

Abstract
The anti-inflammatory phytohormone abscisic acid (ABA) modulates immune and inflammatory responses in mouse models of colitis and obesity. ABA has been identified as a ligand of lanthionine synthetase C-like 2, a novel therapeutic target upstream of the peroxisome proliferator-activated receptor γ (PPAR γ) pathway. The goal of this study was to investigate the immune modulatory mechanisms underlying the anti-inflammatory efficacy of ABA against influenza-associated pulmonary inflammation. Wild type (WT) and conditional knockout mice with defective PPAR γ expression in lung epithelial and hematopoietic cells (cKO) treated orally with or without ABA (100 mg/kg diet) were challenged with Influenza A/Udorn (H3N2) to assess ABA’s impact in disease, lung lesions and gene expression. Dietary ABA ameliorated disease activity, lung inflammatory pathology, accelerated recovery and increased survival in WT mice. ABA suppressed leukocyte infiltration and MCP-1 mRNA expression in WT mice through PPAR γ, since this effect was abrogated in cKO mice. ABA ameliorated disease when administered therapeutically on the same day of the infection to WT but not mice lacking PPAR γ in myeloid cells. We also show that ABA’s greater impact is between days 7 and 10 post-challenge when it regulates the expression of genes involved in resolution, like 5 lipoxygenase and other members of the 5-lipoxygenase pathway. Furthermore, ABA significantly increased the expression of the immunoregulatory cytokine IL-10 in WT mice. Our results show that ABA, given preventively or therapeutically, ameliorates influenza virus-induced pathology by activating PPAR γ in pulmonary immune cells, suppressing initial proinflammatory responses and promoting resolution.
Keywords: influenza, PPAR γ, abscisic acid, pulmonary inflammation, LANCL2
1. Introduction
Studies on the pathogenesis of Influenza A virus infection have highlighted the relevance of disassociating the cytopathic effects caused by the virus from the immunopathology resulting from an exacerbated host response following infection in the better understanding of viral pathogenesis and informed development of prophylactic and therapeutic interventions against flu[1]. The use of immunotherapeutics targeting the immune response and not directly the virus to ameliorate disease severity has been proposed as an alternative approach to reduce flu-associated morbidity[2–4]. Human cases of influenza A (H5N1) have been typically characterized by a predominance of high viral loads and increased levels of inflammatory cytokines and chemokines (IP-10, MIG and MCP-1) in blood, which were even more accentuated in patients that succumbed to the infection [5]. Mice challenged with the reconstructed Influenza A/1918 (H1N1)virus overexpressed inflammatory genes associated with immune cells infiltrating the lungs, mainly neutrophils and macrophages [6]. Moreover, even though the newly emerged Influenza A/2009 (H1N1) virus generally caused mild disease, reported pathologic findings in fatal cases included enhanced bronchiolitis, alveolitis and neutrophilic infiltration, closely resembling those described for the Influenza A/1918 (H1N1) pandemic[7].
The underlying mechanisms by which the dysregulated cytokine and chemokine production contributes to the pathogenesis of influenza are incompletely understood. Experimental infections of mice lacking key cytokines, chemokines or their receptors have yielded inconclusive results possibly due to the built-in redundancies found in the cytokine/chemokine signaling pathways[8–11]. In support of the role of the host inflammatory response in the pathogenesis of influenza, recent reports have shown that the use of immunomodulators that partially block inflammatory cascades improve the outcome of influenza A virus infection. For instance, inhibition of cyclooxygenase 2 combined with the inflammatory bowel disease drug and peroxisome proliferator-activated receptor (PPAR) γ agonist mesalazine[12], the PPAR γ agonist pioglitazone (Actos)[13], and the PPAR α agonist germ fibrozil [14] have shown protective effects against Influenza A H5N1, H1N1 and H2N2, respectively. Interestingly, Cloutier at al demonstrated that rosiglitazone (Avandia) was unable to protect mice against lethal influenza challenge [15]. This along with their adverse side effects raises concerns on the eventual use of thiazolidinediones (TZDs) for treating influenza. Therefore, the development of safer orally active, broad-based, anti-inflammatory, host-targeted therapeutics represents a promising new avenue to decrease the damage at the respiratory tract associated with influenza virus infection.
PPAR γ is a ligand-activated transcription factor that participates in cellular metabolic adaptations to meet structural and energetic needs for differentiation, growth and proliferation during inflammation [16]. PPAR γ in particular regulates lipid uptake and storage and is activated by endogenous ligands, many of which arise during the inflammatory process from the metabolism of fatty acids through the lipoxygenase and cyclooxygenase pathways. The cellular metabolic control exerted by activated PPAR γ occurs through transactivation and requires direct binding of the receptor to DNA in promoter regions of its target genes. In parallel, activated PPAR γ antagonizes signal-activated transcription factors that regulate the expression of pro-inflammatory genes such as NF-κB, STAT-1 and AP1[17,18]. Ligand-activated PPAR γ has the ability to sequester co-activator complexes on the promoter regions of pro-inflammatory genes making them inaccessible to these transcription factors. In addition, the transcriptional and transprepressional activities of PPAR γ are modulated by pos-transcriptional modifications, such as phosphorylation. In this regard, cAMP has been shown to increase ligand-activated PPAR γ transcriptional activity by modulating its ability to bind to DNA.
We have identified the phytohormone abscisic acid (ABA) as a novel PPAR γ agonist that activates the receptor through an alternative mode of action [19]. ABA mimicked other PPAR γ agonists like TZDs in their ability to improve insulin resistance and suppress systemic inflammation in obese mice, and upregulated PPAR γ target genes. However, ligand binding assays and molecular modeling demonstrated that ABA does not bind to the PPAR γ ligand-binding domain. It was also shown that ABA binds to the G-protein coupled receptor lanthionine synthetase component C-like 2 (LANCL2), which results in cAMP elevation. Thus, the molecular mechanism of action of ABA differs from that of the TZD. Results of pre-clinical efficacy studies demonstrate that ABA ameliorates experimental IBD through a mechanism that requires expression of PPAR γ in T cells[20].
The objectives of this study were to 1) determine whether dietary ABA prevents or ameliorates influenza virus-related pulmonary immunopathology, 2) characterize the mechanisms of immunoregulation by ABA in the respiratory mucosa, and 3) examine whether the deletion of PPAR γ in immune and epithelial cells or myeloid cells abrogates the preventive or therapeutic effects of ABA on influenza.
2. Materials and Methods
2.1 Animal Procedures
Hematopoietic and epithelial cell-specific PPAR γ-deficient mice were obtained by crossing PPAR γ fl/fl mice with mice expressing cre under the control of the MMTV-LTR promoter (PPAR γ fl/fl; MMTV-Cre+, referred to as conditional KO-cKO). Myeloid-specific PPAR γ-deficient mice were obtained by crossing PPAR γ fl/fl mice with mice expressing cre under the control of the LysM promoter (PPAR γ fl/fl; LysM-Cre+, referred to as Myeloid KO). Homozygous PPAR γ fl/fl littermate mice lacking the MMTV-Cre transgene were used as wild type (WT). All mice were in a C57BL/6 background. Screening of PPARγ gene deletion was done by PCR on tail DNA using previously published protocols[21,22]. In experiments where ABA was use preventively, mice were fed purified AIN-93G rodent diets (Harlan Teklad) with or without ABA (Supplementary Table 1). Based on previous findings, a dose of 100 mg ABA/kg of diet was determined to be optimal for down-modulating systemic inflammation and glucose tolerance in mouse models of obesity and diabetes [23] and decreasing intestinal inflammation in mouse models of IBD [24,25]. Four week-old mice were administered the experimental diets supplemented for 36 days prior to an intranasal challenge with influenza A/Udorn (H3N2), and throughout the challenge period equivalent to an optimal prophylactic dosage of 0.2 mg ABA daily per mouse, based on average feed intake of 2 g food/mouse/day. In experiments where ABA was administered therapeutically, mice received 100 mg/kg of ABA given by oral gavage from day 0 to day 10 post-infection. Body weights were monitored daily following challenge. Mice were housed at the animal facilities at Virginia Tech. All experimental procedures were approved by the Institutional Animal Care and Use Committee of Virginia Tech and met or exceeded requirements of the Public Health Service/National Institutes of Health and the Animal Welfare Act.
2.2 Influenza virus challenge
Mice were challenged intranasally with 5×104 tissue culture infectious dose 50 (TCID)50 of Influenza A/Udorn/72 (H3N2) given in 50 μl of sterile PBS under anesthesia with xylazine and ketamine (50–150 mg/kg). Mock-infected mice received the same volume of PBS.
2.3 Quantification of viral loads
Viral loads in lung homogenates were determined as described[26]. Briefly, serial 10-fold sample dilutions of lung homogenates were incubated with MDCK cells for 1 hour at 37°C to allow for virus adsorption. Subsequently, cells were washed and incubated for 3 days at 37°C in the presence of 1.5 μg/ml TPCK-treated trypsin (Sigma) and cytopathic effects were recorded. Viral loads are reported as 50% tissue culture infectious dose units (TCID50/ml) per gram lung tissue as determined by the Reed-Muench method[27].
2.4 Pulmonary Histopathology
Lung specimens were fixed in formalin and processed for hematoxylin and eosin staining. Lesions were graded based on a compounded histology score including the extent of 1) epithelial necrosis/regeneration, 2) presence of desquamated cells and inflammatory cellular infiltrates within the airways, 3) presence of leukocytic infiltrates in the mucosa and submucosa of airways, 4) presence of marginated leukocytes and inflammatory cells surrounding blood vessels (i.e., perivascular cuffing), 5) presence of edema, fibrin deposits or hyaline membranes and 6) terminal airway infiltration. The sections were graded with a score of 0–4 for each of the previous categories and data were analyzed as a normalized compounded score.
2.5 Bronchoalveolar lavage (BAL)
To obtain cells from the bronchoalveolar space, the trachea was cannulated postmortem by using a gavage needle and lungs were washed three times with 1 ml of room temperature PBS that were subsequently combined. Approximately 90% of the total instilled volume was consistently recovered.
2.6 Western blot
Cells obtained by lavage and lung specimens were homogenized in RIPA buffer (150 mM NaCl, 1.0% IGEPAL® CA-630, 0.5% sodium deoxycholate, 0.1% SDS, 50 mM Tris, pH 8.0) containing protease and phosphatase inhibitors and incubated for 30 min on ice. Whole lysates were cleared by centrifugation (10,000 rpm for 10 min) and protein concentration was measured using a DC protein assay kit (Bio-Rad Laboratories). Proteins were separated on a 10% SDS-PAGE gel and transferred to polyvinylidene difluoride (PVDF) membrane. The membrane was blocked with 1x TBS-T (20 mM Tris-HCl pH 7.6, 8.5% NaCl, 0.1% Tween-20) containing 3% Bovine Serum Albumin (BSA, Sigma-Aldrich) 30 min at room temperature. Membranes were incubated overnight at 4°C with anti-PPAR γ antibody diluted in 1x TBS-T 3% BSA. After 3 washes with 1x PBS-T, membrane was incubated for 45min at room temperature with anti-mouse IgG conjugated to horseradish peroxidase (HRP). The antigen detection was performed with the ECL (Bio-Rad Laboratories) chemiluminescent detection system.
2.7 Real-time RT-PCR
Total RNA was isolated from the lung homogenates or BAL-derived cells using the RNA isolation Minikit (Qiagen) according to the manufacturer’s instructions. Total RNA (0.5 to 1 μg) was used to generate cDNA template using the iScript cDNA Synthesis Kit (Bio-Rad, Hercules, CA). Starting cDNA concentrations for genes of interest were examined by quantitative PCR using an iCycler IQ System and the iQ SYBR green supermix (Bio-Rad) as previously described [21]. A standard curve was generated for each gene using 10-fold dilutions of purified amplicons starting at 5 pg of cDNA and used later to calculate the starting amount of target cDNA in the unknown samples.
2.8 Statistical analyses
Data were analyzed as a 2 × 2 factorial arrangement of treatments within completely randomized design. The statistical model was: Yijk = μ + Genotypei + Treatmentj + (Genotype ×Treatment)ij + error Aijk, in which μ was the general mean, Genotypei was the main effect of the ith level of the genotypic effect (expression of PPAR γ by immune and epithelial cells), Treatmentj was the main effect of the jth level of the dietary effect (ABA vs control), (Genotype ×Treatment)ij was the interaction effect between genotype and diet, and error A representing the random error. For analyzing the weight loss or histological lesion scores over time a repeated measures ANOVA was utilized (treatment × genotype × time). To determine the statistical significance of the model, analysis of variance (ANOVA) was performed using the general linear model procedure of Statistical Analysis Software (SAS), and probability value (P)< 0.05 was considered to be significant. When the model was significant, ANOVA was followed by Fisher’s Protected Least Significant Difference multiple comparison method.
3. Results
3.1 Influenza virus infection upregulates pulmonary PPARγ expression
PPARγ is expressed in the lungs of healthy mice [28], however the expression of PPARγ during influenza virus infection has not been reported before. Because we were proposing to use a therapeutic intervention targeting PPARγ, we evaluated changes in protein expression overtime following infection. Figure 1 shows that PPARγ is significantly upregulated and reaches its maximum expression in the lungs as early as 24 hours post-infection, it slightly declines on day 3 and again increases between days 5 and 9 post-infection. The kinetics of PPARγ expression in cells obtained from the lung airways by lavage, which correspond mainly to infiltrated immune cells, was very different from the pattern obtained in the lungs. Protein was not detectable until day 3 post-infection and reached a maximum at day 9. These results indeed give support to interventions targeting PPARγ via exogenous activation using orally active compounds to ameliorate influenza virus-associated disease and lung pathology.
Figure 1.
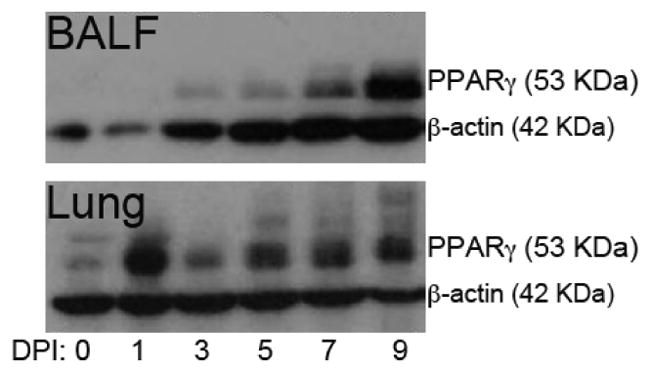
Peroxisome proliferator-activated receptor (PPAR)γ protein expression during influenza virus infection in cells obtained by bronchoalveolar lavage fluid (BALF) (top), and lung (bottom). WT mice were infected with 5 × 104 tissue culture infectious dose 50 (TCID50) of influenza A/Udorn (H3N2) and sacrificed 1,3,5,7 or 9 days post-challenge. Non-infected mice (0) were used to assess baseline PPAR γ expression at day 0. Influenza virus infection upregulated PPAR γ protein, reaching highest expression at day one post-infection in whole lung and on day 9 post-infection in cells obtained from the bronchoalveolar space. Results from BALF correspond to cells pooled from at least 3 mice at each time point. Results from lung correspond to a sample obtained from one mouse at each time point and the picture is representative of 4 replicates with identical results.
3.2 Effect of ABA and conditional PPAR γ deletion on influenza virus infection-associated weight loss and pulmonary pathology
Based on previous work showing that ABA transactivates PPARγ reporter activity in macrophages [19] and mimics the capacity of other PPARγ agonists to suppress inflammation [29,30] we hypothesized that ABA could ameliorate the inflammatory response that takes place during viral pneumonia. To investigate the possible interaction between ABA and PPAR γ during influenza A virus infection, we used conditional KO (cKO) mice that have defective PPAR γ expression in immune and epithelial cells of the lungs due to cre-induced deletion of PPAR γ, and their cre- WT littermates. Mice were fed either a diet containing ABA (100 mg/kg) or an isocaloric control AIN-96G diet without ABA for 36 days prior to an intranasal challenge with 5 × 104 TCID50 of Influenza A/Udorn (H3N2). Weight loss was monitored daily after challenge as an indicator of disease severity. Figure 2 illustrates that following the influenza virus challenge WT mice treated orally with ABA lost a maximum of 5 % of their original body weight compared to 10 % in mice that received control diet. Moreover, WT mice orally treated with ABA recovered their pre-challenge body weight between days 8 and 9 post challenge, while control-fed WT mice did not recover their original body weight until day 14 post-infection. Thus, oral ABA treatment ameliorated the clinical disease associated with influenza virus infection and accelerated the recovery in WT mice. Our data also show that the beneficial effect of ABA on influenza-related weight loss was PPAR γ dependent, since cKO mice behaved similar to WT mice fed the control diet. In addition to minimizing influenza-associated weight loss, oral ABA administration improved survivability rates in WT mice. Specifically, whereas 100% of mice in the WT/ABA group survived the infection, the rate dropped to 85.7 % in the cKO mice in control diet, 71,2 % in cKO mice in ABA diet and to 62.5 % in WT mice in control diet (Figure 2B).
Figure 2.

Effect of dietary abscisic acid (ABA)-supplementation on weight loss (A), survival rates (B), and lung viral loads(C) following infection with influenza virus. Wild-type (WT) or immune/epithelial cell-specific PPAR γ null mice (cKO) mice were fed either a control or an ABA-supplemented diet (100 mg/ABA kg of diet) for 36 days and then challenged with 5 × 104 tissue culture infectious dose 50 (TCID50) of influenza A/Udorn (H3N2). Body weight results indicate that ABA treatment prevented weight loss associated with influenza virus infection in the WT but not in the cKO mice, suggesting a PPAR γ-dependent mechanism of action (A). A similar beneficial pattern was observed in the survival rates for the ABA-treated mice (B). For the weight loss data, Asterisks denote statistically significant differences between the ABA-treated WT and the myeloid KO mice. Pound signs denote statistically significant differences between the ABA-treated WT and the control WT (n=10 mice per treatment and genotype). These effects were not associated to differences in lung viral load due to ABA treatment on day 4 post-infection (C).
3.3 ABA pre-treatment does not suppress virus replication
To evaluate whether ABA had any direct or indirect impact on the virus, we measured virus loads in lungs of mice at day 4 post-infection, coinciding with the end of the phase of maximum replication. Figure 2C shows that there were no significant differences in the amount of virus detected due to ABA treatment or genotype, which indicates that ABA ameliorated the disease through a mechanism independent of suppressing viral replication or enhancing early viral clearance. Of note, ABA also did not influence viral replication or viral yield when in vitro infection of MDCK cells was carried out in the presence of ABA, further suggesting that ABA does not act directly on the virus replication machinery or interfere in virus assembly or release (data not shown). Thus, suggesting an effect on the modulation of the host response to the virus.
3.4 Dietary ABA improves influenza virus-associated lung immunopathology in WT mice by diminishing the recruitment of inflammatory leukocytes
To examine whether the improved clinical findings observed in ABA-treated WT mice correlated with decreased lung pathology, we evaluated microscopic pulmonary lesions at 2, 4 and 7 days post-infection. Dietary ABA treatment did not affect the extent of epithelial necrosis or the amount of necrotic and inflammatory cells in large airways occurring early following infection (Figure 3A–B). However, ABA treatment diminished the extent of vascular infiltrates (Figure 3C) as well as the infiltration of respiratory airways mucosa and submucosa, and alveolar space in WT mice (Figure 3D) when compared to infected WT mice fed the control diet. Moreover, the effect of ABA in decreasing pulmonary damage was PPARγ-dependent, since the beneficial effect of ABA on lung inflammatory cell infiltration observed in WT mice was abrogated in cKO mice fed ABA. These results suggest that the mechanism by which ABA protects from influenza-induced lung pathology is related to diminished recruitment of inflammatory cells into the lung and requires full expression of PPAR γ.
Figure 3.

Effect of dietary ABA-supplementation on lung histopathology. Wild-type (WT) or cKO mice were fed either a control or pre-treated with ABA and infected with influenza 104 TCID50 of A/Udorn (H3N2) virus. ABA treatment did not ameliorate epithelial necrosis (A), but it diminished perivascular infiltration (C) as well as the infiltration of the terminal respiratory airways mucosa and submucosa (D) when compared to control-fed mice. The lower number of inflammatory cells in the lungs of ABA-treated WT mice correlated with downregulation of monocyte chemo attractant protein-1 (MCP-1) mRNA following infection when compared to control-fed mice(E). Data points with an asterisk (P<0.05) are significantly different (n=10 mice per treatment and genotype).
We had previously demonstrated that ABA treatment suppressed macrophage infiltration into the white adipose tissue of obese mice in part by suppressing the expression of MCP-1[30]. The expression of this chemokine in the lungs during infection and inflammation is regulated by NF-κB binding to its promoter region, a process that is sensitive to inhibition by activated PPAR γ. Figure 3E confirms that ABA-treated WT mice had consistently lower levels of pulmonary MCP-1 mRNA compared to WT mice fed the control diet. Our data also shows that by day 7, cKO mice had not down-regulated lung MCP-1 expression whereas in the WT mice, irrespective of the diet, MCP-1 mRNA levels dropped to almost pre-challenge levels by day 7. These results confirm the relevance of PPAR γ expression in the down-regulation of pulmonary inflammation by ABA.
3.5 Effect of ABA on influenza virus infection-associated inflammatory gene expression in BALF-derived cells
Our cKO mice have deficient PPAR γ expression in immune and epithelial cells[21,31]. Because PPARγ is highly expressed in the lungs by different cell types besides immune and epithelial cells, we measured gene expression in cells obtained from bronchoalveolar space at day 4 post-challenge with influenza virus A/Udorn. This compartment is composed mainly of proinflammatory immune and dying or apoptotic epithelial cells, thereby permitting a more cell-specific assessment of the role of PPAR γ in regulating inflammation triggered by influenza virus. First we showed that dietary administration of ABA for 36 days increased the expression of PPAR γ in cells obtained from alveolar space of healthy uninfected WT mice (Figure 4A). Following influenza virus infection PPAR γ mRNA levels were higher in control-fed WT mice (Figure 4B), which correlated with higher expression of MCP-1 and TNFα in BAL-derived cells (Figure 4C and D).
Figure 4.
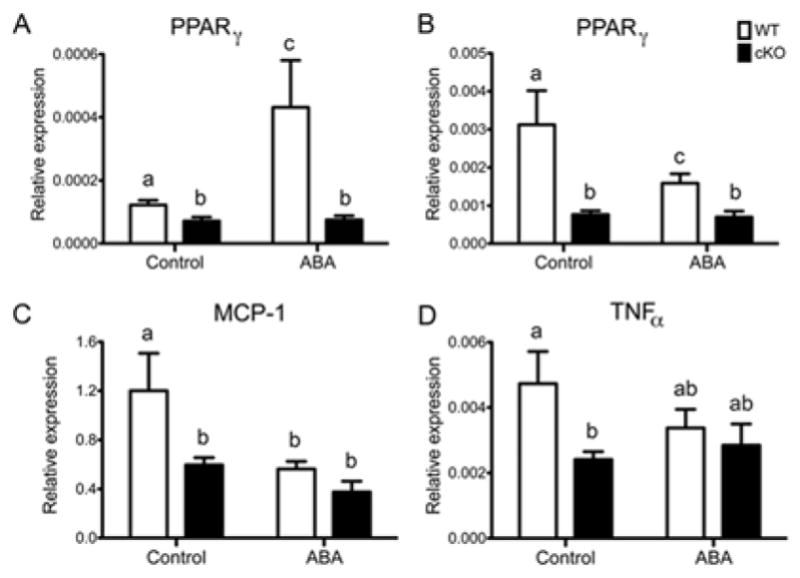
Effect of dietary ABA-supplementation on pulmonary expression of PPAR γ and monocyte chemotactic protein 1 (MCP-1) in broncholveolar lavage-derived cells. Cells were collected from the airway of WT or cKO mice 4 days post-infection. Dietary treatment and infection were as described before. ABA treatment increased PPAR γ mRNA expression in healthy non-infected WT mice (A) although following infection WT mice fed the control diet had significantly higher levels of PPAR γ, which correlated with higher mRNA expression of MCP-1 and TNFα, compared to the rest of the groups. Data points with different letters (P<0.05) are significantly different (n=10 mice per treatment and genotype).
3.6 ABA targets PPAR γ in myeloid cells
In our initial studies, we used mice with deficient PPAR γ expression in epithelial and immune cells (cKO mice) to demonstrate ABA’s dependency on PPAR γ for protection. The results indicate that the loss of PPAR γ broadly in epithelial and immune cells indeed abrogates ABA-induced protection. Based on our data, one of the mechanisms by which ABA suppresses influenza virus-induced pathology is by decreasing the leukocyte recruitment. Neutrophils, exudate macrophages and monocyte-derived dendritic cells are the predominant cell types involved in the inflammatory host response to influenza. To follow on in these initial findings, we specifically addressed whether ABA acts on PPAR γ expressed by myeloid cells using myeloid-specific PPAR γ conditional KO (myeloid KO) mice. In this case we used a therapeutic approach by giving mice 100 mg/kg of ABA by oral gavage on the same day of challenge. Similar to our initial results, WT mice that received ABA recovered their initial condition faster than the WT mice that received vehicle only, or myeloid KO mice, irrespective of the treatment (not shown). The improvement in morbidity was paralleled by amelioration of lung inflammation assessed by histopathology. The WT mice treated with ABA had significantly less leukocytic infiltration in perivascular areas (Figure 5A) and had a dramatic reduction of inflammatory infiltrates associated with the respiratory airways (Figure 5B). The greatest differences between groups were seen at day 10 post-infection. In addition, the effect was abrogated in myeloid KO mice, demonstrating that ABA requires the expression of PPAR γ in myeloid cells (e.g., macrophages, dendritic cells) to suppress the inflammatory response that followed challenge with influenza virus H3N2.
Figure 5.

Effect ABA treatment on vascular infiltration (A) and airway infiltration (B) in the lung following infection with influenza virus. Wild-type (WT) or myeloid-specific PPAR γ null mice (myeloid KO) mice were challenged with 5 x TCID50 of influenza A/Udorn (H3N2) and then treated with either PBS or ABA (100 mg/ABA kg body weight) by oral gavage for 10 days starting on the day of infection. Asterisks denote statistically significant differences between the ABA-treated WT and the myeloid KO mice. Pound signs denote statistically significant differences between the ABA-treated WT and the control WT (n=10 mice per treatment and genotype).
3.7 Impact of ABA in gene expression at the recovery phase
We conducted targeted gene expression analysis in lung specimens collected from the WT and myeloid KO mice described above. Our analysis focused on PPAR γ and target genes, LANCL2, which has been shown to interact with ABA[32], genes that participate in lipid mediators generation and cytokines that participate in inflammatory, regulatory or immune pathways. Unexpectedly, we found that the major impact of ABA on gene expression occurred between days 7 and 10 post-infection. Our results show that there was a significant effect of ABA treatment in WT mice. Specifically, both PPAR γ and LANCL2 were significantly upregulated on day 10 (Figure 6A and B). The PPAR γ target gene angiopoietin like 2 showed two peaks on days 3 and 7. On day 3 the expression was higher in WT mice that received vehicle while on day 7, angiopoietin like 2 expression was significantly higher in WT mice that received ABA (Figure 6C). iPhospholipase A2 (iPLA2), which acts in proximal steps of the lipoxygenase and cyclooxygenase pathways by releasing arachidonic acid from cell membranes, was also significantly upregulated in WT mice treated with ABA (Figure 6D). In parallel to iPLA2 we detected upregulation of 5-lipoxygenase (5-LO) and 5-lipoxygenase activating protein (FLAP), also at day 10 post-infection (Figure 6E and F). TNFα was also upregulated in WT mice treated with ABA only on day 10 post-infection, and followed a similar pattern of expression as iPLA2, 5-LO and FLAP (Figure 6G). Finally, the immunoregulatory cytokine IL-10 showed a peak of expression at day 7 post-infection in WT mice compared to the myeloid KO mice, irrespective of the treatment, although the expression was significantly higher in the WT mice that were treated with ABA (Figure 6H).
Figure 6.

Effect of ABA treatment on pulmonary gene expression following infection with influenza virus.(A) PPAR γ, (B) LANCL2, (C) angiopoietin-like 4, (D) iPLA2, (E) LOX5, (F) FLAP, (G) IL-10 and (H) TNF-α. WT or myeloid KO mice were challenged with 5 × 104 TCID50 of influenza A/Udorn (H3N2) and then treated with either PBS or ABA (100 mg/ABA kg body weight) by oral gavage for 10 days. Asterisks denote statistically significant differences between the ABA-treated WT and the myeloid KO mice. Pound signs denote statistically significant differences between the ABA-treated WT and the control WT (n=10 mice per treatment and genotype).
4. Discussion
Our results show for the first time that dietary ABA ameliorates influenza-virus induced pulmonary pathology through a mechanism that depends on the full expression of PPAR γ in epithelial and immune cells of the lung. The epithelial compartment is the main target of influenza virus and the site where antiviral and subsequent immune responses are initiated[33]. Very recent findings however, point at endothelial cells as major initiators of the cytokine storm early post-infection [34], or as important cellular targets of the cytokines and chemokines released by other cell types during the initial innate immune response [35,36]. Our analyses of microscopic lesions, indicate that dietary ABA does not significantly change influenza-induced pulmonary epithelial cell necrosis. The main effect of ABA in the lung of WT mice is to diminish in filtration of inflammatory leukocytes, which correlates with a sustained down-regulation of MCP-1 mRNA expression in both lung tissue and cells obtained from the bronchoalveolar space. Moreover, these results were confirmed in our second study with myeloid KO mice, in which we found the same effect of ABA. In both cases, the greatest differences in lung pathology were found at later stages of the disease, which coincided with infiltration of the lung by T cells and with the initiation of the resolution phase (discussed below). These results do not rule out a possible impact of ABA on the epithelial, or even endothelial compartments, since they are source of MCP-1 and other chemokines [34,37]. These novel findings are consistent with our previous reports on the ability of ABA to suppress macrophage infiltration into the white adipose tissue and MCP-1 gene expression in the stromal vascular fraction of obese mice[30]. They are also in line with the reduction of pro-inflammatory cell infiltration, MCP-1 and MCP-3 levels in the lungs of mice infected with influenza A virus that received prophylactic treatment with a synthetic PPARγ agonist[13]. More recent data also shows that exogenous administration of 12-deoxy-PGJ2(15d-PGJ2), a metabolite generated through the COX2 pathway that can activate PPAR γ, protects mice from lethal infection with H1N1 influenza A virus [15]. Interestingly, we find that ABA did not reduce viral loads in the lung measured 4 days post-infection. These data indicate that the treatment targets the host and not the virus and are in line too with results obtained with pioglitazone [13]. Interestingly 15-dPGJ2 reduced lung viral loads when mice were treated 24 hours post-infection although no effect on virus replication was noted in vitro[15].
Our data demonstrate for the first time that deficient PPARγ expression in immune and epithelial cells prolongs the MCP-1 response in the lungs during influenza virus infection. More specifically, on day 7 post-infection the pulmonary MCP-1 mRNA concentrations had returned to pre-challenge levels in WT mice, whereas in PPARγ cKO mice MCP-1 lung transcripts remained upregulated. In addition, dietary ABA-supplementation was effective in down regulating lung MCP-1 mRNA expression. This chemokine is required for homing of CCR2+ monocytes, which then differentiate into exudate macrophages and monocyte-derived dendritic cells, two major contributors to the pathogenesis of influenza by producing large amounts of TNFα and iNOS[10,13]. When we analyzed the impact of dietary ABA in cells residing in the alveolar space in the absence of virus (figure 4A), which corresponds to healthy mice that had been in control or ABA diets for 36 days, we found that ABA increased PPARγ mRNA expression in BAL-derived cells from WT mice. About 90 % of the cells extracted from the alveolar space of a healthy mouse are alveolar macrophages. The alveolar macrophage in particular expresses high levels of PPAR γ constitutively[38] thus it is likely that the increase in mRNA observed after 35 days of dietary supplementation corresponds to higher levels of the receptor in these cells. However, the effect that the “preconditioning” of alveolar macrophages by ABA has on the outcome of infection is unknown. In contrast, monocytes express low levels of PPARγ, and the protein is highly induced upon differentiation into macrophages or dendritic cells[39,40]. The distinct pattern of PPARγ expression we observe in figure 4B, where WT mice in control diet have higher expression than WT mice in ABA diet, is likely due to differences in the composition of cells within the bronchoalveolar space between infected and uninfected control mice. FACS profiling results show that at day 4 post-infection alveolar macrophages are significantly reduced and the space is filled with a mixture of GR1+ myeloid cells (data not shown) that include mainly neutrophils and macrophages. Thus the increase in PPAR γ is likely due to the presence of myeloid cells undergoing differentiation and maturation.
Our data on gene expression analysis indicates that ABA has a very significant impact on later phases on the disease, between days 7 and 10, which coincides with the initiation of resolution and of T cell responses. These data suggest that in addition to suppressing infiltration of inflammatory cells, at least in part, by downregulating MCP-1, ABA might accelerate the active process of resolution. This assessment is supported by data on morbidity and mortality shown in figure 2 where 100 % of ABA-treated WT mice survived. In contrast, between 15 and 40 % of mice in the control and cKO groups died between days 10 and 14 post-infection. Thus in addition to suppressing the initial inflammatory response, ABA had a significant impact in the recovery phase. The arachidonic acid-derived eicosanoids lipoxins and resolvins are important mediators of resolution[41]. In a recent study of global gene expression analysis it was concluded that suppression of lipoxin signaling might contribute to the pathogenesis of highly virulent H5N1 influenza virus infection, particularly to its dissemination [42]. We report that ABA significantly increases the expression of 5-LO and FLAP. The former enzyme participates in the process of transcellular biosynthesis of lipoxins, which have anti-inflammatory and pro-resolutory actions. Although 5-LO also participates in the biosynthesis of pro-inflammatory leukotriens, this process is relevant during initial phases of the acute inflammatory response[43]. It was shown that Rosiglita zone has neuroprotective effects during experimental stroke by increasing lipoxin A 4 synthesis by 5-LO. More importantly, 5-LO expression was positively modulated PPAR γ transcriptional activity contributing toits anti-inflammatory/pro-resolutory effects [44]. Our results show that the enhanced expression of 5-LO induced by ABA on day 10 is highly dependent on the expression of PPAR γ in myeloid cells. In addition to the changes in enzymes that participate in the synthesis of lipid mediators, ABA differentially regulates the expression of IL-10. A study mapping IL-10 production in the lung during influenza virus infection, identified CD4 and CD8 effector T cells as the major source of this cytokine [45]. This however doesn’t rule out that other cells and organs secrete this cytokine as well. For instance, splenic macrophages were found to secrete large amounts of IL-10 in burned mice which ameliorated acute lung injury due to endotoxemia[46]. It was also reported in a model of acute lung injury that the IL-10 response was enhanced in the absence of MCP-1 [47]. The increase in IL-10 gene expression occurs in parallel to upregulation of TNF α. Flow cytometry data shows that changes in IL-10 and TNF α occur in parallel to the accumulation of CD8 T cells in the lung (not shown), suggesting that both cytokines are produced locally during the T cell response to the virus. Thus, ABA modulates inflammation without suppressing the anti-viral response.
Results of human studies demonstrate the presence of high levels of proinflammatory cytokines and chemokines in serum specimens of hospitalized patients infected with influenza A virus[5]. These findings provide support for the theory that the extent of the host innate immune response to influenza virus infection contributes to the pathogenesis of the disease [5]. The hypercytokinemia hypothesis was later confirmed in experimental animal models, including mice, non-human primates and ferrets challenged with the highly pathogenic strains H5N1 and the reconstructed 1918 H1N1 [8,48,49]. Although the presence of high levels of inflammatory mediators is common for these two strains of influenza A virus, a report by Garigliani et al. [50] shows that two subtypes of mouse-adapted influenza A virus, an H1N1 and H5N1 of equal pathogenicity induced different pattern of lesions. Specifically, the lungs of mice infected with the H1N1 strain showed extensive epithelial damage and an infiltrative pattern characterized by the presence of inflammatory leukocytes surrounding blood vessels and respiratory airways with little or no edema. On the other hand, lungs from mice infected with the H5N1 subtype showed mild lesions in the airway epithelium and extensive alveolar edema and hemorrhage with low numbers of inflammatory leukocytes[50], suggesting the existence of distinct mechanisms of immunopathogenesis some of which are dominated by the elevated secretion of the same chemokines and cytokines. The influenza model we have used is characterized by an influx of inflammatory cells into the lungs, which were diminished in WT mice treated with ABA. Thus, PPAR γ agonists and anti-inflammatory compounds might be most effective for treating influenza virus infections that produce a leukocytic infiltrative pattern. So far, there have been no reports showing that a PPAR γ agonist alone protect against H5N1 influenza A virus infection, although it was shown that mezalamine, a prescription drug for Ulcerative Colitis which activates PPAR γ, in combination with COX-2 inhibitors and zanamivir protected mice against lethal challenge with this strain [12]. Palamara and colleagues demonstrated that the plant-derived polyphenol resveratrol improved survival and decreased pulmonary viral titers in influenza-infected mice, by blocking nuclear-cytoplasmic translocation of the viral ribonucleoprotein, which affects the synthesis of late proteins. This mechanism is actually dependent on the host and involves the inhibition of protein kinase C [51]. Thus both plant-derived products ABA and resveratrol protect against influenza virus-induced disease through targeting the host, although with different mechanisms. Moreover, the lack of efficacy of rosiglitazone treatment in protecting mice from lethal influenza infection as well as better safety profiles for ABA might suggest differential mechanisms of action for ABA and TZDs.
The results of these studies support the use of ABA as a novel adjunct for influenza that dampens the host inflammatory response in the lungs, ameliorates clinical disease and lung pathology, and improves resolution of lung pathology and survival. We also provide definitive molecular evidence in vivo demonstrating that the beneficial effects of ABA on influenza are mediated through a mechanism involving immune cell PPAR γ both at the initiation and resolution stages of disease. In summary, ABA has the potential to become an effective immune modulatory treatment for respiratory infections that targets the host and not the pathogen and has no known adverse side effects.
Supplementary Material
Acknowledgments
Supported by award number 5R01AT004308 of the National Center for Complementary and Alternative Medicine at the National Institutes of Health awarded to J.B.-R., the Virginia Bioinformatics Institute-Fralin CRI grants program to R.H., funds from the Nutritional Immunology and Molecular Medicine Laboratory and start-up funds from P.C.R.
Footnotes
Disclosures
None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Peiris JS, Hui KP, Yen HL. Host response to influenza virus: Protection versus immunopathology. Curr Opin Immunol. 2010;22:475–81. doi: 10.1016/j.coi.2010.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fedson DS. Pandemic influenza: A potential role for statins in treatment and prophylaxis. Clin Infect Dis. 2006;43:199–205. doi: 10.1086/505116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fedson DS. Confronting an influenza pandemic with inexpensive generic agents: Can it be done? Lancet Infect Dis. 2008;8:571–6. doi: 10.1016/S1473-3099(08)70070-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fedson DS. Confronting the next influenza pandemic with anti-inflammatory and immunomodulatory agents: Why they are needed and how they might work. Influenza Other Respi Viruses. 2009;3:129–42. doi: 10.1111/j.1750-2659.2009.00090.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.de Jong MD, Simmons CP, Thanh TT, et al. Fatal outcome of human influenza a (h5n1) is associated with high viral load and hypercytokinemia. Nat Med. 2006;12:1203–7. doi: 10.1038/nm1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kash JC, Tumpey TM, Proll SC, et al. Genomic analysis of increased host immune and cell death responses induced by 1918 influenza virus. Nature. 2006;443:578–81. doi: 10.1038/nature05181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gill JR, Sheng ZM, Ely SF, et al. Pulmonary pathologic findings of fatal 2009 pandemic influenza a/h1n1 viral infections. Arch Pathol Lab Med. 2010;134:235–43. doi: 10.5858/134.2.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Szretter KJ, Gangappa S, Lu X, et al. Role of host cytokine responses in the pathogenesis of avian h5n1 influenza viruses in mice. J Virol. 2007;81:2736–44. doi: 10.1128/JVI.02336-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Le Goffic R, Balloy V, Lagranderie M, et al. Detrimental contribution of the toll-like receptor (tlr)3 to influenza a virus-induced acute pneumonia. PLoS Pathog. 2006;2:e53. doi: 10.1371/journal.ppat.0020053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lin KL, Suzuki Y, Nakano H, Ramsburg E, Gunn MD. Ccr2+ monocyte-derived dendritic cells and exudate macrophages produce influenza-induced pulmonary immune pathology and mortality. J Immunol. 2008;180:2562–72. doi: 10.4049/jimmunol.180.4.2562. [DOI] [PubMed] [Google Scholar]
- 11.Wareing MD, Lyon A, Inglis C, Giannoni F, Charo I, Sarawar SR. Chemokine regulation of the inflammatory response to a low-dose influenza infection in ccr2−/− mice. J Leukoc Biol. 2007;81:793–801. doi: 10.1189/jlb.0506299. [DOI] [PubMed] [Google Scholar]
- 12.Zheng BJ, Chan KW, Lin YP, et al. Delayed antiviral plus immunomodulator treatment still reduces mortality in mice infected by high inoculum of influenza a/h5n1 virus. Proc Natl Acad Sci U S A. 2008;105:8091–6. doi: 10.1073/pnas.0711942105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Aldridge JR, Jr, Moseley CE, Boltz DA, et al. Tnf/inos-producing dendritic cells are the necessary evil of lethal influenza virus infection. Proc Natl Acad Sci U S A. 2009;106:5306–11. doi: 10.1073/pnas.0900655106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Budd A, Alleva L, Alsharifi M, et al. Increased survival after gemfibrozil treatment of severe mouse influenza. Antimicrob Agents Chemother. 2007;51:2965–8. doi: 10.1128/AAC.00219-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cloutier A, Marois I, Cloutier D, Verreault C, Cantin AM, Richter MV. The prostanoid 15-deoxy-delta12, 14-prostagland in-j2 reduces lung inflammation and protects mice against lethal influenza infection. J Infect Dis. 2012;205:621–30. doi: 10.1093/infdis/jir804. [DOI] [PubMed] [Google Scholar]
- 16.Harmon GS, Lam MT, Glass CK. Ppars and lipid ligands in inflammation and metabolism. Chem Rev. 2011;111:6321–40. doi: 10.1021/cr2001355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Straus DS, Glass CK. Anti-inflammatory actions of ppar ligands: New insights on cellular and molecular mechanisms. Trends Immunol. 2007;28:551–8. doi: 10.1016/j.it.2007.09.003. [DOI] [PubMed] [Google Scholar]
- 18.Straus DS, Pascual G, Li M, et al. 15-deoxy-delta 12,14-prostaglandin j2 inhibits multiple steps in the nf-kappa b signaling pathway. Proc Natl Acad Sci U S A. 2000;97:4844–9. doi: 10.1073/pnas.97.9.4844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bassaganya-Riera J, Guri AJ, Lu P, et al. Abscisic acid regulates inflammation via ligand-binding domain-independent activation of ppar gamma. Journal of Biological Chemistry. 2010;286:2504–16. doi: 10.1074/jbc.M110.160077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guri AJ, Misyak SA, Hontecillas R, et al. Abscisic acid ameliorates atherosclerosis by suppressing macrophage and cd4(+) t cell recruitment into the aortic wall. J Nutr Biochem. 2010 doi: 10.1016/j.jnutbio.2009.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bassaganya-Riera J, Reynolds K, Martino-Catt S, et al. Activation of ppar gamma and delta by conjugated linoleic acid mediates protection from experimental inflammatory bowel disease. Gastroenterology. 2004;127:777–91. doi: 10.1053/j.gastro.2004.06.049. [DOI] [PubMed] [Google Scholar]
- 22.Hontecillas R, Horne WT, Climent M, et al. Immunoregulatory mechanisms of macrophage ppar-gamma in mice with experimental inflammatory bowel disease. Mucosal Immunol. 2011;4:304–13. doi: 10.1038/mi.2010.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guri AJ, Hontecillas R, Si H, Liu D, Bassaganya-Riera J. Dietary abscisic acid ameliorates glucose tolerance and obesity-related inflammation in db/db mice fed high-fat diets. Clin Nutr. 2007;26:107–16. doi: 10.1016/j.clnu.2006.07.008. [DOI] [PubMed] [Google Scholar]
- 24.Guri AJ, Hontecillas R, Bassaganya-Riera J. Abscisic acid ameliorates experimental ibd by downregulating cellular adhesion molecule expression and suppressing immune cell infiltration. Clinical Nutrition. 2010;29:824–31. doi: 10.1016/j.clnu.2010.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guri AJ, Evans NP, Hontecillas R, Bassaganya-Riera J. T cell ppar gamma is required for the anti-inflammatory efficacy of abscisic acid against experimental ibd. Journal of Nutritional Biochemistry. 2010 doi: 10.1016/j.jnutbio.2010.06.011. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Herbert AS, Heffron L, Sundick R, Roberts PC. Incorporation of membrane-bound, mammalian-derived immunomodulatory proteins into influenza whole virus vaccines boosts immunogenicity and protection against lethal challenge. Virol J. 2009;6:42. doi: 10.1186/1743-422X-6-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reed LJ, Muench H. A simple method of estimating fifty percent end points. Am j Hyg. 1938;27:493–7. [Google Scholar]
- 28.Becker J, Delayre-Orthez C, Frossard N, Pons F. Regulation of inflammation by ppars: A future approach to treat lung inflammatory diseases? Fundam Clin Pharmacol. 2006;20:429–47. doi: 10.1111/j.1472-8206.2006.00425.x. [DOI] [PubMed] [Google Scholar]
- 29.Guri AJ, Hontecillas R, Bassaganya-Riera J. Abscisic acid synergizes with rosiglitazone to improve glucose tolerance and down-modulate macrophage accumulation in adipose tissue: Possible action of the camp/pka/ppar gamma axis. Clin Nutr. 2010;29:646–53. doi: 10.1016/j.clnu.2010.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Guri AJ, Hontecillas R, Ferrer G, et al. Loss of ppar gamma in immune cells impairs the ability of abscisic acid to improve insulin sensitivity by suppressing monocyte chemo attractant protein-1 expression and macrophage infiltration into white adipose tissue. J Nutr Biochem. 2008;19:216–28. doi: 10.1016/j.jnutbio.2007.02.010. [DOI] [PubMed] [Google Scholar]
- 31.Cui Y, Miyoshi K, Claudio E, et al. Loss of the peroxisome proliferation-activated receptor gamma (ppargamma) does not affect mammary development and propensity for tumor formation but leads to reduced fertility. J Biol Chem. 2002;277:17830–5. doi: 10.1074/jbc.M200186200. [DOI] [PubMed] [Google Scholar]
- 32.Bassaganya-Riera J, Guri AJ, Lu P, et al. Abscisic acid regulates inflammation via ligand-binding domain-independent activation of ppar gamma. Journal of Biological Chemistry. 2011;286:2504–16. doi: 10.1074/jbc.M110.160077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Garcia-Sastre A, Biron CA. Type 1 interferons and the virus-host relationship: A lesson in detente. Science. 2006;312:879–82. doi: 10.1126/science.1125676. [DOI] [PubMed] [Google Scholar]
- 34.Teijaro JR, Walsh KB, Cahalan S, et al. Endothelial cells are central orchestrators of cytokine amplification during influenza virus infection. Cell. 2011;146:980–91. doi: 10.1016/j.cell.2011.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Steinberg BE, Goldenberg NM, Lee WL. Do viral infections mimic bacterial sepsis? The role of microvascular permeability: A review of mechanisms and methods. Antiviral Res. 2012;93:2–15. doi: 10.1016/j.antiviral.2011.10.019. [DOI] [PubMed] [Google Scholar]
- 36.Goldenberg NM, Steinberg BE, Slutsky AS, Lee WL. Broken barriers: A new take on sepsis pathogenesis. Sci Transl Med. 2011;3:88ps25. doi: 10.1126/scitranslmed.3002011. [DOI] [PubMed] [Google Scholar]
- 37.Adachi M, Matsukura S, Tokunaga H, Kokubu F. Expression of cytokines on human bronchial epithelial cells induced by influenza virus a. Int Arch Allergy Immunol. 1997;113:307–11. doi: 10.1159/000237584. [DOI] [PubMed] [Google Scholar]
- 38.Smith MR, Standiford TJ, Reddy RC. Ppars in alveolar macrophage biology. PPAR Res. 2007;2007:23812. doi: 10.1155/2007/23812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Szatmari I, Torocsik D, Agostini M, et al. Ppargamma regulates the function of human dendritic cells primarily by altering lipid metabolism. Blood. 2007;110:3271–80. doi: 10.1182/blood-2007-06-096222. [DOI] [PubMed] [Google Scholar]
- 40.Gogolak P, Rethi B, Szatmari I, et al. Differentiation of cd1a− and cd1a+ monocyte-derived dendritic cells is biased by lipid environment and ppargamma. Blood. 2007;109:643–52. doi: 10.1182/blood-2006-04-016840. [DOI] [PubMed] [Google Scholar]
- 41.Serhan CN. The resolution of inflammation: The devil in the flask and in the details. FASEB J. 2011;25:1441–8. doi: 10.1096/fj.11-0502ufm. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cilloniz C, Pantin-Jackwood MJ, Ni C, et al. Molecular signatures associated with mx1-mediated resistance to highly pathogenic influenza virus infection: Mechanisms of survival. J Virol. 2012;86:2437–46. doi: 10.1128/JVI.06156-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stables MJ, Gilroy DW. Old and new generation lipid mediators in acute inflammation and resolution. Prog Lipid Res. 2011;50:35–51. doi: 10.1016/j.plipres.2010.07.005. [DOI] [PubMed] [Google Scholar]
- 44.Sobrado M, Pereira MP, Ballesteros I, et al. Synthesis of lipoxin a4 by 5-lipoxygenase mediates ppargamma-dependent, neuroprotective effects of rosiglitazone in experimental stroke. J Neurosci. 2009;29:3875–84. doi: 10.1523/JNEUROSCI.5529-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sun J, Madan R, Karp CL, Braciale TJ. Effector t cells control lung inflammation during acute influenza virus infection by producing il-10. Nat Med. 2009;15:277–84. doi: 10.1038/nm.1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sekine K, Fujishima S, Sasaki J, Ishizaka A, Aiso S, Aikawa N. In vivo il-18 supplementation ameliorates lethal acute lung injury in burn-primed endotoxemic mice: A novel anti-inflammatory role of il-18. Shock. 2009;32:554–62. doi: 10.1097/SHK.0b013e31819e2db6. [DOI] [PubMed] [Google Scholar]
- 47.van Zoelen MA, Verstege MI, Draing C, et al. Endogenous mcp-1 promotes lung inflammation induced by lps and lta. Mol Immunol. 2011;48:1468–76. doi: 10.1016/j.molimm.2011.04.001. [DOI] [PubMed] [Google Scholar]
- 48.Kobasa D, Jones SM, Shinya K, et al. Aberrant innate immune response in lethal infection of macaques with the 1918 influenza virus. Nature. 2007;445:319–23. doi: 10.1038/nature05495. [DOI] [PubMed] [Google Scholar]
- 49.Lipatov AS, Andreansky S, Webby RJ, et al. Pathogenesis of hong kong h5n1 influenza virus ns gene reassortants in mice: The role of cytokines and b- and t-cell responses. J Gen Virol. 2005;86:1121–30. doi: 10.1099/vir.0.80663-0. [DOI] [PubMed] [Google Scholar]
- 50.Garigliany MM, Habyarimana A, Lambrecht B, et al. Influenza a strain-dependent pathogenesis in fatal h1n1 and h5n1 subtype infections of mice. Emerg Infect Dis. 2010;16:595–603. doi: 10.3201/eid1604.091061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Palamara AT, Nencioni L, Aquilano K, et al. Inhibition of influenza a virus replication by resveratrol. J Infect Dis. 2005;191:1719–29. doi: 10.1086/429694. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.