

Abstract
The thrombocytopenia of Wiskott–Aldrich syndrome (WAS) is thought to be due to both reduced platelet production and accelerated platelet consumption. We have previously demonstrated that platelets from WASP-deficient mice are consumed more rapidly in vivo than are WT platelets, and that opsonization accelerates their uptake by bone marrow- derived macrophages more than it does that of WT platelets. Here we asked whether platelets from WAS patients show similar features. We show that ex vivo phagocytosis by activated THP-1 cells of DIO-labeled platelets from a series of WAS or XLT patients is increased in comparison to that of normal control platelets. Using a numerical analysis method, we distinguish this effect from a concurrent effect on the amount of detectable fluorescent signal transferred to the macrophage per phagocytosed platelet. We show that the latter quantity is reduced by platelet WASP deficiency, as might be expected if the fluorescence transferred from these smaller platelets is more rapidly quenched. We are unable to detect a differential effect of opsonization with anti-CD61 antibody on the uptake of WASP(−) vs. WT platelets. However, the high probability of phagocytosis per adsorbed WASP(−) platelet could limit the sensitivity of the assay in this case. We also see no effect of sera from WAS patients on the uptake of normal control platelets, suggesting that in vivo opsonization is not the cause of increased uptake of WASP(−) platelets. Finally, we show little, if any, increase in the reticulated platelet fraction in WAS patients, suggesting that impaired production of reticulated platelets contributes to the thrombocytopenia. Our findings suggest that rapid in vivo platelet consumption contributes significantly to the thrombocytopenia of WAS. They also demonstrate the feasibility of routinely performing functional assays of phagocytosis of small numbers of platelets obtained at remote locations, a method which should be applicable to the study of other types of thrombocytopenia such as ITP.
Keywords: Wiskott–Aldrich syndrome, phagocytosis, numerical analysis, thrombocytopenia
Introduction
The Wiskott–Aldrich syndrome (WAS) is an X-linked recessive condition caused by mutations which reduce the expression of the Wiskott-Aldrich protein (WASP). It is characterized most commonly by immunodeficiency, severe eczema, and thrombocytopenia, and is often complicated by any of several autoimmune conditions. Mutations in the WAS gene which result in a milder reduction in WASP levels typically show a similarly severe thrombocytopenia with little or no evidence of immunodeficiency, and fall under the term X-linked thrombocytopenia (XLT) [1]. While the immunodeficiency of WAS has been well characterized at both cellular and molecular levels [2], there are still some unanswered questions concerning the mechanism responsible for the thrombocytopenia and its unusually small platelets. Ex vivo studies of megakaryopoiesis [3] and thrombopoiesis [4–6] demonstrate impairments in both functions in the absence of WASP. In particular, altered proplatelet formation is thought to result from the absence of WASP’s function in transducing signals that result in actin polymerization [4]. There have been conflicting reports concerning whether the fraction of immature or “reticulated” platelets is normal or increased in WAS patients [7, 8].
Several lines of investigation suggest that rapid platelet consumption also contributes to the thrombocytopenia of WAS. A small number of in vivo turnover studies demonstrate rapid consumption of platelets from WAS patients in normal recipients [9]. Their consumption rate in thrombocytopenic WAS patients has in most cases also been reported as increased [10, 11], although these studies are more difficult to interpret. In a murine model of WAS, we have seen increased platelet consumption in both contexts [12]. Additionally, ex vivo phagocytosis of WASP-deficient murine platelets is more accelerated by opsonization than is that of WT platelets, as is the in vivo platelet consumption rate [12]. While a contribution of autoimmunity to the thrombocytopenia of WAS has not been ruled out, the fact that WASP(−) uMT(−/−) mice, which lack virtually all antibodies, do not show a significantly improved platelet count [13] suggests that rapid WASP(−) platelet consumption is due to an intrinsic platelet defect.
Here we use ex vivo phagocytosis assays to provide evidence for a similar defect in platelets from WAS patients. This type of study with fluorescently labeled platelets presents analytic difficulties due to (A) the problem of quantitatively distinguishing the rates of the sequential processes of platelet adsorption (adhesion to the cell surface) and platelet uptake and (B) the problem of distinguishing an increased rate of phagocytosis from reduced quenching of fluorescence after uptake. We recently described a numerical analysis method which resolves these issues [14]. Here we employ it to study platelets from a series of WAS patients.
Materials and methods
Reagents
Mouse anti-Human CD61 (clone VP-PL2), Mouse anti-Human CD41 (clone HIP8), PE-labeled mouse anti-human CD61, thiazole orange (BD-Retic-count), and PE-labeled Mouse anti-human CD41 were obtained from BD Biosciences. Dimethyl sulfoxide (anhydrous), phorbol 12-myristate 13 acetate (PMA), prostaglandin E1 (PGE1), l-glutamine, poly-l-lysine, and Hank’s balanced salt solution were purchased from Sigma (St. Louis, MO). RPMI media, trypsin (catalog number 25 300-054), beta mercaptoethanol, 3,3′-dioctadecyloxacarbocyanine perchlorate (DIO, catalog number D275), wheat germ agglutinin (Alexa Fluor 555 conjugate), phosphate-buffered saline (PBS), and penicillin/streptomycin were purchased from Invitrogen/Life Technologies. Fico/lite for platelets was from Atlanta Biologicals. THP-1 (TIB-202) cells were purchased from ATCC. Paraformaldehyde was from Electron Microscopy Sciences. Vectashield was from Vector Laboratories Inc.
Human platelet preparation
Procedures were as previously described [14]. All studies were approved by the Institutional Review Boards of the Memphis VA Medical Center and the University of Tennessee Health Science Center. Informed consent of the participants or (for minors) their parent/guardian was obtained in all cases. All clinical investigations were conducted according to the principles expressed in the Declaration of Helsinki. Citrate-anticoagulated blood was obtained from healthy volunteer adults. For studies comparing controls to WAS patients, both specimens were placed on rotating platforms immediately after blood was obtained. WAS patient specimens were shipped overnight on such a platform, while (rotating) control specimens were stored at room temperature. Specimens were layered over a ficoll cushion (Fico/lyte) and centrifuged at 350 g for 15 minutes at room temperature. The platelet layer was diluted in modified tyrode’s buffer (20 mM Hepes, 137 mM NaCl, 13.8 mM NaHCO3, 2.5 mM KCl, 0.36 mM NaH2PO4–H2O, 5.5 mM glucose, 0.25% bovine serum albumin, 1 mM MgCl2) supplemented with 1 μg/ml of PGE1, and centrifuged at 6000g for 5 minutes. Pellets were resuspended in modified tyrode’s buffer supplemented with PGE1, and counted with a Beckman Coulter model Z2 particle analyzer.
Platelet and reticulated platelet counts
Platelet counts were performed on whole blood specimens by a quantitative flow cytometric method, as previously described [12]. Reticulated platelet counts were performed on while blood specimens using thiazole orange (BD-Retic-count), as previously described [13].
Ex vivo phagocytosis assay
Procedures were adapted from those used by Semple et al. [15] and Hoffmeister et al. [16], as described previously [14]. Briefly, 1 × 106 THP-1 cells per ml, grown in an RPMI medium [supplemented with 10% ifbs, penicillin/streptomycin, l-glutamine, and 5 μM beta mecraptoethanol], were activated for 3 hours with PMA (50 ng/ml). These activation conditions were found to optimize the sensitivity of the assay. After activation, cells were centrifuged at 450 g for 10 minutes at room temperature, then resuspended in the above medium at 3 × 105 cells/ml, and distributed in 48-well dishes. All assays were performed in duplicate or triplicate. Platelets (9 × 104 to 8 × 105 per μl) were labeled with DIO (0.5 μM) in the presence of Tween 20 (0.35 μM) and 46% DMSO (46%), for 30 minutes at 37°. After the addition of 5 volumes of modified tyrode’s buffer supplemented with PGE-1, the platelets were centrifuged (6000 g, 5 minutes, room temperature) and resuspended in the same buffer. When used, antibodies or sera were incubated with the platelets for 2 hours at room temperature, followed by a rinse step in modified tyrode’s buffer supplemented with PGE1. After platelets (10 per THP-1 cell) were added to the wells, the plates were centrifuged for 1 minute at 200 g (room temperature), then incubated for 1 hour at 37° (5% CO2). Non-adherent and adherent (trypsinized) cells (unless prepared for microscopy) were exposed to PE-anti-human CD61, then analyzed by flow cytometry (LSR-II, Becton Dickinson). Initial data interpretation was performed with Flowjo software (Treestar, Inc). Numerical analysis was performed using Microsoft Excel, as previously described [14].
Widefield deconvolution microscopy
Glass 12 mm round coverslips (Belco) were treated with 70% ethanol for 1 hour and dried for 1 hour, and covered in 24-well dishes with 0.01% poly-l-lysine for 1 hour at room temperature. Coverslips were rinsed twice with PBS before use. THP-1 cells obtained as above were resuspended in 150 μl of PBS containing 5 μg/ml wheat germ agglutinin (AlexaFluor 555 conjugate) and incubated for 20 minutes in darkness at room temperature. After the addition of 700 ul PBS, cells were placed in 24-well dish wells containing poly-l-lysine coated cover slips, and centrifuged for 10 minutes at 450 RCF at room temperature. The dishes were incubated another 15 minutes at room temperature, then rinsed twice with PBS. They were then treated with PBS containing 4% paraformaldehyde (500 ul) for 15 minutes at room temperature, rinsed once with PBS, and mounted on slides using a Vectashield mounting medium. Microscopy was performed with a Zeiss Axio Observer A1 inverted fluorescence microscope using Zeiss Axiovision software.
Results
Phagocytosis
To detect uptake of WASP(−) and control platelets by activated THP-1 cells, we labeled platelets with the lipophilic fluor DIO before exposing them to the cells. To distinguish uptake from adsorption, we then bound PE-labeled anti-CD61 to the cells. A sketch of the type of data this generates (Figure 1) shows our gating strategy for quantification of the results. The horizontal line of demarcation is placed so as to facilitate the numerical analysis, as previously described [14].
Figure 1.
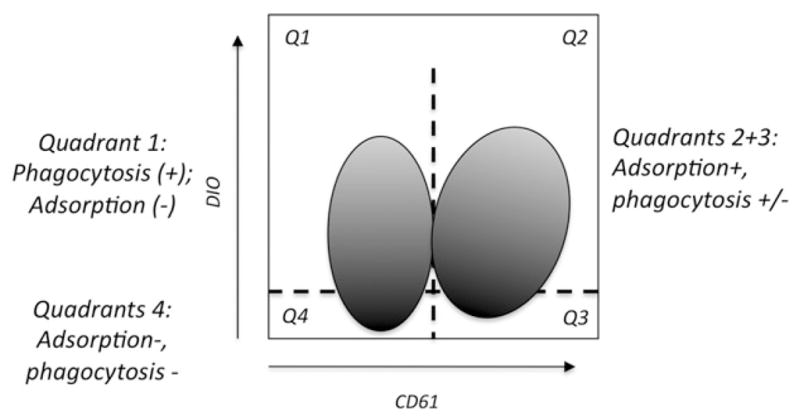
Schematic diagram of the type of data obtained from ex vivo phagocytosis assays.
Clinical data for the patients in the study is shown in Table I. Data demonstrating both uptake and adsorption of WAS and control platelets by activated THP-1 cells is shown in Figure 2. We verified that both uptake and adsorption of DIO-labeled platelets occurs under these conditions with the use of widefield deconvolution microscopy (Figure 3). For five of the six patients, the ratio of cells showing predominant platelet uptake (Figure 2, Quadrant 1) to those showing a combination of uptake and adsorption (Quadrants 2 + 3) is clearly increased relative to the same ratio for WT controls (Mean quadrant values are shown in Table II). While this suggests an increase in the probability of phagocytosis per adsorbed WASP(−) platelet (P) (here we use “P” to avoid confusion with the statistical p value), it is not immediately distinguishable from the possible effects of variation in the labeling intensity of the WASP(−) and control platelets, or variation in the quenching of fluorescence after phagocytosis. Specifically, the smaller WASP(−) platelets tended to label at a higher geometric mean fluorescence intensity (see below). Their smaller volume may also render them more susceptible to quenching after phagocytosis.
Table I.
Clinical and laboratory data on the WAS patients studied.
| Patient | 1 | 2 | 3 | 4 | 5 | 6 |
|---|---|---|---|---|---|---|
| Diagnosis | XLT | WAS | XLT | WAS | WAS | WAS |
| Splenectomy | Yes | No | No | No | No | Yes |
| Platelet count | 78.0 | 13.8 | 16.1 | 18.8 | 26.1 | 70,7 |
| Platelet fsc, % of control | 31.7 | 23.1 | 14.7 | 42.2 | 11.3 | 41.6 |
| Platelet MPV (ft) | 5.6 | 4.1 | 3.9 | 3.4 | na | 5.4 |
Notes: Platelet counts were obtained by quantitative flow cytometry on receipt of the specimens, which occurred either within 1 hour of blood draw (patients 4 and 5) or approximately 24 hours later (patients 1, 2, 3, and 6). MPV values were obtained with a coulter counter. Mean MPV values for WT controls were 8.7 fL (n = 5, standard deviation 0.7).
Figure 2.

Results of phagocytosis data. Activated THP-1 cells which had been exposed to DIO-labeled platelets were analyzed as described in the text. Representative results from duplicate or triplicate assays are shown. Note that patients 2 and 3 were analyzed simultaneously; the control data is identical for these two.
Figure 3.

Visualization of adsorbed (bound) and internalized DIO-labeled platelets. Activated THP-1 cells were exposed to DIO-labeled normal control platelets as for the phagocytosis studies shown in figure 2. The platelets in this case were opsonized with anti-CD61 antibody (150 ng per million platelets). After rinsing, the cells were bound to poly-l-lysine coated cover slips, exposed to AF-555-labeled wheat germ agglutinin, and examined by widefield deconvolution microscopy. Consecutive planes separated by 1 μM are shown. Green: DIO-labeled platelets. Red: wheat germ agglutinin marking the cell membrane.
Table II.
Phagocytosis data.
| Patient | 1 | 2 | 3 | 4 | 5 | 6 |
|---|---|---|---|---|---|---|
| Q(1), % | 22.5 | 69.8 | 70.6 | 73.6 | 71.8 | 34.4 |
| Q(1) control, % | 33.5 | 32.7 | 32.7 | 28.2 | 31.4 | 16.9 |
| Q(2 + 3), % | 76.8 | 21.8 | 18.1 | 22.7 | 21.7 | 43.7 |
| Q(2 + 3) control, % | 65.5 | 63.7 | 63.7 | 67.7 | 67.9 | 77.2 |
Notes: Shown are mean quadrant values, gated as in Figure 2, from duplicate or triplicate assays. These are the “input” values used for numerical analysis.
To resolve this ambiguity, we used numerical analysis to distinguish variation in the fluorescence transfer per phagocytosed platelet (alpha) from variation in P [14]. The method requires consideration of a third variable (m, the ratio of initially adsorbed platelets to macrophages) as well. It predicts the fraction of cells showing adsorbed platelets (“Q2 + 3”, quadrants 2 and 3, Figure 1) and the fraction falling in quadrant 1 (“Q1”), as a function of P, m, and alpha. It also allows prediction of the fluorescence histogram for cells not showing adsorbed platelets (Q1 + 4). The possible combinations of the three parameter values define a hypothetical volume called “parameter space” (PS). The model compares predicted and observed quantities of Q2 + 3 and Q1 for all points in parameter space. We previously showed that only a limited number of such points (those demonstrating “local minima” of residual values) were compatible with each observed Q1, Q2 + 3 pair. We validated the model by showing that it correctly interpreted experimental variation in m, P, and alpha as shifts in the local minima along the m, P, or alpha axes of PS.
Local minima for the data in Figure 2 are shown in Figure 4. Given that the same number of control and WASP(−) platelets were used in each study, we reasoned that comparison of control and WASP(−) minima along a vertical line (i.e. a constant m value) in these plots is valid. Thus for patients 2 and 3, although we do not know which m value best represents the actual ratio of initially adsorbed platelets to macrophages, it is clear that at any such value the WASP(−) platelets show a higher P value than controls. That is also the case for patients 4, 5, and 6.
Figure 4.
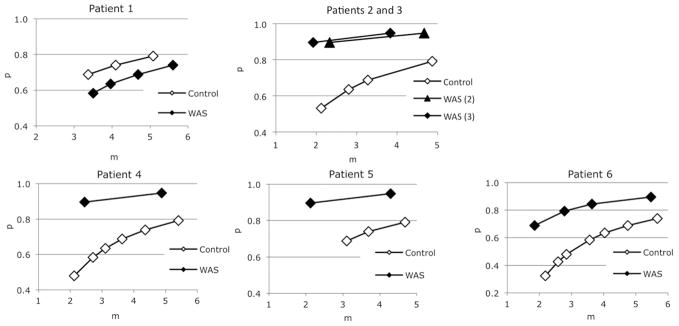
Numerical analysis of phagocytosis data. Mean Q1 and Q2 + 3 values were used to locate the local minima (LM) of residual values in parameter space. The LM points correspond to value sets of P, m, and alpha (i.e. points in parameter space) which predict Q1 and Q2 + 3 values that differ by less than 1% from the observed mean values.
To arrive at a better estimate of the P values, we used a “nearest neighbor” approach in which WASP(−) and control minima differing by no more than 10% of the control m value were compared. In some cases we were also able to omit suboptimal minima (patients 4 and 5, the left most minima) on the basis of a poor fit of their predicted Q1 + 4 histograms to the observed data. Results of this method (Figure 5A) suggest a significant increase in the P value for five of the six cases studied. We performed a repeat study on patient 1 and obtained similar results (data not shown). A nearest neighbor comparison for the associated alpha values (Figure 5B) suggests a reduced alpha value in, again, five of the six cases. This underestimates the reduction in fluorescence transfer for most of the WASP(−) platelets, however, because of their previously noted increased geometric mean fluorescence intensity (gmfi; Table III). This variable, which was difficult to control, likely is a consequence of the distribution of our input DIO reagent into the smaller net membrane volume that it encounters in WAS platelets (vs. an identical number of control platelets labeled in parallel). Normalization of variation in alpha (WAS/control alpha) to the gmfi ratio (WAS/control gmfi) yields the more strikingly reduced gauge of fluorescence transfer shown in Table III (normalized WAS/control alpha), with values well below 1.0 in, again, 5 of the 6 cases. Of those five, the highest normalized alpha value was that of patient 6, who had undergone splenectomy and whose platelet size (estimated by fsc, Table I) was correspondingly higher than that of the most of the other patients.
Figure 5.

Inferred P and alpha values. Nearest neighbor analysis, as described in the text, allowed comparison of a limited number of optimally matched WAS and control local minima in parameter space. The number of minima compared for patients 1–6 were 2, 2, 2, 1, 1, and 4, respectively.
Table III.
Normalization of WAS platelet alpha values using fluorescence intensity data.
| Patient | 1 | 2 | 3 | 4 | 5 | 6 |
|---|---|---|---|---|---|---|
| Platelet fsc, % of control | 31.7 | 23.1 | 14.7 | 42.2 | 11.3 | 41.6 |
| Platelet gmfi | 254 | 1531 | 2150 | 1492 | 920 | 465 |
| Control gmfi | 493 | 604 | 604 | 439 | 734 | 900 |
| WAS/control gmfi | 0.52 | 2.53 | 3.56 | 3.40 | 1.25 | 0.52 |
| Platelet alpha | 4.11 | 2.76 | 2.83 | 2.79 | 2.63 | 1.21 |
| Control alpha | 4.21 | 3.24 | 3.74 | 3.92 | 4.18 | 2.82 |
| WAS/control alpha | 0.98 | 0.85 | 0.76 | 0.71 | 0.63 | 0.43 |
| Normalized WAS/control alpha | 1.89 | 0.34 | 0.21 | 0.21 | 0.50 | 0.83 |
Note: The ratio of WAS and control alpha values was normalized to their relative fluorescence intensities as described in the text.
While the reduced volume of WASP(−) platelets could in theory result in a lower m value in comparison to a study using the same number of control platelets (due to the centrifugation step we use to co-localize platelets and macrophages), we found that this size difference results in at most a 10% reduction in recovery of WASP(−) platelets after centrifugation (data not shown). This suggests that the reduced adsorption seen with most of the WASP(−) platelets is a consequence of increased phagocytosis of adsorbed platelets. (The numerical analysis model takes both processes into account in predicting the fraction of cells in Q1 and Q(2 + 3).) Alternatively, reduced adsorption due to some other mechanism could conceivably mean that valid comparison of control and WASP(−) P values requires comparison of, for example, the left-most WASP(−) minimum to the right-most control minimum (for any of the plots in Figure 4). However, such a comparison also shows a higher P value for WASP(−) platelets from four of the six patients (patients 2, 3, 4, and 5).
Effect of opsonization
Opsonization of control platelets with a mouse anti-human CD61 antibody yielded the expected increase in the probability of phagocytosis per adsorbed platelet (Figure 6, controls). We did not, however, see any increase in P values for three of the four WAS patients we studied (Figure 6; raw flow data is in supplemental Figures S2, S3, and S4). The high P values associated with these three specimens before opsonization may have limited our ability to detect such an effect, since we did see an effect of opsonization (comparable to that seen with normal control platelets) on the uptake of platelets from patient 1. Alternatively, we considered the possibility that the platelets were in these cases already opsonized by auto-antibodies. To test that, we exposed normal (type O) platelets to sera from normal controls, or from WAS patients. Numerical analyses of the results are shown in Figure 7. Here the normal ranges obtained with control sera are represented by vertical bidirectional arrows (the minima themselves are shown in supplemental Figure S1). Mouse anti-CD61 antibody again served as a positive control (data not shown). The results show no evidence for phagocytosis-inducing anti-platelet antibodies in any of the WAS sera.
Figure 6.

Effect of opsonization on phagocytosis of WT vs. WAS platelets. Anti-CD61 antibody was bound at 150 ng per million platelets prior to the phagocytosis assays. Representative flow cytometric data is shown in supplemental figures S2, S3, and S4. Numerical analysis of the resultant mean Q1 and Q2 + 3 values revealed the local minima shown.
Figure 7.
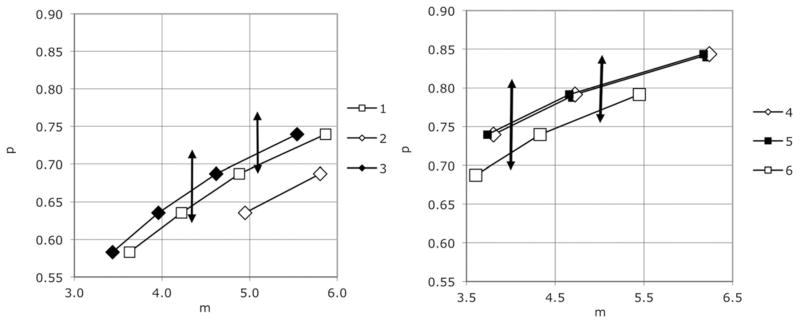
Effect of WAS sera on uptake of normal platelets. Numerical analysis of flow cytometric data was used to detect the local minima shown for sera obtained from six WAS patients. Control sera yielded minima (Supplemental figure S1) falling in the ranges shown by the double headed arrows.
Reticulated platelets
Because there is disagreement in the literature concerning the levels of reticulated platelets in WAS patients, we evaluated this using thiazole orange uptake. An example of the stringent gating method used by us is shown in Figure 8. While we saw a trend toward a higher fraction of reticulated platelets in the WAS patients (1.37% vs. 0.52%), it did not reach significance for the pooled WAS and control specimens we studied (p = 0.06; n = 6 (WAS), n = 5 (control)).
Figure 8.

Platelet and reticulated platelet counts. (A) flow cytometric data. (B) Platelet and reticulated platelet counts for six WAS patients. Asterisks mark duplicate specimens from one of the patients. Open diamonds show results for patients who have been splenectomized. The triangle shows the mean (+ SD) for five normal controls.
Discussion
Our finding of an increased probability of phagocytosis of WASP-deficient platelets by activated THP-1 cells confirms and extends our previous report of this with platelets from a single patient [14]. Consistent with reports of rapid in vivo platelet turnover in WAS patients [9–11], these findings suggest that accelerated phagocytosis of platelets by macrophages contributes to the thrombocytopenia of WAS. While our numerical analysis model does not currently allow calculation of statistical significance for each of the P values obtained, our finding of a reduced P value in 5 of the 6 WAS patients obviates that need. We also find that fluorescence transfer from WASP(−) platelets to phagocytes is reduced compared to control platelets, most likely due to increased quenching. As it can mask the magnitude of phagocytosis, this effect could lead to misinterpretation of the results of phagocytosis studies of platelets or other targets. Additionally, more efficient quenching of fluorescence could indicate that the entire process of endophagocytic digestion is more efficient for WASP(−) platelets – a feature which could in turn contribute to an increased rate of in vivo platelet clearance (see below).
If the reduced fluorescence transfer (alpha) were due to increased quenching of the smaller WASP(−) platelets, splenectomy (which results in a partial normalization of the size of WASP(−) platelets) would be expected to increase alpha. That is what we see (Table II, patient 6). Alternatively, more efficient digestion of WASP-deficient platelets might be expected if platelet WASP activity were normally transferred to the phagosome itself. Something similar occurs following macrophage uptake of Leishmania donovani promastigotes [17]. In that case, a glycoconjugate (LPG) is transferred from the parasite to the surface of phagosomes, and induces a WASP-dependent accumulation of f-actin around the phagosome. This in turn impairs phagosome maturation, and is associated with a “backup” inhibition of phagocytosis itself [18]. If normal platelets induce a similar process, platelet WASP deficiency would be expected to accelerate their uptake and digestion by macrophages.
We cannot, however, explain the one case (patient 1) in which both the P and alpha values are substantially different from the others. While genetic reversion, which occurs at a high rate in WAS patients (over 10%) [19], could explain this, a recent study of this patient’s peripheral blood lymphocytes showed no evidence of this. That finding does not, however, rule out (partial) reversion affecting only the megakaryocyte lineage.
There is general agreement that in the most common of the thrombocytopenias (ITP), concurrent impairment of platelet production and augmentation of platelet destruction are likely to occur in many and perhaps most cases. Here we provide evidence that in WAS and XLT patients something similar occurs; an intrinsic platelet defect appears to augment ex vivo uptake of WASP(−) platelets by phagocytes, and the reticulated platelet (RP) fraction shows little if any compensatory increase in platelet production. The presence of, and requirement for, defects at both ends of the platelet lifespan (production and destruction) is consistent with the excellent response these patients typically get to splenectomy, and with their susceptibility after splenectomy to episodic thrombocytopenias clinically indistinguishable from ITP [20].
Alternatively, the low RP fraction could be misleading, since even thrombocytopenias, due entirely to impaired platelet production, demonstrate a greater elevation of “immature” platelets (via the proprietary assay used by Sysmex hematology analyzers [21]) than we report here. Such an increase is to be expected when the platelet count is sufficiently low to shift the bulk of platelet consumption from a nearly linear (lifespan-dependent) course to a more exponential (random, presumably hemostatic) one. Failure to observe a comparable increase in WAS patients raises the question of whether WASP deficiency has a particular effect on the production of RP. This would require the existence of a (WASP-independent) pathway for production of immature, non-reticulated platelets. That option is supported in principle by our previous finding that RP account for less than one-third of platelet production in WT mice – and for an even lower fraction (−11%) in WASP(−) mice [12].
Although we were unable to detect a differential effect of opsonization on the susceptibility of platelets from WAS patients to phagocytosis, the probability of uptake of non-opsonized WASP(−) platelets is sufficiently high with this assay configuration (activated THP-1 cells, DIO-labeled platelets) that we likely could not detect such an effect if it occurred. Conversely, in the ex vivo phagocytosis system we used to study murine WAS (bone marrow derived macrophages; CMFDA-labeled platelets), the phagocytosis rate of non-opsonized platelets was sufficiently low that opsonization may have been required to detect a difference between WT and WASP(−) platelets. Thus, while we cannot rule out a fundamental difference between the clinical and murine thrombocytopenias, the simplest explanation is that in both cases an intrinsic platelet defect augments phagocytosis of WASP(−) platelets and amplifies the effect of opsonization on the process.
One candidate for this defect would be increased expression, or clustering, of platelet GP1balpha, which has been shown to mediate adherence of platelets to the Mac-1 receptor (CR3; CD11b/CD18; alphambeta2 integrin) on THP-1 cells [22] and on (hepatic) Kupffer cells [16]. While we have not characterized the level and distribution of GP1ba on platelets from WAS patients, we have seen no difference in the distribution or cold-induced clustering of the (GPIb-IX)2V complex on platelets from WASP(−) mice (unpublished). Also, the efficacy of splenectomy in treating the thrombocytopenia of both clinical and murine [12] WAS suggests that clearance by Kupffer cells does not play a major role in the thrombocytopenia. More precise delineation of the cell types responsible for rapid in vivo clearance of WASP(−) platelets from the circulation is needed.
Platelets which have been left at room temperature overnight demonstrate, after DIO labeling, no uptake by activated THP-1 cells (data not shown). This finding suggests that the same level of metabolic activity required for platelets to remain hemostatically functional is required for their (normal) interactions with phagocytes. Here we have shown that this requirement does not prevent the routine analysis of phagocytosis of platelets using small numbers of the latter obtained from routine (4 ml) blood specimens from severely thrombocytopenic patients at remote locations. Application of this method to the study of other types of thrombocytopenia, such as ITP, is therefore feasible.
Supplementary Material
Acknowledgments
We thank Dr Hans Ochs (University of Washington, Seattle, WA) and Dr Mary Ellen Conley (St. Jude Children’s Research Hospital, Memphis, TN) for their help in finding WAS patients for this study. We thank Lidia Gardner (Memphis VA Medical Center) and Dr Michael Levin for their generous assistance with the microscopy. This work was supported by National Institutes of Health (NIH) National Heart, Lung, and Blood Institute (NHLBI) award 1R21AI079757 (TS, PI) and by the Department of Veterans Affairs.
References
- 1.Albert MH, Bittner TC, Nonoyama S, Notarangelo LD, Burns S, et al. X-linked thrombocytopenia (XLT) due to WAS mutations: Clinical characteristics, long-term outcome, and treatment options. Blood. 2010;115:3231–3238. doi: 10.1182/blood-2009-09-239087. [DOI] [PubMed] [Google Scholar]
- 2.Cleland SY, Siegel RM. Wiskott-Aldrich Syndrome at the nexus of autoimmune and primary immunodeficiency diseases. FEBS Lett. 2011;585:3710–3714. doi: 10.1016/j.febslet.2011.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kajiwara M, Nonoyama S, Eguchi M, Morio T, Imai K, et al. WASP is involved in proliferation and differentiation of human haemopoietic progenitors in vitro. Br J Haematol. 1999;107:254–262. doi: 10.1046/j.1365-2141.1999.01694.x. [DOI] [PubMed] [Google Scholar]
- 4.Sabri S, Foudi A, Boukour S, Franc B, Charrier S, et al. Deficiency in the Wiskott-Aldrich protein induces premature proplatelet formation and platelet production in the bone marrow compartment. Blood. 2006;108:134–140. doi: 10.1182/blood-2005-03-1219. [DOI] [PubMed] [Google Scholar]
- 5.Luthi JN, Gandhi MJ, Drachman JG. X-linked thrombocytopenia caused by a mutation in the Wiskott–Aldrich syndrome (WAS) gene that disrupts interaction with the WAS protein (WASP)-interacting protein (WIP) Exp Hematol. 2003;31:150–158. doi: 10.1016/s0301-472x(02)01023-8. [DOI] [PubMed] [Google Scholar]
- 6.Schulze H, Korpal M, Hurov J, Kim SW, Zhang J, et al. Characterization of the megakaryocyte demarcation membrane system and its role in thrombopoiesis. Blood. 2006;107:3868–3875. doi: 10.1182/blood-2005-07-2755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Semple JW, Siminovitch KA, Mody M, Milev Y, Lazarus AH, et al. Flow cytometric analysis of platelets from children with the Wiskott-Aldrich syndrome reveals defects in platelet development, activation and structure. Br J Haematol. 1997;97:747–754. doi: 10.1046/j.1365-2141.1997.1132938.x. [DOI] [PubMed] [Google Scholar]
- 8.Matzdorff A, Kemkes-Matthes B, Pralle H. Microparticles and reticulated platelets in Wiskott–Aldrich syndrome patients. Br J Haematol. 2000;109:673. doi: 10.1046/j.1365-2141.2000.01996.x. [DOI] [PubMed] [Google Scholar]
- 9.Baldini MG. Nature of the platelet defect in the Wiskott-Aldrich syndrome. Ann N Y Acad Sci. 1972;201:437–444. doi: 10.1111/j.1749-6632.1972.tb16316.x. [DOI] [PubMed] [Google Scholar]
- 10.Grottum KA, Hovig T, Holmsen H, Abrahamsen AF, Jeremic M, et al. Wiskott-Aldrich syndrome: Qualitative platelet defects and short platelet survival. Br J Haematol. 1969;17:373–388. doi: 10.1111/j.1365-2141.1969.tb01383.x. [DOI] [PubMed] [Google Scholar]
- 11.Ochs HD, Slichter SJ, Harker LA, Von Behrens WE, Clark RA, et al. The Wiskott–Aldrich syndrome: Studies of lymphocytes, granulocytes, and platelets. Blood. 1980;55:243–252. [PubMed] [Google Scholar]
- 12.Prislovsky A, Marathe B, Hosni A, Bolen AL, Nimmerjahn F, et al. Rapid platelet turnover in WASP(−) mice correlates with increased ex vivo phagocytosis of opsonized WASP(−) platelets. Exp Hematol. 2008;36:906–906. doi: 10.1016/j.exphem.2007.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Marathe BM, Prislovsky A, Astrakhan A, Rawlings DJ, Wan JY, et al. Antiplatelet antibodies in WASP(−) mice correlate with evidence of increased in vivo platelet consumption. Exp Hematol. 2009;37:1353–1363. doi: 10.1016/j.exphem.2009.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Strom TS, Anur P, Prislovsky A. A numerical analysis model for interpretation of flow cytometric studies of ex vivo phagocytosis. PLoS ONE. 2011;6:e26657. doi: 10.1371/journal.pone.0026657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Semple JW, Aslam R, Kim M, Speck ER, Freedman J. Platelet-bound lipopolysaccharide enhances Fc receptor-mediated phagocytosis of IgG-opsonized platelets. Blood. 2007;109:4803–4805. doi: 10.1182/blood-2006-12-062695. [DOI] [PubMed] [Google Scholar]
- 16.Hoffmeister KM, Felbinger TW, Falet H, Denis CV, Bergmeier W, et al. The clearance mechanism of chilled blood platelets. Cell. 2003;112:87–97. doi: 10.1016/s0092-8674(02)01253-9. [DOI] [PubMed] [Google Scholar]
- 17.Lodge R. Descoteaux A Leishmania donovani promastigotes induce periphagosomal F-actin accumulation through retention of the GTPase Cdc42. Cellular Microbiol. 2005;7:1647–1658. doi: 10.1111/j.1462-5822.2005.00582.x. [DOI] [PubMed] [Google Scholar]
- 18.Vinet AF, Jananji S, Turco SJ, Fukuda M, Descoteaux A. Exclusion of synaptotagmin V at the phagocytic cup by Leishmania donovani lipophosphoglycan results in decreased promastigote internalization. Microbiology. 2011;157:2619–2628. doi: 10.1099/mic.0.050252-0. [DOI] [PubMed] [Google Scholar]
- 19.Davis BR, Candotti F. Revertant somatic mosaicism in the Wiskott–Aldrich syndrome. Immunol Res. 2009;44:127–131. doi: 10.1007/s12026-008-8091-4. [DOI] [PubMed] [Google Scholar]
- 20.Mullen C, Anderson KD, Blaese RM. Splenectomy and/or bone marrow transplantation in the management of the wiskott-aldrich syndrome: Long-term followup of 52 cases. Blood. 1993;82:2961–2966. [PubMed] [Google Scholar]
- 21.Pons I, Monteagudo M, Lucchetti G, Munoz L, Perea G, et al. Correlation between immature platelet fraction and reticulated platelets. Usefulness in the etiology diagnosis of thrombocytopenia. Eur J Haematol. 2010;85:158–163. doi: 10.1111/j.1600-0609.2010.01468.x. [DOI] [PubMed] [Google Scholar]
- 22.Simon DI, Chen Z, Xu H, Li CQ, Dong J, et al. Platelet glycoprotein ibalpha is a counterreceptor for the leukocyte integrin Mac-1 (CD11b/CD18) J Exp Med. 2000;192:193–204. doi: 10.1084/jem.192.2.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.