

Potential endpoints for community-acquired bacterial pneumonia and acute bacterial skin and skin structure infections trials were evaluated. Review of historical and modern data led to the conclusion that antimicrobial treatment effects are most apparent early in therapy. Later outcomes provide important supportive information.
Abstract
Efficacy endpoints for previous registrational trials of antimicrobials for acute bacterial skin and skin structure infections (ABSSSIs) and community-acquired bacterial pneumonia (CABP) were based on nonstandardized, clinician-based observations and decisions, as well as on patient reports. More quantifiable, reproducible, and externally verifiable endpoints could improve the design of future noninferiority trials. At the request of the Food and Drug Administration, the Foundation for the National Institutes of Health convened a broadly representative scientific project team to evaluate potential endpoints for such registrational trials. Review of historical and modern data led to the conclusion that antimicrobial treatment effects are most apparent early in therapy; later outcomes provide important supportive information. Although evidence is incomplete, early response endpoints can anchor noninferiority hypotheses in ABSSSI and CABP registrational trials, thereby allowing evidence-based drug development to continue. Further research is underway to establish which short- and long-term outcomes are well-defined, reliable, and reflective of how patients feel, function, or survive.
(See the Editorial by Toerner et al, on pages 1122–3.)
Acute bacterial skin and skin structure infections (ABSSSIs) and community-acquired bacterial pneumonia (CABP) are common infections, successful treatment of which requires administration of safe and efficacious antimicrobials. Regulatory requirements for design and conduct of registrational trials for these indications must be scientifically sound and clearly articulated to facilitate development of new antimicrobials.
In past decades, efficacy endpoints for ABSSSI and CABP registrational trials were based on resolution/improvement of signs and symptoms of infection at a time point after completion of therapy. By design, these endpoints included assessments at earlier time points as an element of outcome. As the science advanced, the Food and Drug Administration (FDA) and others realized that the design of noninferiority trials evaluating antimicrobials could be improved by defining more reliable outcome measures that reduced dependence on subjective elements and by evaluating outcomes at time points for which prior evidence had demonstrated reliable and reproducible drug effects. The ability to measure known treatment effects on well-defined and reliable outcome measures is essential for noninferiority trial designs. In addition, unambiguous standards for measurement are needed to ensure constancy of the effect of antimicrobials from prior trials to current trials. Finally, the FDA stipulates that outcome measures for studies that support drug registration should be direct measures or established surrogates of how patients feel, function, or survive.
These considerations led to publication of new FDA guidance for ABSSSI and CABP trials [1, 2], both of which focus on assessment of efficacy at an earlier time point than previously recommended. For example, the ABSSSI guidance notes historical data demonstrating treatment effects at 48–72 hours after initiation of antimicrobials. However, the data are incomplete and cannot be audited; the outcomes included body temperature, pulse, respiratory rate, and other measures that are biomarkers (ie, not direct measures of how a patient feels, functions, or survives); and there are significant questions regarding the applicability of these historical data to current circumstances (ie, is the constancy assumption met, which is required for noninferiority designs).
The FDA asked the Biomarkers Consortium of the Foundation for the National Institutes of Health (FNIH; available at: http://www.biomarkersconsortium.org/) to convene a project team (PT) to evaluate past evidence on endpoints for these 2 indications and to develop and evaluate proposed endpoints for future ABSSSI and CABP registrational trials. Team membership included broad participation from the National Institutes of Health (NIH), the FDA, the academic research community, and biopharmaceutical companies. Considering the FDA's standards for drug approval, the PT evaluated historical evidence for treatment effects; evaluated outcomes in recent clinical trials; outlined research gaps; proposed interim, bridging endpoints; and began developing future research on outcome assessment in ABSSSI and CABP trials. The PT conclusions were submitted to the relevant FDA dockets [3, 4]. This article summarizes the process, major findings, and planned future research of this intensive, multidisciplinary effort.
REGULATORY STANDARDS FOR APPROVAL OF NEW DRUGS
The US regulatory standard for drug approval is “substantial evidence” from “adequate and well-controlled trials” [5]. One requirement is that methods of assessment of subjects' response must be “well-defined and reliable.” A proposed study should explain (1) what to measure, (2) how the outcome was measured, and (3) how the data were analyzed to define a meaningful outcome; that is, what defines a “responder” [6].
US regulations, as well as a recent Institute of Medicine recommendation [7], define a “clinically meaningful” outcome measure as a direct measure of how patients feel, function, or survive. However, appropriately developed and validated surrogate endpoints can be used as outcome measures to support approval if the “surrogate endpoint … is reasonably likely, based on epidemiologic, therapeutic, pathophysiologic, or other evidence, to predict clinical benefit” [8] and if follow-up studies show that benefit on the surrogate reflects benefit on direct patient-centered outcomes. “Surrogate endpoints” are biomarkers (eg, signs of disease, results of radiological tests, cultures, and laboratory values) that act as substitute indirect measures for how patients feel, function, or survive [9]. In trials of antimicrobials, acute infection symptoms (eg, pain) are direct measures of patient benefit, and signs of disease (eg, erythema) and selected markers of systemic response (eg, leukocytosis) are biomarkers having mechanistic links to infection.
An important consideration for the design of registrational trials for these indications is that they are conducted globally and submitted for marketing authorization globally. Accordingly, harmonization of clinical trial design with requirements of non-US regulatory authorities, such as the European Medicines Agency, is crucial [10].
HISTORICAL EVIDENCE
Two trials published in 1937 compared sulfa drugs to ultraviolet light in the treatment of erysipelas [11, 12]. The studies used alternate assignment and were not blinded. The investigators evaluated outcome measures that included cessation of erysipelas skin lesion spread, durations of pyrexia and toxemia, death, relapse, and complications. The greatest treatment effect on which to base noninferiority hypotheses was cessation of lesion spread at 48 hours (Table 1). In a meta-analysis of the 2 studies, the difference was 24% in favor of sulfa drugs (95% confidence interval [CI], 18.2%–30.0%) (communication from FDA to FNIH, 5 May 2010). If deaths and failures during initial therapy are included in the analysis, the difference was 26% in favor of sulfa drugs (95% CI, 19.0%–33.5%). There was no treatment effect on death, relapse, or complications on which to base future noninferiority hypotheses. The PT agreed that data on control of lesion spread were a useful—albeit imperfect—starting point for further investigation. The PT performed a review of the medical literature evaluating current techniques for measuring skin lesions. No study specifically addressing the measurement of acute cellulitis lesion size was identified; therefore, the characteristics, reliability, and reproducibility of various measurement techniques in the measurement of ABSSSI lesions are unknown. Optimal measurement methods for acute bacterial skin lesions still need to be determined.
Table 1.
Findings From 2 Historical Clinical Studies Involving Erysipelas
| Study 1 |
Study 2 |
|||
|---|---|---|---|---|
| Variable | Ultraviolet Light (n = 104) | Prontosil (n = 106) | Ultraviolet Light (n = 135) | Sulphanilamidea (n = 135) |
| Deaths | 6 | 4 | 4 | 5 |
| Treatment discontinuation | 0 | 0 | 9b | 0 |
| Evaluable for cessation of spread of lesion | 98 | 102 | 122 | 130 |
| Cessation of lesion spread | ||||
| At 48 hours | 75/98 (76.5) | 100/102 (98) | 89/122 (73) | 129/130 (99.2) |
| At 72 hours | 86/98 (87.8) | 101/102 (99) | 103/122 (84.4) | 130/130 (100) |
| At 96 hours | 91/98 (92.9) | 102/102 (100) | 115/122 (94.3) | 130/130 (100) |
| Did not have fever | 9 | 10 | 10 | 5 |
| Evaluable for resolution of fever | 89 | 92 | 112 | 125 |
| Resolution of fever | ||||
| At 48 hours | 43/89 (48.3) | 70/92 (76.1) | 53/112 (47.3) | 94/125 (75.2) |
| At 72 hours | 55/89 (61.8) | 84/92 (91.3) | 67/112 (59.8) | 113/125 (90.4) |
| At 96 hours | 66/89 (74.2) | 86/92 (93.5) | 77/112 (68.8) | 122/125 (97.6) |
| Not “toxemic” at baseline | 11 | 5 | 6 | 2 |
| Evaluable for cessation of “toxemia” at 48 hours | 87 | 98 | 116 | 128 |
| Cessation of “toxemia” at 48 hours | 32/87 (39) | 58/98 (60) | 44/116 (37.9) | 60/128 (46.9) |
| Recurrence of erysipelas | 12 (11.5) | 9 (8.5) | 8 (5.9) | 2 (1.5) |
| Complications | 32 (30) | 23 (21) | 28 (20.7) | 11 (8.1) |
| Average duration of therapy | 2.6 days | (5 g total exposure) | 1.4 days | 2.5 days (high dose exposure) |
Data are No., No. (%), or proportion (%) of subjects, unless otherwise indicated. Data are from [11, 12] and unpublished (communication from the Food and Drug Administration to the Foundation of the National Institutes of Health, 5 May 2010).
a Patients continued to receive sulphanilamide during entire hospitalization, which resulted in numerically lower rates of recurrence and complications for this treatment group.
b “Failure” of ultraviolet light therapy.
In CABP, reviews of work from the preantibiotic era [13, 14] provided illustrations of the typical course of symptoms associated with acute bacterial pneumonia in adults, including cough, dyspnea, chest pain (especially pleuritic pain), and sputum expectoration. In general, steady deterioration of untreated patients' respiratory symptoms continued through days 3–4. These results contrast with those in the early antibiotic era, when the treatment effect was both rapid and large [15–20]: a quantitative estimate of treatment effect for symptom resolution at 48–72 hours was 29% (95% CI, 21%–37%) [18], a quantitative estimate of treatment effect for clinical recovery at day 3 was 72%–77% [14–16], and quantitative estimates of treatment effect for mean days to clinical improvement, fall in temperature, and clinical recovery were 2.5, 3.4, and 4.2 days, respectively [17].
A review of historical data on antimicrobial treatment effect in CABP was presented at a 2009 FDA advisory committee meeting [21]. Figure 1 is illustrative of the FDA's findings.
Figure 1.
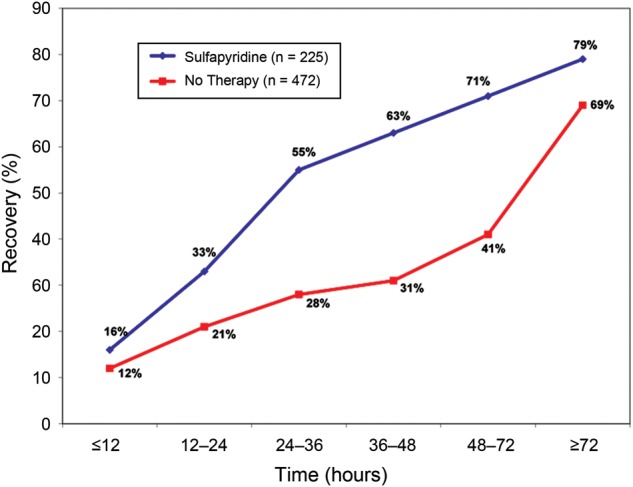
Duration of symptoms in patients with pneumococcal pneumonia [21].
These data do have limitations: they mostly derive from observational or small studies, cross-study comparisons were used to determine treatment effect, and the endpoints were not clearly defined, although they were clinically reasonable. However, collectively, these preantibiotic and early antibiotic era data indicated a significant treatment effect at days 3–4 after initiation of therapy.
RETROSPECTIVE DATA ANALYSES
The PT performed retrospective analyses of data sets from modern clinical studies to (1) refine the FDA-proposed outcome measures by evaluating their operational characteristics, changes over time, and responsiveness to change at specific time point and (2) identify additional relevant endpoints or biomarkers. For ABSSSI, data from registrational trials of ceftaroline fosamil, dalbavancin, and linezolid were used [22–24]. For CABP, data from trials of tigecycline, daptomycin, and ceftaroline fosamil were used [25–27]. Analyses were performed per statistical analysis plans drafted prospectively by qualified PT biostatisticians.
The PT analyzed these data sets in a post hoc subgroup of all randomized patients defined by criteria similar to those used for inclusion in current clinical trials. These analyses aimed to provide descriptive data of potential early response criteria for ABSSSI and CABP. Analyses provided descriptive statistics and graphical depictions of summary data. No inferential statistical analyses (ie, hypothesis tests) were completed. The PT noted that these analyses are relevant to the specific context of use evaluated in these studies and have the potential to confirm observations from the earliest antimicrobial studies. A detailed description of the results, which is beyond the scope of this article, can be found in the FNIH submissions to the ABSSSI and CABP FDA dockets [3, 4] and also in the Supplementary Materials.
INTERIM RECOMMENDATIONS
On the basis of these analyses, the PT proposed outcome measures for use by sponsors for registrational trials involving ABSSSI and CABP. Most active-controlled trials of antimicrobials for these indications use a noninferiority design because of ethical and feasibility concerns. The PT's discussion therefore focused on the outcome measures used in ABSSSI noninferiority trials in adults. These outcome measures are proposed as “bridging” measures for registrational clinical trials planned for submission to the FDA.
In ABSSSI, response should be determined 48–72 hours after randomization, with baseline lesion size measurement and administration of the first dose of study drugs occurring as close to randomization as possible. For adults, success should be defined as control of infection lesion spread (ie, a ≥20% decrease in lesion area [calculated as the longest head-to-toe length times the longest perpendicular width] vs baseline). Absence of elevated body temperature (fever) should not be a component of the primary outcome measure, particularly because it is not on the causal pathway of the disease; furthermore, a requirement for fever as an inclusion criterion would exclude important patient populations (eg, elderly individuals), and frequent temperature measurements cannot be obtained reliably in many clinical trial settings, as confirmed in a subsequent phase 3 study [28]. However, resolution of elevated body temperature is important in clinical practice. Thus, resolution of fever and achievement of stability in other physiological parameters should be included as sensitivity analyses and/or as part of late assessment endpoints.
The proposed early endpoint serves as the link to the historical evidence of drug effect in previous studies, as required in the design of noninferiority trials [29]. The classification of response or failure is also independent of study events other than death; institution of alternative therapy, unplanned surgical drainage, and adverse events are not counted as part of the early response classification, but these measures can be captured as part of supportive information.
Patients may continue to receive the study drug even if they are classified as an early nonresponder, or they may have therapy withdrawn even if they are classified as an early responder. This element of trial conduct is especially important to enable analysis of a traditional later endpoint for both FDA and non-US regulatory authorities. Longer-term clinical outcome measures at a prespecified time point, such as the end of therapy and thereafter, are of relevance to clinicians and patients. These can be measured as secondary outcome measures. Evaluation of sustained control of lesion spread is critical, as are other clearly defined assessments of how patients feel or function from treatment initiation to infection resolution. However, defining components of later time points requires further research. In the meantime, for later endpoints, sponsors should propose to the FDA what they plan to measure and how and when they will measure it. Performing early and late assessments will allow separate analyses that meet differing requirements of FDA and non-US regulatory agencies, thereby allowing global drug development [10].
In CABP, assessment of symptom improvement at treatment day 4 (approximately 72 hours after randomization and initiation of therapy) provides relevant data on how patients feel and function and can provide evidence of a strong treatment effect for antibiotics via its link to assessments of symptom improvement in historical studies. Four biologically relevant symptoms (cough, pleuritic chest pain, dyspnea, and sputum production, scored as points for absent, mild, moderate, and severe) are recommended for adults. The proposed endpoint measure comprises (1) a 1-point improvement in at least 2 symptoms, (2) no worsening of any other symptoms, and (3) assessment on study day 4. Evaluations of the comprehensiveness of the symptoms in the definition should be a focus for future research. The timing of resolution of early symptom endpoints has not been well documented for infants and children.
As for ABSSSI, absence of elevated body temperature is not recommended as part of the early CABP endpoint, in part since it may be confounded by antipyretic therapy.
ALTERNATIVE VIEWPOINTS
Alternative viewpoints were voiced regarding the conclusion that the interim, bridging primary endpoints for registrational trials of ABSSSI and CABP should be based on the proposed early outcome measures. Although team members agreed that these early measurements provide important information, concerns included the lack of rigorous justification of a biomarker (control of lesion spread) as a registrational endpoint, the insufficiency of control of lesion spread as a comprehensive measure of overall improvement and ability to return to full functional status, the implicit inclusion of early response measures in test-of-cure endpoints, and the inability to determine whether the use of test-of-cure endpoints has led to approval of inactive therapies for life-threatening infections such as ABSSSI. Members agreed that well-defined, reliable, later endpoints would provide an important overall perspective. Furthermore, minimal lesion size requirements at baseline, as required by the FDA, exclude certain target populations (eg, infants and children). Finally, recent nonrandomized pharmacometric analyses show a correlation between drug exposure and traditional clinical and microbiological endpoints, although some observed that such evidence does not establish causality. The PT agreed that these issues should be addressed in future research.
ADDITIONAL INFORMATION NEEDED TO ADVANCE TO FINAL RECOMMENDATIONS
Analysis of current data reveals that the evidence base for determining outcomes in ABSSSI and CABP remains incomplete, particularly for certain populations (eg, pediatric patients). Future studies must address research gaps such as determining which outcomes are most important to patients and how to reliably and reproducibly assess them across various disease states, populations, and anatomic sites. The FNIH has initiated studies to address these questions.
DISCUSSION AND CONCLUSIONS
Clinical studies serve many purposes. For registrational trials, FDA regulations state that “the purpose of conducting clinical investigations of a drug is to distinguish the effect of a drug from other influences, such as spontaneous change in the course of the disease, placebo effect, or biased observation” [6]. For valid inferences from noninferiority trials, historical evidence of drug effects under the conditions of prior studies must apply to the active comparator's effect in the noninferiority trial [30], leading to need for consistency in design features, including definitions and timing of outcome measures.
In the process outlined above, members of academia, industry, and governmental agencies proposed interim, bridging outcome measures for future trials involving the ABSSSI and CABP indications. The PT evaluated historical evidence for drug effects—that is, the difference in outcomes between the test and control groups for antimicrobial and nonantimicrobial interventions for ABSSSI and CABP—and found that the largest treatment effects were documented early in the course of disease. This observation aligns with clinician assessment in that therapy is often changed in the absence of response after a few days of treatment. The evidence shows that, as the duration of follow-up increases for patients in the test and control groups, the proportion of patients who appear to achieve a response increases. However, the difference in outcomes between the antimicrobial and nonantimicrobial interventions often decreases over this period, which suggests that patients' responses later in treatment may be due in part to the disease's natural history.
Previous studies have not evaluated response at early time points in a standardized way. Carrying early nonresponses forward and combining them with later nonresponses may lower success rates, making it more challenging to demonstrate noninferiority while also not accurately measuring drug effect.
During the interim bridging period, early time points should be used in primary hypotheses of registrational noninferiority trials, because the choice of outcome measures is constrained by that design's reliance on estimates of the active control's effects from prior studies. However, the PT noted that measurement of effects early on the basis of noninferiority hypotheses does not preclude superiority hypotheses for later effects. This approach conforms to clinical concerns that some drugs may differ on later outcomes. Superiority studies can evaluate other endpoints and timing of outcomes as long as they are clinically meaningful to patients, well-defined, and reliable. Of importance, failure to show superiority at later time point should not preclude approval.
The importance of a late time point is greatest in diseases in which a lack of sustained cure is part of the natural history of the disease. Evaluation of the historical evidence and current trials showed low rates of late failure in both ABSSSI and CABP. This situation is in contrast to conditions such as Clostridium difficile infection, in which up to a quarter of patients may not maintain an initial response [31].
The measurement of early effects addresses clinical practice as well as clinical trials. The historical evidence shows that a few doses of an antimicrobial can have a profound effect on patient outcome. The historical evidence, as well as results of more recent clinical trials, also shows that shorter therapy durations may be appropriate for both ABSSSI and CABP.
The PT recognized the limitations of the proposed interim endpoints. Therefore, the FNIH is proceeding with prospective research to evaluate the content validity and measurement properties of endpoints in both ABSSSI and CABP for adults. The studies on ABSSSI are anticipated to begin in the summer of 2012, with work on CABP to begin in the subsequent winter pneumonia season.
On the basis of these deliberations and input from the FDA, new antimicrobials for ABSSSI have advanced into phase 2 and 3 development [18, 32–36]. In addition, retrospective analyses of other ABSSSI and CABP clinical trial data sets have produced results concordant with those of the PT analyses [37–40].
In summary, these interim, evidence-based outcome measures are proposed on the basis of an analysis of the historical literature, which showed an early treatment effect of antimicrobials for ABSSSI and CABP. A core focus of registrational studies is to determine the presence or absence of a treatment effect. These interim outcome measures allow noninferiority registrational studies to proceed in the United States while future qualitative and quantitative research studies are being conducted. These future studies are critical in addressing knowledge gaps and would help address unanswered questions related to designing noninferiority trials involving these 2 important indications.
Supplementary Data
Supplementary materials are available at Clinical Infectious Diseases online (http://cid.oxfordjournals.org). Supplementary materials consist of data provided by the author that are published to benefit the reader. The posted materials are not copyedited. The contents of all supplementary data are the sole responsibility of the authors. Questions or messages regarding errors should be addressed to the author.
Notes
Acknowledgments. We thank the following members of the CABP ABSSSI Project Team for their contributions: Joseph Toerner, MD, MPH (cochair, FDA; nonvoting); George H. Talbot, MD (cochair, Infectious Diseases Society of America [IDSA]); Jeff Alder, PhD (Bayer); Paul G. Ambrose, PharmD (Institute for Clinical Pharmacodynamics, University of Oxford); Helen Boucher, MD (Tufts University); John Bradley, MD (University of California, San Diego); Laurie Burke, RPh, MPH (FDA; nonvoting); Edward M. Cox (FDA; nonvoting); Aaron Dane, MSc (AstraZeneca); Anita Das, PhD (AxiStat); Dennis Dixon, PhD (NIH/NIAID); Michael Dunne, MD (Durata Therapeutics); Barry Eisenstein, MD (Cubist Pharmaceuticals); Thomas R. Fleming, PhD (University of Washington); Dean Follmann, PhD (NIH/NIAID); David Friedland, MD (Cerexa); Ian Friedland, MD (Cubist Pharmaceuticals); Nicholas Kartsonis, MD (Merck); Achim Kaufhold, MD, PhD (Basilea Pharmaceutica International); Scott Komo DrPH (FDA; nonvoting); Mike Kurilla, MD, PhD (NIH/NIAID); Kim Lindfield, PhD (Cubist Pharmaceuticals); Lily Llorens, PhD (Cerexa); Susan Moriarty, MD (Cempra Pharmaceuticals); Sumati Nambiar, MD (FDA observer; nonvoting); Gary J. Noel, MD (Paratek Pharmaceuticals); David Oldach (Cempra Pharmaceuticals); Elektra Papadopoulos, MD (FDA; nonvoting); Roger Pomerantz, MD (Merck); John Powers, MD (SAIC, in support of NIH/NIAID); Philippe Prokocimer, MD (Trius Therapeutics); John Quinn, MD (Pfizer); John Rex, MD (AstraZeneca); Denise Russo (NIH/NINR); Jennifer Schranz, MD (Cempra Pharmaceuticals); Judith A. Siuciak, PhD (FNIH); William Stubbings, PhD (Basilea Pharmaceutica International); and David Wholley (FNIH).
Disclaimer. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the US Government. The conclusions described within this document represent the work of the FNIH Biomarkers Consortium Project “Developing Endpoints for Clinical Trials of Drugs for Treatment of Acute Bacterial Skin and Skin Structure Infections and Community-Acquired Bacterial Pneumonia (CABP ABSSSI Project).”
Financial support. This work was supported by the National Cancer Institute (contract HHSN261200800001E) and the National Institute of Allergy and Infectious Diseases, National Institutes of Health; Actelion Pharmaceuticals; Basilea Pharmaceutica International; Cempra Pharmaceuticals; Cerexa, a wholly owned subsidiary of Forest Laboratories; Cubist Pharmaceuticals; Merck; Nabriva Therapeutics; and Trius Therapeutics. Clinical trial data were also contributed to the project by Cerexa, Cubist Pharmaceuticals, Durata Therapeutics, and Pfizer.
Potential conflicts of interest. Within the past 36 months, G. H. T., through Talbot Advisors, has received board compensation and/or consultancy fees from Actelion, Basilea, Bayer, Cempra, Cerexa, Durata, Cubist-Calixa, FAB Pharma, J&J, Kalidex, Meiji, Merada, Merck, Nabriva, and Wyeth/Pfizer (data safety monitoring board). G.H.T. also has equity interests in Durata, Calixa, Cerexa, Kalidex, and Nabriva; he did not receive compensation for work on the PT, including manuscript preparation, but travel expenses to PT meetings were reimbursed by the Infectious Diseases Society of America and the FNIH. In past 36 months, J. P. served as consultant for Advanced Life Sciences, Astra-Zeneca, Cardeas, Contrafect, Gilead, Johnson and Johnson, LEO, Methylgene, Novartis, Optimer, Pfizer, Trius, Warner Chilcott, and Wyeth. J.S. is an employee of the FNIH. In the last 3 years, H. B. provided advice or consultation to Basilea, Cerexa, Cubist, Durata, Merck (adjudication committee), J&J, Rib-X, Targanta/The Medicines Company, and Wyeth/Pfizer (data safety monitoring board). All other authors: No reported conflicts.
All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
References
- 1.US Food and Drug Administration, Center for Drug Evaluation. Guidance for industry: acute bacterial skin and skin structure infections: developing drugs for treatment. 2010 Available at: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm071185.pdf . Accessed 4 March 2012. [Google Scholar]
- 2.US Food and Drug Administration, Center for Drug Evaluation. Guidance for industry: community-acquired bacterial pneumonia: developing drugs for treatment. 2009 Available at: http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm123686.pdf . Accessed 4 March 2012. [Google Scholar]
- 3.Foundation for the National Institutes of Health Biomarkers Consortium Project Team. Recommendations to FDA for interim endpoints for clinical trials in acute bacterial skin and skin structure infections. Available at: http://www.regulations.gov/#!docketDetail;rpp=10;po=0;D=FDA-2010-D-0433T . Accessed 4 March 2012. [Google Scholar]
- 4.Foundation for the National Institutes of Health Biomarkers Consortium Project Team. Recommendations to FDA for interim endpoints for clinical trials in community-acquired bacterial pneumonia. Available at: http://www.regulations.gov/#!docketDetail;rpp=10;po=0;D=FDA-2009-D-0136 . Accessed 4 March 2012. [Google Scholar]
- 5.Powers JH. Increasing the efficiency of clinical trials of antimicrobials: the scientific basis of substantial evidence of effectiveness of drugs. Clin Infect Dis. 2007;45:S153–62. doi: 10.1086/519253. [DOI] [PubMed] [Google Scholar]
- 6.US Government Printing Office. Applications for FDA approval to market a new drug: adequate and well controlled studies. US Code of Federal Regulations, Title 21, section 314.126. Available at: http://ecfr.gpoaccess.gov/cgi/t/text/text-idx?c=ecfr&sid=7d9fbc2195c6aaa883251008d0eb45bc&rgn=div8&view=text&node=21:5.0.1.1.4.4.1.14&idno=21 . Accessed 4 March 2012. [Google Scholar]
- 7.Micheel CM, Ball JR, editors. 2010. Evaluation of biomarkers and surrogate endpoints in chronic disease. Institute of Medicine: committee on qualifications of biomarkers and surrogate endpoints in chronic disease. Available at: http://www.nap.edu/catalog/12869.html . Accessed 4 March 2012. [PubMed] [Google Scholar]
- 8.US Government Printing Office. Applications for FDA approval to market a new drug: accelerated approval of new drugs for serious and life-threatening illnesses. US Code of Federal Regulations, Title 21, section 314.510. Available at: http://ecfr.gpoaccess.gov/cgi/t/text/text-idx?c=ecfr&sid=7d9fbc2195c6aaa883251008d0eb45bc&rgn=div8&view=text&node=21:5.0.1.1.4.8.1.2&idno=21 . Accessed 4 March 2012. [Google Scholar]
- 9.Department of Health and Human Services, Food and Drug Administration. New drug, antibiotic, and biological drug regulations; accelerated approval. Available at: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM077130.pdf . Accessed 3 July 2012. [Google Scholar]
- 10.European Medicines Agency. Guideline on the evaluation of medicinal products indicated for treatment of bacterial infections. 2011 CPMP/EWP/558/95 rev 2. Available at: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003417.pdf . Accessed 4 March 2012. [Google Scholar]
- 11.Snodgrass WR, Anderson T. Prontosil in erysipelas. Br Med J. 1937;2:101–4. doi: 10.1136/bmj.2.3993.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Snodgrass WR, Anderson T. Sulphanilamide in the treatment of erysipelas. Br Med J. 1937;2:1156–9. doi: 10.1136/bmj.2.4014.1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Osler W. The principles and practice of medicine. New York: D. Appleton and Company; 1910. Specific infectious diseases: lobar pneumonia; pp. 164–92. [Google Scholar]
- 14.Bullowa JGM. The management of pneumonias. New York, NY: Oxford University Press; 1937. Chapter II. The course, symptoms and physical findings; pp. 36–76. [Google Scholar]
- 15.Flippin HF, Lockwood JS, Pepper DS, Schwartz D. The treatment of pneumococcic pneumonia with sulfapyridine. JAMA-J Am Med Assn. 1939;112:529–34. [Google Scholar]
- 16.Meakins JC, Hanson FR. The treatment of pneumococcic pneumonia with sulfapyridine. Can Med Assoc J. 1939;40:333–6. [PMC free article] [PubMed] [Google Scholar]
- 17.Wilson AT, Spreen HA, Cooper ML, Stevensen FE, Cullen GE, Mitchell AG. Sulfapyridine in the treatment of pneumonia in infancy and childhood. JAMA. 1939;112:1435–9. [Google Scholar]
- 18.Finland M, Spring WC, Lowell FC. Immunological studies on patients with pneumococcic pneumonia treated with sulfapyridine. J Clin Invest. 1940;19:179–99. doi: 10.1172/JCI101110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Petersdorf RG, Cluff LE, Hoeprich PD, Hopkins FT, McCann WP. Pneumoccocal pneumonia treated with penicillin and aspirin. Bull Johns Hopkins Hosp. 1957;101:1–12. [PubMed] [Google Scholar]
- 20.Finland M, Spring WC, Lowell FC. Specific treatment of the pneumococcic pneumonias; an analysis of the results of serum therapy and chemotherapy at the Boston City Hospital from July 1938 through June 1939. Ann Intern Med. 1940;13:1567–93. [Google Scholar]
- 21.Singer M. Treatment effect of antibacterial drugs in community acquired bacterial pneumonia. Paper presented at: Anti-Infective Drugs Advisory Committee Meeting, 9 December 2009. Available at: http://www.fda.gov/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/Anti-InfectiveDrugsAdvisoryCommittee/ucm195619.htm . Accessed 4 March 2012. [Google Scholar]
- 22.Corey GR, Wilcox M, Talbot GH, et al. Integrated analysis of CANVAS 1 and 2: phase 3, multicenter, randomized, double-blind studies to evaluate the safety and efficacy of ceftaroline versus vancomycin plus aztreonam in complicated skin and skin structure infection. Clin Infect Dis. 2010;51:641–50. doi: 10.1086/655827. [DOI] [PubMed] [Google Scholar]
- 23.Jauregui LE, Babazadeh S, Seltzer E, et al. Randomized, double-blind comparison of once-weekly dalbavancin versus twice-daily linezolid therapy for the treatment of complicated skin and skin structure infections. Clin Infect Dis. 2005;41:1407–15. doi: 10.1086/497271. [DOI] [PubMed] [Google Scholar]
- 24.Weigelt J, Itani K, Stevens D, et al. Linezolid versus vancomycin in treatment of complicated skin and soft tissue infections. Antimicrob Agents Chemother. 2005;49:2260–6. doi: 10.1128/AAC.49.6.2260-2266.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tanaseanu C, Bergallo C, Teglia O, et al. Integrated results of 2 phase 3 studies comparing tigecycline and levofloxacin in community-acquired pneumonia. Diagn Microbiol Infect Dis. 2008;61:329–38. doi: 10.1016/j.diagmicrobio.2008.04.009. [DOI] [PubMed] [Google Scholar]
- 26.Pertel PE, Bernardo P, Fogarty C, et al. Effects of prior effective therapy on the efficacy of daptomycin and ceftriaxone for the treatment of community-acquired pneumonia. Clin Infect Dis. 2008;46:1142–51. doi: 10.1086/533441. [DOI] [PubMed] [Google Scholar]
- 27.File TM, Jr, Low DE, Eckburg PB, et al. Integrated Analysis of FOCUS 1 and FOCUS 2: randomized, doubled-blinded, multicenter phase 3 trials of the efficacy and safety of ceftaroline fosamil versus ceftriaxone in patients with community-acquired pneumonia. Clin Infect Dis. 2010;51:1395–405. doi: 10.1086/657313. [DOI] [PubMed] [Google Scholar]
- 28.Trius reports positive results from first phase 3 trial of tedizolid in acute bacterial skin and skin structure infections. 19 December 2011. Available at: http://investor.triusrx.com/releases.cfm . Accessed 4 March 2012. [Google Scholar]
- 29.Guidance for industry antibacterial drug products: use of noninferiority trials to support approval. Available at: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM070951.pdf . Accessed 4 March 2012. [Google Scholar]
- 30.Department of Health and Human Services, Food and Drug Administration. E10 Choice of control group and related issues in clinical trials. Available at: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM073139.pdf . Accessed 3 July 2012. [Google Scholar]
- 31.Louie TJ, Miller MA, Mullane KM, et al. Fidaxomicin versus vancomycin for Clostridium difficile infection. N Engl J Med. 2011;364:422–31. doi: 10.1056/NEJMoa0910812. [DOI] [PubMed] [Google Scholar]
- 32.Rib-X reports positive top-line data from phase 2b study of delafloxacin in patients with acute bacterial skin and skin structure infections. 15 December 2011. Available at: http://www.rib-x.com/investors/press-release_2011_12_15.php . Accessed 4 March 2012. [Google Scholar]
- 33.Covington P, Davenport JM, Andrae D, et al. Randomized, double-blind, phase 2, multicenter study evaluating the safety/tolerability and efficacy of JNJ-Q2, a novel fluoroquinolone, compared with linezolid for treatment of acute bacterial skin and skin structure infection. Antimicrob Agents Chemother. 2011;55:5790. doi: 10.1128/AAC.05044-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Durata Therapeutics initiates enrollment in Discover-2, second phase 3 study of dalbavancin for the treatment of acute bacterial skin and skin structure infections. Available at: http://www.duratatherapeutics.com/prSEPT292011.php . Accessed 4 March 2012. [Google Scholar]
- 35.Prince WT, Obermayr F, Ivezic-Schoenfeld Z, et al. Washington, DC: American Society for Microbiology; 2011. A phase 2 study comparing the safety and efficacy of two doses of BC-3781 versus vancomycin in acute bacterial skin and skin structure infections (ABSSSI). [abstract L-966] Program and Abstracts of the 51st Interscience Conference on Antimicrobial Agents and Chemotherapy (Chicago) [Google Scholar]
- 36.Bhavnani SM, Hammel JP, Rubino CM, et al. Washington, DC: American Society for Microbiology; 2011. Pharmacokinetic-pharmacodynamic analysis for efficacy of BC-3781 using new clinical trial endpoints in patients with acute bacterial skin and skin structure infection [abstract A2-025] Program and Abstracts of the 51st Interscience Conference on Antimicrobial Agents and Chemotherapy (Chicago) [Google Scholar]
- 37.De Anda C, Das A, Fang E, Prokocimer P. Washington, DC: American Society for Microbiology; 2011. Acute bacterial skin and skin structure infection (ABSSSI) dose-ranging phase 2 tedizolid phosphate (TR-701) study; assessment of efficacy and safety with new FDA guidance [abstract L1-1496] Program and Abstracts of the 51st Interscience Conference on Antimicrobial Agents and Chemotherapy (Chicago) [Google Scholar]
- 38.Dunne MW, Talbot GH, Das AF. Dalbavancin vs linezolid for treatment of acute bacterial infections of the skin: a comparison of early and standard outcome measures in Study Ver001-9 [abstract P1530] Abstracts of 21st ECCMID/27th ICC, Milan, Italy, 7–10 May 2011. [Google Scholar]
- 39.Hait H, Arbeit R, Molnar D, Noel GJ, Tanaka SK. In a phase 2 complicated skin and soft tissue infections trial, outcomes assessed early in the course of therapy were consistent with outcomes 1017 days after completing therapy with either omadacycline (OMC; PTK796) or linezolid [abstract P1528] Abstracts of the 21st ECCMID/27th ICC, Milan, Italy, 7–10 May 2011. [Google Scholar]
- 40.Stryjewski M, Corey RG, Yu-Ping Li, Barriere SL. Washington, DC: American Society for Microbiology; 2011. Post-hoc analysis of efficacy of telavancin with complicated skin and skin structure infections—applying the new FDA guidance [abstract L1-1493] Program and Abstracts of the 51st Interscience Conference on Antimicrobial Agents and Chemotherapy (Chicago) [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.