

Abstract
Introduction
Diabetes mellitus impacts almost 200 million individuals worldwide and leads to debilitating complications. New avenues of drug discovery must target the underlying cellular processes of oxidative stress, apoptosis, autophagy, and inflammation that can mediate multi-system pathology during diabetes mellitus.
Areas Covered
We examine novel directions for drug discovery that involve the β-nicotinamide adenine dinucleotide (NAD+) precursor nicotinamide, the cytokine erythropoietin, the NAD+-dependent protein histone deacetylase SIRT1, the serine/threonine-protein kinase mammalian target of rapamycin (mTOR), and the wingless pathway. Implications for the targeting of these pathways that oversee gluconeogenic genes, insulin signaling and resistance, fatty acid beta-oxidation, inflammation, and cellular survival are presented.
Expert Opinion
Nicotinamide, erythropoietin, and the downstram pathways of SIRT1, mTOR, forkhead transcription factors, and wingless signaling offer exciting prospects for novel directions of drug discovery for the treatment of metabolic disorders. Future investigations must dissect the complex relationship and fine modulation of these pathways for the successful translation of robust reparative and regenerative strategies against diabetes mellitus and the complications of this disorder.
Keywords: Akt, apoptosis, autophagy, β-cell, Beclin 1, β-catenin, CCN family, diabetes mellitus, erythropoietin, forkhead transcription factors, FoxO, FRAP1, glycogen synthase kinase-3β (GSK-3β), insulin, mammalian target of rapamycin (mTOR), nicotinamide, nicotinamide adenine dinucleotide (NAD+), oxidative stress, peroxisome proliferators activated receptor (PPAR), peroxisome proliferators-activated receptor-γ coactivator (PGC), phosphoinositide, 3–kinase (PI 3-K), poly (ADP-ribose) polymerase-1 (PARP-1), programmed cell death (PCD), protein tyrosine phosphatase, SIRT1, sirtuin, wingless, Wnt1 inducible signaling pathway protein 1 (WISP1)
1. Introduction
The incidence of obesity in the population throughout the world is increasing at an alarming rate that ultimately leads to metabolic disease and diabetes mellitus (DM) 1. Recent studies have shown that the duration of obese-years rather than body mass index (BMI) translates into a strong risk for developing diabetes mellitus 2. Increased weight gain also leads to other disorders that may be a result of metabolic disease, such as coronary artery calcifications and the loss of cognition 3, 4. Yet, despite significant improvements over the prior thirty years in the management and care of individuals who suffer from DM, life expectancy in highly developed countries such as the United States and the United Kingdom continues to fall behind other developed countries such as Japan. This in part may be a result of the growing level of obesity in countries such as the United States that is not occurring at the same pace in other countries, but also signals the critical need for further understanding of the cellular pathways that may foster complications in debilitating disorders such as DM and the necessity to develop new directions to prevent and limit the course of this disease.
In the United States alone, DM has reached proportions of the population affecting almost 26 million individuals in all age groups. An additional 7 million individuals are estimated to be undiagnosed with DM 3. In younger individuals, impaired glucose tolerance is of significant concern, since those with impaired glucose tolerance have a greater than twice the risk for the development of diabetic complications than individuals with normal glucose tolerance. Worldwide, more than 165 million individuals are afflicted with DM and this number could reach close to 400 million individuals by the year of 2030.
DM is defined by two categories that are either insulin dependent or non-insulin dependent. Type 2 non-insulin dependent DM is the most prominent and occurs in approximately eighty percent of all diabetics in individuals greater than 40 years of age. Type 2 DM represents a progressive deterioration of glucose tolerance with early β-cell compensation through cell hyperplasia followed by a decrease in β-cell mass 5. Insulin resistance or defective insulin action occurs when physiological levels of insulin result in a subnormal physiologic response. Although insulin resistance forms the basis for the development of Type 2 DM, elevated serum glucose levels also occur from impairment in insulin secretion. Poor insulin secretion may be a result of defective β-cell function, chronic exposure to free fatty acids and hyperglycemia, or the absence of inhibitory feedback through plasma glucagon levels. Type 1 insulin dependent DM is present in a much smaller group of individuals in approximately 5–10 percent of diabetic patients 1, 3, 4. Type 1 DM is an autoimmune disorder with the presence of alleles of the Human leukocyte antigen (HLA) class II genes within the major histocompatibility complex (MHC). Inflammatory infiltration of the islets of Langerhans and the selective destruction of β-cells in the pancreas that leads to insulin loss are considered to be significant components of Type 1 DM. Almost 90 percent of individuals with Type 1 DM have increased titers of autoantibodies (Type 1A DM). The remaining ten percent of Type 1 DM individuals do not have serum autoantibodies. These individuals are considered to have maturity-onset diabetes of the young (MODY) that can be a result of β-cell dysfunction with autosomal-dominant inheritance (Type IB DM). Some individuals with Type 1 DM also may suffer from insulin resistance that is found in Type 2 DM. In addition, approximately 10 percent of individuals with Type 2 DM may have elevated serum autoantibodies similar to Type 1 DM.
2. Diabetes Mellitus, Oxidative Stress, and Cell Injury
Given the systemic manifestations of metabolic disorders, DM can lead to complications in multiple areas of the body. DM can result in hepatic dysfunction, mood disorders, cognitive loss, renal disease, platelet disorders, sympathetic nervous system dysfunction, and cardiac injury 1, 3. Each of these complications of DM can be a manifestation of oxidative stress.
Oxidative stress occurs following the production of reactive oxygen species (ROS) that are formed through superoxide free radicals, hydrogen peroxide, singlet oxygen, nitric oxide (NO), and peroxynitrite 5–7. ROS can be maintained at non-toxic levels by antioxidant systems in the body that include catalase, superoxide dismutase, glutathione peroxidase, and vitamins C, D, E, and K. However, excessive production of ROS or deficits in the endogenous antioxidant system can lead to oxidative stress and cell death through DNA degradation, mitochondrial dysfunction, and protein misfolding. Oxidative stress can result in mitochondrial injury, nutritional impairment, and DNA damage in diabetic patients 1, 3.
Oxidative stress during DM can lead to cell injury through pathways of programmed cell death (PCD) that involve either autophagy or apoptosis 8. Autophagy leads to the destruction of organelles and the preservation of cytoskeleton structures, allowing cells to recycle cytoplasmic components. Autophagy consists of three different categories known as microautophagy, macroautophagy, and chaperone-mediated autophagy. Macroautophagy represents the degradation of cytoplasmic material and the sequestration of the cytoplasmic protein and organelles into autophagosomes. Autophagosomes fuse with lysosomes for degradation and can regenerate for other cellular processes. Microautophagy consists of the sequestration of cytoplasmic components by invagination of the lysosomal membrane. Vesicles that are formed are transferred to the lumen of the lysosomes for digestion. In chaperone mediated autophagy, the cytoplasmic component is delivered by cytosolic chaperones to the receptors on the lysosomal membranes for translocation across lysosomal membranes into the lumen. During oxidative stress, autophagy can lead to cardiac injury, astrocytic cell death, endothelial cell loss, dopaminergic cell injury, and spinal cord injury 9, 10. Yet, some studies support a protective role for the induction of autophagy during hypoxia-ischemia in the brain, in models of Huntington’s disease, and prion neurotoxicity 11, 12. During DM, autophagy may be necessary to remove misfolded proteins and eliminate non-functioning mitochondria in β-cells to prevent β-cell dysfunction and the onset of DM 13. Exercise in mice has been shown to initiate autophagy and regulate glucose homeostasis 14. However, the generation of advanced glycation end products (AGEs) during DM also may lead to autophagy that contributes to vascular smooth muscle proliferation, atherosclerosis 15, and potential cardiomyopathy 16. The elevation of free fatty acids in cell models of DM suggest that fatty acids may be necessary to activate autophagy in beta cells through endoplasmic reticulum stress 17. During endoplasmic reticulum stress and elevated low-density lipoproteins, autophagy and oxidative stress has been associated with the progression of diabetic retinopathy in animal and human tissue18. In other scenarios, autophagy may not have a significant influence upon cell death 8,19.
Apoptosis also is considered important for tissue re-modeling especially during development, but apoptosis also can lead to cell demise during DM 1. Apoptosis consists of two distinct components that involve genomic DNA degradation and the loss of plasma membrane lipid asymmetry. The cleavage of genomic DNA into fragments during apoptosis occurs during later stages once a cell has been committed to die. In contrast, the loss of asymmetry of membrane phosphatidylserine (PS) distribution is an early reversible feature of apoptosis. Both membrane PS exposure and genomic DNA degradation are considered to be the outcomes of nuclease and protease activation that occurs during apoptosis 6, 20. During DM, apoptosis can lead to vascular complications, impair endothelial cell survival, destroy immune mediated cells, affect cardiomyocyte function, inhibit wound repair, and injure neurons 6, 9, 20. It is important to note that the pathways of autophagy and apoptosis also may intersect to control cell survival. Induction of autophagy may delay the onset of apoptotic cell injury 21. Under other circumstances, apoptosis may lead to the inhibition of autophagy during cell death 22. Cell demise also can be a result of the combined effects of autophagy and apoptosis 23.
3. Novel Considerations for Drug Discovery
Nicotinamide
Given that the pathways of oxidative stress, apoptosis, and autophagy can be significant contributors to the pathogenesis and the subsequent complications of DM, development of strategies that can modulate these pathways may prove fruitful for both the prevention and treatment of DM. Nicotinamide is one agent that has become highly relevant for the treatment of metabolic disorders (Figure 1). Nicotinamide is the amide form of vitamin B3 (niacin) and is generated through the conversion of nicotinic acid in the liver or through the hydrolysis of NAD+. After nicotinamide is obtained in the body, it becomes the precursor for the coenzyme β-nicotinamide adenine dinucleotide (NAD+) 24. Nicotinamide also is essential for the synthesis of nicotinamide adenine dinucleotide phosphate (NADP+).
Figure 1. Nicotinamide, erythropoietin, and wingless modulation of diabetes mellitus.

During diabetes mellitus (DM), elevated glucose leads to oxidative stress and results in the activation of “pro-apoptotic” proteins Bad, Bax, and forkhead transcription factors (FoxOs). Activation of these pathways subsequently impairs mitochondrial (Mito) membrane potential to result in cytochrome c release and caspase activation. Nicotinamide (NIC) prevents cell injury by maintaining mitochondrial membrane potential and modulating Bad, Bax, and FoxO to prevent cytochrome c release from mitochondria and block caspase activation. NIC also may be beneficial during DM through the protection of β-cells and promoting insulin secretion. The cytokine EPO is another therapeutic target that can limit cell injury during DM. EPO can promote the activation of protein kinase B (Akt) that inhibits the activity of FoxO and glycogen synthase kinase-3β (GSK-3β) by phosphorylating (p) these entities. EPO maintains the integrity of wingless pathways, such as Wnt1, during elevated glucose and relies upon Wnt1 to inactivate GSK-3β through phosphorylation (p) that ultimately protects the “anti-apoptotic” properties of β-catenin and prevents its degradation.
Nicotinamide affects both early apoptotic changes with membrane PS exposure and late apoptotic injury with DNA degradation. Nicotinamide prevents membrane PS exposure in endovascular cells that can prevent cardiovascular injury and potentially block thrombotic disease 25. Nicotinamide can control the activation of inflammatory cells as well as early PS membrane exposure in neurons during oxidative stress 26, 27. During the later stages of apoptotic DNA degradation, nicotinamide can limit traumatic brain injury, reduce cortical damage during ischemic and ketamine injury, control cytokine release during sepsis, and prevent apoptotic cell injury during oxidative stress 25–29. Interestingly during tumorigenesis and unchecked cellular proliferation, nicotinamide may block cell growth and prevent tumor progression through the induction of apoptotic pathways 28. Nicotinamide also may promote autophagy induction that can affect mitochondrial membrane potential and lead to mitochondrial fragmentation 30. Under other conditions, nicotinamide may alter apoptosis rather than autophagy during oxidative stress 31, 32.
Nicotinamide may maintain cellular energy homeostasis during DM through mitochondrial pathways 24, 33. During DM, lipid toxicity and oxidative stress can lead to mitochondrial damage and impaired pancreatic β-cell function 34. Preservation of mitochondrial integrity may improve immune and cardiac function during DM. Nicotinamide can maintain mitochondrial membrane polarization during oxidative stress and prevent the release of cytochrome c 25, 27, 29. Nicotinamide can control mitochondrial function and prevent induction of the apoptotic cascade through a number of pathways that involve Bad, modulation of caspase activity, reduction in Bax expression, and control of forkhead transcription factors 25–29.
In clinical studies, oral nicotinamide administration protects β-cell function and prevents clinical disease in islet-cell antibody-positive first-degree relatives of Type 1 DM 35. Nicotinamide administration in patients with recent onset Type 1 DM and intensive insulin therapy can significantly reduced HbA1c levels 36. Nicotinamide also has been shown to reduce intestinal absorption of phosphate and prevent the development of hyperphosphatemia and progressive renal dysfunction 37. In non-clinical studies, nicotinamide can prevent peripheral nerve injury during elevated glucose 38, result in the remission of Type 1 DM in mice in combination therapy with acetyl-l-carnitine 39, and improve glucose utilization and prevent excessive lactate production in ischemic animal models 40. During models of diabetic neuropathy in rats, nicotinamide treatment can reduce poly (ADP-ribose) polymerase-1 (PARP-1) activity to partially restore vital NAD+ and ATP levels 41. Nicotinamide also has been reported to reduce the production of ROS and enhance regulatory T cell immune function in animal models of DM 42. However, some reports suggest that nicotinamide may have reduced protective function in older animal subjects 43 and excessive nicotinamide levels may promote progression of Type 2 DM 44. Nicotinamide also may reduce embryonic stem cell proliferation, but assist with the production of insulin secreting cells 45. Since increased levels of nicotinamide also can limit cytoprotective sirtuin activity that may be detrimental to cell survival 27, 46, 47, determination of the degree of nicotinamide exposure may be a significant component for effective cellular protection during DM. These observations fall in line with the need to determine effective and safe concentrations of nicotinamide, since prior clinical trails that examined nicotinamide application to prevent Type I DM onset did not identify an effective concentration 48.
Erythropoietin
Similar to nicotinamide, erythropoietin (EPO) is a novel agent that also can modulate a number of components in the apoptotic cascade to potentially prevent the devastating effects of DM 49 (Figure 1). EPO is a 30.4 kDa protein with four glycosylated chains that include three N-linked and one O-linked acidic oligosaccharide side chains 50, 51. The function and integrity of EPO depend upon the N- and O-linked chains. Biological activity of EPO is dependent upon the two disulfide bonds formed between cysteine7 and cysteine160 as well as between cysteine29 and cysteine33. Alkylation of the sulfhydryl groups results in the loss of the biological activity of EPO. EPO is produced in multiple organs of the body that include the brain, liver, and uterus. The expression of EPO can be regulated by hypoxia as well as other stimuli. Hypoxia-dependent expression of EPO and its receptor, the EPO receptor (EPOR), are modulated through hypoxia-inducible factor 1 (HIF-1). Gene transcription of EPO and EPOR are tied to the activation of HIF-1 50, 51. In addition to hypoxia, free radical exposure can alter EPO and EPOR expression in vascular cells and in neurons to result in increased HIF-1 expression and subsequent increase in EPO expression. EPO and the EPOR are present in renal tubular cells during high glucose-induced oxidative stress 52. Anemic stress, insulin release, and several cytokines, including insulin-like growth factor, tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β), and interleukin-6 (IL-6) also can result in increased expression of EPO and the EPOR 50, 51. Additional changes in the cellular environment, such as hypoglycemia, cadmium exposure, raised intracellular calcium, or strong neuronal depolarizations also can alter the expression of EPO.
EPO can block cell injury during DM through reduction in cellular oxidative stress and limiting apoptotic cell injury. EPO can limit the generation of ROS, may prevent oxidative stress at high altitudes, and is cytoprotective against oxidative stress during a variety of insults such as hypoxia, TNF-α, glutamate toxicity, and amyloid 50, 51. EPO can preserve cellular survival in neurons, vascular cells, and inflammatory cells during oxidative stress exposure. In regards to preventing apoptotic cell injury, EPO blocks mitochondrial depolarization and the release of cytochrome c. EPO can control mitochondrial signaling through Bad, Bax, Puma. EPO also blocks Apaf-1 activation and prevents the early activation of several caspases such as caspase 1, caspase 3, and caspase 9 that can lead to both early apoptotic membrane PS exposure and late genomic DNA degradation 50–55. Recent work suggests that EPO may control caspase activation through downstream pathways of the proline rich Akt substrate 40 kDa (PRAS40), an inhibitor of the mammalian target of rapamaycin (mTOR) pathway 56, 57.
In clinical studies, EPO plasma levels may be depressed in diabetic patients with anemia or without anemia. Reports that patients with DM cannot produce EPO in response to low hemoglobin levels indicate that an impaired EPO response during DM may be present. Interestingly, increased EPO secretion during diabetic pregnancies occurs and may represent an endogenous protection response against the complications of DM. For example, EPO can promote neurite outgrowth that may prevent cognitive decline 50, 51. Treatment with EPO has been shown in individuals with DM with severe congestive heart failure to decrease fatigue, increase left ventricular ejection fraction, and lessen hospitalization duration 58. In addition, EPO can serve to reverse the complications of anemia during DM 59.
During studies with elevated glucose and experimental models of diabetes, EPO can reduce ROS generation and prevent high glucose-induced renal cell apoptosis 52, promote wound repair in diabetic mice 60, and maintain vascular cell integrity in cellular models of DM through pathways that involve the sirtuin SIRT1 53. Intravitreal EPO treatment in diabetic rat retinas has been shown to modulate the expression of pro-apoptotic genes and limit inflammation 61. EPO also may have a role in the prevention of diabetic neuropathy 62. EPO in streptozotocin-induced and db/db mouse models of Type 1 and Type 2 DM also can prevent apoptotic pancreatic β-cell loss through aβ-cell specific EPOR 63. Yet, the protective effects of EPO may not always require a direct association with the EPOR. Some studies suggest that although mRNA for the EPOR is present in numerous tissues, the presence of the EPOR protein in the tissue samples examined may be functionally low in non-hematopoeitc cell types 64. Prevention of the complications of DM may be closely linked to the effects of EPO on vascular cells. EPO can block vascular injury through activation of wingless pathways, by limiting apoptotic forkhead subcellular trafficking to the nucleus, and through the modulation of downstream β-catenin and glycogen synthase kinase-3β (GSK-3β) pathways 65–67.
Sirtuins
Both nicotinamide and EPO are closely linked to the metabolic pathways of the sirtuin SIRT1 that can influence cellular metabolism during DM. Sirtuins are class III NAD+-dependent protein histone deacetylases that are the mammalian homologues of Sir2 of the yeast silent information regulator-2 (Sir2) 6. Of the seven mammalian homologues of Sir2, SIRT1 is present in the brain, heart, liver, pancreas, skeletal muscle, spleen, and adipose tissues. During DM, SIRT1 is protective against cellular injury. For example, SIRT1 activation prevents endothelial senescence during hyperglycemia, blocks oxidative stress injury in cardiomyocytes, limits atherosclerotic lesions during elevated lipid states, and prevents endothelial cell apoptosis during experimental diabetes 7, 68.
Activation of SIRT1 can increase lifespan in higher organisms such as Drosophila and protect cells from oxidative stress 27, 46. Loss of SIRT1 is associated with insulin resistance. Gene deletion or inhibition of SIRT1 impairs insulin signaling by interfering with insulin stimulated insulin receptor phosphorylation and glycogen synthase 69. In contrast, over-expression of SIRT1 decreases hepatic steatosis and improves insulin sensitivity that leads to improved glucose homeostasis 70. SIRT1 can increase insulin signaling in insulin-sensitive organs through protein kinase B (Akt) and phosphotidylinositide 3-kinase (PI 3-K) 71. SIRT1 also can stimulate glucose-dependent insulin secretion from pancreatic β cells by repressing the uncoupling protein (UCP) gene UCP2 72. In addition, SIRT1 controls insulin sensitivity through tyrosine phosphatase1B (PTP1B). PTP1B deficiency leads to improved insulin sensitivity and glycemic control. SIRT1 over-expression or SIRT1 activation can reduce both PTP1B mRNA and protein levels during insulin-resistance. However, an increase in PTP1B expression prevents SIRT1 mediated glucose uptake and insulin receptor phosphorylation in response to insulin stimulation 69. SIRT1 also may improve insulin sensitivity through the regulation of fat mobilization, gluconeogenesis, and inflammation 6, 7, 68.
SIRT1 can regulate food intake, cellular metabolism, and lipid homeostasis (Figure 2). SIRT1 is expressed in anorexigenic proopiomelanocortin (POMC) neurons and orexigenic agouti-related peptide (AgRP) neurons in the arcuate nucleus of the hypothalamus that control food intake 73. Over-expression of SIRT1 in theses regions of the hypothalamus prevents the forkhead transcription factor FoxO1 from promoting hyperphagia and body weight gain 73. In contrast, absence of SIRT1 in POMC neurons leads to obesity due to reduced energy expenditure. During the regulation of hepatic glucose output and lipid homeostasis, SIRT1 controls peroxisome proliferators-activated receptor-γ coactivator (PGC)-1α via deacetylation in the liver to transcribe gluconeogenic genes and increase hepatic glucose output. As a transcriptional coactivator, PGC-1α interacts with transcription factors to activate transcription and increase the expression of genes that regulate mitochondrial functions and fatty acid oxidation. Increased PGC-1α activity may function to protect against some metabolic diseases and improve mitochondrial biogenesis. SIRT1 also controls the ability of PGC-1α to repress glycolytic genes in response to fasting and pyruvate 74. SIRT1 modulates fatty acid metabolism and cellular stress through peroxisome proliferators activated receptor-α (PPAR-α). Liver-specific SIRT1 knockout mice develop hepatic steatosis, hepatic inflammation, and endoplasmic reticulum stress when challenged with a high fat diet since hepatocyte-specific deletion of SIRT1 impairs PPAR-α signaling and decreases fatty acid beta-oxidation 75. In addition, SIRT1 requires PPAR-α to protect cardiac cells from hypertrophy, metabolic dysfunction, and inflammation 76.
Figure 2. The intimate relationship among SIRT1, mTOR, and FoxOs during diabetes mellitus.

Following the induction of oxidative stress during diabetes mellitus (DM), insulin resistance can ensue. Activation of SIRT1 can alter insulin sensitivity and increase the secretion of insulin by repressing the mitochondrial uncoupling protein 2 (UCP2), promote lypolysis by mediating peroxisome proliferators-activated receptor-γ (PPAR-α), and increase gluconeogenesis and lipid homeostasis by regulating proliferators-activated receptor-γ coactivator (PGC)-1α. In addition, increased activity of forkhead transcription factors (FoxOs) can increase gene transcription of PGC-1α and also SIRT1 to regulate insulin sensitivity and glucose metabolism. Yet, FoxOs can have an inverse relationship with SIRT1 and may prevent lipolysis through inhibition of PPAR-α. During DM, the activity of the serine/threonine protein kinase mammalian target of rapamaycin (mTOR) may be blocked. Activation of mTOR and its downstream pathways of p70S6K promote the secretion of insulin and increase insulin sensitivity. If mTOR activity is inhibited, such as during rapamycin application, insulin sensitivity and glucose uptake are reduced although obesity may be averted.
SIRT1 is one pathway that allows EPO to prevent cell injury during DM. During vascular cell protection, EPO increases endogenous SIRT1 activity and promotes the subcellular trafficking of SIRT1 to the nucleus 53. SIRT1 also can increase Akt activity that is considered to be a principle pathway for cellular proliferation and survival, for the cytoprotective capacity of EPO and nicotinamide, and for mediating insulin signaling 24, 50, 53, 77–79. SIRT1 can block the activity of forkhead transcription factors such as FoxO1, FoxO3a, and FoxO4. SIRT1 activates Akt1 to control the phosphorylation and subcellular trafficking of the forkhead transcription factor FoxO3a 53. Mammalian forkhead transcription factors of the O class (FoxO1, FoxO3, FoxO4, and FoxO6), such as FoxO3a also have been tied to the cellular pathways of EPO as well as nicotinamide and are intimately involved with cellular metabolism, insulin sensitivity, and oxidative stress (Figure 2) 80, 81. Phosphorylation of FoxOs leads to the retention of these transcription factors in the cytoplasm and the subsequent inhibition of their transcriptional activity. SIRT1 relies upon the Akt pathway for cytoprotection and the subsequent phosphorylation of target genes such as FoxOs 53, 71. However, SIRT1 may control a fine balance over FoxO activity since acetylation of FoxOs also can limit their transcriptional activity. SIRT1 can lead to the deacetylation of FoxOs to increase the activity of these transcription factors 82, 83. Yet, the deacetylation of FoxOs may ultimately lead to degradation of these transcription factors 84. SIRT1 also may rely upon the regulation of FoxO transcription factors to protect against inflammation, to promote β-cell function during DM, and to maintain endothelial survival during oxidative stress 7,83. In addition, an increase in FoxO3a and SIRT1 activity can occur in the heart during exercise 85, suggesting that beneficial physical activity for the cardiovascular system and for patients with DM may be mediated through SIRT1 and FoxO proteins. FoxOs also exert a positive feedback mechanism regulating SIRT1 expression. FoxO1 can directly bind to the SIRT1 promoter region containing a cluster of five putative FoxO1 core binding repeat motifs (IRS-1) and a forkhead-like consensus-binding site (FKHD-L). This results in FoxO1 modulating SIRT1 transcription and leads to an increase in the expression of SIRT1 86. FoxO3a also can regulate the expression of SIRT1 by binding to two p53 binding sites within the SIRT1 promoter to foster SIRT1 transcription during acute nutrient withdrawal 80, 81.
Unlike EPO, a more complex relationship appears to exist between nicotinamide and SIRT1 in regards to cell survival and longevity. Nicotinamide can block sirtuin activity by intercepting an ADP-ribosyl-enzyme-acetyl peptide intermediate with the regeneration of NAD+ (transglycosidation). As a result, reduction in nicotinamide levels during nicotinamidase expression can increase SIRT1 activity and foster cellular protection 27, 46. Although nicotinamide provides cellular protection in millimole concentrations against early apoptotic membrane exposure and late DNA degradation, physiological concentrations of nicotinamide noncompetitively inhibit sirtuins such as SIRT1, indicating that nicotinamide is a physiologically relevant regulator of sirtuin activity. In addition, sirtuins may regulate nicotinamide by preventing nicotinamide from assisting with DNA repair by altering the accessibility of DNA damaged sites for repair enzymes 87.
Serine/threonine-protein kinase mTOR
Metabolic signaling that is mediated through Akt not only relies upon pathways with nicotinamide, EPO, and SIRT1, but also controls metabolic cell pathways through the serine/threonine protein kinase mTOR that has other alternative names including mechanistic target of rapamycin and FK506-binding protein 12-rapamycin complex-associated protein 1 (FRAP1) 88 (Figure 2). As part of the PI 3-K related kinase family that is activated through the PI 3-K and Akt, mTOR is a 289-kDa serine/threonine protein kinase that can control transcription, cytoskeleton organization, cellular survival, and cellular metabolism 9, 89. mTOR signaling is dependent upon the protein complexes mTOR Complex 1 (mTORC1) or mTOR Complex 2 (mTORC2) that each contain mTOR. p70 ribosomal S6 kinase (p70S6K) and eukaryotic initiation factor 4E (eIF4E)-binding protein 1 (4EBP1) are downstream targets of mTORC1 that are relevant in DM pathways. Phosphorylation of p70S6K promotes mRNA biogenesis, translation of ribosomal proteins, and cell growth. In contrast, phosphorylation of 4EBP1 results in its inactivation. Hypophosphorylated 4EBP1 is active and binds competitively with eukaryotic translation initiation factor 4 gamma (eIF4G) to eukaryotic translation initiation factor 4 epsilon (eIF4E) that regulate translation initiation by interacting with the 5′-mRNA cap structure. The phosphorylation of 4EBP1 by mTORC1 results in its dissociation from eIF4E allowing eIF4G to interact with eIF4E and promote protein translation.
The signaling pathways of mTOR can be closely tied to pathways associated with cell protection and longevity, such as those with EPO and SIRT1. EPO uses mTOR signaling for the neuronal differentiation of post-mortem neural precursors 90. Retinal progenitor cells are resistant to hypoxia when exposed to EPO that leads to mTOR activation 91. EPO regulates bone homeostasis with osteoblastogenesis and osteoclastogenesis through mTOR activation 92. EPO can activate mTOR to block apoptotic cell death during oxidative stress in inflammatory cells 93. In cellular models of Alzheimer’s disease, amyloid degeneration of microglia is limited by EPO through combined activation of PI 3-K and mTOR pathways 55. In regards to SIRT1 and mTOR, loss of SIRT1 expression leads to hepatic glucose overproduction, hyperglycemia, products of oxidative stress, and inhibition of the gene encoding Rictor that lead to impaired TORC2 and Akt signaling 94. Under some conditions, SIRT1 and mTOR may have an inverse relationship. For example, SIRT1 attenuates hepatic steatosis, ameliorates insulin resistance, and restores glucose homeostasis primarily through the inhibition of mTORC1 70. SIRT1 also may promote neuronal growth through pathways that inhibit mTOR signaling 95. With nicotinamide, mTOR activity can be increased that may be dependent upon blockade of SIRT1 activity 96. During high glucose exposure to mesangial cells, loss of SIRT1 activity, such as with the application of nicotinamide, is necessary for mTOR to arrest mesangial cell senescence 97.
During metabolic disorders, mTOR signaling may be necessary for maintenance of insulin function. Loss of mTOR signaling with inhibition of p70S6K can result in hypoinsulinemia, glucose intolerance, insulin insensitivity to glucose secretion, and a decrease in pancreatic β-cell size 9, 89. Conversely, activation of p70S6K and inhibition of 4EBP1 in pancreatic β-cells in murine models of DM results in improved insulin secretion and resistance to β-cell streptozotocin toxicity and obesity 98. However, mTOR inhibition with rapamycin application leads to insulin resistance, reduces β-cell function and mass, limits insulin secretion, and results in DM 99. Although inhibition of mTOR reduces food intake and prevents fat-diet induced obesity in mice, loss of mTOR activity also attenuates glucose uptake and metabolism in skeletal muscle through the prevention of insulin generated Akt activation and alteration in the translocation of glucose transporters to the plasma membrane 100. Other studies support a role for components of mTORC1 and mTORC2 in insulin signaling. Loss of the mTORC1 substrate p70S6K activity with combined loss of the mTORC2 substrate Akt2 results in defects in both insulin action and β-cell function, suggesting that both mTOR components are required to maintain insulin signaling and prevent DM 101.
Wingless Signaling
Cytoprotective signaling through mTOR during metabolic disease has recently been associated with the wingless pathway. The wingless pathway consists of the Wnt family of cysteine-rich glycosylated proteins that control stem cell development, angiogenesis, tumorigenesis, and metabolism 102, 103. Genetic variations in Wnt signaling pathways, such as with transcription factor 7-like 2 gene, may impart an increased risk for Type 2 DM in some populations 104. Intronic variants of the low-density lipoprotein receptor-related protein 5 (LRP5) gene, a member of the Wnt signaling pathway, are also markedly associated with obesity 105. Impaired Wnt signaling through a missense mutation in LRP6 has been reported in patients with coronary artery disease and the combined metabolic syndrome with hypertension, hyperlipidemia, and DM 106.
Wnt family members can provide improved glucose tolerance, increased insulin sensitivity, weight control, protect glomerular mesangial cells from apoptotic cell injury during elevated glucose, and prevent vascular cell injury in models of DM (Figure 2) 102. Wnt signaling requires mTOR to prevent cell injury during models of cardiac ischemia, oxidative stress, and during amyloid exposure 102, 107. Interestingly, a significant crosstalk between mTOR and Wnt exists to promote insulin signaling and cellular proliferation 108.
As a component of the Wnt signaling pathway, Wnt1 inducible signaling pathway protein 1 (WISP1) also is cytoprotective and may represent a new target for metabolic disorders. WISP1 is a member of the CCN family of proteins and is known as CCN4. The CCN family of proteins is termed by the first three members of the family that include Cysteine-rich protein 61, Connective tissue growth factor, and Nephroblastoma over-expressed gene and consists of six secreted extracellular matrix associated proteins. WISP1 initially was shown to prevent p53 mediated DNA damage and apoptosis in kidney fibroblasts 109. A reparative and regenerative role for WISP1 may exist since WISP1 has been shown to have elevated expression during cardiac ischemia, neuronal exposure to oxidative stress, lung epithelial damage, and cellular repair of fractured bone 19, 109–111. Subsequent work has demonstrated that WISP1 can initiate cardiac remodeling after myocardial infarction, stimulate lung tissue repair, promote cardiomyocyte proliferation, and foster vascular smooth muscle growth during cytokine exposure. WISP1 also can prevent cell death during bone fractures and block oxygen-glucose deprivation injury in primary neuronal cells 19, 110, 111. In relation to DM, WISP1 expression has been shown to be up-regulated during pancreatic regeneration in rats following partial pancreatectomy 112, suggesting that pancreatic and β-cell regeneration may require WISP1 expression similar to the role mTOR has recently been shown to play during β-cell proliferation 101.
4. Conclusion
DM impacts a significant proportion of the world’s population with a large number of individuals that continue to remain undiagnosed and without treatment. In addition, incidence in young individuals with DM continues to grow. The complications of DM involve multiple systems of the body and can rapidly lead to disability as well as death. New avenues of discovery that can address the complications of DM and the underlying cellular processes of oxidative stress, apoptosis, and autophagy may foster the development of new therapeutic strategies for DM. Novel considerations for drug discovery for the treatment of metabolic disorders include nicotinamide, EPO, and the targeted pathways of SIRT1, mTOR, and wingless signaling. These pathways have an intricate relationship that requires a fine modulation to achieve desired biological and clinical outcomes that can be both reparative and regenerative against the multi-systemic complications of DM.
5. Expert Opinion
Our understanding of the biological processes that lead to DM and the complications of this disorder have grown remarkably over the prior decades. Yet despite these advances, the incidence of DM continues to grow worldwide especially in young adults. The increased complications that are observed with DM in both a young and an aging population can negatively affect life expectancy. Recent studies also point to new concerns. Although intensive therapy to control plasma glucose levels may promote long-term benefits, studies note that intensive glycemic control in patients with Type 2 DM may not improve renal outcomes or prevent the development of renal disease 113. In addition, current therapies for plasma glucose control are not without risk and may lead to hypoglycemia, weight gain, or increased mortality 114.
As a result, development of novel strategies for DM and its complications are highly warranted and met with enthusiasm. Yet, development of new strategies will require thorough analysis of appropriate clinical applications and the elucidation of cellular pathways that can foster reparative and regenerative mechanisms but lack detrimental clinical outcomes. For example, recent work has suggested that in some injury models nicotinamide may offer greater benefit for cytoprotection in young rather than aged subjects 43. Temporal administration with nicotinamide also may affect expected clinical outcome since prolonged administration of nicotinamide may lead to β-cell dysfunction and limit P450 and hepatic metabolism 24. Nicotinamide treatment during DM also requires further investigations that can assess effective treatment concentrations. Although other NAD+ precursors similar to nicotinamide, such as nicotinamide riboside, can increase NAD+ levels and activate SIRT1 resulting in protection against high-fat diet-induced metabolic abnormalities 115, NAD+ precursors such as nicotinamide also can lead to cellular energy depletion that is detrimental to cell survival. Nicotinamide is a substrate for PARP-1 and maintains the integrity of PARP-1 to prevent its cleavage. At elevated concentrations, nicotinamide can promote PARP-1 activation that can deplete NAD+ stores, lower ATP production, and lead to cell death 87. During inflammatory disorders that activate TNFβ-1, nicotinamide also can block hepatic cell proliferation and lead to apoptosis with caspase 3 activation 24. In addition, increased levels of nicotinamide may negate the benefits of SIRT1 during DM thereby limiting treatment efficacy 27, 46, 47. Similar to nicotinamide, EPO administration also requires careful consideration in regards to duration of administration and the co-morbidity factors present in patients with DM. For example, increased expression of EPO may lead to progressive hepatic cell ischemic injury as well as the development of thrombotic and hypertensive complications in patients with DM 50, 51. EPO also has been associated with tumorigenesis that may complicate administration of EPO 116–118. In addition, EPO may be contraindicated during severe hypertension that can occur in diabetic patients since EPO may raise mean arterial blood pressure 51, 119, 120.
Vital to moderating potential adverse effects and maximizing clinical efficacy is the further investigation of the novel pathways of SIRT1, mTOR, and wingless that can mediate the cytoprotective pathways of nicotinamide and EPO. Although offering potential efficacy for the treatment of DM and the complications of this disorder, the pathways of SIRT1, mTOR and wingless are cellular proliferative and can have the potential to lead to unchecked cell growth. Under some conditions, loss of the protein Deleted in Breast Cancer 1 (DBC1), a negative regulator of SIRT1, can result in excessive cell growth, inhibition of apoptosis, and increased risk for tumorigenesis 121. Activation of mTOR signaling may prevent insulin resistance during diabetes mellitus, but long-term mTOR activity may worsen conditions such as diabetic retinopathy and diabetic cardiac disease given the ability of mTOR to promote angiogenesis 10, 89. In addition, mTOR activation can lead to tumorigenesis and epilepsy 122, 123 as well as impairment in neuronal stem cell maturation 124 and dyskinesias 125. In regards to wingless signaling, pathways such as Wnt and WISP1 may promote vascular smooth muscle growth that has the potential to lead to vascular complications in diabetic patients already at risk for adverse vascular events 126.
In consideration of cell repair and the potential for cell regeneration during DM, new studies also must address the complex relationship between apoptosis and autophagy during DM. For example, cytoprotection against some toxins in the nervous system such as methamphatamine requires the induction of “anti-apoptotic” pathways that block autophagy 10, 89, suggesting that under some conditions cellular protection may require the inhibition of both apoptosis and autophagy. However, under other conditions, cytoprotection may require the promotion of autophagy but the inhibition of apoptosis to limit apoptotic cell death 21. Furthermore, other scenarios suggest that autophagy may have a limited role in controlling cell death during DM even though pathways that are responsible for cell protection such as PI 3-K and Akt may be necessary for modulating autophagy 19, 23. Over the next several years, future investigations that can address a number of parameters that include clinical appropriateness for new therapeutic strategies for a diabetic population that suffers from multiple disease complications, duration and temporal administration of treatments that can ultimately impact multiple cellular pathways, and the further dissection of the complex and intricate cellular pathways that determine biological outcome will be crucial for the successful translation of novel strategies for DM and its complications into realistic and effective treatments.
Figure 3. Novel clinical pathways for drug development during diabetes mellitus.
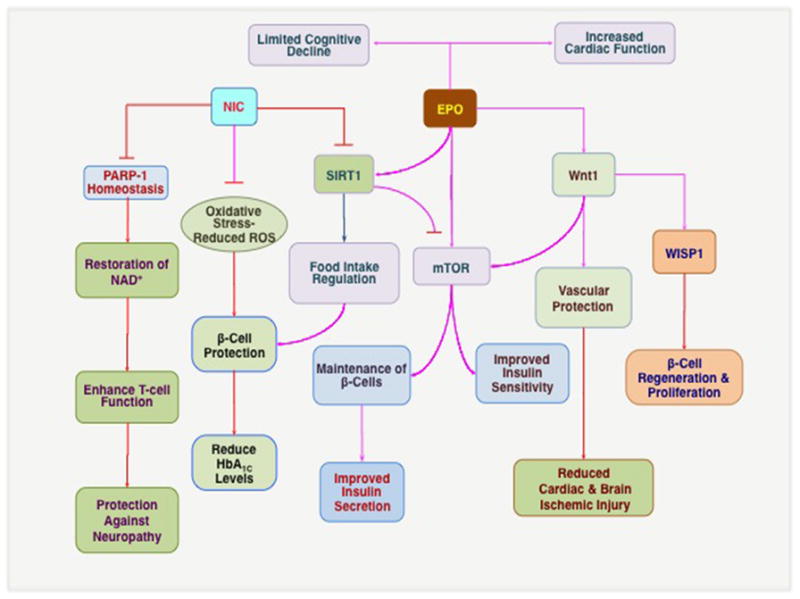
Nicotinamide (NIC) can maintain poly (ADP-ribose) polymerase-1 (PARP-1) homeostasis, restore NAD+ and ATP levels, enhance T-cell immune function, and lead to the prevention of diabetic neuropathy. NIC can limit reactive oxygen species (ROS) generation and oxidative stress during diabetes mellitus (DM) to assist with β-cell protection and also reduce HbA1C levels. NIC can function as a feedback mechanism to inhibit SIRT1 and activate the mammalian target of rapamycin (mTOR) to promote insulin sensitivity and maintain β-cell function. However, high concentrations of NIC may be detrimental to cell survival by limiting the activity of SIRT1 that is necessary for food intake regulation and cellular metabolism. Erythropoietin (EPO) can limit cognitive decline, promote neurite outgrowth, and increase cardiac function during DM that may function through the ability of EPO to modulate SIRT1 activity and foster cellular protection. EPO also directly or through Wnt1 increases mTOR activity to maintain β-cell survival and insulin sensitivity. Following Wnt1 activation, Wnt1 can promote vascular protection and reduce complications of DM, such as cardiac and brain ischemic injury. Wnt1 also governs Wnt1 inducible signaling pathway protein 1 (WISP1) that may assist with β-cell regeneration and proliferation.
Highlights.
Oxidative stress during diabetes mellitus can lead to cell injury through the pathways of programmed cell death that involve either autophagy or apoptosis
Nicotinamide can maintain cellular energy homeostasis during diabetes mellitus but may have an inverse relationship with the sirtuin SIRT1
The cytokine erythropoietin can offer cellular protection during diabetes mellitus by controlling inflammation and preventing the induction of apoptotic mitochondrial pathways
The sirtuin SIRT1 controls insulin sensitivity, food intake, and lipid homeostasis through pathways that involve UCP, PTP1B, PGC-1α, and PPAR-α signaling
Forkhead transcription factors (FoxOs) are intimately tied to nicotinamide, erythropoietin, and SIRT1 to inflammation, β-cell function, and cellular survival during diabetes mellitus
Wingless (Wnt) family members can promote improved glucose tolerance, increased insulin sensitivity, and protection in neuronal, vascular, and renal cells during diabetes mellitus
Acknowledgments
This research was supported by the following grants to Kenneth Maiese: American Diabetes Association, American Heart Association (National), Bugher Foundation Award, Janssen Neuroscience Award, LEARN Foundation Award, NIH NIEHS, NIH NIA, NIH NINDS, and NIH ARRA (NS059346-04 and NS059346-03S1).
Bibliography
- 1.Maiese K, Chong ZZ, Shang YC, et al. Novel Avenues of Drug Discovery and Biomarkers for Diabetes Mellitus. Journal of clinical pharmacology. 2011 Mar 10;51:128–52. doi: 10.1177/0091270010362904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Abdullah A, Wolfe R, Mannan H, et al. Epidemiologic Merit of Obese-Years, the Combination of Degree and Duration of Obesity. Am J Epidemiol. 2012 Jun 28; doi: 10.1093/aje/kwr522. [DOI] [PubMed] [Google Scholar]
- 3.Maiese K, Shang YC, Chong ZZ, et al. Diabetes mellitus: channeling care through cellular discovery. Curr Neurovasc Res. 2010 Feb 1;7:59–64. doi: 10.2174/156720210790820217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Reagan LP. Diabetes as a chronic metabolic stressor: causes, consequences and clinical complications. Exp Neurol. 2012 Jan;233:68–78. doi: 10.1016/j.expneurol.2011.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maiese K, Chong ZZ, Hou J, et al. Oxidative stress: Biomarkers and novel therapeutic pathways. Exp Gerontol. 2010 Mar;45:217–34. doi: 10.1016/j.exger.2010.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chong ZZ, Shang YC, Wang S, et al. SIRT1: New avenues of discovery for disorders of oxidative stress. Expert opinion on therapeutic targets. 2012 Feb;16:167–78. doi: 10.1517/14728222.2012.648926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Maiese K, Chong ZZ, Shang YC, et al. Translating cell survival and cell longevity into treatment strategies with SIRT1. Rom J Morphol Embryol. 2011;52:1173–85. [PMC free article] [PubMed] [Google Scholar]
- 8.Maiese K, Chong ZZ, Shang YC, et al. Targeting disease through novel pathways of apoptosis and autophagy. Expert opinion on therapeutic targets. 2012 Aug 27; doi: 10.1517/14728222.2012.719499. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chong ZZ, Maiese K. Mammalian Target of Rapamycin Signaling in Diabetic Cardiovascular Disease. Cardiovasc Diabetol. 2012 Apr 30;11:45. doi: 10.1186/1475-2840-11-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chong ZZ, Shang YC, Maiese K. Cardiovascular Disease and mTOR Signaling. Trends Cardiovasc Med. 2011 Jul;21:151–5. doi: 10.1016/j.tcm.2012.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Balduini W, Carloni S, Buonocore G. Autophagy in hypoxia-ischemia induced brain injury. J Matern Fetal Neonatal Med. 2012 Apr;25( Suppl 1):30–4. doi: 10.3109/14767058.2012.663176. [DOI] [PubMed] [Google Scholar]
- 12.Hyrskyluoto A, Reijonen S, Kivinen J, et al. GADD34 mediates cytoprotective autophagy in mutant huntingtin expressing cells via the mTOR pathway. Exp Cell Res. 2012 Jan 1;318:33–42. doi: 10.1016/j.yexcr.2011.08.020. [DOI] [PubMed] [Google Scholar]
- 13• *.Liu Y, Shi S, Gu Z, et al. Impaired autophagic function in rat islets with aging. Age (Dordr) 2012 Jul 28; doi: 10.1007/s11357-012-9456-0. The study provides evidence that aging can impair autophagy that subsequently leads to islet cell dysfunction and the development of Type e DM. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14• *.He C, Bassik MC, Moresi V, et al. Exercise-induced BCL2-regulated autophagy is required for muscle glucose homeostasis. Nature. 2012 Jan 26;481:511–5. doi: 10.1038/nature10758. The investigation offers a mechanistic rationale for the benefits of exercise to prevent the progression of DM through the regulation of autophagy. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hu P, Lai D, Lu P, et al. ERK and Akt signaling pathways are involved in advanced glycation end product-induced autophagy in rat vascular smooth muscle cells. Int J Mol Med. 2012 Apr;29:613–8. doi: 10.3892/ijmm.2012.891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee Y, Hong Y, Lee SR, et al. Autophagy contributes to retardation of cardiac growth in diabetic rats. Lab Anim Res. 2012 Jun;28:99–107. doi: 10.5625/lar.2012.28.2.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Martino L, Masini M, Novelli M, et al. Palmitate activates autophagy in INS-1E beta-cells and in isolated rat and human pancreatic islets. PLoS ONE. 2012;7:e36188. doi: 10.1371/journal.pone.0036188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fu D, Fau-Wu M, Wu M, Fau-Zhang J, Zhang J, Fau-Du M, et al. Mechanisms of modified LDL-induced pericyte loss and retinal injury in diabetic retinopathy. doi: 10.1007/s00125-012-2692-0. 20120831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang S, Chong ZZ, Shang YC, et al. WISP1 (CCN4) autoregulates its expression and nuclear trafficking of beta-catenin during oxidant stress with limited effects upon neuronal autophagy. Curr Neurovasc Res. 2012 Apr 4;9:89–99. doi: 10.2174/156720212800410858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Troy CM, Akpan N, Jean YY. Regulation of caspases in the nervous system implications for functions in health and disease. Prog Mol Biol Transl Sci. 2011;99:265–305. doi: 10.1016/B978-0-12-385504-6.00007-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Viola G, Bortolozzi R, Hamel E, et al. MG-2477, a new tubulin inhibitor, induces autophagy through inhibition of the Akt/mTOR pathway and delayed apoptosis in A549 cells. Biochem Pharmacol. 2012 Jan 1;83:16–26. doi: 10.1016/j.bcp.2011.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Luo S, Rubinsztein DC. Apoptosis blocks Beclin 1-dependent autophagosome synthesis: an effect rescued by Bcl-xL. Cell Death Differ. 2010 Feb;17:268–77. doi: 10.1038/cdd.2009.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kapoor V, Zaharieva MM, Das SN, et al. Erufosine simultaneously induces apoptosis and autophagy by modulating the Akt-mTOR signaling pathway in oral squamous cell carcinoma. Cancer Lett. 2012 Jun 1;319:39–48. doi: 10.1016/j.canlet.2011.12.032. [DOI] [PubMed] [Google Scholar]
- 24.Maiese K, Chong ZZ, Hou J, et al. The vitamin nicotinamide: translating nutrition into clinical care. Molecules. 2009;14:3446–85. doi: 10.3390/molecules14093446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chong ZZ, Lin SH, Maiese K. Nicotinamide Modulates Mitochondrial Membrane Potential and Cysteine Protease Activity during Cerebral Vascular Endothelial Cell Injury. J Vasc Res. 2002;39:131–47. doi: 10.1159/000057762. [DOI] [PubMed] [Google Scholar]
- 26.Chong ZZ, Lin SH, Maiese K. The NAD+ precursor nicotinamide governs neuronal survival during oxidative stress through protein kinase B coupled to FOXO3a and mitochondrial membrane potential. J Cereb Blood Flow Metab. 2004 Jul;24:728–43. doi: 10.1097/01.WCB.0000122746.72175.0E. [DOI] [PubMed] [Google Scholar]
- 27.Chong ZZ, Maiese K. Enhanced Tolerance against Early and Late Apoptotic Oxidative Stress in Mammalian Neurons through Nicotinamidase and Sirtuin Mediated Pathways. Curr Neurovasc Res. 2008 Aug;5:159–70. doi: 10.2174/156720208785425666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Audrito V, Vaisitti T, Rossi D, et al. Nicotinamide Blocks Proliferation and Induces Apoptosis of Chronic Lymphocytic Leukemia Cells through Activation of the p53/miR-34a/SIRT1 Tumor Suppressor Network. Cancer Res. 2011 Jul 1;71:4473–83. doi: 10.1158/0008-5472.CAN-10-4452. [DOI] [PubMed] [Google Scholar]
- 29.Ullah N, Ullah I, Lee HY, et al. Protective Function of Nicotinamide Against Ketamine-induced Apoptotic Neurodegeneration in the Infant Rat Brain. J Mol Neurosci. 2012 May;47:67–75. doi: 10.1007/s12031-011-9685-1. [DOI] [PubMed] [Google Scholar]
- 30.Jang Sy, Fau-Kang HT, Kang Ht, Fau-Hwang ES, Hwang ES. Nicotinamide-induced mitophagy: event mediated by high NAD+/NADH ratio and SIRT1 protein activation. doi: 10.1074/jbc.M112.363747. 0120604 DCOM- 20120820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lin SH, Vincent A, Shaw T, et al. Prevention of nitric oxide-induced neuronal injury through the modulation of independent pathways of programmed cell death. J Cereb Blood Flow Metab. 2000;20:1380–91. doi: 10.1097/00004647-200009000-00013. [DOI] [PubMed] [Google Scholar]
- 32.Maiese K. Triple play: Promoting neurovascular longevity with nicotinamide, WNT, and erythropoietin in diabetes mellitus. Biomed Pharmacother. 2008 Apr-May;62:218–32. doi: 10.1016/j.biopha.2008.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li F, Chong ZZ, Maiese K. Cell Life Versus Cell Longevity: The Mysteries Surrounding the NAD(+) Precursor Nicotinamide. Curr Med Chem. 2006;13:883–95. doi: 10.2174/092986706776361058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu Z, Stanojevic V, Brindamour LJ, et al. GLP1-derived nonapeptide GLP1(28-36)amide protects pancreatic beta-cells from glucolipotoxicity. J Endocrinol. 2012 May;213:143–54. doi: 10.1530/JOE-11-0328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Olmos PR, Hodgson MI, Maiz A, et al. Nicotinamide protected first-phase insulin response (FPIR) and prevented clinical disease in first-degree relatives of type-1 diabetics. Diabetes Res Clin Pract. 2006 Mar;71:320–33. doi: 10.1016/j.diabres.2005.07.009. [DOI] [PubMed] [Google Scholar]
- 36.Crino A, Schiaffini R, Ciampalini P, et al. A two year observational study of nicotinamide and intensive insulin therapy in patients with recent onset type 1 diabetes mellitus. J Pediatr Endocrinol Metab. 2005 Aug;18:749–54. doi: 10.1515/jpem.2005.18.8.749. [DOI] [PubMed] [Google Scholar]
- 37.Eto N, Miyata Y, Ohno H, et al. Nicotinamide prevents the development of hyperphosphataemia by suppressing intestinal sodium-dependent phosphate transporter in rats with adenine-induced renal failure. Nephrol Dial Transplant. 2005 Jul;20:1378–84. doi: 10.1093/ndt/gfh781. [DOI] [PubMed] [Google Scholar]
- 38.Stevens MJ, Li F, Drel VR, et al. Nicotinamide reverses neurological and neurovascular deficits in streptozotocin diabetic rats. J Pharmacol Exp Ther. 2007 Jan;320:458–64. doi: 10.1124/jpet.106.109702. [DOI] [PubMed] [Google Scholar]
- 39.Cresto JC, Fabiano de Bruno LE, Cao GF, et al. The association of acetyl-l-carnitine and nicotinamide remits the experimental diabetes in mice by multiple low-dose streptozotocin. Pancreas. 2006 Nov;33:403–11. doi: 10.1097/01.mpa.0000236740.07854.b1. [DOI] [PubMed] [Google Scholar]
- 40.Tam D, Tam M, Maynard KI. Nicotinamide modulates energy utilization and improves functional recovery from ischemia in the in vitro rabbit retina. Ann N Y Acad Sci. 2005 Aug;1053:258–68. doi: 10.1196/annals.1344.023. [DOI] [PubMed] [Google Scholar]
- 41.Kuchmerovska T, Shymanskyy I, Donchenko G, et al. Poly(ADP-ribosyl)ation enhancement in brain cell nuclei is associated with diabetic neuropathy. J Diabetes Complications. 2004 Jul-Aug;18:198–204. doi: 10.1016/S1056-8727(03)00039-4. [DOI] [PubMed] [Google Scholar]
- 42.John CM, Ramasamy R, Al Naqeeb G, et al. Enhanced CD4+CD25+ regulatory T cells with splenic proliferation and protection against oxidative stress by nicotinamide in gestational diabetes. Curr Med Chem. 2012 Aug 16; [PubMed] [Google Scholar]
- 43• **.Swan AA, Chandrashekar R, Beare J, et al. Preclinical efficacy testing in middle-aged rats: nicotinamide, a novel neuroprotectant, demonstrates diminished preclinical efficacy after controlled cortical impact. J Neurotrauma. 2011 Mar;28:431–40. doi: 10.1089/neu.2010.1519. The study suggests that aged subjects may have a different biological response to treatment than young subjects; highlighting the need to address potential activation or inhibition of cellular pathways with the aging process during drug development. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhou SS, Li D, Sun WP, et al. Nicotinamide overload may play a role in the development of type 2 diabetes. World J Gastroenterol. 2009 Dec 7;15:5674–84. doi: 10.3748/wjg.15.5674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vaca P, Berna G, Araujo R, et al. Nicotinamide induces differentiation of embryonic stem cells into insulin-secreting cells. Exp Cell Res. 2008 Mar 10;314:969–74. doi: 10.1016/j.yexcr.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 46.Balan V, Miller GS, Kaplun L, et al. Life span extension and neuronal cell protection by Drosophila nicotinamidase. J Biol Chem. 2008 Oct 10;283:27810–9. doi: 10.1074/jbc.M804681200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Siegel C, McCullough LD. NAD+ depletion or PAR polymer formation: which plays the role of executioner in ischaemic cell death? Acta Physiol (Oxf) 2011 Sep;203:225–34. doi: 10.1111/j.1748-1716.2010.02229.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gale EA, Bingley PJ, Emmett CL, et al. European Nicotinamide Diabetes Intervention Trial (ENDIT): a randomised controlled trial of intervention before the onset of type 1 diabetes. Lancet. 2004 Mar 20;363:925–31. doi: 10.1016/S0140-6736(04)15786-3. [DOI] [PubMed] [Google Scholar]
- 49.Maiese K, Chong ZZ, Shang YC, et al. Erythropoietin: New Directions for the Nervous System. Int J Mol Sci. 2012;13:11102–29. doi: 10.3390/ijms130911102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Maiese K, Chong ZZ, Li F, et al. Erythropoietin: elucidating new cellular targets that broaden therapeutic strategies. Prog Neurobiol. 2008 Jun;85:194–213. doi: 10.1016/j.pneurobio.2008.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Maiese K, Li F, Chong ZZ. New avenues of exploration for erythropoietin. Jama. 2005 Jan 5;293:90–5. doi: 10.1001/jama.293.1.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dang J, Jia R, Tu Y, et al. Erythropoietin prevents reactive oxygen species generation and renal tubular cell apoptosis at high glucose level. Biomed Pharmacother. 2010 Dec;64:681–5. doi: 10.1016/j.biopha.2010.06.011. [DOI] [PubMed] [Google Scholar]
- 53.Hou J, Wang S, Shang YC, et al. Erythropoietin Employs Cell Longevity Pathways of SIRT1 to Foster Endothelial Vascular Integrity During Oxidant Stress. Curr Neurovasc Res. 2011 Aug 1;8:220–35. doi: 10.2174/156720211796558069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pankratova S, Gu B, Kiryushko D, et al. A new agonist of the erythropoietin receptor, Epobis, induces neurite outgrowth and promotes neuronal survival. J Neurochem. 2012 Jun;121:915–23. doi: 10.1111/j.1471-4159.2012.07751.x. [DOI] [PubMed] [Google Scholar]
- 55.Shang YC, Chong ZZ, Wang S, et al. Prevention of beta-amyloid degeneration of microglia by erythropoietin depends on Wnt1, the PI 3-K/mTOR pathway, Bad, and Bcl-xL. Aging (Albany NY) 2012 Mar 3;4:187–201. doi: 10.18632/aging.100440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chong ZZ, Shang YC, Wang S, et al. PRAS40 Is an Integral Regulatory Component of Erythropoietin mTOR Signaling and Cytoprotection. PLoS ONE. 2012;7:e45456. doi: 10.1371/journal.pone.0045456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shang YC, Chong ZZ, Wang S, et al. WNT1 Inducible Signaling Pathway Protein 1 (WISP1) Targets PRAS40 to Govern beta-Amyloid Apoptotic Injury of Microglia. Curr Neurovasc Res. 2012 Aug 6; doi: 10.2174/156720212803530618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Silverberg DS, Wexler D, Iaina A, et al. The interaction between heart failure and other heart diseases, renal failure, and anemia. Semin Nephrol. 2006 Jul;26:296–306. doi: 10.1016/j.semnephrol.2006.05.006. [DOI] [PubMed] [Google Scholar]
- 59.Singh DK, Winocour P, Farrington K. Erythropoietic stress and anemia in diabetes mellitus. Nat Rev Endocrinol. 2009 Apr;5:204–10. doi: 10.1038/nrendo.2009.17. [DOI] [PubMed] [Google Scholar]
- 60.Hamed S, Ullmann Y, Egozi D, et al. Fibronectin potentiates topical erythropoietin-induced wound repair in diabetic mice. J Invest Dermatol. 2011 Jun;131:1365–74. doi: 10.1038/jid.2011.15. [DOI] [PubMed] [Google Scholar]
- 61.Chu Q, Zhang J, Wu Y, et al. Differential gene expression pattern of diabetic rat retinas after intravitreal injection of erythropoietin. Clin Experiment Ophthalmol. 2011 Mar;39:142–51. doi: 10.1111/j.1442-9071.2010.02437.x. [DOI] [PubMed] [Google Scholar]
- 62.Chattopadhyay M, Walter C, Mata M, et al. Neuroprotective effect of herpes simplex virus-mediated gene transfer of erythropoietin in hyperglycemic dorsal root ganglion neurons. Brain. 2009 Apr;132:879–88. doi: 10.1093/brain/awp014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Choi D, Schroer SA, Lu SY, et al. Erythropoietin protects against diabetes through direct effects on pancreatic beta cells. J Exp Med. 2010 Dec 20;207:2831–42. doi: 10.1084/jem.20100665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sinclair AM, Coxon A, McCaffery I, et al. Functional erythropoietin receptor is undetectable in endothelial, cardiac, neuronal, and renal cells. Blood. 2010 May 27;115:4264–72. doi: 10.1182/blood-2009-10-248666. [DOI] [PubMed] [Google Scholar]
- 65• *.Chong ZZ, Hou J, Shang YC, et al. EPO Relies upon Novel Signaling of Wnt1 that Requires Akt1, FoxO3a, GSK-3beta, and beta-Catenin to Foster Vascular Integrity During Experimental Diabetes. Curr Neurovasc Res. 2011 May 1;8:103–20. doi: 10.2174/156720211795495402. The investigation highlights the complex and intimate role that multiple cellular pathways have in cytoprotection that require consideration in the development of agents for the treatment of DM. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chong ZZ, Shang YC, Maiese K. Vascular injury during elevated glucose can be mitigated by erythropoietin and Wnt signaling. Curr Neurovasc Res. 2007 Aug;4:194–204. doi: 10.2174/156720207781387150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ghaboura N, Tamareille S, Ducluzeau PH, et al. Diabetes mellitus abrogates erythropoietin-induced cardioprotection against ischemic-reperfusion injury by alteration of the RISK/GSK-3beta signaling. Basic Res Cardiol. 2011 Jan;106:147–62. doi: 10.1007/s00395-010-0130-3. [DOI] [PubMed] [Google Scholar]
- 68.Kelly GS. A review of the sirtuin system, its clinical implications, and the potential role of dietary activators like resveratrol: part 2. Altern Med Rev. 2010 Dec;15:313–28. [PubMed] [Google Scholar]
- 69.Sun C, Zhang F, Ge X, et al. SIRT1 improves insulin sensitivity under insulin-resistant conditions by repressing PTP1B. Cell Metab. 2007 Oct;6:307–19. doi: 10.1016/j.cmet.2007.08.014. [DOI] [PubMed] [Google Scholar]
- 70.Li Y, Xu S, Giles A, et al. Hepatic overexpression of SIRT1 in mice attenuates endoplasmic reticulum stress and insulin resistance in the liver. Faseb J. 2011 Feb 14; doi: 10.1096/fj.10-173492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Frojdo S, Durand C, Molin L, et al. Phosphoinositide 3-kinase as a novel functional target for the regulation of the insulin signaling pathway by SIRT1. Mol Cell Endocrinol. 2011 Mar 30;335:166–76. doi: 10.1016/j.mce.2011.01.008. [DOI] [PubMed] [Google Scholar]
- 72.Bordone L, Motta MC, Picard F, et al. Sirt1 regulates insulin secretion by repressing UCP2 in pancreatic beta cells. PLoS Biol. 2006 Feb;4:e31. doi: 10.1371/journal.pbio.0040031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sasaki T, Kitamura T. Roles of FoxO1 and Sirt1 in the central regulation of food intake. Endocrine journal. 2010 Nov 30;57:939–46. doi: 10.1507/endocrj.k10e-320. [DOI] [PubMed] [Google Scholar]
- 74• **.Rodgers JT, Lerin C, Haas W, et al. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature. 2005 Mar 3;434:113–8. doi: 10.1038/nature03354. The study outlines a significant role for SIRT1 for energy and glucose homeostasis that are relevant for the development of agents to treat DM. [DOI] [PubMed] [Google Scholar]
- 75.Purushotham A, Schug TT, Xu Q, et al. Hepatocyte-specific deletion of SIRT1 alters fatty acid metabolism and results in hepatic steatosis and inflammation. Cell Metab. 2009 Apr;9:327–38. doi: 10.1016/j.cmet.2009.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Planavila A, Iglesias R, Giralt M, et al. Sirt1 acts in association with PPAR{alpha} to protect the heart from hypertrophy, metabolic dysregulation, and inflammation. Cardiovasc Res. 2010 Dec 22; doi: 10.1093/cvr/cvq376. [DOI] [PubMed] [Google Scholar]
- 77.Chong ZZ, Maiese K. The Src homology 2 domain tyrosine phosphatases SHP-1 and SHP-2: diversified control of cell growth, inflammation, and injury. Histol Histopathol. 2007 Nov;22:1251–67. doi: 10.14670/hh-22.1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hou J, Chong ZZ, Shang YC, et al. Early apoptotic vascular signaling is determined by Sirt1 through nuclear shuttling, forkhead trafficking, bad, and mitochondrial caspase activation. Curr Neurovasc Res. 2010 May;7:95–112. doi: 10.2174/156720210791184899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sundaresan NR, Pillai VB, Wolfgeher D, et al. The deacetylase SIRT1 promotes membrane localization and activation of Akt and PDK1 during tumorigenesis and cardiac hypertrophy. Science signaling. 2011;4:ra46. doi: 10.1126/scisignal.2001465. [DOI] [PubMed] [Google Scholar]
- 80.Maiese K, Chong ZZ, Shang YC, et al. A “FOXO” in sight: targeting Foxo proteins from conception to cancer. Med Res Rev. 2009 May;29:395–418. doi: 10.1002/med.20139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Storz P. Forkhead homeobox type O transcription factors in the responses to oxidative stress. Antioxid Redox Signal. 2011 Feb 15;14:593–605. doi: 10.1089/ars.2010.3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kousteni S. FoxO1, the transcriptional chief of staff of energy metabolism. Bone. 2012 Feb;50:437–43. doi: 10.1016/j.bone.2011.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Maiese K, Chong ZZ, Shang YC. OutFOXOing disease and disability: the therapeutic potential of targeting FoxO proteins. Trends Mol Med. 2008 May;14:219–27. doi: 10.1016/j.molmed.2008.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wang F, Chan CH, Chen K, et al. Deacetylation of FOXO3 by SIRT1 or SIRT2 leads to Skp2-mediated FOXO3 ubiquitination and degradation. Oncogene. 2012 Mar 22;31:1546–57. doi: 10.1038/onc.2011.347. [DOI] [PubMed] [Google Scholar]
- 85.Ferrara N, Rinaldi B, Corbi G, et al. Exercise Training Promotes SIRT1 Activity in Aged Rats. Rejuvenation Res. 2008 Dec 10;11(1):139–50. doi: 10.1089/rej.2007.0576. [DOI] [PubMed] [Google Scholar]
- 86.Xiong S, Salazar G, Patrushev N, et al. FoxO1 Mediates an Autofeedback Loop Regulating SIRT1 Expression. J Biol Chem. 2011 Feb 18;286:5289–99. doi: 10.1074/jbc.M110.163667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Surjana D, Halliday GM, Damian DL. Role of nicotinamide in DNA damage, mutagenesis, and DNA repair. J Nucleic Acids. 2010 doi: 10.4061/2010/157591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Chong ZZ, Shang YC, Wang S, et al. Shedding new light on neurodegenerative diseases through the mammalian target of rapamycin. Prog Neurobiol. 2012 Aug 15; doi: 10.1016/j.pneurobio.2012.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Chong ZZ, Shang YC, Zhang L, et al. Mammalian target of rapamycin: hitting the bull’s-eye for neurological disorders. Oxid Med Cell Longev. 2010 Nov-Dec;3:374–91. doi: 10.4161/oxim.3.6.14787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Marfia G, Madaschi L, Marra F, et al. Adult neural precursors isolated from post mortem brain yield mostly neurons: an erythropoietin-dependent process. Neurobiol Dis. 2011 Jul;43:86–98. doi: 10.1016/j.nbd.2011.02.004. [DOI] [PubMed] [Google Scholar]
- 91.Sanghera KP, Mathalone N, Baigi R, et al. The PI3K/Akt/mTOR pathway mediates retinal progenitor cell survival under hypoxic and superoxide stress. Mol Cell Neurosci. 2011 Jun;47:145–53. doi: 10.1016/j.mcn.2011.03.010. [DOI] [PubMed] [Google Scholar]
- 92.Kim J, Jung Y, Sun H, et al. Erythropoietin mediated bone formation is regulated by mTOR signaling. J Cell Biochem. 2012 Jan;113:220–8. doi: 10.1002/jcb.23347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Shang YC, Chong ZZ, Wang S, et al. Erythropoietin and Wnt1 Govern Pathways of mTOR, Apaf-1, and XIAP in Inflammatory Microglia. Curr Neurovasc Res. 2011 Oct 19;8:270–85. doi: 10.2174/156720211798120990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wang RH, Kim HS, Xiao C, et al. Hepatic Sirt1 deficiency in mice impairs mTorc2/Akt signaling and results in hyperglycemia, oxidative damage, and insulin resistance. J Clin Invest. 2011 Nov;121:4477–90. doi: 10.1172/JCI46243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Guo W, Qian L, Zhang J, et al. Sirt1 overexpression in neurons promotes neurite outgrowth and cell survival through inhibition of the mTOR signaling. J Neurosci Res. 2011 Nov;89:1723–36. doi: 10.1002/jnr.22725. [DOI] [PubMed] [Google Scholar]
- 96.Ghosh HS, McBurney M, Robbins PD. SIRT1 negatively regulates the mammalian target of rapamycin. PLoS ONE. 2010;5:e9199. doi: 10.1371/journal.pone.0009199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Zhang S, Cai G, Fu B, et al. SIRT1 is required for the effects of rapamycin on high glucose-inducing mesangial cells senescence. Mech Ageing Dev. 2012 Jun;133:387–400. doi: 10.1016/j.mad.2012.04.005. [DOI] [PubMed] [Google Scholar]
- 98.Hamada S, Hara K, Hamada T, et al. Upregulation of the mammalian target of rapamycin complex 1 pathway by Ras homolog enriched in brain in pancreatic beta-cells leads to increased beta-cell mass and prevention of hyperglycemia. Diabetes. 2009 Jun;58:1321–32. doi: 10.2337/db08-0519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Fraenkel M, Ketzinel-Gilad M, Ariav Y, et al. mTOR inhibition by rapamycin prevents beta-cell adaptation to hyperglycemia and exacerbates the metabolic state in type 2 diabetes. Diabetes. 2008 Apr;57:945–57. doi: 10.2337/db07-0922. [DOI] [PubMed] [Google Scholar]
- 100.Deblon N, Bourgoin L, Veyrat-Durebex C, et al. Chronic mTOR inhibition by rapamycin induces muscle insulin resistance despite weight loss in rats. Br J Pharmacol. 2012 Apr;165:2325–40. doi: 10.1111/j.1476-5381.2011.01716.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101• *.Treins C, Alliouachene S, Hassouna R, et al. The combined deletion of S6K1 and Akt2 deteriorates glycaemic control in high fat diet. Mol Cell Biol. 2012 Jul 30; doi: 10.1128/MCB.00514-12. The study suggests that multiple pathways of mTOR signaling should be targeted for the development of new therapeutic strategies against DM. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Maiese K, Li F, Chong ZZ, et al. The Wnt signaling pathway: Aging gracefully as a protectionist? Pharmacol Ther. 2008 Apr;118:58–81. doi: 10.1016/j.pharmthera.2008.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Noguti J, DEM CF, Hossaka TA, et al. The Role of Canonical WNT Signaling Pathway in Oral Carcinogenesis: A Comprehensive Review. Anticancer Res. 2012 Mar;32:873–8. [PubMed] [Google Scholar]
- 104.Lehman DM, Hunt KJ, Leach RJ, et al. Haplotypes of Transcription Factor 7-Like 2 (TCF7L2) Gene and Its Upstream Region Are Associated With Type 2 Diabetes and Age of Onset in Mexican Americans. Diabetes. 2007 Feb;56:389–93. doi: 10.2337/db06-0860. [DOI] [PubMed] [Google Scholar]
- 105.Guo YF, Xiong DH, Shen H, et al. Polymorphisms of the low-density lipoprotein receptor-related protein 5 (LRP5) gene are associated with obesity phenotypes in a large family-based association study. J Med Genet. 2006 Oct;43:798–803. doi: 10.1136/jmg.2006.041715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Mani A, Radhakrishnan J, Wang H, et al. LRP6 mutation in a family with early coronary disease and metabolic risk factors. Science. 2007 Mar 2;315:1278–82. doi: 10.1126/science.1136370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.L’Episcopo F, Serapide MF, Tirolo C, et al. A Wnt1 regulated Frizzled-1/beta-Catenin signaling pathway as a candidate regulatory circuit controlling mesencephalic dopaminergic neuron-astrocyte crosstalk: Therapeutical relevance for neuron survival and neuroprotection. Molecular neurodegeneration. 2011;6:49. doi: 10.1186/1750-1326-6-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Sun J, Jin T. Both Wnt and mTOR signaling pathways are involved in insulin-stimulated proto-oncogene expression in intestinal cells. Cell Signal. 2008 Jan;20:219–29. doi: 10.1016/j.cellsig.2007.10.010. [DOI] [PubMed] [Google Scholar]
- 109.Berschneider B, Konigshoff M. WNT1 inducible signaling pathway protein 1 (WISP1): a novel mediator linking development and disease. Int J Biochem Cell Biol. 2010 Mar;43:306–9. doi: 10.1016/j.biocel.2010.11.013. [DOI] [PubMed] [Google Scholar]
- 110.Macsai CE, Georgiou KR, Foster BK, et al. Microarray expression analysis of genes and pathways involved in growth plate cartilage injury responses and bony repair. Bone. 2012 May;50:1081–91. doi: 10.1016/j.bone.2012.02.013. [DOI] [PubMed] [Google Scholar]
- 111.Wang S, Chong ZZ, Shang YC, et al. Wnt1 inducible signaling pathway protein 1 (WISP1) blocks neurodegeneration through phosphoinositide 3 kinase/Akt1 and apoptotic mitochondrial signaling involving Bad, Bax, Bim, and Bcl-xL. Curr Neurovasc Res. 2012 Feb;9:20–31. doi: 10.2174/156720212799297137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Lim HW, Lee JE, Shin SJ, et al. Identification of differentially expressed mRNA during pancreas regeneration of rat by mRNA differential display. Biochem Biophys Res Commun. 2002 Dec 20;299:806–12. doi: 10.1016/s0006-291x(02)02741-9. [DOI] [PubMed] [Google Scholar]
- 113.Coca SG, Ismail-Beigi F, Haq N, et al. Role of intensive glucose control in development of renal end points in type 2 diabetes mellitus: systematic review and meta-analysis intensive glucose control in type 2 diabetes. Arch Intern Med. 2012 May 28;172:761–9. doi: 10.1001/archinternmed.2011.2230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kruger DF. Managing diabetes from first diagnosis: choosing well-tolerated therapies with durability. Diabetes Educ. 2012 Jul;38:4S–11S. doi: 10.1177/0145721712450619. [DOI] [PubMed] [Google Scholar]
- 115.Canto C, Houtkooper RH, Pirinen E, et al. The NAD(+) Precursor Nicotinamide Riboside Enhances Oxidative Metabolism and Protects against High-Fat Diet-Induced Obesity. Cell Metab. 2012 Jun 6;15:838–47. doi: 10.1016/j.cmet.2012.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Hedley BD, Allan AL, Xenocostas A. The role of erythropoietin and erythropoiesis-stimulating agents in tumor progression. Clin Cancer Res. 2011 Oct 15;17:6373–80. doi: 10.1158/1078-0432.CCR-10-2577. [DOI] [PubMed] [Google Scholar]
- 117.Maiese K, Chong ZZ, Hou J, et al. The “O” class: crafting clinical care with FoxO transcription factors. Adv Exp Med Biol. 2009;665:242–60. doi: 10.1007/978-1-4419-1599-3_18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Maiese K, Li F, Chong ZZ. Erythropoietin and cancer. JAMA. 2005;293:1858–9. doi: 10.1001/jama.293.1.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Miyashita K, Tojo A, Kimura K, et al. Blood pressure response to erythropoietin injection in hemodialysis and predialysis patients. Hypertens Res. 2004 Feb;27:79–84. doi: 10.1291/hypres.27.79. [DOI] [PubMed] [Google Scholar]
- 120.Novak BL, Force RW, Mumford BT, et al. Erythropoietin-induced hypertensive urgency in a patient with chronic renal insufficiency: case report and review of the literature. Pharmacotherapy. 2003 Feb;23:265–9. doi: 10.1592/phco.23.2.265.32077. [DOI] [PubMed] [Google Scholar]
- 121.Kim JE, Chen J, Lou Z. DBC1 is a negative regulator of SIRT1. Nature. 2008 Jan 31;451:583–6. doi: 10.1038/nature06500. [DOI] [PubMed] [Google Scholar]
- 122.Curran MP. Everolimus: in patients with subependymal giant cell astrocytoma associated with tuberous sclerosis complex. Paediatr Drugs. 2012 Feb 1;14:51–60. doi: 10.2165/11207730-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 123.Krueger DA, Care MM, Holland K, et al. Everolimus for subependymal giant-cell astrocytomas in tuberous sclerosis. N Engl J Med. 2010 Nov 4;363:1801–11. doi: 10.1056/NEJMoa1001671. [DOI] [PubMed] [Google Scholar]
- 124.Magri L, Cambiaghi M, Cominelli M, et al. Sustained activation of mTOR pathway in embryonic neural stem cells leads to development of tuberous sclerosis complex-associated lesions. Cell Stem Cell. 2011 Nov 4;9:447–62. doi: 10.1016/j.stem.2011.09.008. [DOI] [PubMed] [Google Scholar]
- 125.Santini E, Heiman M, Greengard P, et al. Inhibition of mTOR signaling in Parkinson’s disease prevents L-DOPA-induced dyskinesia. Science signaling. 2009;2:ra36. doi: 10.1126/scisignal.2000308. [DOI] [PubMed] [Google Scholar]
- 126.Marchand A, Atassi F, Gaaya A, et al. The Wnt/beta-catenin pathway is activated during advanced arterial aging in humans. Aging Cell. 2011 Apr;10:220–32. doi: 10.1111/j.1474-9726.2010.00661.x. [DOI] [PubMed] [Google Scholar]