | 92:8 | 99:1 | 92:8 |
| 2 |
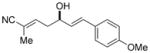 9b
9b
|
79 | 92:8 | 98:2 | 93:7 |
| 3 |
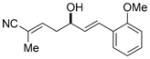 9c
9c
|
79 | 90:10 | 94:6 | 94:6 |
| 4 |
 9d
9d
|
83 | 92:8 | 99:1 | 92:8 |
1.0 mmol scale reaction.
Yields are for the isolated, constitutionally and analytically pure material.
γ/α and E/Z ratios determined by 1H NMR analysis of crude reaction mixtures.
Enantiomeric ratios determined by CSP-SFC analysis.
To investigate the generality of the vinylogous aldol reaction with respect to aromatic aldehydes, various electron-rich, electron-poor, and hindered aromatic aldehydes were investigated (Table 3). Initial optimization studies revealed that the catalyst loading could be reduced without significant loses in the reaction rate or enantioselectivity. Hence, the addition of 4 to various aromatic aldehydes was investigated using 2.5 mol% of the Lewis base catalyst (R,R)-6. Electronically neutral aromatic aldehydes such as benzaldehyde and 2-naphthaldehyde underwent addition in high yields and good enantioselectivities (Table 3, entries 1–2). Electron-rich aromatic aldehydes exhibited the highest enantiomeric ratios, for example the addition of 4 to 4-methoxybenzaldehyde afforded nitrile product 11e in 97:3 e.r. (Table 3, entry 5). In contrast, electron-poor aromatic aldehydes reacted with attenuated enantioselectivities (Table 3, entry 9–10), thus suggesting a competitive achiral background reaction is ensuing for these more reactive substrates. The lowest enantioselectivity observed in the survey was for the addition to the sterically hindered aldehyde 1-naphthaldehyde (Table 3, entry 3). Interestingly, in this case a significant disparity was observed in the enantiomeric ratios of the resulting E and Z nitriles. Previous studies with this catalyst system have also shown higher observed enantioselectivities in minor diastereomers, but the difference is typically not as dramatic. Importantly, additions to slightly less hindered aromatic aldehydes, such as 2-methyl benzaldehyde and 2-methoxybenzaldehyde, yielded nitrile products in good diastereo- and enantioselectivity (Table 3, entries 4 and 7). In general, unsaturated nitriles derived from selective γ addition of 4 to aromatic aldehydes were obtained in excellent yields and with good to high enantioselectivities.
Table 3.
Addition of N-silyl vinylketene imine 4 to aromatic aldehydes.[a]

| ||||
|---|---|---|---|---|
| Entry | Product | Yield [%][b] | γ/α[c] | Stereo-selectivity[c,d] |
| 1 |
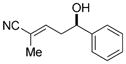 11a
11a
|
97 | 98:2 | 99:1 E/Z 93.5:6.5 e.r. |
| 2 |
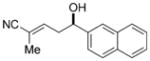 11b
11b
|
97 | 99:1 | 99:1 E/Z 93:7 e.r. |
| 3 |
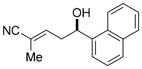 11c
11c
|
85 | 95:5 | 93:7 E/Z 60:40 e.r. (E) 94:6 e.r. (Z) |
| 4 |
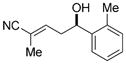 11d
11d
|
87 | 96:4 | 98:2 E/Z 92:8 e.r. |
| 5 |
 11e
11e
|
93 | 99:1 | 99:1 E/Z 97:3 e.r. |
| 6 |
 11f
11f
|
92 | 99:1 | 97:3 E/Z 93:7 e.r. |
| 7 |
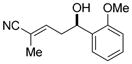 11g
11g
|
92 | 95:5 | 98:2 E/Z 94.5:5.5 e.r. |
| 8 |
 11h
11h
|
95 | 98:2 | 98:2 E/Z 92.5:7.5 e.r. |
| 9 |
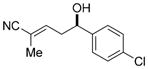 11i
11i
|
91 | 97:3 | 98:2 E/Z 91.5:8.5 e.r. |
| 10 |
 11j
11j
|
93 | 92:8 | 99:1 E/Z 77:23 e.r. |
1.0 mmol scale reaction.
Yields are for the isolated, constitutionally and analytically pure material.
γ/α and E/Z ratios determined by 1H NMR analysis of crude reaction mixtures.
Enantiomeric ratios determined by CSP-SFC analysis.
The double-bond geometry of the alkene subunit was unambiguously established to be E by single-crystal X-ray analysis of compound 11b,[18] which resulted from addition of 4 to 2-naphthaldehyde. Correlation of the E-configured double bond to nitriles derived from other classes of aldehydes was confirmed by comparison of the diagnostic 13C and 1H NMR spectra. The absolute configuration of the hydroxy-bearing stereogenic center in the nitrile products was determined by chemical derivatization to known compounds and comparison of their optical rotations (see the Supporting Information). The absolute and relative configurations of the nitrile products confirm that the N-silyl vinylketene imine 4 undergoes addition to the Re face of the aldehyde in an s-trans conformation and is congruent with previous studies with this catalyst system.[16,19] The high selectivity for formation of the E-configured double bond likely results from unfavorable steric interactions encountered in the approach of 4 to the activated Lewis acid/aldehyde complex in an s-cis orientation (Scheme 4).
Scheme 4.
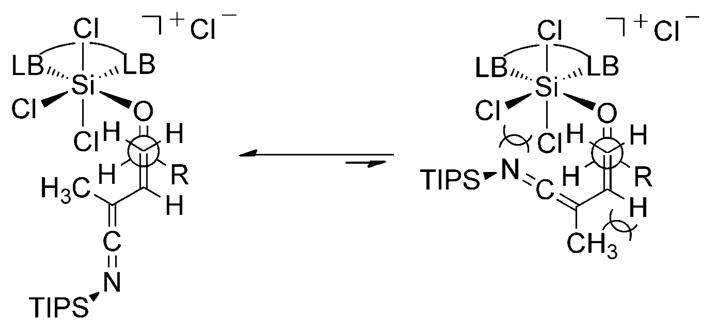
Proposed open transition structures for rationalization of the E-configured double bond observed in the nitrile products. LB =Lewis base.
In conclusion, a novel Lewis base catalyzed, Lewis acid mediated vinylogous aldol addition of the N-silyl vinylketene imine 4 has been described. This stable and storable silylated nucleophile is easily prepared in a single step from 2-methyl-3-butenenitrile. The Lewis base catalyzed reactions of 4 are characterized by exceptionally high site selectivity for γ addition to a diverse range of aldehyde acceptors. The resulting α,β-unsaturated nitriles are obtained in good to high yield and with excellent selectivity for formation of the E-configured double-bond isomer. Furthermore, the products could be prepared in good to excellent enantioselectivity by employing Lewis base catalyst (R,R)-6 with loadings as low as 2.5 mol%. Importantly, this method provides an alternative strategy to existing methods for accessing nucleophilic allylic nitriles, which allows addition to aldehydes.[3] Ongoing studies are focused on expanding the scope of this reaction with respect to the N-silyl vinylketene imine and examining their reactivity with other classes of electrophiles such as ketones and imines.
Supplementary Material
Footnotes
Financial support was provided by the National Science Foundation (NSF CHE-0717989 and 1012663). The authors acknowledge Jeremy Henle for the preparation of benzyloxy aldehydes.
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/anie.201108795.
References
- 1.Kisanga PB, Verkade JG. J Org Chem. 2002;67:426–430. doi: 10.1021/jo0106492. [DOI] [PubMed] [Google Scholar]
- 2.Aydin J, Szabo KJ. Org Lett. 2008;10:2881–2884. doi: 10.1021/ol801070n. [DOI] [PubMed] [Google Scholar]
- 3.a) Yazaki R, Nitabaru T, Kumagai N, Shibasaki M. J Am Chem Soc. 2008;130:14477–14478. doi: 10.1021/ja806572b. [DOI] [PubMed] [Google Scholar]; b) Yazaki R, Kumagai N, Shibasaki M. J Am Chem Soc. 2009;131:3195–3196. doi: 10.1021/ja900001u. [DOI] [PubMed] [Google Scholar]; c) Yazaki R, Kumagai N, Shibasaki M. J Am Chem Soc. 2010;132:5522–5531. doi: 10.1021/ja101687p. [DOI] [PubMed] [Google Scholar]
- 4.We are aware of a single example where aliphatic aldehydes are utilized in the copper-catalyzed addition of acetonitrile. Syringe pump addition was used to avoid self-condensation; see: Suto Y, Riichiro T, Kanai M, Shibasaki M. Org Lett. 2005;7:3757–3760. doi: 10.1021/ol051423e.
- 5.Fleming FF, Wang QZ. Chem Rev. 2003;103:2035–2077. doi: 10.1021/cr020045d. [DOI] [PubMed] [Google Scholar]
- 6.Verdegem PJE, Monnee MCF, Lugtenburg J. J Org Chem. 2001;66:1269–1282. doi: 10.1021/jo0009595. [DOI] [PubMed] [Google Scholar]
- 7.a) Casiraghi G, Battistini L, Curti C, Rassu G, Zanardi F. Chem Rev. 2011;111:3076–3154. doi: 10.1021/cr100304n. [DOI] [PubMed] [Google Scholar]; b) Denmark SE, Heemstra JR, Beutner GL. Angew Chem. 2005;117:4760–4777. doi: 10.1002/anie.200462338. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2005;44:4682–4698. doi: 10.1002/anie.200462338. [DOI] [PubMed] [Google Scholar]; c) Kalesse M. In: Topics In Current Chemistry. Mulzer J, editor. Vol. 244. Springer; Berlin: 2005. pp. 43–76. [Google Scholar]; d) Casiraghi G, Zanardi F, Appendino G, Rassu G. Chem Rev. 2000;100:1929–1972. doi: 10.1021/cr990247i. [DOI] [PubMed] [Google Scholar]
- 8.a) West R, Gornowicz GA. J Am Chem Soc. 1971;93:1714–1720. [Google Scholar]; b) Watt DS. Synth Commun. 1974;4:127–131. [Google Scholar]
- 9.a) Denmark SE, Wilson TW, Burk MT, Heemstra JR., Jr J Am Chem Soc. 2007;129:14864–14865. doi: 10.1021/ja077134y. [DOI] [PubMed] [Google Scholar]; b) Denmark SE, Wilson TW. Synlett. 2010:1723–1728. [Google Scholar]
- 10.a) Mermerian AH, Fu GC. Angew Chem. 2005;117:971–974. doi: 10.1002/anie.200461886. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2005;44:949–952. doi: 10.1002/anie.200461886. [DOI] [PubMed] [Google Scholar]; b) Notte GT, Vu JMB, Leighton JL. Org Lett. 2011;13:816–818. doi: 10.1021/ol103096u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Differding E, Vandevelde O, Roekens B, Van TT, Ghosez L. Tetrahedron Lett. 1987;28:397–400. [Google Scholar]
- 12.a) Watt DS. J Org Chem. 1974;39:2799–2800. [Google Scholar]; b) Cunico RF, Kuan CP. J Org Chem. 1992;57:1202–1205. [Google Scholar]
- 13.2-Methyl-3-butenenitrile, 70% was obtained from TCI America ($0.14/g) and was used without further purification. The remainder is a mixture of consitutional and geometrical isomers. All isomers converge to the product through either α or γ deprotonation.
- 14.Denmark SE, Beutner GL. Angew Chem. 2008;120:1584–1663. [Google Scholar]; Angew Chem Int Ed. 2008;47:1560–1638. doi: 10.1002/anie.200604943. [DOI] [PubMed] [Google Scholar]
- 15.Denmark SE, Wilson TW. Nat Chem. 2010;2:937–943. doi: 10.1038/nchem.857. [DOI] [PubMed] [Google Scholar]
- 16.In the presense of silicon tetrachloride and Lewis base, aliphatic aldehydes form trichlorosilyl chlorohydrins that do not react with enoxysilane nucleophiles, see: Denmark SE, Beutner GL, Wynn T, Eastgate MD. J Am Chem Soc. 2005;127:3774–3789. doi: 10.1021/ja047339w.
- 17.a) Denmark SE, Beutner GL. J Am Chem Soc. 2003;125:7800–7801. doi: 10.1021/ja035448p. [DOI] [PubMed] [Google Scholar]; b) Denmark SE, Heemstra JR. J Am Chem Soc. 2006;128:1038–1039. doi: 10.1021/ja056747c. [DOI] [PubMed] [Google Scholar]; c) Denmark SE, Heemstra JR. J Org Chem. 2007;72:5668–5688. doi: 10.1021/jo070638u. [DOI] [PubMed] [Google Scholar]
- 18.CCDC 852351 (11b) contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
- 19.Denmark SE, Eklov BM, Yao PJ, Eastgate MD. J Am Chem Soc. 2009;131:11770–11787. doi: 10.1021/ja902474j. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.