

Abstract
A deeper understanding of the mechanisms that control responses to inflammation is critical to the development of effective therapies. We sought to define the most proximal regulators of the Cullin-Ring-Ligases (CRL), which play a central role in the stabilization of NF-κB and HIF. In these studies, we identify the human deneddylase-1 (SENP8) as a key regulator of Cullin neddylation response in vitro and in vivo. Using human microvascular endothelial cells (HMEC), we examined inflammatory responses to LPS or TNF-α by assessing Cullin-neddylation status, NF-κB and HIF-1α stabilization and inflammatory cytokine secretion. HMEC with an intact neddylation pathway showed a time-dependent induction of Cullin-1 (Cul-1) neddylation, nuclear translocation of NF-κB, stabilization of HIF-1α and increased NF-κB/HIF-α promoter activity in response to LPS. HMEC cells lacking SENP8 were unable to neddylate Cul-1 and subsequently were unable to activate NF-κB or HIF-1α. Pharmacological targeting of neddylation (MLN4924) significantly abrogated NF-κB responses, induced HIF-1α promoter activity and reduced secretion of TNF-α-elicited pro-inflammatory cytokines. MLN4924 stabilized HIF and abrogated pro-inflammatory responses while maintaining anti-inflammatory IL-10 response in vivo, following LPS administration. These studies identify SENP8 as a proximal regulator of Cullin neddylation and provide an important role for SENP8 in fine-tuning of the inflammatory response. Moreover, our findings provide feasibility for therapeutic targeting of the Cullins during inflammation.
Introduction
Sepsis remains one of the leading causes of death in developed countries (1, 2). The underlying mechanisms of sepsis severity are incompletely understood, and therefore, treatment options beyond those available today are needed. One of the hallmarks of sepsis is loss of endothelial function and subsequent activation of the immune system. (3, 4). The NF-κB (Nuclear factor κB) pathway has been well established as one of the key pathways for intracellular pro-inflammatory signaling (5–7). Furthermore, there is evidence that NF-κB can influence hypoxia-inducible factor (HIF), an oxygen homeostasis transcription factor, levels and vice versa (8, 9). NF-κB is regulated through IkappaB kinases (IKK) and its activation relies on IKK degradation through the 26S proteasome (10). HIF is also tightly regulated in a posttranslational fashion. Under normoxic conditions, HIF is hydroxylated by prolylhydroxylases, followed by ubiquitination and ultimately degradation by the proteasome (11). Hypoxia is often detectable at sites of inflammation (12). Under such conditions, hypoxia-inducible factor (HIF) is stabilized, translocates to the nucleus and enhances transcription of a number of anti-inflammatory genes (13).
The ubiquitination of IκB-kinase and the alpha subunit of HIF-1 and HIF-2 is mediated through Cullin-proteins which, to be active, require conjugation to the small ubiquitin-like modifier NEDD8 (Neural precursor cell expressed, developmentally downregulated)(14). The conjugation of NEDD8 to its target proteins is a multistep process. Initially, the NEDD8 precursor is cleaved at a carboxy-terminal glycine residue and conjugated to the UBA3-APPB1 E1 ligase, also referred to as NEDD8-activating enzyme, NAE (15–17). The mature NEDD8 moiety is transferred to its E2 ligase UBC12 (ubiquitin converting enzyme) (18) and then linked to its E3 ligase, a Cullin protein, thereby activating it (19, 20). NEDD8 conjugation to Cul-1 is necessary to process the NF-κB p50 subunit precursor p105 through the Skp-Cul1-FboxβTrCP (SCFβTrCP)(21). Based on their findings, Amir et al. conclude that NEDDylation of Cullin proteins is essential for induced processing of p105 and NF-κB activity (21). Apart from NF-κB, Cullin-neddylation also impacts the transcription factor hypoxia-inducible-factor (HIF). The alpha subunit of HIF, under normoxic conditions, is hydroxylated through prolyl-hydroxylases (PHD)(22). This hydroxylation facilitates recognition and binding of HIFα by the Von-Hippel-Lindau protein (pVHL), which is part of an E3-Ligase complex containing Cullin-2(23), thereby mediating the proteasomal degradation of hydroxylated HIF(11). In its non-hydroxylated form, HIFα translocates to the nucleus, binds to the constitutively expressed HIF1β subunit and transcriptionally regulates a wide variety of genes important for the inflammatory response(24, 25).
Lately some interest has been generated in understanding the mechanisms that regulate Cullin-neddylation, more specifically the reverse process, deneddylation. It has been shown that adenosine generated by hypoxic preconditioning mediates deneddylation of Cul-1. This process is dependent on the COP9 signalosome and offers a potential protective mechanism against hypoxia induced pro-inflammatory stimuli (26). The discovery of SENP8, an isopeptidase capable of directly deneddylating Cullin-proteins (17, 27) provides new insights into how Cullin-neddylation might be regulated. SENP8 appears to be able to deneddylate hyper-neddylated Cullin-proteins, thus offering a cleavage pathway beyond the COP9 signalosome (28, 29).
In this study, we define the role of SENP8 mediated deneddylation during inflammation using several approaches including knockdown and overexpression of SENP8. Furthermore we use the small molecule inhibitor of neddylation, MLN4924 (30), which has been shown to impact NF-κB inhibition via Cullin-mediated IκBα degradation (31), to evaluate its benefits for modulating the inflammatory response following LPS or TNF-α stimulation in vitro and in vivo. We identify here a previously unappreciated role for SENP8 as a central regulator of the inflammatory process. Furthermore, we show that under physiological conditions, SENP8 functions to make mature NEDD8 available for conjugation to Cullins, impacting on two transcription factors, i.e. NF-κB and HIF, important for balancing the inflammatory response.
Material and Methods
Cell culture
Human microvascular endothelial cells (HMEC-1) were a kind gift of Francisco Candal (Centers for Disease Control, Atlanta, GA). HMEC-1 cells were cultured in molecular, cellular, and developmental biology (MCDB)-131 medium, supplemented with heat-inactivated fetal bovine serum, penicillin, streptomycin, l-glutamine, epidermal growth factor, and hydrocortisone, as described previously (32). Stable knockdown of SENP8 was performed using shRNA with the following sequences: CCGGCCTAACTTCATTCAAGACCTACTCGAGTAGGTCTTGAATGAAGTTAGGTTTTTG (TRCN0000073338, referred to as clone 38 in figures) or CCGGGACTGTGGGATGTACGTGATACTCGAGTATCACGTACATCCCACAGTCTTTTTG (TRCN0000073342, referred to as clone 42 in figures, Sigma, St. Louis, USA) introduced using lentiviral particles. Cells transfected with scrambled shRNA were used as control. Knockdown cells were kept in cell culture medium with a maintenance dose of 0.25μg/ml puromycin. Transient overexpression of SENP8 was achieved by transfecting 1μg of a FLAG-tagged SENP8 [Addgene plasmid 18066, (33)] construct under a CMV-promoter into control HMEC-1 cells. Cells were cultured at 37°C in an atmosphere of 95% air and 5% CO2 in a humidified incubator. Where indicated, human umbilical vein endothelial cells (HUVEC) were obtained and cultured according to manufacturers recommendations (Promocell, Heidelberg, Germany).
Real-time PCR
To initially assess the level of SENP8 knockdown by lentiviral knockdown, SENP8 transcript levels were measured by relative realtime PCR. mRNA was isolated using the thiocyanate-phenol-chloroform method. Relative realtime PCR was then performed using Power SYBR Mastermix (Applied Biosystems, Carlsbad, CA, USA) and the following primer sequences:
| Sense 5′-3′ | Antisense 5′-3′ | |
|---|---|---|
| SENP8 | CAACACAGAGGTGCCTGAAG | CGGGGTCCATCTTGTACTGA |
| IL-1β | TTCGACACATGGGATAACGA | TCTTTCAACACGCAGGACAG |
| IL-6 | ATGCAATAACCACCCCTGAC | GAGGTGCCCATGCTACATTT |
| IL-8 | ATGACTTCCAAGCTGGCCGTGG | CATAATTTCTGTGTTGGCGCAGTGTGG |
| TNF-α | TCCTTCAGACACCCTCAACC | AGGCCCCAGTTTGAATTCTT |
| ICAM | TTTTCTATCGGCACAAAAGC | AATGCAAACAGGACAAGAGG |
| Actin | CGACTTCGAGCAAGAGATGG | AGCACTGTGTTGGCGTACAG |
Cell treatments
For western blot experiments and luciferase assays the following treatment regimen was used: Six hours prior to harvesting, cells were treated with 10μg/ml medium of E.coli derived LPS (E. coli 055:B5, List Biological Laboratories, Campbell, CA, USA), LPS+MLN4924 (0.33μM, Millennium Pharmaceuticals, Boston, MA, USA), TNF-α (1ng/ml Medium), TNF-α+MLN4924 or vehicle (DMSO, Sigma, St. Louis, USA) for 0.5, 1, 2, 4 and 6 hours under normoxic conditions. Adenosine (Calbiochem) was used as a 30 minute pretreatment at concentrations from 100μM to 100nM prior to LPS stimulation.
Western Blotting
Cytosolic and nuclear proteins were isolated using the Nuclear and Cytoplasmic Protein extraction kit (NE-PER, ThermoFisher Scientific, Rockford, IL, USA. Protein concentration was determined by Lowry assay. Proteins were resolved under reducing conditions on SDS-PAGE gels followed by transfer onto polyvinylidene difluoride (PVDF) membranes (Millipore; Billerica, MA, USA). PVDF membranes were blocked and probed at 4°C overnight with the following primary antibodies: rabbit anti-SENP8 (1:1000, Abnova, Taiwan), murine anti-Cul-1 (1:500, Invitrogen, USA), rabbit anti-Nedd8, NF-κB p50, p60 (1:500, Cell Signaling, Danvers, MA, USA), murine HIF-1α (1:500, BD Transduction Labs, San Diego, CA, USA), rabbit OH-HIF-1α (1:500, Novus Biologicals, USA), murine anti-TATA-Box Protein (1:1000, Abcam, USA), rabbit anti-β-actin 1:10000, and subsequently with a 1:10,000 dilution of horseradish peroxidase-linked anti-rabbit or mouse IgG (MP Biomedicals, Solon, OH, USA). Antibody staining was detected using LumiGlo chemiluminescence detection system (KPL, Gaithersburg, MD, USA).
Immunoprecipitation
100μg of protein lysate was precipitated on Protein A μMACS protein beads (Miltenyi Biotec, Auburn, CA, USA) precoated with 2μl of rabbit anti-SENP8 (Abnova, Taiwan). Following incubation, protein beads were washed and protein eluted according to the manufacturers protocol. 40ug of protein were then used as input on a 10% SDS-PAGE gel and Western blot analysis using murine anti-SENP8 (1:500, Abcam USA) performed as described above.
Immunofluorescence
Control and SENP8-knockdown cells were seeded on collagen-coated coverslips in a 24 well dish format. After 24 hours of growth, cells were fixed and stained with an anti-SENP8 primary antibody (Anti-SENP8, 1:100, Abnova, Taiwan) overnight, washed and stained with secondary antibody (Alexa Flour 555, donkey anti-rabbit, 1:500, Invitrogen, USA). Cells were washed and counterstained with DAPI (1:25000, Invitrogen, USA). Images were recorded from coverslips using a Zeiss Axioimager A.1 microscope with an AxioCam MRc5 at 20x magnification (Zeiss EC-Plan/NEOFLUAR 20x/0.5) utilizing Zeiss Axiovision Software Version 4.6.3. SENP8 expression was quantified by assessing the fluorimetric ratio of SENP8-to-DAPI.
Luciferase assay of HIF and NF-κB promoter activity
The promotor constructs for pNF-kB-Luc and pHRE-Luc (34) were described previously. Transfection of HMEC-1 cells was performed using Fugene 6 (Roche Diagnostics, Indianapolis, IN, USA) in addition to 0.5 μg (HRE) or 1.5μg (NF-κB) DNA. Following overnight transfection, Luciferase activity was measured after LPS for 6 hours (for NF-κB activity) or 24 hours (for HRE response) using the luciferase reporter-assay (Promega, Madison, WI, USA). Observed firefly luciferase activity was normalized to the total protein amount of each sample determined by Lowry assay.
Endothelial permeability assay
FITC-dextrance flux over an endothelial monolayer was measured as previously described by Lennon et al (35).
Animal experiments
12 week old ΔODD mice (FVB-background) of both genders received intraperitoneal LPS doses of 100μg/kg bodyweight (BW). A respective control cohort received vehicle i.p. only. 6 hrs post treatment mice were sacrificed under Tribromoethanol anesthesia and blood samples were collected for measuring systemic cytokine release. All animal experiments have been reviewed by the Institutional Animal Care and Use Committee (IACUC) and are in compliance with the U.S. Department of Health and Human Services Guide for the Care and Use of Laboratory Animals.
Cytokine measurements in serum and HIF luciferase in kidney tissue
Pro-inflammatory cytokines in murine blood and organ samples were harvested following intraperitoneal 6h treatment with either 100μg/kg LPS, vehicle, 3mg/kg MLN4924 or LPS+MLN4924 (one hour MLN4924 pretreatment), snap frozen in liquid nitrogen and stored at −80°C until further analysis. Serum was collected by centrifugation and organ proteins isolated using a tissue homogenizer. Serum cytokines were measured using the murine inflammatory 7-Plex assay (Mesoscale, Gaithersburg, MD, USA). Luciferase activity in the kidney was measured using the luciferase reporter-assay (Promega, Madison, WI, USA).
Data analysis
All raw and calculated data are expressed as mean±standard error of the mean (SEM) of n observations, n being the number of biological replicates, and analyzed using Prism 5.0 (GraphPad San Diego, CA, USA). Changes in transcript and protein levels and changes in NF-κB and HIF promoter activity levels were compared using Student’s unpaired t-test or one-way ANOVA with Newman-Keuls post-hoc test where appropriate. P-values <0.05 were considered significant.
Results
Model to study Cullin neddylation
Neddylation of Cul-1, an E3 ligase critical for the negative regulation of NF-κB through ubiquitination of IκB (36, 37), has been strongly implicated in inflammation (21). To establish a model of inflammation-induced Cullin neddylation, we determined the ability of LPS to modulate endothelial Cul-1. As shown in Figure 1A, LPS increased endothelial Cul-1 neddylation (reflected as an increase in Nedd8-associated band shift upward in the gel) in a time dependent manner. Densitometry analysis of Nedd8:loading control ratios revealed a 1±0.07, 1.3±.54, 2.7±0.51, 2.1±0.52. 1.7±0.38 and 3±0.67-fold increase for 0, 0.5, 1, 2, 4 and 6 hr stimulation, respectively (n=3 experiments).
Figure 1. Influence of LPS and adenosine on Cul-1 neddylation status.
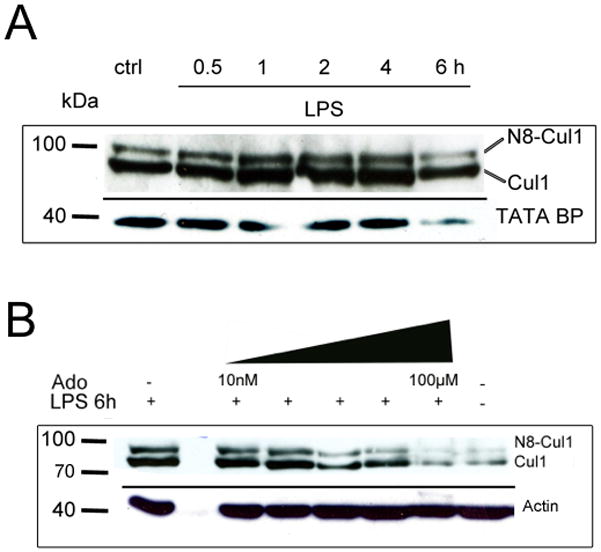
A) LPS-stimulation (10μg/ml medium) of human microvascular endothelial cells increases neddylation of Cul-1 protein in time dependent manner. B) Similar to previous findings in mice treated with hypoxic preconditioning, adenosine reduced the LPS induced Cul-1 neddylation in a dose dependent manner. Representative blots of 3 experiments per group.
Our previous work showed that extracellular adenosine promotes the deneddylation of Cul-1 (26). We therefore determined whether adenosine might influence LPS induced Cul-1 neddylation. As depicted in Figure 1B, the addition of extracellular adenosine (range 10nM-100μM) attenuated LPS-induced Cul-1 neddylation in a concentration-dependent manner with nearly complete loss of LPS induced Cul-1 neddylation at concentrations >1μM compared to untreated control. Densitometry analysis revealed a 41±3.3, 75±8.2, and 86±6.7% decrease relative to LPS alone at 10nM, 1μM and 100μM adenosine, respectively (n=3 experiments).
SENP8 as a critical regulator of Cullin neddylation
Having profiled endothelial LPS-induced Cul-1 neddylation, we turned our attention to defining molecular regulation. Our previous studies provided a role for the COP9 signalosome in the deneddylation of Cul-1 (26), but revealed little with regard to inflammation-associated neddylation of the Cullins. Moreover, our previous work implicated targets upstream of the COP9 signalosome in control of Cullin de-neddylation. This turned our attention to SENP8 and here we demonstrate a role for isopeptidase in LPS mediated Cul-1 neddylation. To demonstrate this point, we used lentiviral-mediated shRNA knockdown to generate stable endothelial cell lines with reduced SENP8 expression. Efficiency of gene silencing was determined by real-time PCR and indicated a >50% loss of transcript in cells transfected with clone 42 shRNA targeting SENP8 compared to non-target control (ctrl) and a less efficient clone 38 (Figure 2A). Analysis of SENP8 protein levels by a combination of immunoprecipitation/western blotting revealed an approximate 85% loss of SENP8 in clone 42 knockdown cells compared to control and clone 38 (Figure 2A, insert). Likewise, localization of SENP8 by immunofluorescence (Figure 2B) revealed perinuclear cytoplasmic distribution of SENP8 and a significant loss in SENP8 clone 42 cells (SENP8/DAPI ratio in empty vector controls 2.5±0.42 vs SENP8 knockdowns 1.3±0.07, p<0.03). The secondary Ab-only control showed no detectable staining for SENP8 (data not shown).
Figure 2. Generation and validation of lentiviral SENP8 knockdown in endothelia.
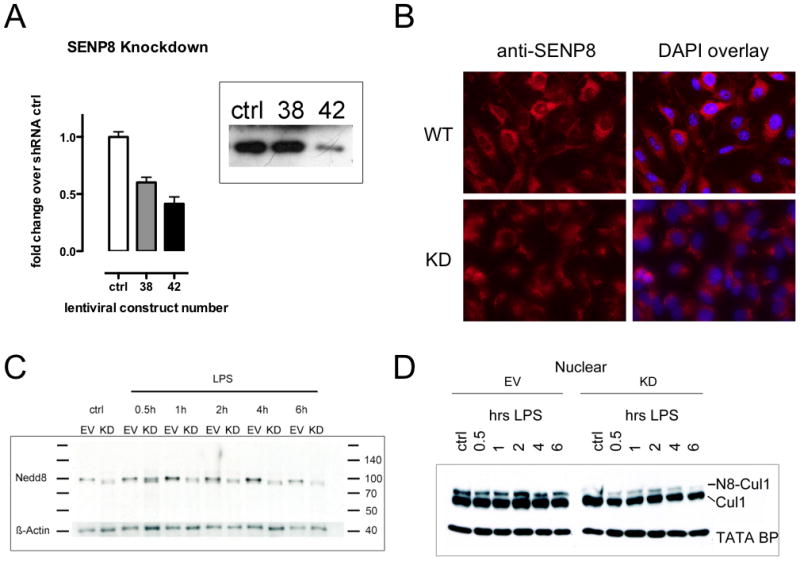
A Efficiency of lentiviral gene silencing on SENP8 transcript and protein levels was assessed by analysis of two different functional hairpin vectors (clones 38 and 42) compared to a scrambled vector (ctrl). Real-time PCR analysis showed different efficiency levels for both functional vectors, mirrored by immunoprecipitation/western blot analysis of SENP8. B Knockdown of SENP8 was further ascertained in the #42 clone by immunofluorescence, indicating loss of positive staining in the knockdown cell line. C Direct detection of Cullin-conjugated Nedd8 in HMEC SENP8-KD cells in response to LPS over the time course of 6 hours is indicated by western blot against NEDD8. Cells lacking functional SENP8 (KD) show a diminished neddylation response to LPS compared to empty vector (EV) transfected controls. D This impaired general neddylation response was mirrored by abrogated neddylation of Cul-1 in SENP8 knockdown cells. Representative blots of ≥3 experiments.
To further confirm altered neddylation status of Cullins, we assessed the conjugation of NEDD8 to Cullin by probing directly with anti-Nedd8 Ab. LPS increased the abundance of NEDD8 protein in EV cells but not in SENP8 knockdown cells (Fig 2C). Nedd8 detected in this manner (i.e. directly) likely reflects Nedd8 conjugation to several Cullin proteins (e.g. Cullin-1 and Cullin-2 which have different molecular masses), and thus, the diffuse nature of the bands on the blots shown in Figure 2C. These same cells, when treated with LPS in a similar fashion to control cells, showed a blunted neddylation response of nuclear Cul-1 protein, indicating that SENP8 is necessary for LPS induced Cul-1 neddylation (Fig 2D).
To define the influence of endothelial SENP8 on functional inflammatory responses, we evaluated the NF-κB response in control and SENP8 knockdown cells following LPS exposure. As shown in Figure 3A, LPS induced a profound translocation of the p50 and p65 subunit of NF-κB from the cytoplasm to the nucleus. Within 30min, control cells showed a robust increase in the nuclear fraction of p50 and p65, which persisted to time points beyond 4hr (Figure 3A). Both p50 and p65 responses were markedly reduced in cells lacking SENP8 (Figure 3A). In SENP8 knockdown cells, both the kinetics and the magnitude of the p50/p65 response, i.e. the nuclear translocation of said heterodimer, was attenuated compared to those observed in control cells, indicating a central role for SENP8 for the immediate NF-κB response following LPS exposure.
FIGURE 3. Functional influence of SENP8 on LPS-induced activation of NF-κB and HIF.
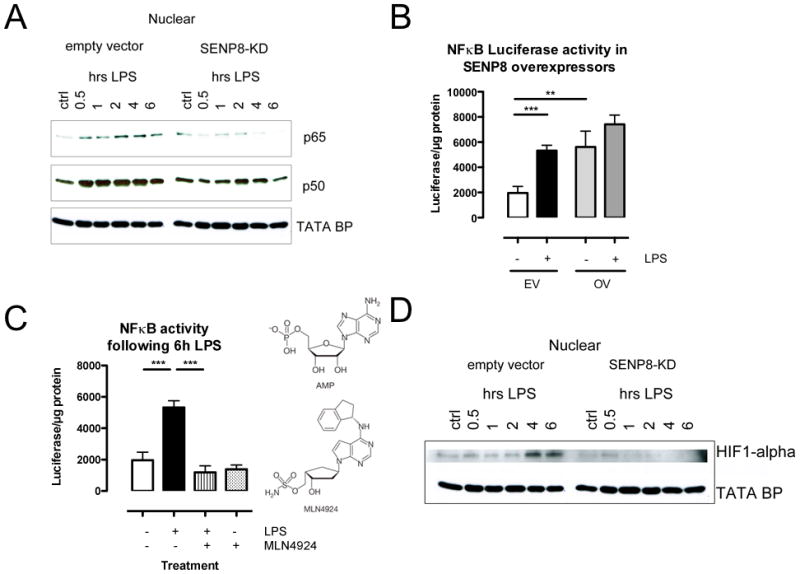
A Following LPS stimulation, control HMEC cells exhibit an increased translocation of the NFκB subunits p50 and p65 to the nucleus. This effect is abrogated in cells lacking SENP8. B Cells transfected with a constitutive SENP8 overexpressing (OE) vector showed baseline NFκB-luciferase levels comparable to LPS treated control cells transfected with empty vector (EV) only. C MLN4924 (structure homologue of adenosine-monophosphate, AMP) significantly quenched LPS induced NFκB-Luciferase response. D LPS induced HIF-1α stabilization in the nucleus of empty vector cells after 4 and 6 hours of stimulation. This effect is not observable in SENP8 knockdown cells. All numerical data are mean±SEM, n≥3 experiments, **p<0.01, ***p<0.001.
Conversely, overexpression of SENP8 in HMEC-1 using a SENP8 plasmid on a heterologous promoter significantly increased NF-κB responses. Indeed, as shown in Figure 3B, compared to empty plasmid transfection controls, overexpression of SENP8 increased basal NF-κB reporter plasmid activity by as much as 2.8±0.5-fold (p<0.01), approaching that of maximal LPS-induced NF-κB activity. LPS treatment of SENP8 overexpressing cells did not further increase NF-κB activity compared to empty plasmid controls, suggesting that a maximum level of NF-κB activity had been achieved with these conditions. Nearly identical results were observed in primary endothelial cell cultures (HUVEC), where over-expression of SENP8 increased baseline and LPS-induced TNF-α transcript (Supplemental Figure 1). Thus, both loss and gain of function studies strongly implicate SENP8 as a critical component to vascular endothelial NF-κB responses.
Influence of the neddylation inhibitor MLN4924 on NF-κB and HIF activity
Recently, a small molecule inhibitor (MLN4924) targeting Nedd8 conjugation to the Cullins became commercially available. Given our findings that adenosine (via metabolism from AMP) deneddylates Cul-1 (26), it is interesting that MLN4924 is a structural AMP analog (Figure 4C) that functions to inhibit Nedd8 activating enzyme (NAE) and results in the de-neddylation of Cul1 and Cul2 (30, 38). As shown in Figure 3C, the NF-κB activity elicited by LPS was quenched to nearly control levels in HMEC-1 cells that had been pretreated with MLN4924 (p<0.01). Similar results were obtained in cultured HUVECs, where MLN4924 abrogated LPS-induced Cul-1 neddylation and LPS-induced TNF-α transcript (Supplemental Figure 1).
FIGURE 4. Influence of neddylation inhibition on HIF stabilization, HIF hydroxylation, Cul-1 neddylation and HIF activity.
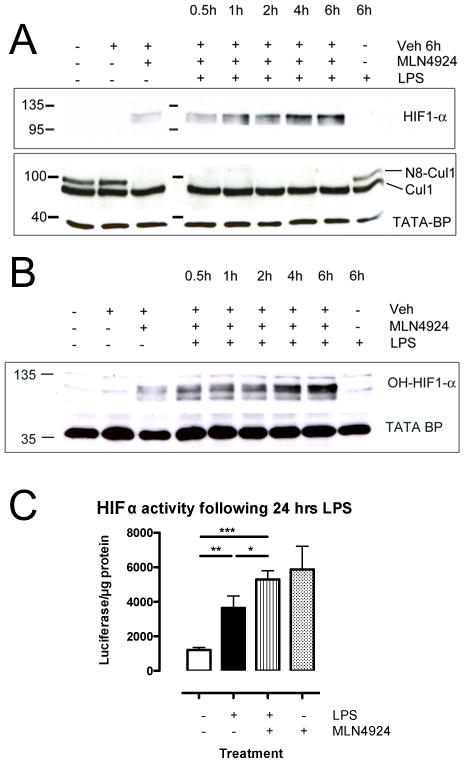
A Pretreatment with MLN4924 increases HIF-1α protein in nuclear lysates of HMEC cells stimulated with LPS to higher levels than MLN4924 alone, indicative of synergistic effects of both compounds. LPS induced Cullin-1 neddylation was lost when cells were pretreated with MLN4924. B Treatment with MLN4924 stabilized HIF-1α in its hydroxylated form and allowed for its translocation to the nucleus, implicating effects of MLN4924 on Cullin-2 neddylation. C Luciferase-activity of HRE-Luciferase following 24hr of LPS stimulation mirrored protein results. All numerical data are mean±SEM, n≥3 experiments. *p<0.05, **p<0.01, ***p<0.001.
Given the importance of Cul-2 neddylation for pVHL activity and thus in the degradation of HIF protein (39), we next addressed the impact of MLN4924 on HIF function. As shown in Figure 4, in stark contrast to the inhibition of NFκB (Figure 3D), MLN4924 stabilized HMEC-1 HIF-1α protein (Figure 4A), and more specifically the hydroxylated isoform of HIF-1α (Figure 4B). This observation was significantly enhanced by the presence of LPS, which is consistent with previous studies indicating that LPS stabilizes HIF activity (40, 41). Moreover, HIF activity (as measured using an hypoxia-response element reporter plasmid) was significantly increased by MLN4924 (p<0.05). These findings indicate that the status of Cul-2 neddylation strongly influences HIF activity.
We next addressed whether Cullin neddylation in vascular inflammatory responses was specific for LPS. For these purposes, we selected TNF-α-elicited cytokine/chemokine induction in the presence and absence of MLN4924 as primary endpoints. As shown in Figure 5, TNF-α potently induced HMEC-1 IL-1β, IL-6 and IL-8 mRNA and protein (all p<0.05 or p<0.01). In each case, the neddylation inhibitor MLN4924 significantly inhibited mRNA induction (p<0.01 or p<0.05) as well as IL-1β and IL-6 protein induction (p<0.01). Moreover, MLN4924 significantly inhibited TNF-α-elicited IFNγ protein (83±8% inhibition, p<0.05) and IL-12p70 protein (76±5% inhibition, p<0.01). Furthermore, MLN4924 inhibited TNF-α-mediated induction of ICAM-1 transcript (Figure 5D, 79±9% decrease p<0.05) and completely abrogated barrier dysfunction elicited by LPS (measured by FITC-dextran flux, see Figure 5E). Such findings place Cullin neddylation as a central regulator of the vascular inflammatory response and clearly implicate Cullin deneddylation as a target to inhibit this phenotype.
FIGURE 5. Influence of neddylation inhibition on transcription and translation of inflammatory cytokines and endothelial function.

6 hours TNFα treatment of endothelial cells results in the significant upregulation of proinflammatory cytokine transcript and protein, which is absent when cells were pretreated with MLN4924 (A–C). MLN4924 also prevents transcriptional upregulation of ICAM-1 (D) and LPS induced increases of endothelial permeability (E). All numerical data are mean±SEM, n≥3 experiments, *p<0.05, **p<0.01.
Cullin neddylation and LPS-induced inflammation in vivo
Reagents to define specific aspects of Nedd8/Cullin conjugation pathways in vivo have been limiting due to the embryonic lethality of the gene-targeted mouse lines (42). Thus, we elected to employ MLN4924, which is well tolerated for use in murine tumor models (30). Initially, we profiled whether MLN4924 influenced HIF expression in vivo. For these purposes, we used the HIF reporter ΔODD-Luc mouse model in combination with LPS (100μg/kg bodyweight) exposure in the presence and absence of MLN4924 pretreatment. As shown in Figure 6A, the combination of LPS and MLN4924 synergistically increased HIF stabilization (reflected as luciferase activity) in kidneys from ΔODD-Luc mice (p<0.01). Further analysis of serum cytokines in these mice revealed prominent increases in pro-inflammatory cytokines and chemokines compared to vehicle treated mice (Figure 6B–H). LPS also induced the anti-inflammatory cytokine IL-10 (Figure 6E). Administration of MLN4924 30min prior to LPS significantly attenuated the induction of each of the pro-inflammatory cytokines, including IL-1β, IL-6,, TNF-α, IL-12p70 and IFNγ (Figure 6B–H). Interestingly, mice that received MLN4924 and LPS showed a further, albeit not statistically significant, increase in the anti-inflammatory cytokine IL-10 (Figure 6F). Taken together, these in vivo observations reveal a potent anti-inflammatory role for MLN4924.
Figure 6. Impact of MLN4924 on LPS-mediated HIF activity and cytokine production in vivo.

ΔODD mice received vehicle, MLN4924 (3mg/kg BW), LPS (100μg/kg BW) or MLN4924+LPS i.p. for 6hrs. Inflammatory cytokines were measured in serum and HIF-luciferase was measured in renal lysates. A LPS+MLN4924 induced a significant increase of renal HIF cytokines. B–E, G–H Mice pretreated with the neddylation inhibitor MLN4924 prior to LPS showed no significant upregulation of all pro-inflammatory serum cytokines, while maintaining increased levels of the anti-inflammatory cytokine IL-10 (F). All values are mean±SEM with ≥4 animals per group, *p<0.05, **p<0.01, ***p<0.001.
Discussion
In productive inflammatory responses, a number of transcription factors, including NF-κB and HIF, are tightly regulated by posttranslational modifications that control the kinetics of expression via proteasomal degradation pathways (reviewed in (43)). The polyubiquitination of components within these pathways (e.g. IκB and HIF) is mediated through a multi-protein E3 ligase complex, for which one of these components is a member of the Cullin family of proteins (44). E3 ligase activity is controlled through its neddylation status, i.e. the conjugation of a NEDD8 moiety to the Cullin subunit (45). Regulation of Cullin-neddylation is achieved through the COP9 signalosome and/or the human deneddylase SENP8 (27, 29). Work by Mendoza and colleagues identified the isopeptidase SENP8 to primarily deneddylate Cullins, thereby inactivating the E3 ligase (17).
Limited information is available regarding the direct role of Cullin regulation in inflammation. Amir et al., demonstrated that lack of functional E2 or E3 ligases results in reduced breakdown of the NF-κB precursor p105, thereby limiting the induced inflammatory response (21). Their work, however, focused on the E1 and E2 ligases of the neddylation pathway, whereas the present studies address the function of upstream targets of said ligases. To address the role of Cullin-neddylation in inflammation, we initially established a model using HMEC and LPS to evaluate the biochemical features of Cul-1 neddylation. This model demonstrated prominent Cul-1 neddylation that was inhibited by adenosine, which we have previously shown to potently deneddylate the Cullins (26).
Based on these observations and our previous work suggesting that targets upstream of the COP9 signalosome should control Cullin neddylation (26), we targeted SENP8 as a central regulator for the pro-inflammatory phenotype. Loss and gain of function studies strongly implicated SENP8 as a central regulator of the inflammatory response. Indeed, lentiviral-mediated shRNA knockdown of SENP8 resulted in a loss of the neddylated Cul-1 that was reversed by constitutive overexpression of SENP8 (33). These observations are likely due to the dual protease actions of SENP8. For instance, Wu et al. have described SENP8 as capable of not only removing NEDD8 from target proteins, but also cleaving the precursor of NEDD8 to its mature form (27). Immature NEDD8 is incapable of coupling to the E1 and E2 ligases, thus making it unavailable for conjugation to the E3 ligase. Thus, our observations in the SENP8 knockdown line are likely due to a lack of mature NEDD8 available for conjugation. The molecular mechanism of Cullin-Ring-Ligase activity relies on the conjugation of mature NEDD8 to the CRL.
We have shown in the past that adenosine actively deneddylates Cul1 and results in an inhibition of NF-κB signaling (26). Likewise, we have recently demonstrated that adrenomedullin deneddylates Cul-2 (46), a critical component of the HIFα E3 SCF ubiquitin ligase (47). Cul-2 neddylation should result in diminished pVHL activity and subsequently an accumulation of HIF-1α protein, an effect we did not observe with SENP8 knockdown. It is therefore likely that permanent loss of SENP8 triggers adaptive pathways for HIF-1α control independent of pVHL. By contrast, MLN4924 increased HIF-1α levels and HIF activity, suggesting that short-term inhibition of neddylation influences both Cul-1 and Cul-2 activity. This was also indicated by our in vivo data revealing that HIF levels increase in the kidney in response to LPS and that MLN4924 synergistically increases this response. Important in this regard, HIF stabilization has been proven beneficial in a number of murine disease models (12), including the kidney (48). The underlying mechanisms for HIF mediated renoprotection remain unresolved but HIF-mediated induction of the ectonucleotidase CD73 (49), a transmembrane protein critical for the generation of extracellular adenosine, has been strongly implicated. For example, Grenz and colleagues showed that renal CD73 is upregulated by ischemic preconditioning and protective against ischemic injury (50). Additionally, work by Song et al. demonstrated protective influences of pharmacological HIF induction in the kidney using DMOG, a known HIF stabilizer (51). Likewise, constitutive activation of HIF dampens glomerulonephritis progression, providing further evidence for beneficial influences of HIF for renal injury (52). Our results extend these findings to a murine model of septic renal injury, emphasizing the importance of renal HIF stabilization during kidney injury and hinting at potential beneficial influences of MLN4924 in various nephropathies.
Pro-inflammatory cytokine responses were quenched in cells and animals treated with the AMP analog MLN4924. Upregulation of circulating cytokines is one of the hallmarks of sepsis and also serves as a surrogate predictor of disease outcome. For instance, significant evidence suggests that IL-6 serum levels greater than 2000 pg/ml are associated with higher mortality in mice suffering from polymicrobial sepsis (53). These results from a murine model are supported by data from human neonates. Cernada and colleagues showed that cord blood from septic neonates displayed higher IL-6 levels and that IL-6 served as a good predictor for early onset neonatal sepsis (54). Our current in vitro following LPS stimulation and in vivo data from experimental endotoxemia, a model of gram-negative sepsis, show significantly attenuated pro-inflammatory cytokine levels, including IL-6, with MLN4924 treatment, indicating some degree of therapeutic potential. The anti-inflammatory properties of MLN4924 are further exemplified by the observation that LPS-induced IL-10 is not influenced by MLN4924. At present, differences in the mechanism(s) of cytokine-specific regulation are not known.
We conclude that: a) SENP8 is necessary for inflammatory activation of NF-κB, b) inflammation induced stabilization of HIF is in part relying on functional SENP8, and c) that pharmacological inhibition of SENP8 using the structural AMP analog MLN4924 might serve as a suitable therapeutic target for modulating inflammatory diseases.
Supplementary Material
Figure 7. Neddylation pathways influencing NF-κB and HIFα.

NFκB pathway (left). Proinflammatory stimuli, such as LPS, facilitate the phosphorylation of IκB and result in the recognition of P-IκB by the Cullin-1-Nedd8-Skp-βTRCP complex, culminating in its polyubiquitination and proteasomal degradation. The rate-limiting step for this is the conjugation of NEDD8 to Cullin-1. Neddylation is achieved through a multi-enzyme process, wherein SENP8 enables cleavage of the NEDD8-precursor and promotes NEDD8 conjugation to the Cullins. Loss of SENP8, or pharmacological inhibition of NEDD8 conjugation by MLN4924, prevents activation of Cullin-1 and thus prevents liberation of NFκB from IκB, resulting in quenched pro-inflammatory signaling. HIFα pathway (right). In contrast to NFκB, HIFα in its hydroxylated form is degraded by the proteasome after ubiquitination through the von Hippel Lindau protein (pVHL). pVHL in its activated form contains neddylated Cullin-2, thereby controlling cellular HIFα levels. Use of MLN4924 prevents Cullin-2 neddylation, and as shown in this study leads to higher levels of the hydroxylated HIFα isoform.
Acknowledgments
Grant support: Funding from the Deutsche Forschungsgemeinschaft DFG (EH 371/1-1), National Institute of Health (DK50189, HL60569, DK095491) and the Crohn’s and Colitis Foundation of America.
Footnotes
Authorship Contributiona: Ehrentraut SF, Kominsky DJ, Glover LE, Campbell EL, Kelly CJ, Bowers BE, Bayless AJ performed research, Ehrentraut SF and Colgan SP analyzed data and wrote the manuscript, Ehrentraut SF and Colgan SP designed research
Conflict of Interest Disclosures: The authors declare no conflicts of interest.
References
- 1.Angus DC, Linde-Zwirble WT, Lidicker J, Clermont G, Carcillo J, Pinsky MR. Epidemiology of severe sepsis in the United States: analysis of incidence, outcome, and associated costs of care. Crit Care Med. 2001;29:1303–1310. doi: 10.1097/00003246-200107000-00002. [DOI] [PubMed] [Google Scholar]
- 2.Martin GS, Mannino DM, Eaton S, Moss M. The epidemiology of sepsis in the United States from 1979 through 2000. N Engl J Med. 2003;348:1546–1554. doi: 10.1056/NEJMoa022139. [DOI] [PubMed] [Google Scholar]
- 3.Paulus P, Jennewein C, Zacharowski K. Biomarkers of endothelial dysfunction: can they help us deciphering systemic inflammation and sepsis? Biomarkers. 2011;16(Suppl 1):S11–21. doi: 10.3109/1354750X.2011.587893. [DOI] [PubMed] [Google Scholar]
- 4.Shapiro NI, Schuetz P, Yano K, Sorasaki M, Parikh SM, Jones AE, Trzeciak S, Ngo L, Aird WC. The association of endothelial cell signaling, severity of illness, and organ dysfunction in sepsis. Crit Care. 2010;14:R182. doi: 10.1186/cc9290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baker RG, Hayden MS, Ghosh S. NF-kappaB, inflammation, and metabolic disease. Cell Metab. 2011;13:11–22. doi: 10.1016/j.cmet.2010.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hayden MS, Ghosh S. Shared principles in NF-kappaB signaling. Cell. 2008;132:344–362. doi: 10.1016/j.cell.2008.01.020. [DOI] [PubMed] [Google Scholar]
- 7.Barnes PJ, Karin M. Nuclear factor-kappaB: a pivotal transcription factor in chronic inflammatory diseases. N Engl J Med. 1997;336:1066–1071. doi: 10.1056/NEJM199704103361506. [DOI] [PubMed] [Google Scholar]
- 8.Walmsley SR, Print C, Farahi N, Peyssonnaux C, Johnson RS, Cramer T, Sobolewski A, Condliffe AM, Cowburn AS, Johnson N, Chilvers ER. Hypoxia-induced neutrophil survival is mediated by HIF-1alpha-dependent NF-kappaB activity. J Exp Med. 2005;201:105–115. doi: 10.1084/jem.20040624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Belaiba RS, Bonello S, Zahringer C, Schmidt S, Hess J, Kietzmann T, Gorlach A. Hypoxia up-regulates hypoxia-inducible factor-1alpha transcription by involving phosphatidylinositol 3-kinase and nuclear factor kappaB in pulmonary artery smooth muscle cells. Mol Biol Cell. 2007;18:4691–4697. doi: 10.1091/mbc.E07-04-0391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hacker H, Karin M. Regulation and function of IKK and IKK-related kinases. Sci STKE. 2006;2006:re13. doi: 10.1126/stke.3572006re13. [DOI] [PubMed] [Google Scholar]
- 11.Semenza GL. Oxygen sensing, homeostasis, and disease. N Engl J Med. 2011;365:537–547. doi: 10.1056/NEJMra1011165. [DOI] [PubMed] [Google Scholar]
- 12.Colgan SP, Taylor CT. Hypoxia: an alarm signal during intestinal inflammation. Nat Rev Gastroenterol Hepatol. 2010;7:281–287. doi: 10.1038/nrgastro.2010.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Grenz A, Clambey E, Eltzschig HK. Hypoxia signaling during intestinal ischemia and inflammation. Current opinion in critical care. 2012;18:178–185. doi: 10.1097/MCC.0b013e3283514bd0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kumar S, Yoshida Y, Noda M. Cloning of a cDNA which encodes a novel ubiquitin-like protein. Biochem Biophys Res Commun. 1993;195:393–399. doi: 10.1006/bbrc.1993.2056. [DOI] [PubMed] [Google Scholar]
- 15.Wada H, Kito K, Caskey LS, Yeh ET, Kamitani T. Cleavage of the C-terminus of NEDD8 by UCH-L3. Biochem Biophys Res Commun. 1998;251:688–692. doi: 10.1006/bbrc.1998.9532. [DOI] [PubMed] [Google Scholar]
- 16.Huang DT, Miller DW, Mathew R, Cassell R, Holton JM, Roussel MF, Schulman BA. A unique E1-E2 interaction required for optimal conjugation of the ubiquitin-like protein NEDD8. Nat Struct Mol Biol. 2004;11:927–935. doi: 10.1038/nsmb826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mendoza HM, Shen LN, Botting C, Lewis A, Chen J, Ink B, Hay RT. NEDP1, a highly conserved cysteine protease that deNEDDylates Cullins. J Biol Chem. 2003;278:25637–25643. doi: 10.1074/jbc.M212948200. [DOI] [PubMed] [Google Scholar]
- 18.Liakopoulos D, Doenges G, Matuschewski K, Jentsch S. A novel protein modification pathway related to the ubiquitin system. EMBO J. 1998;17:2208–2214. doi: 10.1093/emboj/17.8.2208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jones J, Wu K, Yang Y, Guerrero C, Nillegoda N, Pan ZQ, Huang L. A targeted proteomic analysis of the ubiquitin-like modifier nedd8 and associated proteins. J Proteome Res. 2008;7:1274–1287. doi: 10.1021/pr700749v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Parry G, Estelle M. Regulation of cullin-based ubiquitin ligases by the Nedd8/RUB ubiquitin-like proteins. Semin Cell Dev Biol. 2004;15:221–229. doi: 10.1016/j.semcdb.2003.12.003. [DOI] [PubMed] [Google Scholar]
- 21.Amir RE, Iwai K, Ciechanover A. The NEDD8 pathway is essential for SCF(beta -TrCP)-mediated ubiquitination and processing of the NF-kappa B precursor p105. J Biol Chem. 2002;277:23253–23259. doi: 10.1074/jbc.M200967200. [DOI] [PubMed] [Google Scholar]
- 22.Semenza GL. Oxygen homeostasis. Wiley Interdiscip Rev Syst Biol Med. 2010;2:336–361. doi: 10.1002/wsbm.69. [DOI] [PubMed] [Google Scholar]
- 23.Sufan RI, Ohh M. Role of the NEDD8 modification of Cul2 in the sequential activation of ECV complex. Neoplasia. 2006;8:956–963. doi: 10.1593/neo.06520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Giatromanolaki A, Sivridis E, Maltezos E, Papazoglou D, Simopoulos C, Gatter KC, Harris AL, Koukourakis MI. Hypoxia inducible factor 1alpha and 2alpha overexpression in inflammatory bowel disease. J Clin Pathol. 2003;56:209–213. doi: 10.1136/jcp.56.3.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mariani F, Sena P, Marzona L, Riccio M, Fano R, Manni P, Gregorio CD, Pezzi A, Leon MP, Monni S, Pol AD, Roncucci L. Cyclooxygenase-2 and Hypoxia-Inducible Factor-1alpha protein expression is related to inflammation, and up-regulated since the early steps of colorectal carcinogenesis. Cancer Lett. 2009;279:221–229. doi: 10.1016/j.canlet.2009.02.001. [DOI] [PubMed] [Google Scholar]
- 26.Khoury J, Ibla JC, Neish AS, Colgan SP. Antiinflammatory adaptation to hypoxia through adenosine-mediated cullin-1 deneddylation. J Clin Invest. 2007;117:703–711. doi: 10.1172/JCI30049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wu K, Yamoah K, Dolios G, Gan-Erdene T, Tan P, Chen A, Lee CG, Wei N, Wilkinson KD, Wang R, Pan ZQ. DEN1 is a dual function protease capable of processing the C terminus of Nedd8 and deconjugating hyper-neddylated CUL1. J Biol Chem. 2003;278:28882–28891. doi: 10.1074/jbc.M302888200. [DOI] [PubMed] [Google Scholar]
- 28.Cope GA, Deshaies RJ. COP9 signalosome: a multifunctional regulator of SCF and other cullin-based ubiquitin ligases. Cell. 2003;114:663–671. doi: 10.1016/s0092-8674(03)00722-0. [DOI] [PubMed] [Google Scholar]
- 29.Lyapina S, Cope G, Shevchenko A, Serino G, Tsuge T, Zhou C, Wolf DA, Wei N, Deshaies RJ. Promotion of NEDD-CUL1 conjugate cleavage by COP9 signalosome. Science. 2001;292:1382–1385. doi: 10.1126/science.1059780. [DOI] [PubMed] [Google Scholar]
- 30.Soucy TA, Smith PG, Milhollen MA, Berger AJ, Gavin JM, Adhikari S, Brownell JE, Burke KE, Cardin DP, Critchley S, Cullis CA, Doucette A, Garnsey JJ, Gaulin JL, Gershman RE, Lublinsky AR, McDonald A, Mizutani H, Narayanan U, Olhava EJ, Peluso S, Rezaei M, Sintchak MD, Talreja T, Thomas MP, Traore T, Vyskocil S, Weatherhead GS, Yu J, Zhang J, Dick LR, Claiborne CF, Rolfe M, Bolen JB, Langston SP. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature. 2009;458:732–736. doi: 10.1038/nature07884. [DOI] [PubMed] [Google Scholar]
- 31.Milhollen MA, Traore T, Adams-Duffy J, Thomas MP, Berger AJ, Dang L, Dick LR, Garnsey JJ, Koenig E, Langston SP, Manfredi M, Narayanan U, Rolfe M, Staudt LM, Soucy TA, Yu J, Zhang J, Bolen JB, Smith PG. MLN4924, a NEDD8-activating enzyme inhibitor, is active in diffuse large B-cell lymphoma models: rationale for treatment of NF-{kappa}B-dependent lymphoma. Blood. 2010;116:1515–1523. doi: 10.1182/blood-2010-03-272567. [DOI] [PubMed] [Google Scholar]
- 32.Ades EW, Candal FJ, Swerlick RA, George VG, Summers S, Bosse DC, Lawley TJ. HMEC-1: establishment of an immortalized human microvascular endothelial cell line. J Invest Dermatol. 1992;99:683–690. doi: 10.1111/1523-1747.ep12613748. [DOI] [PubMed] [Google Scholar]
- 33.Gao F, Cheng J, Shi T, Yeh ET. Neddylation of a breast cancer-associated protein recruits a class III histone deacetylase that represses NFkappaB-dependent transcription. Nat Cell Biol. 2006;8:1171–1177. doi: 10.1038/ncb1483. [DOI] [PubMed] [Google Scholar]
- 34.Sheta EA, Trout H, Gildea JJ, Harding MA, Theodorescu D. Cell density mediated pericellular hypoxia leads to induction of HIF-1alpha via nitric oxide and Ras/MAP kinase mediated signaling pathways. Oncogene. 2001;20:7624–7634. doi: 10.1038/sj.onc.1204972. [DOI] [PubMed] [Google Scholar]
- 35.Lennon PF, Taylor CT, Stahl GL, Colgan SP. Neutrophil-derived 5′-adenosine monophosphate promotes endothelial barrier function via CD73-mediated conversion to adenosine and endothelial A2B receptor activation. J Exp Med. 1998;188:1433–1443. doi: 10.1084/jem.188.8.1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kumar A, Wu H, Collier-Hyams LS, Kwon YM, Hanson JM, Neish AS. The bacterial fermentation product butyrate influences epithelial signaling via reactive oxygen species-mediated changes in cullin-1 neddylation. J Immunol. 2009;182:538–546. doi: 10.4049/jimmunol.182.1.538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kumar A, Wu H, Collier-Hyams LS, Hansen JM, Li T, Yamoah K, Pan ZQ, Jones DP, Neish AS. Commensal bacteria modulate cullin-dependent signaling via generation of reactive oxygen species. EMBO J. 2007;26:4457–4466. doi: 10.1038/sj.emboj.7601867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Boh BK, Smith PG, Hagen T. Neddylation-Induced Conformational Control Regulates Cullin RING Ligase Activity In Vivo. J Mol Biol. 2011;409:136–145. doi: 10.1016/j.jmb.2011.03.023. Epub 2011 Apr 2012. [DOI] [PubMed] [Google Scholar]
- 39.Liakopoulos D, Busgen T, Brychzy A, Jentsch S, Pause A. Conjugation of the ubiquitin-like protein NEDD8 to cullin-2 is linked to von Hippel-Lindau tumor suppressor function. Proc Natl Acad Sci U S A. 1999;96:5510–5515. doi: 10.1073/pnas.96.10.5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brooks AC, Menzies-Gow N, Bailey SR, Cunningham FM, Elliott J. Endotoxin-induced HIF-1alpha stabilisation in equine endothelial cells: synergistic action with hypoxia. Inflamm Res. 2010;59:689–698. doi: 10.1007/s00011-010-0180-x. [DOI] [PubMed] [Google Scholar]
- 41.Kuschel A, Simon P, Tug S. Functional regulation of HIF-1alpha under normoxia - is there more than posttranslational regulation? J Cell Physiol. 2011 doi: 10.1002/jcp.22798. [DOI] [PubMed] [Google Scholar]
- 42.Tateishi K, Omata M, Tanaka K, Chiba T. The NEDD8 system is essential for cell cycle progression and morphogenetic pathway in mice. J Cell Biol. 2001;155:571–579. doi: 10.1083/jcb.200104035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ehrentraut SF, Colgan SP. Implications of protein posttranslational modifications in IBD. Inflammatory bowel diseases. 2012 doi: 10.1002/ibd.22859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Guardavaccaro D, Pagano M. Oncogenic aberrations of cullin-dependent ubiquitin ligases. Oncogene. 2004;23:2037–2049. doi: 10.1038/sj.onc.1207413. [DOI] [PubMed] [Google Scholar]
- 45.Merlet J, Burger J, Gomes JE, Pintard L. Regulation of cullin-RING E3 ubiquitin-ligases by neddylation and dimerization. Cell Mol Life Sci. 2009;66:1924–1938. doi: 10.1007/s00018-009-8712-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.MacManus CF, Campbell EL, Keely S, Burgess A, Kominsky DJ, Colgan SP. Anti-inflammatory actions of adrenomedullin through fine tuning of HIF stabilization. FASEB J. 2011;25:1856–1864. doi: 10.1096/fj.10-170316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mikus P, Zundel W. COPing with hypoxia. Semin Cell Dev Biol. 2005;16:462–473. doi: 10.1016/j.semcdb.2005.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sitkovsky M, Lukashev D. Regulation of immune cells by local-tissue oxygen tension: HIF1 alpha and adenosine receptors. Nature reviews Immunology. 2005;5:712–721. doi: 10.1038/nri1685. [DOI] [PubMed] [Google Scholar]
- 49.Synnestvedt K, Furuta GT, Comerford KM, Louis N, Karhausen J, Eltzschig HK, Hansen KR, Thompson LF, Colgan SP. Ecto-5′-nucleotidase (CD73) regulation by hypoxia-inducible factor-1 mediates permeability changes in intestinal epithelia. J Clin Invest. 2002;110:993–1002. doi: 10.1172/JCI15337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Grenz A, Zhang H, Eckle T, Mittelbronn M, Wehrmann M, Kohle C, Kloor D, Thompson LF, Osswald H, Eltzschig HK. Protective role of ecto-5′-nucleotidase (CD73) in renal ischemia. J Am Soc Nephrol. 2007;18:833–845. doi: 10.1681/ASN.2006101141. [DOI] [PubMed] [Google Scholar]
- 51.Song YR, You SJ, Lee YM, Chin HJ, Chae DW, Oh YK, Joo KW, Han JS, Na KY. Activation of hypoxia-inducible factor attenuates renal injury in rat remnant kidney. Nephrology, dialysis, transplantation : official publication of the European Dialysis and Transplant Association - European Renal Association. 2010;25:77–85. doi: 10.1093/ndt/gfp454. [DOI] [PubMed] [Google Scholar]
- 52.Theilig F, Enke AK, Scolari B, Polzin D, Bachmann S, Koesters R. Tubular deficiency of von Hippel-Lindau attenuates renal disease progression in anti-GBM glomerulonephritis. The American journal of pathology. 2011;179:2177–2188. doi: 10.1016/j.ajpath.2011.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Remick DG, Bolgos GR, Siddiqui J, Shin J, Nemzek JA. Six at six: interleukin-6 measured 6 h after the initiation of sepsis predicts mortality over 3 days. Shock. 2002;17:463–467. doi: 10.1097/00024382-200206000-00004. [DOI] [PubMed] [Google Scholar]
- 54.Cernada M, Badia N, Modesto V, Alonso R, Mejias A, Golombek S, Vento M. Cord blood interleukin-6 as a predictor of early-onset neonatal sepsis. Acta Paediatr. 2011 doi: 10.1111/j.1651-2227.2011.02577.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.