

Abstract
Herpes Simplex Virus-1 (HSV-1) infects the majority of the world’s population. These infections are often asymptomatic, but ocular HSV-1 infections cause multiple pathologies with perhaps the most destructive being Herpes Stromal Keratitis (HSK). HSK lesions, which are immunoinflammatory in nature, can recur throughout life and often cause progressive corneal scaring resulting in visual impairment. Current treatment involves broad local immunosuppression with topical steroids along with antiviral coverage. Unfortunately, the immunopathologic mechanisms defined in animal models of HSK have not yet translated into improved therapy. Herein, we review the clinical epidemiology and pathology of the disease and summarize the large amount of basic research regarding the immunopathology of HSK. We examine the role of the innate and adaptive immune system in the clearance of virus and the destruction of the normal corneal architecture that is typical of HSK. Our goal is to define current knowledge of the pathogenic mechanisms and recurrent nature of HSK and identify areas that require further study.
1 Herpetic Infection
1.1 Types of herpetic disease
Herpes simplex virus (HSV) is a prevalent viral pathogen infecting the majority the of world’s population. While oral and genital lesions are the most common manifestations of infection, HSV type 1 (HSV-1) can also cause disease in all of the major ocular tissues, including the lids, conjunctiva, cornea, uveal tract, and retina. HSV-1 infection can be categorized into primary and recurrent disease. Following initial primary infection of the oral facial region including the cornea, the virus travels to the innervating trigeminal ganglia (TG) where it establishes a state of latency in which viral DNA is maintained within neuronal nuclei but no infectious virus particles are produced. Thereafter, the virus may undergo cycles of reactivation causing recurrent viral or immune pathology at the initial site of infection.
1.2 Epidemiology
Recent studies show an HSV-1 seroprevalence of >50%, >75% and >90% in the general adult populations of the United States, Germany and Tanzania respectively (Rabenau et al., 2002; Xu et al., 2006). Thus, the majority of the world’s population has been infected with HSV-1 and likely carries a latent viral load. Clinical manifestations of primary ocular HSV-1 infections tend to manifest in youths or young adults. Long term studies of ocular HSV-1 in the United States and Britain found the mean age for the first occurrence to be 37.4 years of age and 25 years of age respectively (Darougar et al., 1978; Liesegang, 1988, 1989). The annual incidence of all types of new ocular HSV infections has recently been estimated at 11.8 per 100,000 people in the United States (Young et al., 2010), similar to the incidence of 8.4 per 100,000 people found over two decades ago in a similar population (Liesegang et al., 1989). In France, the incidence of herpetic keratitis was estimated at 31.5 per 100,000 person-years, with 13.2 per 100,000 being new cases and 18.3 per 100,000 being recurrences (Labetoulle et al., 2005). Epithelial dendritic lesions represented the most frequent type of recurrent keratitis (56.3%), followed by herpes stromal keratitis (29.5%), and geographic epithelial lesions (9.8%) (Labetoulle et al., 2005). In the Herpetic Eye Disease Study, herpes stromal keratitis (HSK) represented 44% of recurrences with 18% of patients diagnosed with HSV-1 ocular disease experiencing a recurrence involving the corneal stroma (1998; Liesegang, 2001; Mikloska et al., 2001). Furthermore, previous bouts of HSK significantly increased the risk of future recurrences. Therefore, HSK represents a significant burden of ocular disease caused by HSV-1 infection.
1.3 Clinical symptoms
Clinical manifestations of primary ocular HSV-1 infections are rare and usually occur early in life. They typically present as conjunctivitis that can involve inflammation of the eyelids (blepharoconjunctivitis), marked by inflammatory vesicles and ulcers, and can include dendritic lesions in the corneal epithelium (Darougar et al., 1985). More often HSV-1 ocular infections result from reactivation of virus that originally established a latent infection in the TG following a non-ocular route of infection. Reactivation of latent virus in the ophthalmic branch of the TG can result in shedding at the corneal surface (Shimeld et al., 1990a, b). HSV-1 corneal lesions can either be restricted to the corneal epithelium, a disease referred to as infectious epithelial keratitis (Darougar et al., 1985), or have stromal involvement with or without damage to the overlying epithelium, known as HSK.
Patients with epithelial keratitis may present with pain, photophobia, blurred vision, tearing, and redness (Jones, 1958). Epithelial lesions are caused by the virus replicating in and destroying epithelial cells (Liesegang, 1999). The lesions start as punctuate vesicular eruptions in the corneal epithelium, but quickly coalesce into dendritic shaped lesions (Chang and Dreyer, 1996; Green and Pavan-Langston, 2006). The shape of the epithelial lesion is visualized by staining the basement membrane with fluorescein dye or staining the damaged cells at the outer limits of the lesion with rose bengal dye (Chodosh et al., 1992; Spencer and Hayes, 1970). The lesions can progress to enlarged non-linear lesions referred to as geographic lesions. These lesions can be distinguished from the neurotrophic keratopathy that can develop in patients with recurrent herpes infectious epithelial keratitis by virtue of the swollen epithelial cells and scalloped borders at the margins of herpetic lesions. Because herpetic epithelial lesions are caused by viral cytopathic effect, the lesions can be self-limiting (i.e., controlled by innate and/or adaptive immunity), but heal more rapidly when treated with antiviral drugs, such as trifluorothymidine (TFT) and acyclovir (ACV) (Pavan-Langston and Foster, 1977; Porter et al., 1990).
HSK may occur as a progression from infectious epithelial keratitis, or can be the primary manifestation of keratitis (Knickelbein et al., 2009a). Clinical signs of HSK include stromal opacity, edema, and neovascularization that are triggered by recurrent bouts of HSV-1 reactivation and shedding into the cornea (Fukuda et al., 2008; Kumaraguru et al., 1999; Ohashi et al., 1991) (Fig. 1). The direct effect of the virus, and more importantly, the potent immune response to the viral proteins trigger ingrowth of blood vessels, infiltration of leukocytes, and damage to the corneal stroma and endothelium that combine to promote corneal opacity and edema. The inflammation clears, usually aided by topical corticosteroids, but often with some degree of scar tissue deposition (Jones, 1958; Wilhelmus, 1987). Repeated bouts of HSK can lead to progressive irreversible corneal scarring and blindness.
Figure 1.
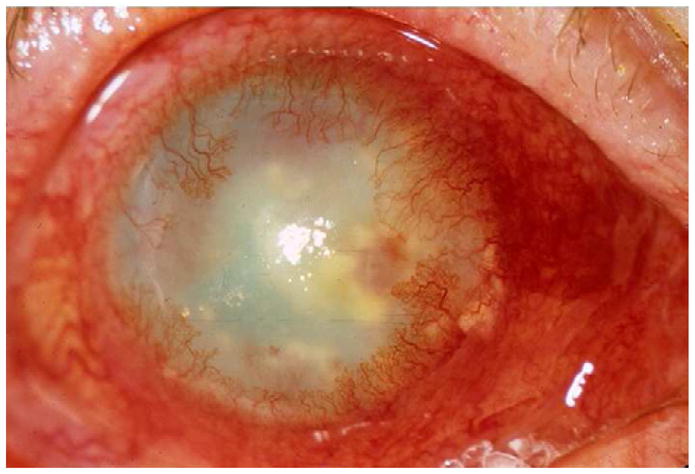
A) Slit lamp photograph of a patient with herpes stromal keratitis demonstrating opacity and neovascularization of the cornea.
HSK is often subdivided into two clinical categories: necrotizing and non-necrotizing disease. In necrotizing HSK, an epithelial defect overlies the stromal opacity, and active viral replication as well as immune-mediated tissue damage are thought to contribute to corneal injury (Holland and Schwartz, 1999; Liesegang, 1999). Necrotizing HSK is an eye-threatening emergency requiring aggressive management to prevent corneal melting and perforation. Non-necrotizing, also referred to as immune HSK involves stromal inflammation without an associated epithelial defect (Wilhelmus, 1987). As the name implies, immune-mediated destruction of the corneal stroma is the presumed cause of non-necrotizing HSK, without obligate involvement of active viral replication.
2 Animal models of infection
HSV-1 corneal infections have been studied in a variety of animal models. While all animal models are inherently imperfect representations of human disease, the broad species specificity of HSV-1 permits the development of animal models that display many of the features of HSV-1 corneal disease in humans. The most popular animal models have been developed in rabbits and mice. In both models primary HSV-1 corneal infection with appropriate HSV-1 strains results in epithelial lesions in most if not all animals, with subsequent development of HSK in a portion of animals. In both models, the initial epithelial lesions resemble those in human corneas with their punctuate, dendritic, or geographic morphology. In both models HSK primarily resembles immune HSK in humans. In both models the virus gains access to the termini of sensory neurons while replicating in the corneal epithelium, is transported by retrograde axonal transport to the neuronal nuclei where after a brief period of replication the virus established a latent infection. During latency the virus is retained in neuronal nuclei as circular episomal DNA (Gardella et al., 1984), but no infectious virions are produced.
However, each model has advantages and disadvantages. The rabbit model has the advantage of a large eye and spontaneous HSV-1 reactivation from latency and shedding at the ocular surface. Since HSK in humans represents recurrent herpetic disease resulting from HSV-1 reactivation from latency in the TG and shedding at the cornea, the capacity to induce recurrent HSK is a useful feature of the rabbit model. In addition, infectious epithelial keratitis typically persists longer in the rabbit, which can be helpful in testing the efficacy of antiherpetic drugs. The main disadvantages of the rabbit model are the lack of inbred strains, and the relative lack of reagents for dissecting the immune response. These represent significant disadvantages in attempting to understand an immunopathologic process such as HSK with a genetic contribution from the host. The mouse has the advantage of being small and relatively inexpensive with a plethora of genetically defined strains including transgenic strains with immunologically important genes knocked in or knocked out, and available reagents for dissecting the immune response to HSV-1. The main disadvantage of the mouse model is that the virus does not spontaneously reactivate and shed at the corneal surface as it does in rabbits and humans. It is possible to induce HSV-1 reactivation from latency, shedding, and recurrent HSK in the mouse, but the protocols involve raising the body temperature to dangerous levels or exposing the cornea to ultra violet light (refs). These procedures can have unintended consequences, only work in certain mouse strains, and reactivation tends to be inefficient and difficult to reproduce in different laboratories. Moreover, the data obtained in mouse models of recurrent HSK do not suggest a marked difference in the immunopathology compared to HSK that develops following primary infection (Miller et al., 1996). Thus, most of what we know about the immune response to HSV-1 and HSK-associated immunopathology derived from mouse primary infection models. Given the immunologic orientation of the authors, this review will concentrate largely on information gleaned from studies of HSV-1 corneal infections in mice.
2.1 Primary infection
Infection of the mouse corneal epithelium with most strains of HSV-1 requires some degree of scarification. The increased infectivity on abraded corneas probably reflects loss of the protective effect of an intact tear film, access to the basal lateral side of the epithelial cells, access to the less differentiated epithelial cells in the basal layer of the corneal epithelium, and a depot effect of viral binding to the denuded basement membrane. Corneal epithelial cells express HSV-1 entry receptors nectin 1, herpes virus entry mediator (HVEM), and paired immunoglobulin-like type 2 receptor-alpha (PILR-α) rendering them susceptible to viral infection (Shah et al., 2010; Valyi-Nagy et al., 2004). In mice and rabbits the virus replicates in the corneal epithelium producing lesions that are punctuate, dendritic, or geographic in shape when stained with fluorescein or rose bengal. It has been suggested that the dendritic shape of epithelial lesions reflects movement of virus in corneal neurons, though to our knowledge this theory has not been confirmed experimentally. In mice the epithelial lesion typically heals by around 3 days post infection (dpi), although clearance of replicating virus from the cornea typically occurs at around 6–8 dpi (Mott et al., 2007; Tumpey et al., 1996).
3. Immune control of primary corneal infection
3.1 Role of Innate immunity in controlling HSV-1 replication in the corneal epithelium
It is convenient to divide immune responses into innate and adaptive responses although extensive overlap between the two systems is now apparent. The principal difference between the lymphocytes that mediate adaptive immunity and the various cells that comprise the innate immune system is that the former employ receptors that are highly variable being formed through random rearrangement of gene segments during maturation, whereas the latter use invariant receptors that are encoded in the germ line genes (Janeway, 2005). The invariant nature of pathogen recognition receptors on innate cells permits retention of large numbers of these cells in the body at any given time, enabling rapid deployment upon infection. In contrast, the highly variable nature of receptors on lymphocytes of the adaptive immune system prohibits retention of large numbers of cells of any given specificity. Therefore, upon infection a small number of lymphocytes with receptors specific for pathogen proteins must be expanded into a population of sufficient size to effectively deal with the pathogen. Thus, the adaptive response is delayed resulting in preferential involvement of innate immune cells in the early response to primary infection. Accordingly, HSV-1 clearance from the cornea during primary infection is largely through an innate immune response.
In mice and presumably humans, HSV-1 invades a cornea that is avascular and largely devoid of blood-derived leukocytes. In fact, it was only fairly recently that resident leukocytes were demonstrated to be present in normal corneas of mice (Hamrah et al., 2003a; Hamrah et al., 2003b; Nakamura et al., 2005). It is now apparent that the normal mouse cornea contains some components of the innate immune system. Dendritic cells (DC) are present in the basal layer of the epithelium and possibly in the stroma, and the corneal stroma is endowed with a network of macrophages. However, direct involvement of these resident corneal leukocytes in controlling the initial HSV-1 infection remains uncertain.
Corneal epithelial cells can initiate the innate immune response to HSV-1 through expression of a family of receptors called toll like receptors (TLR). HSV-1 DNA alone is capable of inducing a strong TLR response in the cornea by ligating TLR-9 (Krug et al., 2004; Lundberg et al., 2003). Other TLR ligands that appear to be generated in HSV-1 infected corneas include viral double-stranded RNA (TLR3 ligand), single-stranded RNA (TLR7 ligand), heat shock protein 70 (TLR2 ligand) and β defensin 2 (TLR4 ligand). In addition to TLRs, a recent study demonstrated a particularly important role for a DNA sensor IFI-16/p204 in HSV-1 clearance from the corneal epithelium (Conrady et al., 2012). The ligation of TLRs and IFI-16/p204 by HSV-1 induced ligands causes an immediate up-regulation of chemokines (molecules that attract and activate immune cells) and proinflammatory cytokines in the cornea (Hayashi et al., 2006; Takeda et al.; Yan et al., 1998). Among the induced cytokines, type 1 interferons (IFN-α/β) are particularly important in limiting the replication of HSV-1 in the cornea and preventing systemic spread (Conrady et al., 2011; Luker et al., 2003). The TLR-induced chemokines and cytokines are detectable at the protein level within hours of HSV-1 infection. HSV-1 infection induces a variety of chemokines that attract a range of immune cells. These immediate chemokines, their ligands, and the cells expressing these ligands are summarized in Table 1. HSV-1 infection attracts several components of the innate immune system including DC, monocytes/macrophages, neutrophils, and natural killer (NK) cells (Carr et al., 2008; Cheng et al., 2000; Kassim et al., 2009; Pollara et al., 2004; Tumpey et al., 1996). All of these cells reportedly contribute to the clearance of HSV-1 from the cornea during primary infection.
Table 1.
This table summarizes the chemokines found to play a role in the pathologies associated with ocular HSV-1 infections in mice. The second column lists binding partners or receptors for these chemokines. Studies which have blocked these chemokines or employed specific knockout mice have found that these chemokines are responsible or associated with the influx of specific cell types into the infected cornea. Those specific cell types are listed in column three and the studies which identified the association between the chemokines and specific cell types are listed in column four.
NK cells are a component of the innate immune system that is specialized in the detection and elimination of virus infected cells. As their name implies, NK cells are always armed and are capable of destroying aberrant cells within minutes of contact. The activation status of an NK cell is dependent on integrated signals delivered by germ-line-encoded activation and inhibitory receptors for ligands on cellular targets. The stochastic expression of these activation and inhibitory NK receptors can determine the net outcome of interaction with a target cell (Elliott and Yokoyama, 2011). Typically, inhibitory receptors on NK cells recognize self-MHC class I molecules resulting in self tolerance. However, tumor cells or virus-infected cells often have reduced or no expression of surface MHC class I molecules, and this “missing self” triggers NK cell activation. The activated NK cells can then inhibit viral infections through the directed release lytic granules that are stored in their cytoplasm and whose granzyme components can induce apoptosis of the infected cell. Alternatively, activated NK cells can limit virus replication within the target cell through the production of antiviral cytokines such as interferon gamma (IFN-γ) and tumor necrosis factor alpha (TNF-α), and attract other inflammatory cells through the production of a variety of chemokines including CCL2, CCL3, CCL4, CCL5, CXCL-1, and IL-8 (Walzer et al., 2005).
Studies employing mouse models of HSV-1 corneal infection have demonstrated an important role for NK cells in controlling HSV-1 replication in the cornea (Carr et al., 2008; Frank et al., 2012). Absent from the normal cornea, NK cells migrate into the cornea as early as 3 days after HSV-1 corneal infection. This early NK cell migration is impaired in mice that are deficient in the CXCR3 chemokine receptor, suggesting a role for CXCR3 and its ligands CXCL9 and CXCL10 in directing the early migration of NK cells into infected corneas (Carr et al., 2008).
Named by Metchnikoff in 1892, macrophages are “large eaters” with potent phagocytic activity that is crucial for eliminating extracellular pathogens. These members of the innate immune system are activated through recognition of Pathogen Associated Molecular Pattern (PAMPs) by TLRs and through exposure to inflammatory cytokines such as interferon gamma (IFN-γ) and tumor necrosis alpha (TNF-α) to produce reactive oxygen species (ROS) and nitric oxide (NO).
There is a substantial population of CD11b+ macrophages in the normal mouse corneal stroma (Brissette-Storkus et al., 2002). These cells exhibit a unique stratification in which those expressing MHC II+ are found primarily in the anterior stroma directly beneath the corneal epithelium, while those lacking MHC II expression are found evenly throughout the corneal stroma (Knickelbein et al., 2009b). Although the role of these resident corneal macrophages is not clear, they are connected by intercellular membrane nano-tube bridges that seem to be important for cell-to-cell communication during inflammation (Chinnery et al., 2008). Following HSV-1 infection, macrophages are activated through TLRs to produce anti-viral cytokines such as the type I interferons, IFN-α and IFN-β (Mogensen, 1979). These cytokines are crucial for controlling the initial HSV-1 replication as shown by studies using IFN-α/β receptor−/− mice (Leib et al., 1999) and RAG−/− mice treated with IFN-α/β (Vollstedt et al., 2004). In addition to producing type I IFNs, macrophages also are the main source of Interleukin-12 (IL-12) during the first 8–12 hours after HSV-1 corneal infection (Kanangat et al., 1996). IL-12, in synergy with IFN-α/β, can enhance the production of IFN-γ by NK cells (Hunter et al., 1997). IFN-γ released by NK cells subsequently activates macrophages to produce inducible nitric oxide synthase (iNOS), which produces NO to limit HSV replication (Croen, 1993; Gamba et al., 2004).
Neutrophils represent a major component of the initial infiltrate in HSV-1 infected corneas, leading to the assumption that these cells had a major role in viral clearance. Indeed, studies by Tumpey et al (Tumpey et al., 1996) demonstrated that depletion of neutrophils using the Gr-1 antibody significantly delayed clearance of HSV-1 from the corneas of mice. However, the interpretation of results from this and other studies employing the Gr-1 antibody to deplete neutrophils was called into question when it was discovered that the Gr-1 antigen is expressed on subpopulations of a variety of cells including monocyte/macrophages, CD8+ T cells, and DC (Geissmann et al., 2003; Matsuzaki et al., 2003; Nakano et al., 2001; Palamara et al., 2004). Indeed, more recent studies in an HSV-1 lung infection model demonstrated increased virus replication in mice that were depleted of cells expressing the Gr-1 marker, but not in those selectively depleted of neutrophils using the Ly6G antibody (Wojtasiak et al., 2010). Moreover, the delayed clearance of HSV-1 from the cornea resulting from DC depletion was associated with decreased migration of NK cells and inflammatory monocytes, but normal migration of neutrophils into the HSV-1 lesion in the central cornea (Frank et al., 2012). Based on these studies it would appear that neutrophils are neither necessary nor sufficient for optimal clearance of HSV-1 following primary infection.
DCs were originally identified by Steinman and colleagues (Steinman and Cohn, 1973; Steinman et al., 1983) as cells that play a critical role in activating naïve T cells of the adaptive immune system (discussed below). However, more recent studies have identified an additional role for DCs in regulating innate immune responses through licensing of NK cells (Fernandez et al., 1999; Walzer et al., 2005). A population of DCs that are referred to as plasmacytoid DCs (pDC) appear to be a major source of type 1 interferon following HSV-1 infection (Donaghy et al., 2009; Pollara et al., 2004). Though there is evidence of a small population of pDC in normal corneal stroma, their involvement in eliminating HSV-1 from the infected cornea has not been established. There is evidence that cornea resident CD11c+ DC have an indirect role in HSV-1 clearance (Frank et al., 2012). These studies employed mice that express a high affinity diphtheria toxin receptor (DTR) from the CD11c promoter. Since mice do not normally express high affinity DTR, DC are selectively depleted in these mice following DT treatment.
Depletion of resident corneal DCs during the first 24 hours after HSV-1 corneal infection resulted in a delay of NK cell and inflammatory monocyte migration into the HSV-1 epithelial lesions in the central cornea, resulting in delayed viral clearance. Interestingly, DC depletion did not influence NK cell extravasation from the blood, but only prevented their directed migration into the central cornea. The reduced migration of NK cells and inflammatory monocytes into the central cornea that was observed following DC depletion was associated with a significant reduction in several chemokines that are known to direct the migration of these cell types (Frank et al., 2012). These include the chemokines CCL5 (activates NK cells), CCL7 (attracts and activates macrophages), CCL8 (attracts and activates monocytes and NK cells), CXCL9 (attracts NK cells) CXCL10 (attracts macrophages and NK cells), and CXCL11 (attracts NK cells). Depletion of NK cells resulted in a similar delay in clearance of HSV-1 from the cornea. These data suggest a scenario in which DC that are resident in the normal cornea or those that migrate into the cornea within the first 24 hours after HSV-1 infection are stimulated by the virus (likely through ligation of TLR3, 7, and 9) to produce chemokines that direct the migration of NK cells and inflammatory monocytes into the cornea. The DCs further activate the NK cell cytotoxic activity and IFN-γ production, potentiating their involvement in viral clearance. Interestingly, DC depletion did not alter neutrophils migration into the peripheral or central cornea, despite the delayed viral clearance (Frank et al., 2012). This finding is consistent with the growing evidence that neutrophils may not play a major role in HSV-1 clearance from the cornea.
3.2 Role of adaptive immunity in HSV-1 clearance from the cornea
The above findings support a predominant role for innate immunity in clearing HSV-1 from the cornea following primary infection. This is not surprising given the rapid kinetics of viral clearance, which begins around 2 dpi with complete clearance by around 6–7 dpi. However, two caveats bear mentioning. First, most HSV-1 corneal infections represent recurrent disease that occurs in the face of an already primed adaptive immune response. Little is known about the relative contribution of innate and adaptive immunity in viral clearance from the cornea during recurrent lesions. One study demonstrated reduced importance of NK cells for controlling HSV-1 replication following HSV-1 vaccination, suggesting a likely contribution of adaptive immunity (especially anti-HSV-1 antibodies) to viral clearance during recurrent disease (Ghiasi et al., 2000). Second, even primary infections of the cornea and TG are not completely cleared in the absence of adaptive immunity. For instance, severe combined immunodeficiency (SCID) mice that have a normal innate immune system, but lack adaptive immunity never completely clear HSV-1 from their corneas (Banerjee et al., 2004b). These mice do control HSV-1 replication in the cornea following transfer of CD8+ T cells from HSV-1 infected mice (Banerjee et al., 2004b). Interestingly, the transferred CD8+ T cells were found in the lymphoid organs and in the TG, but not in the infected corneas. The authors suggested that CD8+ T cells might control HSV-1 replication in the cornea by preventing transport via infected TG neurons and shedding at the cornea.
4. Herpes stormal keratitis (HSK)
4.1 Theories of HSK pathogenesis
Several groups have demonstrated that CD4+ T cells are the principal mediators of HSK (discussed in Section 4.3). A logical conclusion from these observations would be that HSK represents an immunopathological process in which HSV-specific CD4+ T cells are stimulated by HSV-1 antigens presented by corneal antigen presenting cells following viral clearance. However, this interpretation was called into question by two teams of investigators, each advocating a different form of CD4+ T cells involvement in HSK. A team led by Dr. Harvey Cantor was investigating the differential susceptibility of two strains of BALB/c mice to HSK. The two strains differed only with respect to an IgG2A heavy chain allotype. In a series of very elegant experiments his group established that the susceptible strain could be rendered resistant by tolerizing them to peptides corresponding to IgG2A sequences of the resistant strain (Avery et al., 1995). Conversely, transferring CD4+ T cell clones reactive to the IgG2A of the resistant strain increased the susceptibility of the resistant strain to HSK. They went on to demonstrate that the IgG2A-specific CD4+ T cell clones also reacted to the HSV-1 UL16 protein, and concluded that the HSV-1 protein represented an antigenic mimic capable of activating an autoimmune CD4+ T cell response to corneal antigens (Zhao et al., 1998). The theory was that the IgG2A of the resistant mouse strain contained a peptide sequence that was also found in the cornea and in the UL16 protein of the virus. The HSK resistant mouse strain that expressed this IgG sequence was tolerant to the peptide, and failed to respond to it when infected. In contrast, the HSK susceptible mouse strain lacked this peptide sequence in their IgG2A and thus were not tolerant. The corneas of mice that did not endogenously express the IgG2A motif would mount a CD4 T cell response to UL16 and these UL16 specific CD4 cells also reacted to cornea antigens. The inflammation and neovascularization resulting from HSV-1 infection exposed the cornea to autoimmune attack by these cells.
These studies led to a widely held perception that HSK is an autoimmune disease. However, these interesting findings have not been broadly extrapolated to other animal models of HSK or to human HSK. For instance, other investigators found that immunization of BALB/c mice with UL6 did not induce a response to the peptide mimic, and immunization with the peptide did not result in cross-reactivity to HSV-1 or to the G2a 292–308 corneal peptide that was targeted in the congenic mice (Deshpande et al., 2001). A survey of HSK patients found no associative correlation or trends with polymorphisms in the IgG2A genes of HSK afflicted individuals (Kasman et al., 2006). Moreover, several investigators have isolated HSV-specific CD4+ T cells from corneas of mice and humans with HSK, but have not detected cells responsive to corneal antigens (Verjans et al., 2000; Verjans et al., 1998). Thus, a universal role for autoimmunity in HSK remains unproven.
A second theory that has been advanced and supported by data is that CD4+ T cells of irrelevant specificity mediate HSK when exposed to a cytokine storm unleashed by HSV-1 corneal infection (Gangappa et al., 1999). Support for this view came from studies in which transgenic DO11.10 mice whose CD4+ T cells can only express a receptor for a chicken ovalbumin peptide (Ova) were shown to develop HSK symptoms following HSV-1 corneal infection. The corneal inflammation was dependent on CD4+ T cells, which in these DO11.10 mice are incapable of responding to HSV-1 antigens. The authors concluded that HSK results from bystander activation of CD4+ T cells by cytokines (Huster et al., 2002). However, two caveats need to be considered. First, the intensity of the cytokine storm in these studies was likely influenced by the persistence of replicating virus in corneas of DO11.10 resulting from their inability to generate HSV-specific CD4+ and CD8+ T lymphocytes. Indeed, HSK failed to develop when these mice were provided with a source of CD8+ T cells that controlled virus replication in the cornea with normal kinetics (Gangappa et al., 1999). Second, CD4 independent HSK has been described, and is mediated by CD8 T cells. This HSK is transient and less severe than that observed in CD4 replete mice (Lepisto et al., 2006). CD4 independent HSK in mice also appears to be viral strain dependant (Huster et al., 2002). It is worth noting that in these models which demonstrated CD4 independent or dependent HSK that the addition of transgenic HSV-1 nonspecific CD4 cells would increase the severity of pathology without independently inducing it in situation with effective viral control (Gangappa et al., 1998; Gangappa et al., 1999, 2000; Huster et al., 2002; Russell et al., 1984). Thus, it appears that bystander activation cannot initiate HSK immunologically normal mice, although it might contribute to the severity of existing HSK.
Given the uncertainties surrounding the contributions of autoimmunity and bystander activation, CD4+ T cell reaction to viral antigens in the cornea remains the most likely pathogenic mechanism of HSK. Although proof of the involvement of HSV-1 specific CD4+ T cells would seem to be relatively easy to acquire, it remains elusive. Studies have revealed that T cells obtained from HSV-1 immunized mice transfer HSK more efficiently to SCID mice than those obtained from non-infected mice (Mercadal et al., 1993). However, the transferred cells also contain CD4+ T cells that are not HSV-specific, rendering the results suggestive of, but not proof of a role for HSV-specific CD4+ T cells in HSK. The main problem is the difficulty of obtaining a population of CD4+ T cells that is exclusively reactive to HSV-1 antigens. Typically this problem is resolved by generating mice with a transgenic T cell receptor (TCR) specific for a viral peptide. When these mice are backcrossed to RAG−/− mice that are incapable of TCR gene rearrangement, all T cells express the transgenenic TCR. Generating mice with an HSV-specific CD4+ TCR eluded researchers until recently when Dr. Anthony Brooks at the University of Melbourne developed a mouse with a transgenic TCR specific for HSV-1 glycoprotein D (gDT-II mice) (Bedoui et al., 2009). The availability of these mice will enable future studies investigating an obligate role for HSV-specific CD4+ T cells in HSK.
4.2 Role of vascularization in HSK
Corneal neovascularization is a prominent feature of HSK. Evidence suggests that corneal angiogenesis results from production of a variety growth factors (the most prominent being vascular endothelial growth factor [VEGF]) and metaloproteinases (particularly MMP 9). A healthy cornea is avascular and this helps maintain the corneal immune privilege. It appears that corneal avascularity is maintained in part by constitutive production of a soluble VEGF receptor 1 (sVEGFR-1) that binds to and inactivates VEGF (Ambati et al., 2006). HSV-1 infection results in a rapid reduction of sVEGFR-1 in the cornea that persists during HSK (Suryawanshi et al., 2012). The reduction of sVR-1 requires MMP-9 production by neutrophils that infiltrate cornea rapidly after HSV-1 infection. Neutrophils are also an important source of VEGF. Studies have shown a significant palliative effect of VEGF inhibition on HSK (Kim et al., 2004; Zheng et al., 2001a; Zheng et al., 2002). Blood vessels can be seen encroaching on the normally avascular cornea within a few days of HSV-1 infection (Kumaraguru et al., 1999; Wuest et al., 2011a; Zheng et al., 2001b). This is consistent with recent evidence that infected corneal epithelial cells are a source of VEGF-A (Wuest and Carr, 2010). Typically blood and lymphatic vessels co-migrate into tissue, and this is the case in HSV-1 infected corneas. Both types of vessels are important for HSK development since lymph vessels transport viral antigens to the lymph nodes to activate HSV-1 specific T cells and B cells, while blood vessels carry the activated T cells to the cornea. (Suryawanshi et al., 2012). Under most inflammatory conditions blood angiogenesis is primarily mediated by VEGF-A signaling through VEGF receptor 2 (VEGFR-2), while lymphangiogenesis is primarily regulated by VEGF-C and – D binding to VEGFR-3 (Hirakawa et al., 2007; Skobe and Dana, 2009; Skobe et al., 2001). However, in HSV-1 infected corneas it appears that both blood and lymph angiogenesis is regulated primarily through VEGF-A binding to VEGFR-2. Thus, HSV-1 infection induces corneal neovascularization at least in part by inducing VEGF production by corneal epithelial cells and infiltrating neutrophils, and by reducing the concentration of the VEGF inhibitor sVEGFR-I in the cornea. Since blocking neovascularization effectively inhibits the development of HSK, inhibiting angiogenic factors would seem to represent a novel therapeutic approach to treating HSK.
4.3 Leukocytic involvement in HSK
During HSV-1 replication in the corneal epithelium, viral antigens are thought to be transported to the draining lymph nodes (DLN) by DC that are resident in the normal cornea and those that infiltrate following infection; although transport of free virus to the DLN in lymph is also likely. Transport of viral antigens via DC from skin and other mucosal sites of HSV-1 infection to the DLN has been demonstrated (Allan et al., 2003; Allan et al., 2006; Hendricks et al., 1992a; Lee et al., 2009; Sprecher and Becker, 1989; Zhao et al., 2003), but a similar role for corneal DC has not been established. Elimination of resident corneal DC by ultraviolet B irradiation prior to HSV-1 infection was shown to reduce by 50% the HSV-specific CD4+ T cell-mediated delayed type hypersensitivity (DTH) response, suggesting a likely role for cornea derived DC in expanding HSV-1 specific CD4+ T cells in the DLN (57). Expansion of CD4+ and CD8+ T cells in the DLN and spleen peak around 7–8 days after corneal infection. This corresponds to the time of HSK onset in mice (Bouley et al., 1995; Doymaz and Rouse, 1992), consistent with a role for HSV-specific T cells in mediating the inflammation associated with HSK. Indeed, mice lacking adaptive immunity or those lacking only CD4+ T cells fail to develop HSK (Doymaz and Rouse, 1992; Lepisto et al., 2006).
In mice HSK development is associated with a progressive inflammatory infiltration of the cornea (Divito and Hendricks, 2008). Neutrophils are the predominant infiltrating cell population, comprising 80–90% of the infiltrate (Divito and Hendricks, 2008). The neutrophils infiltrate in two distinct waves. The first wave of neutrophil infiltration coincides with virus replication in the cornea, and subsides following viral clearance (Chen et al., 1996). A second wave of neutrophil infiltration coincides with the onset of HSK, and neutrophils appear to be the proximal mediators of corneal damage and scarring associated with HSK (Osorio et al., 2002). The second wave of neutrophil infiltration into HSV-1 infected corneas fails to occur in mice that lack T cells (Chen et al., 1996). It appears that CD4+ T cells that produce a Th1 cytokine pattern (IFN-γ and IL-2) have a dominant role in inducing neutrophilic infiltration into the cornea (Tang et al., 1997; Thomas et al., 1997). Indeed, neutralizing the cytokines IFN-γ and IL-2 was shown to inhibit the progression of HSK (Hendricks et al., 1992b; Tang et al., 1997). More recent studies demonstrated a role for IL-17 in the pathogenesis of HSK (Molesworth-Kenyon et al., 2008; Suryawanshi et al., 2011). The latter study demonstrated biphasic expression of IL-17 in infected corneas. The first phase was observed early after infection and the primary source was non-traditional T cells that express a TCR comprised of a gamma and delta chain and belong to the innate immune system. The second phase of IL-17 production coincided with HSK development and the source of IL-17 was Th17 cells (differentiated CD4+ T cells with traditional αβ TCR that produce IL-17 and IL-22 as their signature cytokines). Interestingly, the Th17 cells appear to require the prior presence of Th1 cells to infiltrate the infected cornea. Inhibition of IL-17 during either the early or late phase was found to reduce the severity of HSK (Suryawanshi et al., 2011).
It appears, therefore, that HSK represents a complex inflammatory process requiring cooperation between Th1 and Th17 differentiated CD4+ T cells and neutrophils. Accordingly, mice that are genetically deficient in CD4+ T cells or are depleted of these cells as adults are highly resistant to HSK following HSV-1 corneal infection (Lepisto et al., 2006; Newell et al., 1989a; Newell et al., 1989b). This is consistent with the large excess of CD4+ T cells over CD8+ T cells in corneas with HSK (Doymaz and Rouse, 1992). However, CD4+ T cell-deficient mice that are infected with a high dose of HSV-1 can develop HSK that is apparently mediated by CD8+ T cells (Lepisto et al., 2006). The CD8+ T cell-regulated HSK is characterized by a predominantly neutrophilic infiltrate similar to that regulated by CD4+ T cells, but tends to be less severe and more transient. HSV-specific CD8+ T cells produce IFN-γ when stimulated, and thus likely mediate HSK by the same mechanisms used by CD4+ T cells, but less efficiently.
5. HSV-1 latency
As noted above, most cases of HSK represent recrudescent disease resulting from HSV-1 reactivation from latency in TG neurons. For this reason, understanding the mechanisms that control the establishment and maintenance of HSV-1 latency has long been the holy grail of herpes virologists. Unfortunately the virus has very grudgingly shared its secrets. During primary infection the virus gains access to the termini of sensory neurons within the infected tissue. The HSV-1 capsid is then transported to the neuronal nucleus using the transport mechanisms of the host cell. The viral capsid attaches to nuclear pores and inserts its DNA into the neuronal nucleus. The virus then either directly enters into a latent state in which the genome is retained as a non-integrated neucleosome-associated episome in the neuronal nucleus; or does so following a brief lytic cycle in which viral genes are expressed and infectious viral particles are produced. The mechanisms that influence the decision about which life style the virus assumes in a particular neuron remain unclear. However, differential susceptibility of neuronal subpopulations within the TG to lytic or latent infection by HSV-1 has been proposed (Margolis et al., 1992; Margolis et al., 2007).
In animal models and presumably humans, latency is associated with the silencing of most viral gene transcription and the abundant expression of latency associated transcripts (LATs) in some latently infected neurons. The discovery that LATs are the only abundantly expressed viral transcripts during latency raised considerable excitement about their potential role in establishing and maintaining HSV-1 latency. LATs lie partly complementary to the 3′ end of the ICP0 gene and are stable, nuclear RNAs, without polyadenylation or 5′ caps, with structural features of stable lariats of introns. They do not encode protein and may be byproducts of a larger unstable primary transcript that is processed into microRNAs (Umbach et al., 2008) that inhibit HSV-1 lytic gene expression. Although viral mutants lacking LATs are still able to establish, maintain and reactivate from latency, LATs increase the efficiency of latency establishment and of reactivation. Recent findings from the laboratory of Dr. Todd Margolis (Bertke et al., 2011; Yang et al., 2000) demonstrate that LATs also influence the life style decision of HSV-1 and HSV-2 in neuronal subpopulations. The A5+ subpopulation of TG neurons is selectively resistant to HSV-1 replication and preferentially retains latent virus. In contrast, the KH10+ neuronal subpopulation is permissive to HSV-1 replication and tends not to retain latent virus. The opposite pattern is seen with HSV-2, and the differential establishment of lytic or latent infections in the two neuronal subpopulations is governed by differences in the HSV-1 and HSV-2 LAT regions. These studies were performed in mice, and it remains unclear the two HSV serotypes exhibit differential tropism for human neuronal subpopulations. LATs may also enhance latency establishment through their ability to block neuronal apoptosis, thus increasing the survival of latently infected neurons (Perng et al., 2000). Several HSV 1 micro RNAs (miRNAs) that localize to the LAT transcript (Umbach et al., 2008) may regulate gene expression during latency, though exactly how is not clear. A recent study has suggested that siRNAs regulating the neurovirulence factor γ34.5 may contribute to latency and virulence (Tang et al., 2009).
The TG and brainstem appear to constitute important checkpoints in HSV-1 transport from peripheral sites of infection to the brain. In mice, HSV-1 can be detected in the TG and brainstem by 2 – 3 days after corneal infection. Failure to control HSV-1 replication in the TG and progression of HSV-1 beyond the brainstem is associated with lethal encephalitis (Kastrukoff et al., 2010; Sciammas et al., 1997). Mechanisms of viral control have been most thoroughly investigated in the TG. In mice, HSV-1 can be seen replicating in TG neurons as early as 3 days after corneal infection, and virus replication is typically terminated and latency established by 6–8 days post infection. Virus replication is associated with significant leukocytic infiltration. Interestingly, neutrophils that dominate the leukocytic infiltrate in HSV-1 infected corneas, are virtually absent from the acutely infected TG. Instead, the initial infiltrate in HSV-1 infected TG is dominated by macrophages and γδ T cells. γδ T cells are non-conventional T cells that seem to bridge the gap between innate and adaptive immunity. They can be included in the adaptive camp in that they rearrange gene segments to form γ and δ TCR chains with junctional diversity. However, they also fit into the innate immune camp in that they express highly restricted TCR specificity that can serve as pattern recognition receptors. γδ T cells are also unconventional in that they can recognize nominal antigens without processing and presentation in the groves of major histocompatibility complex (MHC) molecules, a feature that might be important in dealing with infected neurons that express little or no MHC. Clearance of the bulk of replicating HSV-1 from the TG involves an interaction between macrophages and δ T cells in which IL-12 produced by macrophages induces IFN-γ production by γδ T cells. The IFN-γ both directly blocks HSV-1 replication, and induces NO production by macrophages, further inhibiting HSV-1 replication (Kodukula et al., 1999).
Although the bulk of HSV-1 replication in the TG is controlled by an innate immune response, mice lacking adaptive immunity never completely control virus replication and succumb to lethal encephalitis following corneal infection with most strains of HSV-1 (Mercadal et al., 1993). CD8+ T cells appear to play a particularly important role in the final eradication of replicating virus from the TG (Lang and Nikolich-Zugich, 2005; Liu et al., 2000). Data suggest that the chemokines CXCL10 (IP-10) and CCL2 (MCP-1) induced largely through TLR9 signaling are primarily responsible for infiltration of T cells and macrophages that mediate viral clearance from the TG (Lima et al., 2010; Wuest et al., 2011b).
5.1 Maintenance and reactivation from latency
HSV-1 lytic gene expression is severely repressed during viral latency. The fact that the latent viral genome has a transcriptionally active LAT region and transcriptionally inactive lytic gene regions suggests epigenetic regulation. Indeed, the LAT region is enriched in marks of transcriptional permissiveness such as H3 K4 and acetyl H3K9, K14, while the lytic genes are enriched for facultative heterochromatin marks (Bloom et al., 2010; Cliffe et al., 2009; Umbach et al., 2008). Current data suggest that rather than being transcriptonally repressed through methylation as is seen with non-expressed host cell genes, the latent HSV-1 genome appears to be in a state of transcriptional equilibrium with micro-RNAs and LATs maintaining gene silencing (Roizman et al., 2011). The LATs contribute to viral lytic gene silencing by influencing facultative heterochromatin assembly and possibly silencing the ICP0 gene (required for efficient HSV-1 reactivation from latency) through an anti-sense mechanism (Farrell et al., 1991; Wang et al., 2005). The LAT region that is actively transcribed during latency overlaps and is anti-sense to that encoding the immediate early protein ICP0 (required for efficient reactivation from latency). The LAT transcripts are predominantly nuclear and there are no known protein products.
Exposure to a variety of environmental and physiologic stimuli can lead to the emergence of the virus from latency, expression of lytic genes, and virion formation. The resulting virions are then transported down nerve axons for release at innervated sites such as the cornea, in some cases resulting in keratitis. Whether complete virions are transported down nerve axons or virion capsids and glycoproteins are separately transported for assembly and release at the nerve termini remains a contentious issue (Ohara et al., 2000; Wisner et al., 2011). Recent findings have suggested models of HSV-1 reactivation from latency involving disordered expression of viral genes rather than the sequential expression observed during lytic infection of cells in culture. For instance, Roizman’s group (Du et al., 2011) has demonstrated that during reactivation from latency in explants cultures the LATs and miRNAs that maintain latency are rapidly degraded by a mechanism requiring protein synthesis, and all classes of viral genes are rapidly expressed by a mechanism that does not require protein synthesis. These findings suggest total derepression of the viral genome very early during reactivation. A more recent study proposed an extension of Roizman’s group’s theory in which HSV-1 gene expression occurs in two stages. The first stage is characterized by a generalized derepression of the viral lytic genes as suggested by Roizman’s group, while in the second stage gene expression is amplified by the viral protein VP16. This second level of amplification might be necessary for formation of viral particles (Kim et al., 2012). These findings have important implications not only for understanding gene regulation in stressed neurons, but also for potential immune control of viral latency. Expression of all classes of viral genes early in the reactivation process would potentiate intervention in the reactivation process by HSV-specific immune cells regardless of the class of viral protein they target.
5.2 Immune control of HSV-1 reactivation from latency
The environmental and physiologic stimuli that induce HSV-1 reactivation from latency include exposure to UV light, stress, changes in sex hormones, and immune suppression (Blyth et al., 1976; Bonneau et al., 1993; Openshaw et al., 1979; Padgett et al., 1998; Sainz et al., 2001; Vicetti Miguel et al., 2010). All of these stressors can impact T cell function (Bonneau, 1996; Bonneau et al., 1991; Bonneau et al., 1998; Glaser and Kiecolt-Glaser, 2005; Leo and Bonneau, 2000; Padgett and Glaser, 2003), suggesting a possible role for T cells in preventing viral reactivation. Studies in rabbits (Gebhardt and Hill, 1988) and mice (Liu et al., 1996) demonstrated that T cells do infiltrate the TG around 8–10 days after corneal infection, associate closely with neurons within the ophthalmic branch, and remain in close apposition to neurons long after latency is established. HSV-1 does not spontaneously reactivate from latency in mice or does so very rarely (Feldman et al., 2002; Gebhardt and Halford, 2005). For many years the dogmatic view of HSV-1 latency held that the latent virus did not produce viral proteins, and could thus effectively hide from the immune system. Therefore it was unclear what maintained the attraction of CD8+ T cells for latently infected neurons in mice. At this juncture a definition of the terms “latency” and “reactivation” is important. One can define reactivation from latency as that point at which viral lytic gene expression commences. By that definition, the virus is able to hide from the host immune system during latency, since the immune system can only respond to viral protein synthesis. Alternatively, one can define reactivation from latency as the point at which infectious virions are produced. That definition accommodates immune control of viral latency by blocking an early step in reactivation that occurs prior to viron formation. This latter definition of reactivation from latency is often used in vivo and in in vitro TG cultures, and will be used in this review.
The observation that CD8+ T cells are juxtaposed to neurons in TG with no apparent replicating virus was consistent with their role in maintaining the virus in a latent state as defined by a lack of virion formation. Subsequent studies established that at least half of the TG resident CD8+ T cells were HSV-1 specific, and many of the HSV-1 specific cells formed an immunologic synapse with neurons (Khanna et al., 2003). In an immunological synapse, the TCR is focused to the middle of the junction with the target cell by binding to MHC-peptide complexes. The observation of immunological synapse formation between HSV-specific CD8+ T cells and neurons in latently infected TG demonstrated that the latent virus is not “invisible” to T cells. Instead, the latently infected neurons were apparently producing enough viral peptide and MHC class I to ligate the TCR molecules on the juxtaposed CD8+ T cells. Subsequent studies revealed that a large percentage of TG resident CD8+ T cells expressed an activation phenotype including the activation markers CD69 and granzyme B (GrB) (Sheridan et al., 2009). Importantly, activated CD8+ T cells were also found surrounding HSV-1 latently infected neurons in human TG (Theil et al., 2003; Verjans et al., 2007).
An important question was whether or not these TG resident CD8+ T cells are capable of blocking HSV-1 reactivation from latency. To test this possibility, TG were removed from latently infected mice, the cells were dispersed with collagenase, and cultured in the presence or absence of anti-CD8 antibody. TG excision and culturing provides a level of neuronal stress that is sufficient to induce HSV-1 reactivation. These experiments clearly demonstrated that the TG resident CD8+ T cells can block HSV-1 reactivation, and additional studies established that protection required HSV-specific CD8+ T cells (Khanna et al., 2003; Liu et al., 2000). CD8+ T cells (also referred to as cytotoxic T lymphocytes) use two main mechanisms to control virus infections: inhibition of viral gene expression by the antiviral cytokine interferon gamma (IFN-γ); and killing the virus infected cell through the release of lytic granules. Lytic granules are stored in the cytoplasm of CD8+ T cells and can be rapidly released into virally infected cells. Lytic granules contain perforin that polymerizes to form pores in cell membranes, and serine proteases called granzymes including GrB that activate the caspases of the infected cell causing it to undergo apoptosis. When stimulated directly ex vivo with HSV-1 antigens, TG resident CD8+ T cells were shown to produce IFN-γ and release lytic granules, suggesting the potential use of both of these mechanisms in preventing HSV-1 reactivation from latency. Moreover, as noted above a significant proportion of TG resident CD8+ T cells express GrB suggesting functional lytic granules. Because neurons do not readily regenerate we favored a non-lytic means of protection and initially investigated a role for IFN-γ in blocking HSV-1 reactivation in TG neurons. Indeed, the TG resident CD8+ T cells were able to block reactivation through IFN-γ (Decman et al., 2005; Liu et al., 2001). However, IFN-γ was only able to block reactivation in about 50% of latently infected neurons, whereas CD8+ T cells could block reactivation in 100% of neurons, suggesting the use of another effector mechanism to block reactivation.
The combined observations that CD8+ T cells in latently infected TG are armed with lytic granules and release them when stimulated directly ex vivo, and that patients with herpes keratitis exhibit a loss of corneal sensation suggested the possible use of lytic granules by CD8+ T cells during immune surveillance of latently infected ganglia. Indeed it was found that mice that lacked the lytic granule components perforin or GrB failed to control HSV-1 latency (Knickelbein et al., 2008). Moreover, HSV-specific CD8+ T cells that lacked these lytic granule components were impaired in their ability to block HSV-1 reactivation in ex vivo cultures of latently infected TG. A perplexing observation was that CD8+ T cells can use lytic granules to block HSV-1 reactivation from latency without killing the neurons. This raised two interesting questions: 1) why don’t lytic granule components activate the caspase system of neurons leading to apoptosis; and 2) how do lytic granules prevent viral replication without killing the neurons. We hypothesized that latently infected neurons were protected from granzyme activation of caspases by their intrinsic expression of anti-apoptotic proteins, and by the presence of LATs with known anti-apoptotic properties. Indeed, the ability of LATs to inhibit caspase activation by CD8+ T cells was subsequently confirmed (Henderson et al., 2002; Perng et al., 2000). A likely answer to the second question arose from an analysis of HSV-1 proteins for putative GrB cleavage sites. Among the viral proteins with predicted cleavage sites was infected cell protein (ICP)4. ICP4 is a viral regulatory protein that is required for expression of HSV-1 early and late proteins. It was established that CD8+ T cell-derived GrB does cleave ICP4 in infected cells, likely short circuiting the HSV-1 replicative cycle.
As noted above, latently infected TG of mice do not exhibit “spontaneous” reactivation of HSV-1 as is seen in humans, although reactivation is probably not truly “spontaneous” in humans. It is more likely that reactivation is a constant process that is more effectively blocked in mice, that mice are less susceptible to the stimuli that induce reactivation, or both. The fact that latently infected human neurons are surrounded by activated CD8+ T cells suggests the possibility that TG resident mouse CD8+ T cells might be more effective at blocking HSV-1 reactivation. An HSV-1 protein ICP47 was shown to inhibit binding of peptides to the transporters of antigen processing (TAPs) for transport into the endoplasmic reticulum and loading on MHC class I molecules. Since the TCR of CD8+ T cells recognize antigenic peptides in the cleft of MHC class I molecules, this function of ICP47 can reduce CD8+ T cell recognition of HSV-1 infected cells (Goldsmith et al., 1998). Moreover, ICP47 binds with much higher affinity of human than mouse TAPs. Therefore, one could evoke the immune evasion function of ICP47 to explain a possible difference in CD8+ T cell function in latently infected TG of mice and humans. This function might be particularly important in CD8+ T cell immunosurveillance of neurons that express low levels of MHC class I. However, the CD8+ T cells surrounding latently infected neurons in human TG exhibit an activation phenotype that is similar to that seen in mice (Theil et al., 2003; Verjans et al., 2007). Moreover, CD8+ T cells play an important role in controlling HSV-2 replication in the human genital tract (Zhu et al., 2007). These observations argue that CD8+ T cells retain function in HSV-1 infected humans in the face of immune evasion by ICP47.
Another possible explanation for the increased ability of HSV-1 to reactivate from latency in humans relative to mice might lie in differential exposure or responsiveness to environmental and physiologic stimuli that are associated with reactivation. As noted previously, all of the stimuli that have been associated with HSV-1 reactivation in humans tend to inhibit T cell function. One such stimulus is exposure to stress. When mice with latent HSV-1 infections were exposed to restraint stress, a dramatic reduction of functionality was observed in their TG resident CD8+ T cells (Freeman et al., 2007). The TG resident CD8+ T cells of stressed mice exhibited a significant reduction in production of IFN-γ and lytic granule release that persisted for approximately three days. An increased HSV-1 genome copy number in the TG of stressed mice suggested that the CD8+ T cell functional compromise was associated with HSV-1 reactivation from latency. These findings led to the hypothesis that transient functional compromise of TG resident CD8+ T cells resulting from intermittent exposure to stress, ultraviolet irradiation, hormonal imbalances, etc might permit the virus to temporarily escape immune surveillance and produce infectious virions. An as yet undefined direct effect of these stimuli on neuronal function might also enhance the capacity of the virus to enter the lytic cycle.
5.3 Can CD8+ T cell control of HSV-1 latency be exploited to reduce HSV-1 recurrent disease?
Given the apparently important role for CD8+ T cells in preventing HSV-1 reaction from latency, approaches designed to enhance the size or functionality of the TG resident CD8+ T cell population could have therapeutic efficacy. This approach has been tested by giving HSV-1 latently infected mice a live attenuated HSV-1 therapeutic vaccine prior to subjecting the cornea to UV-B irradiation to induce HSV-1 reactivation. The therapeutic vaccine reduced detectable HSV-1 shedding at the corneal surface, and elevated the anti-HSV antibody titer in the mice (Keadle et al., 2002; Shimeld et al., 1990b). It was unclear from the assay performed in these studies if the vaccine actually inhibited reactivation of the virus from latency in TG neurons, or inhibited expansion and/or detection of live virus in the tear film. Moreover, it was not determined if the vaccine boosted the CD8 resident population in the TG.
Although vaccines tested to date in clinical trials have lacked efficacy in preventing recurrent disease or viral transmission, these vaccines were designed primarily to augment antibody responses (Corey et al., 1999; Cremer et al., 1985; Ghiasi et al., 1995; Ghiasi et al., 1998; Koelle and Corey, 2003; McLean et al., 1996). Anti-HSV-1 antibodies can inhibit initial infection and reduce extracellular spread of virus, but their efficacy in preventing reactivation and cell-to-cell spread of virus through membrane fusion appears minimal. Thus, a vaccine that targets T cells might be more efficacious. However, the results of a recent study provide an important caveat (Himmelein et al., 2011). HSV-specific CD8+ T cells bearing the Thy 1.1 marker were adoptively transferred into Thy1.2 mice harboring latent virus infections. This was done to mimic the increased level of circulating HSV-specific CD8+ T cells that might result from a therapeutic vaccine, and to trace the migration of these cells in vivo. The transferred CD8+ T cells were retained at high levels in the blood of recipient mice for at least 8.5 weeks, but none of the transferred cells were detected in the latently infected TG. In another study, latently infected TG were transplanted under the kidney capsule of recipient mice with congenically marked CD8+ T cells (Gebhardt et al., 2009; Wakim et al., 2008). The CD8+ T cell population in the transplanted TG declined dramatically following transplant, accompanied by HSV-1 reactivation from latency. Interestingly, the CD8+ T cell population was quickly replenished in the TG, but with no contribution from host CD8+ T cells. These findings suggest that the memory CD8+ T cell population in the TG, once established is compartmentalized and maintained without observable replenishment from the blood. This apparent compartmentalization of the TG resident CD8+ T cell population would seem to present a significant obstacle to vaccine approaches designed to increase CD8+ T cells in the TG. Nonetheless, exploiting the ability of CD8+ T cells to block HSV-1 reactivation from latency seems to hold great promise as a means of preventing recurrent disease.
6. The final recourse for scarred corneas
Treatment of blindness due to HSK-associated corneal opacification is surgical transplantation of a clear donor cornea. However, such grafts are considered “high-risk” as they frequently fail (Williams et al., 1995, 1997). This high-risk phenotype is attributed to a number of causes including the extensive corneal vascularization and leukocytic infiltration associated with HSK (Biswas and Rouse, 2005). However, corneal transplants are usually attempted after inflammation has largely subsided. Therefore, a contribution of factors other than pre-existing inflammation and vascularization cannot be ruled out. Latently infected TG neurons are severed during excision of the host cornea, which might induce HSV-1 reactivation and transport to the graft site. HSV-1 shedding at the graft interface could result in direct viral damage or immunopathology due to an HSV-specific recall response (Kaye and Choudhary, 2006; Kaye et al., 2000). Indirect evidence supporting a contribution of HSV-1 to corneal transplant rejection derives from human clinical studies showing that HSV-1 DNA and even virus particles are detectable in host cornea, donor rejected corneas, and aqueous samples from patients with rejecting corneal grafts (Remeijer et al., 2009). Such data has led to the common practice of antiviral prophylaxis and postoperative treatment in HSK patients preparing for corneal transplantation (1992; Ficker et al., 1989). In support of a viral reactivation mechanism, experimental studies in rabbits have shown an increase in viral shedding in tear fluid after lamellar corneal grafts with some epithelial lesions but no development of HSK (Openshaw et al., 1995; Zheng et al., 1999). In contrast, there did not seem to be a correlation between virus shedding after grafting and eventual allograft rejection in rats (Nicholls et al., 1996). Clarification of the mechanism(s) resulting in the high risk status of HSV-1 infected corneas is an important issue that will require additional investigation. In most cases, corneal transplantation involves replacement of the scarred cornea with a full thickness donor cornea in a surgical procedure called penetrating keratoplasty. A more recently developed procedure called Deep Anterior Lamellar Keratoplasty (DALK) is an alternative approach that can be used in cases where the host corneal endothelium is healthy. Since visual impairment associated with HSK results from corneal stromal scarring, DLAK offers the option of replacing only the corneal stroma, while maintaining host endothelium, which is often the focus of transplant rejection. Whether or not this new surgical procedure will improve the outcome of corneal transplantation in patients with a history of HSK remains to be determined. Moreover, even if the corneal graft is successful, HSK can recur within the transplanted cornea (Fig. 2). Presumably, once the transplanted cornea becomes reinnervated by sensory neurons, the cycle of recurrent infection and inflammation can resume.
Figure 2.

A) Slit lamp photograph of a patient with HSK demonstrating central stromal opacity causing significant visual impairment.
B) Slit lamp photograph of the same patient approximately two years after undergoing deep anterior lamellar keratoplasty demonstrating recurrence of HSV with large geographic ulcer stained with fluorescein dye and deep stromal vessels. Of note, the patient was noncompliant with post-operative medications and stopped all topical steroids and systemic antiviral therapy against medical advice
7. Concluding thoughts
It emerges from the above discussion that our understanding of herpes keratitis has advanced significantly through many years of research. However, much discovery lies ahead. For instance, additional research is required to explain the seemingly stochastic nature of the disease. Many humans harbor latent HSV-1 in their trigeminal ganglia and even shed the virus at the corneal surface, but never develop epithelial or stromal keratitis (Hill and Clement, 2009; Kaufman et al., 2005). Also, many people suffer recurrent HSV-1 epithelial lesions, but never develop HSK. Moreover, even genetically identical mice that are infected with the same dose of the same virus strain exhibit a variable incidence of HSK. A more complete understanding of the complex relationship between HSV-1 and its host is needed. Only a full understanding of the function of viral genes, host cell regulation of viral gene expression, and the contribution of viral and host factors to the antiviral immune response will permit delineation of the unique pathological state of infection in each individual. The journey has been long and arduous, but frequently interrupted with the excitement of discoveries. Recent advances in immunology and molecular virology are bringing the goal of eradicating herpes keratitis into focus.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- The collaborative corneal transplantation studies (CCTS). Effectiveness of histocompatibility matching in high-risk corneal transplantation. The Collaborative Corneal Transplantation Studies Research Group. Arch Ophthalmol. 1992;110:1392–1403. [PubMed] [Google Scholar]
- Acyclovir for the prevention of recurrent herpes simplex virus eye disease. Herpetic Eye Disease Study Group. N Engl J Med. 1998;339:300–306. doi: 10.1056/NEJM199807303390503. [DOI] [PubMed] [Google Scholar]
- Allan RS, Smith CM, Belz GT, van Lint AL, Wakim LM, Heath WR, Carbone FR. Epidermal viral immunity induced by CD8alpha+ dendritic cells but not by Langerhans cells. Science. 2003;301:1925–1928. doi: 10.1126/science.1087576. [DOI] [PubMed] [Google Scholar]
- Allan RS, Waithman J, Bedoui S, Jones CM, Villadangos JA, Zhan Y, Lew AM, Shortman K, Heath WR, Carbone FR. Migratory dendritic cells transfer antigen to a lymph node-resident dendritic cell population for efficient CTL priming. Immunity. 2006;25:153–162. doi: 10.1016/j.immuni.2006.04.017. [DOI] [PubMed] [Google Scholar]
- Ambati BK, Anand A, Joussen AM, Kuziel WA, Adamis AP, Ambati J. Sustained inhibition of corneal neovascularization by genetic ablation of CCR5. Invest Ophthalmol Vis Sci. 2003;44:590–593. doi: 10.1167/iovs.02-0685. [DOI] [PubMed] [Google Scholar]
- Ambati BK, Nozaki M, Singh N, Takeda A, Jani PD, Suthar T, Albuquerque RJ, Richter E, Sakurai E, Newcomb MT, et al. Corneal avascularity is due to soluble VEGF receptor-1. Nature. 2006;443:993–997. doi: 10.1038/nature05249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avery AC, Zhao ZS, Rodriguez A, Bikoff EK, Soheilian M, Foster CS, Cantor H. Resistance to herpes stromal keratitis conferred by an IgG2a-derived peptide. Nature. 1995;376:431–434. doi: 10.1038/376431a0. [DOI] [PubMed] [Google Scholar]
- Banerjee K, Biswas PS, Kim B, Lee S, Rouse BT. CXCR2−/− mice show enhanced susceptibility to herpetic stromal keratitis: a role for IL-6-induced neovascularization. J Immunol. 2004a;172:1237–1245. doi: 10.4049/jimmunol.172.2.1237. [DOI] [PubMed] [Google Scholar]
- Banerjee K, Biswas PS, Kumaraguru U, Schoenberger SP, Rouse BT. Protective and pathological roles of virus-specific and bystander CD8+ T cells in herpetic stromal keratitis. J Immunol. 2004b;173:7575–7583. doi: 10.4049/jimmunol.173.12.7575. [DOI] [PubMed] [Google Scholar]
- Bedoui S, Whitney PG, Waithman J, Eidsmo L, Wakim L, Caminschi I, Allan RS, Wojtasiak M, Shortman K, Carbone FR, et al. Cross-presentation of viral and self antigens by skin-derived CD103+ dendritic cells. Nat Immunol. 2009;10:488–495. doi: 10.1038/ni.1724. [DOI] [PubMed] [Google Scholar]
- Bertke AS, Swanson SM, Chen J, Imai Y, Kinchington PR, Margolis TP. A5-positive primary sensory neurons are nonpermissive for productive infection with herpes simplex virus 1 in vitro. J Virol. 2011;85:6669–6677. doi: 10.1128/JVI.00204-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswas PS, Rouse BT. Early events in HSV keratitis--setting the stage for a blinding disease. Microbes and infection/Institut Pasteur. 2005;7:799–810. doi: 10.1016/j.micinf.2005.03.003. [DOI] [PubMed] [Google Scholar]
- Bloom DC, Giordani NV, Kwiatkowski DL. Epigenetic regulation of latent HSV-1 gene expression. Biochim Biophys Acta. 2010;1799:246–256. doi: 10.1016/j.bbagrm.2009.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blyth WA, Hill TJ, Field HJ, Harbour DA. Reactivation of herpes simplex virus infection by ultraviolet light and possible involvement of prostaglandins. J Gen Virol. 1976;33:547–550. doi: 10.1099/0022-1317-33-3-547. [DOI] [PubMed] [Google Scholar]
- Bonneau RH. Stress-induced effects on integral immune components involved in herpes simplex virus (HSV)-specific memory cytotoxic T lymphocyte activation. Brain Behav Immun. 1996;10:139–163. doi: 10.1006/brbi.1996.0014. [DOI] [PubMed] [Google Scholar]
- Bonneau RH, Sheridan JF, Feng N, Glaser R. Stress-induced modulation of the primary cellular immune response to herpes simplex virus infection is mediated by both adrenal-dependent and independent mechanisms. J Neuroimmunol. 1993;42:167–176. doi: 10.1016/0165-5728(93)90007-l. [DOI] [PubMed] [Google Scholar]
- Bonneau RH, Sheridan JF, Feng NG, Glaser R. Stress-induced effects on cell-mediated innate and adaptive memory components of the murine immune response to herpes simplex virus infection. Brain Behav Immun. 1991;5:274–295. doi: 10.1016/0889-1591(91)90023-4. [DOI] [PubMed] [Google Scholar]
- Bonneau RH, Zimmerman KM, Ikeda SC, Jones BC. Differential effects of stress-induced adrenal function on components of the herpes simplex virus-specific memory cytotoxic T-lymphocyte response. J Neuroimmunol. 1998;82:191–199. doi: 10.1016/s0165-5728(97)00200-2. [DOI] [PubMed] [Google Scholar]
- Bouley DM, Kanangat S, Wire W, Rouse BT. Characterization of herpes simplex virus type-1 infection and herpetic stromal keratitis development in IFN-gamma knockout mice. J Immunol. 1995;155:3964–3971. [PubMed] [Google Scholar]
- Brissette-Storkus CS, Reynolds SM, Lepisto AJ, Hendricks RL. Identification of a novel macrophage population in the normal mouse corneal stroma. Invest Ophthalmol Vis Sci. 2002;43:2264–2271. [PMC free article] [PubMed] [Google Scholar]
- Carr DJ, Ash J, Lane TE, Kuziel WA. Abnormal immune response of CCR5-deficient mice to ocular infection with herpes simplex virus type 1. J Gen Virol. 2006;87:489–499. doi: 10.1099/vir.0.81339-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr DJ, Wuest T, Ash J. An increase in herpes simplex virus type 1 in the anterior segment of the eye is linked to a deficiency in NK cell infiltration in mice deficient in CXCR3. J Interferon Cytokine Res. 2008;28:245–251. doi: 10.1089/jir.2007.0110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang EJ, Dreyer EB. Herpesvirus infections of the anterior segment. Int Ophthalmol Clin. 1996;36:17–28. doi: 10.1097/00004397-199603630-00004. [DOI] [PubMed] [Google Scholar]
- Chen W, Tang Q, Hendricks RL. Ex vivo model of leukocyte migration into herpes simplex virus-infected mouse corneas. J Leukoc Biol. 1996;60:167–173. doi: 10.1002/jlb.60.2.167. [DOI] [PubMed] [Google Scholar]
- Cheng H, Tumpey TM, Staats HF, van Rooijen N, Oakes JE, Lausch RN. Role of macrophages in restricting herpes simplex virus type 1 growth after ocular infection. Invest Ophthalmol Vis Sci. 2000;41:1402–1409. [PubMed] [Google Scholar]
- Chinnery HR, Pearlman E, McMenamin PG. Cutting edge: Membrane nanotubes in vivo: a feature of MHC class II+ cells in the mouse cornea. J Immunol. 2008;180:5779–5783. doi: 10.4049/jimmunol.180.9.5779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chodosh J, Banks MC, Stroop WG. Rose bengal inhibits herpes simplex virus replication in vero and human corneal epithelial cells in vitro. Invest Ophthalmol Vis Sci. 1992;33:2520–2527. [PubMed] [Google Scholar]
- Cliffe AR, Garber DA, Knipe DM. Transcription of the herpes simplex virus latency-associated transcript promotes the formation of facultative heterochromatin on lytic promoters. J Virol. 2009;83:8182–8190. doi: 10.1128/JVI.00712-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrady CD, Jones H, Zheng M, Carr DJ. A Functional Type I Interferon Pathway Drives Resistance to Cornea Herpes Simplex Virus Type 1 Infection by Recruitment of Leukocytes. J Biomed Res. 2011;25:111–119. doi: 10.1016/S1674-8301(11)60014-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrady CD, Zheng M, Fitzgerald KA, Liu C, Carr DJ. Resistance to HSV-1 infection in the epithelium resides with the novel innate sensor, IFI-16. Mucosal Immunol. 2012;5:173–183. doi: 10.1038/mi.2011.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corey L, Langenberg AG, Ashley R, Sekulovich RE, Izu AE, Douglas JM, Jr, Handsfield HH, Warren T, Marr L, Tyring S, et al. Recombinant glycoprotein vaccine for the prevention of genital HSV-2 infection: two randomized controlled trials. Chiron HSV Vaccine Study Group. JAMA. 1999;282:331–340. doi: 10.1001/jama.282.4.331. [DOI] [PubMed] [Google Scholar]
- Cremer KJ, Mackett M, Wohlenberg C, Notkins AL, Moss B. Vaccinia virus recombinant expressing herpes simplex virus type 1 glycoprotein D prevents latent herpes in mice. Science. 1985;228:737–740. doi: 10.1126/science.2986288. [DOI] [PubMed] [Google Scholar]
- Croen KD. Evidence for antiviral effect of nitric oxide. Inhibition of herpes simplex virus type 1 replication. J Clin Invest. 1993;91:2446–2452. doi: 10.1172/JCI116479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darougar S, Hunter PA, Viswalingam M, Gibson JA, Jones BR. Acute follicular conjunctivitis and keratoconjunctivitis due to herpes simplex virus in London. Br J Ophthalmol. 1978;62:843–849. doi: 10.1136/bjo.62.12.843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darougar S, Wishart MS, Viswalingam ND. Epidemiological and clinical features of primary herpes simplex virus ocular infection. Br J Ophthalmol. 1985;69:2–6. doi: 10.1136/bjo.69.1.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decman V, Kinchington PR, Harvey SA, Hendricks RL. Gamma interferon can block herpes simplex virus type 1 reactivation from latency, even in the presence of late gene expression. J Virol. 2005;79:10339–10347. doi: 10.1128/JVI.79.16.10339-10347.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshpande SP, Lee S, Zheng M, Song B, Knipe D, Kapp JA, Rouse BT. Herpes simplex virus-induced keratitis: evaluation of the role of molecular mimicry in lesion pathogenesis. J Virol. 2001;75:3077–3088. doi: 10.1128/JVI.75.7.3077-3088.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Divito SJ, Hendricks RL. Activated inflammatory infiltrate in HSV-1-infected corneas without herpes stromal keratitis. Invest Ophthalmol Vis Sci. 2008;49:1488–1495. doi: 10.1167/iovs.07-1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donaghy H, Bosnjak L, Harman AN, Marsden V, Tyring SK, Meng TC, Cunningham AL. Role for plasmacytoid dendritic cells in the immune control of recurrent human herpes simplex virus infection. J Virol. 2009;83:1952–1961. doi: 10.1128/JVI.01578-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doymaz MZ, Rouse BT. Herpetic stromal keratitis: an immunopathologic disease mediated by CD4+ T lymphocytes. Invest Ophthalmol Vis Sci. 1992;33:2165–2173. [PubMed] [Google Scholar]
- Du T, Zhou G, Roizman B. HSV-1 gene expression from reactivated ganglia is disordered and concurrent with suppression of latency-associated transcript and miRNAs. Proc Natl Acad Sci U S A. 2011;108:18820–18824. doi: 10.1073/pnas.1117203108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott JM, Yokoyama WM. Unifying concepts of MHC-dependent natural killer cell education. Trends Immunol. 2011;32:364–372. doi: 10.1016/j.it.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrell MJ, Dobson AT, Feldman LT. Herpes simplex virus latency-associated transcript is a stable intron. Proc Natl Acad Sci U S A. 1991;88:790–794. doi: 10.1073/pnas.88.3.790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman LT, Ellison AR, Voytek CC, Yang L, Krause P, Margolis TP. Spontaneous molecular reactivation of herpes simplex virus type 1 latency in mice. Proc Natl Acad Sci U S A. 2002;99:978–983. doi: 10.1073/pnas.022301899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenton RR, Molesworth-Kenyon S, Oakes JE, Lausch RN. Linkage of IL-6 with neutrophil chemoattractant expression in virus-induced ocular inflammation. Invest Ophthalmol Vis Sci. 2002;43:737–743. [PubMed] [Google Scholar]
- Fernandez NC, Lozier A, Flament C, Ricciardi-Castagnoli P, Bellet D, Suter M, Perricaudet M, Tursz T, Maraskovsky E, Zitvogel L. Dendritic cells directly trigger NK cell functions: cross-talk relevant in innate anti-tumor immune responses in vivo. Nat Med. 1999;5:405–411. doi: 10.1038/7403. [DOI] [PubMed] [Google Scholar]
- Ficker LA, Kirkness CM, Rice NS, Steele AD. The changing management and improved prognosis for corneal grafting in herpes simplex keratitis. Ophthalmology. 1989;96:1587–1596. doi: 10.1016/s0161-6420(89)32668-6. [DOI] [PubMed] [Google Scholar]
- Frank GM, Buela KA, Maker DM, Harvey SA, Hendricks RL. Early responding dendritic cells direct the local NK response to control herpes simplex virus 1 infection within the cornea. J Immunol. 2012;188:1350–1359. doi: 10.4049/jimmunol.1101968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman ML, Sheridan BS, Bonneau RH, Hendricks RL. Psychological stress compromises CD8+ T cell control of latent herpes simplex virus type 1 infections. J Immunol. 2007;179:322–328. doi: 10.4049/jimmunol.179.1.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda M, Deai T, Higaki S, Hayashi K, Shimomura Y. Presence of a large amount of herpes simplex virus genome in tear fluid of herpetic stromal keratitis and persistent epithelial defect patients. Semin Ophthalmol. 2008;23:217–220. doi: 10.1080/08820530802111366. [DOI] [PubMed] [Google Scholar]
- Gamba G, Cavalieri H, Courreges MC, Massouh EJ, Benencia F. Early inhibition of nitric oxide production increases HSV-1 intranasal infection. J Med Virol. 2004;73:313–322. doi: 10.1002/jmv.20093. [DOI] [PubMed] [Google Scholar]
- Gangappa S, Babu JS, Thomas J, Daheshia M, Rouse BT. Virus-induced immunoinflammatory lesions in the absence of viral antigen recognition. J Immunol. 1998;161:4289–4300. [PubMed] [Google Scholar]
- Gangappa S, Deshpande SP, Rouse BT. Bystander activation of CD4(+) T cells can represent an exclusive means of immunopathology in a virus infection. Eur J Immunol. 1999;29:3674–3682. doi: 10.1002/(SICI)1521-4141(199911)29:11<3674::AID-IMMU3674>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- Gangappa S, Deshpande SP, Rouse BT. Bystander activation of CD4+ T cells accounts for herpetic ocular lesions. Invest Ophthalmol Vis Sci. 2000;41:453–459. [PubMed] [Google Scholar]
- Gardella T, Medveczky P, Sairenji T, Mulder C. Detection of circular and linear herpesvirus DNA molecules in mammalian cells by gel electrophoresis. J Virol. 1984;50:248–254. doi: 10.1128/jvi.50.1.248-254.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gebhardt BM, Halford WP. Evidence that spontaneous reactivation of herpes virus does not occur in mice. Virol J. 2005;2:67. doi: 10.1186/1743-422X-2-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gebhardt BM, Hill JM. T lymphocytes in the trigeminal ganglia of rabbits during corneal HSV infection. Invest Ophthalmol Vis Sci. 1988;29:1683–1691. [PubMed] [Google Scholar]
- Gebhardt T, Wakim LM, Eidsmo L, Reading PC, Heath WR, Carbone FR. Memory T cells in nonlymphoid tissue that provide enhanced local immunity during infection with herpes simplex virus. Nat Immunol. 2009;10:524–530. doi: 10.1038/ni.1718. [DOI] [PubMed] [Google Scholar]
- Geissmann F, Jung S, Littman DR. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity. 2003;19:71–82. doi: 10.1016/s1074-7613(03)00174-2. [DOI] [PubMed] [Google Scholar]
- Ghiasi H, Cai S, Perng GC, Nesburn AB, Wechsler SL. The role of natural killer cells in protection of mice against death and corneal scarring following ocular HSV-1 infection. Antiviral Res. 2000;45:33–45. doi: 10.1016/s0166-3542(99)00075-3. [DOI] [PubMed] [Google Scholar]
- Ghiasi H, Cai S, Slanina S, Nesburn AB, Wechsler SL. Vaccination of mice with herpes simplex virus type 1 glycoprotein D DNA produces low levels of protection against lethal HSV-1 challenge. Antiviral Res. 1995;28:147–157. doi: 10.1016/0166-3542(95)00045-n. [DOI] [PubMed] [Google Scholar]
- Ghiasi H, Wechsler SL, Cai S, Nesburn AB, Hofman FM. The role of neutralizing antibody and T-helper subtypes in protection and pathogenesis of vaccinated mice following ocular HSV-1 challenge. Immunology. 1998;95:352–359. doi: 10.1046/j.1365-2567.1998.00602.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glaser R, Kiecolt-Glaser JK. Stress-induced immune dysfunction: implications for health. Nat Rev Immunol. 2005;5:243–251. doi: 10.1038/nri1571. [DOI] [PubMed] [Google Scholar]
- Goldsmith K, Chen W, Johnson DC, Hendricks RL. Infected cell protein (ICP)47 enhances herpes simplex virus neurovirulence by blocking the CD8+ T cell response. J Exp Med. 1998;187:341–348. doi: 10.1084/jem.187.3.341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green LK, Pavan-Langston D. Herpes simplex ocular inflammatory disease. Int Ophthalmol Clin. 2006;46:27–37. doi: 10.1097/00004397-200604620-00005. [DOI] [PubMed] [Google Scholar]
- Groom JR, Luster AD. CXCR3 in T cell function. Exp Cell Res. 2011a;317:620–631. doi: 10.1016/j.yexcr.2010.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groom JR, Luster AD. CXCR3 ligands: redundant, collaborative and antagonistic functions. Immunol Cell Biol. 2011b;89:207–215. doi: 10.1038/icb.2010.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamrah P, Huq SO, Liu Y, Zhang Q, Dana MR. Corneal immunity is mediated by heterogeneous population of antigen-presenting cells. J Leukoc Biol. 2003a;74:172–178. doi: 10.1189/jlb.1102544. [DOI] [PubMed] [Google Scholar]
- Hamrah P, Liu Y, Zhang Q, Dana MR. The corneal stroma is endowed with a significant number of resident dendritic cells. Invest Ophthalmol Vis Sci. 2003b;44:581–589. doi: 10.1167/iovs.02-0838. [DOI] [PubMed] [Google Scholar]
- Hayashi K, Hooper LC, Chin MS, Nagineni CN, Detrick B, Hooks JJ. Herpes simplex virus 1 (HSV-1) DNA and immune complex (HSV-1-human IgG) elicit vigorous interleukin 6 release from infected corneal cells via Toll-like receptors. J Gen Virol. 2006;87:2161–2169. doi: 10.1099/vir.0.81772-0. [DOI] [PubMed] [Google Scholar]
- Henderson G, Peng W, Jin L, Perng GC, Nesburn AB, Wechsler SL, Jones C. Regulation of caspase 8- and caspase 9-induced apoptosis by the herpes simplex virus type 1 latency-associated transcript. J Neurovirol. 2002;8(Suppl 2):103–111. doi: 10.1080/13550280290101085. [DOI] [PubMed] [Google Scholar]
- Hendricks RL, Janowicz M, Tumpey TM. Critical role of corneal Langerhans cells in the CD4- but not CD8-mediated immunopathology in herpes simplex virus-1-infected mouse corneas. J Immunol. 1992a;148:2522–2529. [PubMed] [Google Scholar]
- Hendricks RL, Tumpey TM, Finnegan A. IFN-gamma and IL-2 are protective in the skin but pathologic in the corneas of HSV-1-infected mice. J Immunol. 1992b;149:3023–3028. [PubMed] [Google Scholar]
- Hill JM, Clement C. Herpes simplex virus type 1 DNA in human corneas: what are the virological and clinical implications? J Infect Dis. 2009;200:1–4. doi: 10.1086/599330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Himmelein S, St Leger AJ, Knickelbein JE, Rowe A, Freeman ML, Hendricks RL. Circulating herpes simplex type 1 (HSV-1)-specific CD8+ T cells do not access HSV-1 latently infected trigeminal ganglia. Herpesviridae. 2011;2:5. doi: 10.1186/2042-4280-2-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirakawa S, Brown LF, Kodama S, Paavonen K, Alitalo K, Detmar M. VEGFC-induced lymphangiogenesis in sentinel lymph nodes promotes tumor metastasis to distant sites. Blood. 2007;109:1010–1017. doi: 10.1182/blood-2006-05-021758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland EJ, Schwartz GS. Classification of herpes simplex virus keratitis. Cornea. 1999;18:144–154. doi: 10.1097/00003226-199903000-00002. [DOI] [PubMed] [Google Scholar]
- Hunter CA, Gabriel KE, Radzanowski T, Neyer LE, Remington JS. Type I interferons enhance production of IFN-gamma by NK cells. Immunol Lett. 1997;59:1–5. doi: 10.1016/s0165-2478(97)00091-6. [DOI] [PubMed] [Google Scholar]
- Huster KM, Panoutsakopoulou V, Prince K, Sanchirico ME, Cantor H. T cell-dependent and -independent pathways to tissue destruction following herpes simplex virus-1 infection. Eur J Immunol. 2002;32:1414–1419. doi: 10.1002/1521-4141(200205)32:5<1414::AID-IMMU1414>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- Janeway C. Immunobiology: the immune system in health and disease. 6. New York: Garland Science; 2005. [Google Scholar]
- Jones BR. The clinical features of viral keratitis and a concept of their pathogenesis. Proc R Soc Med. 1958;51:917–924. [PMC free article] [PubMed] [Google Scholar]
- Kanangat S, Thomas J, Gangappa S, Babu JS, Rouse BT. Herpes simplex virus type 1-mediated up-regulation of IL-12 (p40) mRNA expression. Implications in immunopathogenesis and protection. J Immunol. 1996;156:1110–1116. [PubMed] [Google Scholar]
- Kasman LM, Biber JM, Vroman DT. Immunoglobulin gene implicated in murine herpes stromal keratitis is not associated with the human disease. Cornea. 2006;25:1069–1071. doi: 10.1097/01.ico.0000220774.86228.c6. [DOI] [PubMed] [Google Scholar]
- Kassim SH, Rajasagi NK, Ritz BW, Pruett SB, Gardner EM, Chervenak R, Jennings SR. Dendritic cells are required for optimal activation of natural killer functions following primary infection with herpes simplex virus type 1. J Virol. 2009;83:3175–3186. doi: 10.1128/JVI.01907-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kastrukoff LF, Lau AS, Takei F, Smyth MJ, Jones CM, Clarke SR, Carbone FR. Redundancy in the immune system restricts the spread of HSV-1 in the central nervous system (CNS) of C57BL/6 mice. Virology. 2010;400:248–258. doi: 10.1016/j.virol.2010.02.013. [DOI] [PubMed] [Google Scholar]
- Kaufman HE, Azcuy AM, Varnell ED, Sloop GD, Thompson HW, Hill JM. HSV-1 DNA in tears and saliva of normal adults. Invest Ophthalmol Vis Sci. 2005;46:241–247. doi: 10.1167/iovs.04-0614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaye S, Choudhary A. Herpes simplex keratitis. Progress in retinal and eye research. 2006;25:355–380. doi: 10.1016/j.preteyeres.2006.05.001. [DOI] [PubMed] [Google Scholar]
- Kaye SB, Baker K, Bonshek R, Maseruka H, Grinfeld E, Tullo A, Easty DL, Hart CA. Human herpesviruses in the cornea. The British journal of ophthalmology. 2000;84:563–571. doi: 10.1136/bjo.84.6.563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keadle TL, Laycock KA, Morris JL, Leib DA, Morrison LA, Pepose JS, Stuart PM. Therapeutic vaccination with vhs(−) herpes simplex virus reduces the severity of recurrent herpetic stromal keratitis in mice. J Gen Virol. 2002;83:2361–2365. doi: 10.1099/0022-1317-83-10-2361. [DOI] [PubMed] [Google Scholar]
- Khanna KM, Bonneau RH, Kinchington PR, Hendricks RL. Herpes simplex virus-specific memory CD8+ T cells are selectively activated and retained in latently infected sensory ganglia. Immunity. 2003;18:593–603. doi: 10.1016/s1074-7613(03)00112-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim B, Tang Q, Biswas PS, Xu J, Schiffelers RM, Xie FY, Ansari AM, Scaria PV, Woodle MC, Lu P, et al. Inhibition of ocular angiogenesis by siRNA targeting vascular endothelial growth factor pathway genes: therapeutic strategy for herpetic stromal keratitis. Am J Pathol. 2004;165:2177–2185. doi: 10.1016/S0002-9440(10)63267-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JY, Mandarino A, Chao MV, Mohr I, Wilson AC. Transient reversal of episome silencing precedes VP16-dependent transcription during reactivation of latent HSV-1 in neurons. PLoS Pathog. 2012;8:e1002540. doi: 10.1371/journal.ppat.1002540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knickelbein JE, Hendricks RL, Charukamnoetkanok P. Management of herpes simplex virus stromal keratitis: an evidence-based review. Surv Ophthalmol. 2009a;54:226–234. doi: 10.1016/j.survophthal.2008.12.004. [DOI] [PubMed] [Google Scholar]
- Knickelbein JE, Khanna KM, Yee MB, Baty CJ, Kinchington PR, Hendricks RL. Noncytotoxic lytic granule-mediated CD8+ T cell inhibition of HSV-1 reactivation from neuronal latency. Science. 2008;322:268–271. doi: 10.1126/science.1164164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knickelbein JE, Watkins SC, McMenamin PG, Hendricks RL. Stratification of Antigen-presenting Cells within the Normal Cornea. Ophthalmol Eye Dis. 2009b;1:45–54. doi: 10.4137/oed.s2813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kodukula P, Liu T, Rooijen NV, Jager MJ, Hendricks RL. Macrophage control of herpes simplex virus type 1 replication in the peripheral nervous system. J Immunol. 1999;162:2895–2905. [PubMed] [Google Scholar]
- Koelle DM, Corey L. Recent progress in herpes simplex virus immunobiology and vaccine research. Clin Microbiol Rev. 2003;16:96–113. doi: 10.1128/CMR.16.1.96-113.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu K, Miyazaki D, Morohoshi K, Kuo CH, Kakimaru-Hasegawa A, Komatsu N, Namba S, Haino M, Matsushima K, Inoue Y. Pathogenesis of herpetic stromal keratitis in CCR5- and/or CXCR3-deficient mice. Curr Eye Res. 2008;33:736–749. doi: 10.1080/02713680802344716. [DOI] [PubMed] [Google Scholar]
- Krug A, Luker GD, Barchet W, Leib DA, Akira S, Colonna M. Herpes simplex virus type 1 activates murine natural interferon-producing cells through toll-like receptor 9. Blood. 2004;103:1433–1437. doi: 10.1182/blood-2003-08-2674. [DOI] [PubMed] [Google Scholar]
- Kumaraguru U, Davis I, Rouse BT. Chemokines and ocular pathology caused by corneal infection with herpes simplex virus. J Neurovirol. 1999;5:42–47. doi: 10.3109/13550289909029744. [DOI] [PubMed] [Google Scholar]
- Labetoulle M, Auquier P, Conrad H, Crochard A, Daniloski M, Bouee S, El Hasnaoui A, Colin J. Incidence of herpes simplex virus keratitis in France. Ophthalmology. 2005;112:888–895. doi: 10.1016/j.ophtha.2004.11.052. [DOI] [PubMed] [Google Scholar]
- Lang A, Nikolich-Zugich J. Development and migration of protective CD8+ T cells into the nervous system following ocular herpes simplex virus-1 infection. J Immunol. 2005;174:2919–2925. doi: 10.4049/jimmunol.174.5.2919. [DOI] [PubMed] [Google Scholar]
- Lee HK, Zamora M, Linehan MM, Iijima N, Gonzalez D, Haberman A, Iwasaki A. Differential roles of migratory and resident DCs in T cell priming after mucosal or skin HSV-1 infection. J Exp Med. 2009;206:359–370. doi: 10.1084/jem.20080601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee I, Wang L, Wells AD, Ye Q, Han R, Dorf ME, Kuziel WA, Rollins BJ, Chen L, Hancock WW. Blocking the monocyte chemoattractant protein-1/CCR2 chemokine pathway induces permanent survival of islet allografts through a programmed death-1 ligand-1-dependent mechanism. J Immunol. 2003;171:6929–6935. doi: 10.4049/jimmunol.171.12.6929. [DOI] [PubMed] [Google Scholar]
- Lee SK, Choi BK, Kang WJ, Kim YH, Park HY, Kim KH, Kwon BS. MCP-1 derived from stromal keratocyte induces corneal infiltration of CD4+ T cells in herpetic stromal keratitis. Mol Cells. 2008;26:67–73. [PubMed] [Google Scholar]
- Leib DA, Harrison TE, Laslo KM, Machalek MA, Moorman NJ, Virgin HW. Interferons regulate the phenotype of wild-type and mutant herpes simplex viruses in vivo. J Exp Med. 1999;189:663–672. doi: 10.1084/jem.189.4.663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leo NA, Bonneau RH. Mechanisms underlying chemical sympathectomy-induced suppression of herpes simplex virus-specific cytotoxic T lymphocyte activation and function. J Neuroimmunol. 2000;110:45–56. doi: 10.1016/s0165-5728(00)00336-2. [DOI] [PubMed] [Google Scholar]
- Lepisto AJ, Frank GM, Xu M, Stuart PM, Hendricks RL. CD8 T cells mediate transient herpes stromal keratitis in CD4-deficient mice. Invest Ophthalmol Vis Sci. 2006;47:3400–3409. doi: 10.1167/iovs.05-0898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liesegang TJ. Epidemiology and natural history of ocular herpes simplex virus infection in Rochester, Minnesota, 1950–1982. Trans Am Ophthalmol Soc. 1988;86:688–724. [PMC free article] [PubMed] [Google Scholar]
- Liesegang TJ. Epidemiology of ocular herpes simplex. Natural history in Rochester, Minn, 1950 through 1982. Arch Ophthalmol. 1989;107:1160–1165. doi: 10.1001/archopht.1989.01070020226030. [DOI] [PubMed] [Google Scholar]
- Liesegang TJ. Classification of herpes simplex virus keratitis and anterior uveitis. Cornea. 1999;18:127–143. doi: 10.1097/00003226-199903000-00001. [DOI] [PubMed] [Google Scholar]
- Liesegang TJ. Herpes simplex virus epidemiology and ocular importance. Cornea. 2001;20:1–13. doi: 10.1097/00003226-200101000-00001. [DOI] [PubMed] [Google Scholar]
- Liesegang TJ, Melton LJ, 3rd, Daly PJ, Ilstrup DM. Epidemiology of ocular herpes simplex. Incidence in Rochester, Minn, 1950 through 1982. Arch Ophthalmol. 1989;107:1155–1159. doi: 10.1001/archopht.1989.01070020221029. [DOI] [PubMed] [Google Scholar]
- Lima GK, Zolini GP, Mansur DS, Freire Lima BH, Wischhoff U, Astigarraga RG, Dias MF, das Gracas Almeida Silva M, Bela SR, do Valle Antonelli LR, et al. Toll-like receptor (TLR) 2 and TLR9 expressed in trigeminal ganglia are critical to viral control during herpes simplex virus 1 infection. Am J Pathol. 2010;177:2433–2445. doi: 10.2353/ajpath.2010.100121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin M, Carlson E, Diaconu E, Pearlman E. CXCL1/KC and CXCL5/LIX are selectively produced by corneal fibroblasts and mediate neutrophil infiltration to the corneal stroma in LPS keratitis. J Leukoc Biol. 2007;81:786–792. doi: 10.1189/jlb.0806502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T, Khanna KM, Carriere BN, Hendricks RL. Gamma interferon can prevent herpes simplex virus type 1 reactivation from latency in sensory neurons. J Virol. 2001;75:11178–11184. doi: 10.1128/JVI.75.22.11178-11184.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T, Khanna KM, Chen X, Fink DJ, Hendricks RL. CD8(+) T cells can block herpes simplex virus type 1 (HSV-1) reactivation from latency in sensory neurons. J Exp Med. 2000;191:1459–1466. doi: 10.1084/jem.191.9.1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T, Tang Q, Hendricks RL. Inflammatory infiltration of the trigeminal ganglion after herpes simplex virus type 1 corneal infection. J Virol. 1996;70:264–271. doi: 10.1128/jvi.70.1.264-271.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luker GD, Prior JL, Song J, Pica CM, Leib DA. Bioluminescence imaging reveals systemic dissemination of herpes simplex virus type 1 in the absence of interferon receptors. J Virol. 2003;77:11082–11093. doi: 10.1128/JVI.77.20.11082-11093.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundberg P, Welander P, Han X, Cantin E. Herpes simplex virus type 1 DNA is immunostimulatory in vitro and in vivo. J Virol. 2003;77:11158–11169. doi: 10.1128/JVI.77.20.11158-11169.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maertzdorf J, Osterhaus AD, Verjans GM. IL-17 expression in human herpetic stromal keratitis: modulatory effects on chemokine production by corneal fibroblasts. J Immunol. 2002;169:5897–5903. doi: 10.4049/jimmunol.169.10.5897. [DOI] [PubMed] [Google Scholar]
- Margolis TP, Dawson CR, LaVail JH. Herpes simplex viral infection of the mouse trigeminal ganglion. Immunohistochemical analysis of cell populations. Invest Ophthalmol Vis Sci. 1992;33:259–267. [PubMed] [Google Scholar]
- Margolis TP, Imai Y, Yang L, Vallas V, Krause PR. Herpes simplex virus type 2 (HSV-2) establishes latent infection in a different population of ganglionic neurons than HSV-1: role of latency-associated transcripts. J Virol. 2007;81:1872–1878. doi: 10.1128/JVI.02110-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuzaki J, Tsuji T, Chamoto K, Takeshima T, Sendo F, Nishimura T. Successful elimination of memory-type CD8+ T cell subsets by the administration of anti-Gr-1 monoclonal antibody in vivo. Cell Immunol. 2003;224:98–105. doi: 10.1016/j.cellimm.2003.08.009. [DOI] [PubMed] [Google Scholar]
- McLean CS, Ni Challanain D, Duncan I, Boursnell ME, Jennings R, Inglis SC. Induction of a protective immune response by mucosal vaccination with a DISC HSV-1 vaccine. Vaccine. 1996;14:987–992. doi: 10.1016/0264-410x(95)00259-4. [DOI] [PubMed] [Google Scholar]
- Mercadal CM, Bouley DM, DeStephano D, Rouse BT. Herpetic stromal keratitis in the reconstituted scid mouse model. J Virol. 1993;67:3404–3408. doi: 10.1128/jvi.67.6.3404-3408.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikloska Z, Bosnjak L, Cunningham AL. Immature monocyte-derived dendritic cells are productively infected with herpes simplex virus type 1. J Virol. 2001;75:5958–5964. doi: 10.1128/JVI.75.13.5958-5964.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JK, Laycock KA, Umphress JA, Hook KK, Stuart PM, Pepose JS. A comparison of recurrent and primary herpes simplex keratitis in NIH inbred mice. Cornea. 1996;15:497–504. [PubMed] [Google Scholar]
- Miyazaki D, Haruki T, Takeda S, Sasaki S, Yakura K, Terasaka Y, Komatsu N, Yamagami S, Touge H, Touge C, et al. Herpes simplex virus type 1-induced transcriptional networks of corneal endothelial cells indicate antigen presentation function. Invest Ophthalmol Vis Sci. 2011;52:4282–4293. doi: 10.1167/iovs.10-6911. [DOI] [PubMed] [Google Scholar]
- Mogensen SC. Role of macrophages in natural resistance to virus infections. Microbiol Rev. 1979;43:1–26. doi: 10.1128/mr.43.1.1-26.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molesworth-Kenyon SJ, Yin R, Oakes JE, Lausch RN. IL-17 receptor signaling influences virus-induced corneal inflammation. J Leukoc Biol. 2008;83:401–408. doi: 10.1189/jlb.0807571. [DOI] [PubMed] [Google Scholar]
- Mott K, Brick DJ, van Rooijen N, Ghiasi H. Macrophages are important determinants of acute ocular HSV-1 infection in immunized mice. Invest Ophthalmol Vis Sci. 2007;48:5605–5615. doi: 10.1167/iovs.07-0894. [DOI] [PubMed] [Google Scholar]
- Nakamura T, Ishikawa F, Sonoda KH, Hisatomi T, Qiao H, Yamada J, Fukata M, Ishibashi T, Harada M, Kinoshita S. Characterization and distribution of bone marrow-derived cells in mouse cornea. Invest Ophthalmol Vis Sci. 2005;46:497–503. doi: 10.1167/iovs.04-1154. [DOI] [PubMed] [Google Scholar]
- Nakano H, Yanagita M, Gunn MD. CD11c(+)B220(+)Gr-1(+) cells in mouse lymph nodes and spleen display characteristics of plasmacytoid dendritic cells. J Exp Med. 2001;194:1171–1178. doi: 10.1084/jem.194.8.1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newell CK, Martin S, Sendele D, Mercadal CM, Rouse BT. Herpes simplex virus-induced stromal keratitis: role of T-lymphocyte subsets in immunopathology. J Virol. 1989a;63:769–775. doi: 10.1128/jvi.63.2.769-775.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newell CK, Sendele D, Rouse BT. Effects of CD4+ and CD8+ T-lymphocyte depletion on the induction and expression of herpes simplex stromal keratitis. Reg Immunol. 1989b;2:366–369. [PubMed] [Google Scholar]
- Nicholls SM, Shimeld C, Easty DL, Hill TJ. Recurrent herpes simplex after corneal transplantation in rats. Investigative ophthalmology & visual science. 1996;37:425–435. [PubMed] [Google Scholar]
- Oakes JE, Monteiro CA, Cubitt CL, Lausch RN. Induction of interleukin-8 gene expression is associated with herpes simplex virus infection of human corneal keratocytes but not human corneal epithelial cells. J Virol. 1993;67:4777–4784. doi: 10.1128/jvi.67.8.4777-4784.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohara PT, Chin MS, LaVail JH. The spread of herpes simplex virus type 1 from trigeminal neurons to the murine cornea: an immunoelectron microscopy study. J Virol. 2000;74:4776–4786. doi: 10.1128/jvi.74.10.4776-4786.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohashi Y, Yamamoto S, Nishida K, Okamoto S, Kinoshita S, Hayashi K, Manabe R. Demonstration of herpes simplex virus DNA in idiopathic corneal endotheliopathy. Am J Ophthalmol. 1991;112:419–423. doi: 10.1016/s0002-9394(14)76251-8. [DOI] [PubMed] [Google Scholar]
- Openshaw H, Asher LV, Wohlenberg C, Sekizawa T, Notkins AL. Acute and latent infection of sensory ganglia with herpes simplex virus: immune control and virus reactivation. J Gen Virol. 1979;44:205–215. doi: 10.1099/0022-1317-44-1-205. [DOI] [PubMed] [Google Scholar]
- Openshaw H, McNeill JI, Lin XH, Niland J, Cantin EM. Herpes simplex virus DNA in normal corneas: persistence without viral shedding from ganglia. Journal of medical virology. 1995;46:75–80. doi: 10.1002/jmv.1890460116. [DOI] [PubMed] [Google Scholar]
- Osorio Y, Wechsler SL, Nesburn AB, Ghiasi H. Reduced severity of HSV-1-induced corneal scarring in IL-12-deficient mice. Virus Res. 2002;90:317–326. doi: 10.1016/s0168-1702(02)00249-6. [DOI] [PubMed] [Google Scholar]
- Padgett DA, Glaser R. How stress influences the immune response. Trends Immunol. 2003;24:444–448. doi: 10.1016/s1471-4906(03)00173-x. [DOI] [PubMed] [Google Scholar]
- Padgett DA, Sheridan JF, Dorne J, Berntson GG, Candelora J, Glaser R. Social stress and the reactivation of latent herpes simplex virus type 1. Proc Natl Acad Sci U S A. 1998;95:7231–7235. doi: 10.1073/pnas.95.12.7231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palamara F, Meindl S, Holcmann M, Luhrs P, Stingl G, Sibilia M. Identification and characterization of pDC-like cells in normal mouse skin and melanomas treated with imiquimod. J Immunol. 2004;173:3051–3061. doi: 10.4049/jimmunol.173.5.3051. [DOI] [PubMed] [Google Scholar]
- Pavan-Langston D, Foster CS. Trifluorothymidine and idoxuridine therapy of ocular herpes. Am J Ophthalmol. 1977;84:818–825. doi: 10.1016/0002-9394(77)90504-9. [DOI] [PubMed] [Google Scholar]
- Perng GC, Jones C, Ciacci-Zanella J, Stone M, Henderson G, Yukht A, Slanina SM, Hofman FM, Ghiasi H, Nesburn AB, et al. Virus-induced neuronal apoptosis blocked by the herpes simplex virus latency-associated transcript. Science. 2000;287:1500–1503. doi: 10.1126/science.287.5457.1500. [DOI] [PubMed] [Google Scholar]
- Pollara G, Jones M, Handley ME, Rajpopat M, Kwan A, Coffin RS, Foster G, Chain B, Katz DR. Herpes simplex virus type-1-induced activation of myeloid dendritic cells: the roles of virus cell interaction and paracrine type I IFN secretion. J Immunol. 2004;173:4108–4119. doi: 10.4049/jimmunol.173.6.4108. [DOI] [PubMed] [Google Scholar]
- Porter SM, Patterson A, Kho P. A comparison of local and systemic acyclovir in the management of herpetic disciform keratitis. Br J Ophthalmol. 1990;74:283–285. doi: 10.1136/bjo.74.5.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin S, Rottman JB, Myers P, Kassam N, Weinblatt M, Loetscher M, Koch AE, Moser B, Mackay CR. The chemokine receptors CXCR3 and CCR5 mark subsets of T cells associated with certain inflammatory reactions. J Clin Invest. 1998;101:746–754. doi: 10.1172/JCI1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabenau HF, Buxbaum S, Preiser W, Weber B, Doerr HW. Seroprevalence of herpes simplex virus types 1 and type 2 in the Frankfurt am Main area, Germany. Med Microbiol Immunol. 2002;190:153–160. doi: 10.1007/s00430-001-0102-1. [DOI] [PubMed] [Google Scholar]
- Remeijer L, Duan R, van Dun JM, Wefers Bettink MA, Osterhaus AD, Verjans GM. Prevalence and clinical consequences of herpes simplex virus type 1 DNA in human cornea tissues. The Journal of infectious diseases. 2009;200:11–19. doi: 10.1086/599329. [DOI] [PubMed] [Google Scholar]
- Roizman B, Zhou G, Du T. Checkpoints in productive and latent infections with herpes simplex virus 1: conceptualization of the issues. J Neurovirol. 2011;17:512–517. doi: 10.1007/s13365-011-0058-x. [DOI] [PubMed] [Google Scholar]
- Russell RG, Nasisse MP, Larsen HS, Rouse BT. Role of T-lymphocytes in the pathogenesis of herpetic stromal keratitis. Invest Ophthalmol Vis Sci. 1984;25:938–944. [PubMed] [Google Scholar]
- Sainz B, Loutsch JM, Marquart ME, Hill JM. Stress-associated immunomodulation and herpes simplex virus infections. Med Hypotheses. 2001;56:348–356. doi: 10.1054/mehy.2000.1219. [DOI] [PubMed] [Google Scholar]
- Schall TJ, Bacon K, Toy KJ, Goeddel DV. Selective attraction of monocytes and T lymphocytes of the memory phenotype by cytokine RANTES. Nature. 1990;347:669–671. doi: 10.1038/347669a0. [DOI] [PubMed] [Google Scholar]
- Sciammas R, Kodukula P, Tang Q, Hendricks RL, Bluestone JA. T cell receptor-gamma/delta cells protect mice from herpes simplex virus type 1-induced lethal encephalitis. J Exp Med. 1997;185:1969–1975. doi: 10.1084/jem.185.11.1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah A, Farooq AV, Tiwari V, Kim MJ, Shukla D. HSV-1 infection of human corneal epithelial cells: receptor-mediated entry and trends of re-infection. Mol Vis. 2010;16:2476–2486. [PMC free article] [PubMed] [Google Scholar]
- Sheridan BS, Cherpes TL, Urban J, Kalinski P, Hendricks RL. Reevaluating the CD8 T-cell response to herpes simplex virus type 1: involvement of CD8 T cells reactive to subdominant epitopes. J Virol. 2009;83:2237–2245. doi: 10.1128/JVI.01699-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimeld C, Hill TJ, Blyth WA, Easty DL. Passive immunization protects the mouse eye from damage after herpes simplex virus infection by limiting spread of virus in the nervous system. J Gen Virol. 1990a;71(Pt 3):681–687. doi: 10.1099/0022-1317-71-3-681. [DOI] [PubMed] [Google Scholar]
- Shimeld C, Hill TJ, Blyth WA, Easty DL. Reactivation of latent infection and induction of recurrent herpetic eye disease in mice. J Gen Virol. 1990b;71(Pt 2):397–404. doi: 10.1099/0022-1317-71-2-397. [DOI] [PubMed] [Google Scholar]
- Skobe M, Dana R. Blocking the path of lymphatic vessels. Nat Med. 2009;15:993–994. doi: 10.1038/nm0909-993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skobe M, Hawighorst T, Jackson DG, Prevo R, Janes L, Velasco P, Riccardi L, Alitalo K, Claffey K, Detmar M. Induction of tumor lymphangiogenesis by VEGF-C promotes breast cancer metastasis. Nat Med. 2001;7:192–198. doi: 10.1038/84643. [DOI] [PubMed] [Google Scholar]
- Spencer WH, Hayes TL. Scanning and transmission electron microscopic observations of the topographic anatomy of dendritic lesions in the rabbit cornea. Invest Ophthalmol. 1970;9:183–195. [PubMed] [Google Scholar]
- Sprecher E, Becker Y. Langerhans cell density and activity in mouse skin and lymph nodes affect herpes simplex type 1 (HSV-1) pathogenicity. Arch Virol. 1989;107:191–205. doi: 10.1007/BF01317916. [DOI] [PubMed] [Google Scholar]
- Steinman RM, Cohn ZA. Identification of a novel cell type in peripheral lymphoid organs of mice. I. Morphology, quantitation, tissue distribution. J Exp Med. 1973;137:1142–1162. doi: 10.1084/jem.137.5.1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinman RM, Gutchinov B, Witmer MD, Nussenzweig MC. Dendritic cells are the principal stimulators of the primary mixed leukocyte reaction in mice. J Exp Med. 1983;157:613–627. doi: 10.1084/jem.157.2.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su YH, Yan XT, Oakes JE, Lausch RN. Protective antibody therapy is associated with reduced chemokine transcripts in herpes simplex virus type 1 corneal infection. J Virol. 1996;70:1277–1281. doi: 10.1128/jvi.70.2.1277-1281.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suryawanshi A, Veiga-Parga T, Rajasagi NK, Reddy PB, Sehrawat S, Sharma S, Rouse BT. Role of IL-17 and Th17 cells in herpes simplex virus-induced corneal immunopathology. J Immunol. 2011;187:1919–1930. doi: 10.4049/jimmunol.1100736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suryawanshi A, Veiga-Parga T, Reddy PB, Rajasagi NK, Rouse BT. IL-17A Differentially Regulates Corneal Vascular Endothelial Growth Factor (VEGF)-A and Soluble VEGF Receptor 1 Expression and Promotes Corneal Angiogenesis after Herpes Simplex Virus Infection. J Immunol. 2012;188:3434–3446. doi: 10.4049/jimmunol.1102602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda S, Miyazaki D, Sasaki S, Yamamoto Y, Terasaka Y, Yakura K, Yamagami S, Ebihara N, Inoue Y. Roles played by toll-like receptor-9 in corneal endothelial cells after herpes simplex virus type 1 infection. Invest Ophthalmol Vis Sci. 52:6729–6736. doi: 10.1167/iovs.11-7805. [DOI] [PubMed] [Google Scholar]
- Tang Q, Chen W, Hendricks RL. Proinflammatory functions of IL-2 in herpes simplex virus corneal infection. J Immunol. 1997;158:1275–1283. [PubMed] [Google Scholar]
- Tang S, Patel A, Krause PR. Novel less-abundant viral microRNAs encoded by herpes simplex virus 2 latency-associated transcript and their roles in regulating ICP34.5 and ICP0 mRNAs. J Virol. 2009;83:1433–1442. doi: 10.1128/JVI.01723-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terasaka Y, Miyazaki D, Yakura K, Haruki T, Inoue Y. Induction of IL-6 in transcriptional networks in corneal epithelial cells after herpes simplex virus type 1 infection. Invest Ophthalmol Vis Sci. 2009;51:2441–2449. doi: 10.1167/iovs.09-4624. [DOI] [PubMed] [Google Scholar]
- Theil D, Derfuss T, Paripovic I, Herberger S, Meinl E, Schueler O, Strupp M, Arbusow V, Brandt T. Latent herpesvirus infection in human trigeminal ganglia causes chronic immune response. Am J Pathol. 2003;163:2179–2184. doi: 10.1016/S0002-9440(10)63575-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas J, Gangappa S, Kanangat S, Rouse BT. On the essential involvement of neutrophils in the immunopathologic disease: herpetic stromal keratitis. J Immunol. 1997;158:1383–1391. [PubMed] [Google Scholar]
- Tumpey TM, Chen SH, Oakes JE, Lausch RN. Neutrophil-mediated suppression of virus replication after herpes simplex virus type 1 infection of the murine cornea. J Virol. 1996;70:898–904. doi: 10.1128/jvi.70.2.898-904.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tumpey TM, Cheng H, Cook DN, Smithies O, Oakes JE, Lausch RN. Absence of macrophage inflammatory protein-1alpha prevents the development of blinding herpes stromal keratitis. J Virol. 1998a;72:3705–3710. doi: 10.1128/jvi.72.5.3705-3710.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tumpey TM, Cheng H, Yan XT, Oakes JE, Lausch RN. Chemokine synthesis in the HSV-1-infected cornea and its suppression by interleukin-10. J Leukoc Biol. 1998b;63:486–492. doi: 10.1002/jlb.63.4.486. [DOI] [PubMed] [Google Scholar]
- Umbach JL, Kramer MF, Jurak I, Karnowski HW, Coen DM, Cullen BR. MicroRNAs expressed by herpes simplex virus 1 during latent infection regulate viral mRNAs. Nature. 2008;454:780–783. doi: 10.1038/nature07103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valyi-Nagy T, Sheth V, Clement C, Tiwari V, Scanlan P, Kavouras JH, Leach L, Guzman-Hartman G, Dermody TS, Shukla D. Herpes simplex virus entry receptor nectin-1 is widely expressed in the murine eye. Curr Eye Res. 2004;29:303–309. doi: 10.1080/02713680490516756. [DOI] [PubMed] [Google Scholar]
- Verjans GM, Hintzen RQ, van Dun JM, Poot A, Milikan JC, Laman JD, Langerak AW, Kinchington PR, Osterhaus AD. Selective retention of herpes simplex virus-specific T cells in latently infected human trigeminal ganglia. Proc Natl Acad Sci U S A. 2007;104:3496–3501. doi: 10.1073/pnas.0610847104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verjans GM, Remeijer L, Mooy CM, Osterhaus AD. Herpes simplex virus-specific T cells infiltrate the cornea of patients with herpetic stromal keratitis: no evidence for autoreactive T cells. Invest Ophthalmol Vis Sci. 2000;41:2607–2612. [PubMed] [Google Scholar]
- Verjans GM, Remeijer L, van Binnendijk RS, Cornelissen JG, Volker-Dieben HJ, Baarsma SG, Osterhaus AD. Identification and characterization of herpes simplex virus-specific CD4+ T cells in corneas of herpetic stromal keratitis patients. J Infect Dis. 1998;177:484–488. doi: 10.1086/517382. [DOI] [PubMed] [Google Scholar]
- Vicetti Miguel RD, Sheridan BS, Harvey SA, Schreiner RS, Hendricks RL, Cherpes TL. 17-beta estradiol promotion of herpes simplex virus type 1 reactivation is estrogen receptor dependent. J Virol. 2010;84:565–572. doi: 10.1128/JVI.01374-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vollstedt S, Arnold S, Schwerdel C, Franchini M, Alber G, Di Santo JP, Ackermann M, Suter M. Interplay between alpha/beta and gamma interferons with B, T, and natural killer cells in the defense against herpes simplex virus type 1. J Virol. 2004;78:3846–3850. doi: 10.1128/JVI.78.8.3846-3850.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakim LM, Gebhardt T, Heath WR, Carbone FR. Cutting edge: local recall responses by memory T cells newly recruited to peripheral nonlymphoid tissues. J Immunol. 2008;181:5837–5841. doi: 10.4049/jimmunol.181.9.5837. [DOI] [PubMed] [Google Scholar]
- Walzer T, Dalod M, Robbins SH, Zitvogel L, Vivier E. Natural-killer cells and dendritic cells: “l’union fait la force”. Blood. 2005;106:2252–2258. doi: 10.1182/blood-2005-03-1154. [DOI] [PubMed] [Google Scholar]
- Wang QY, Zhou C, Johnson KE, Colgrove RC, Coen DM, Knipe DM. Herpesviral latency-associated transcript gene promotes assembly of heterochromatin on viral lytic-gene promoters in latent infection. Proc Natl Acad Sci U S A. 2005;102:16055–16059. doi: 10.1073/pnas.0505850102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber C, Weber KS, Klier C, Gu S, Wank R, Horuk R, Nelson PJ. Specialized roles of the chemokine receptors CCR1 and CCR5 in the recruitment of monocytes and T(H)1-like/CD45RO(+) T cells. Blood. 2001;97:1144–1146. doi: 10.1182/blood.v97.4.1144. [DOI] [PubMed] [Google Scholar]
- Wilhelmus KR. Diagnosis and management of herpes simplex stromal keratitis. Cornea. 1987;6:286–291. doi: 10.1097/00003226-198706040-00011. [DOI] [PubMed] [Google Scholar]
- Williams KA, Muehlberg SM, Lewis RF, Coster DJ. How successful is corneal transplantation? A report from the Australian Corneal Graft Register. Eye. 1995;9(Pt 2):219–227. doi: 10.1038/eye.1995.43. [DOI] [PubMed] [Google Scholar]
- Williams KA, Muehlberg SM, Lewis RF, Coster DJ. Long-term outcome in corneal allotransplantation. The Australian Corneal Graft Registry. Transplant Proc. 1997;29:983. doi: 10.1016/s0041-1345(96)00335-1. [DOI] [PubMed] [Google Scholar]
- Wisner TW, Sugimoto K, Howard PW, Kawaguchi Y, Johnson DC. Anterograde transport of herpes simplex virus capsids in neurons by both separate and married mechanisms. J Virol. 2011;85:5919–5928. doi: 10.1128/JVI.00116-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojtasiak M, Pickett DL, Tate MD, Londrigan SL, Bedoui S, Brooks AG, Reading PC. Depletion of Gr-1+, but not Ly6G+, immune cells exacerbates virus replication and disease in an intranasal model of herpes simplex virus type 1 infection. J Gen Virol. 2010;91:2158–2166. doi: 10.1099/vir.0.021915-0. [DOI] [PubMed] [Google Scholar]
- Wuest T, Farber J, Luster A, Carr DJ. CD4+ T cell migration into the cornea is reduced in CXCL9 deficient but not CXCL10 deficient mice following herpes simplex virus type 1 infection. Cell Immunol. 2006;243:83–89. doi: 10.1016/j.cellimm.2007.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wuest T, Zheng M, Efstathiou S, Halford WP, Carr DJ. The herpes simplex virus-1 transactivator infected cell protein-4 drives VEGF-A dependent neovascularization. PLoS Pathog. 2011a;7:e1002278. doi: 10.1371/journal.ppat.1002278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wuest TR, Carr DJ. Dysregulation of CXCR3 signaling due to CXCL10 deficiency impairs the antiviral response to herpes simplex virus 1 infection. J Immunol. 2008;181:7985–7993. doi: 10.4049/jimmunol.181.11.7985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wuest TR, Carr DJ. VEGF-A expression by HSV-1-infected cells drives corneal lymphangiogenesis. J Exp Med. 2010;207:101–115. doi: 10.1084/jem.20091385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wuest TR, Thapa M, Zheng M, Carr DJ. CXCL10 expressing hematopoietic-derived cells are requisite in defense against HSV-1 infection in the nervous system of CXCL10 deficient mice. J Neuroimmunol. 2011b;234:103–108. doi: 10.1016/j.jneuroim.2011.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu F, Sternberg MR, Kottiri BJ, McQuillan GM, Lee FK, Nahmias AJ, Berman SM, Markowitz LE. Trends in herpes simplex virus type 1 and type 2 seroprevalence in the United States. Jama. 2006;296:964–973. doi: 10.1001/jama.296.8.964. [DOI] [PubMed] [Google Scholar]
- Yan XT, Tumpey TM, Kunkel SL, Oakes JE, Lausch RN. Role of MIP-2 in neutrophil migration and tissue injury in the herpes simplex virus-1-infected cornea. Invest Ophthalmol Vis Sci. 1998;39:1854–1862. [PubMed] [Google Scholar]
- Yang L, Voytek CC, Margolis TP. Immunohistochemical analysis of primary sensory neurons latently infected with herpes simplex virus type 1. J Virol. 2000;74:209–217. doi: 10.1128/jvi.74.1.209-217.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young RC, Hodge DO, Liesegang TJ, Baratz KH. Incidence, recurrence, and outcomes of herpes simplex virus eye disease in Olmsted County, Minnesota, 1976–2007: the effect of oral antiviral prophylaxis. Arch Ophthalmol. 2010;128:1178–1183. doi: 10.1001/archophthalmol.2010.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Deak E, Soderberg K, Linehan M, Spezzano D, Zhu J, Knipe DM, Iwasaki A. Vaginal submucosal dendritic cells, but not Langerhans cells, induce protective Th1 responses to herpes simplex virus-2. J Exp Med. 2003;197:153–162. doi: 10.1084/jem.20021109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao ZS, Granucci F, Yeh L, Schaffer PA, Cantor H. Molecular mimicry by herpes simplex virus-type 1: autoimmune disease after viral infection. Science. 1998;279:1344–1347. doi: 10.1126/science.279.5355.1344. [DOI] [PubMed] [Google Scholar]
- Zheng M, Deshpande S, Lee S, Ferrara N, Rouse BT. Contribution of vascular endothelial growth factor in the neovascularization process during the pathogenesis of herpetic stromal keratitis. J Virol. 2001a;75:9828–9835. doi: 10.1128/JVI.75.20.9828-9835.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng M, Klinman DM, Gierynska M, Rouse BT. DNA containing CpG motifs induces angiogenesis. Proc Natl Acad Sci U S A. 2002;99:8944–8949. doi: 10.1073/pnas.132605599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng M, Schwarz MA, Lee S, Kumaraguru U, Rouse BT. Control of stromal keratitis by inhibition of neovascularization. Am J Pathol. 2001b;159:1021–1029. doi: 10.1016/S0002-9440(10)61777-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng X, Loutsch JM, Shimomura Y, Gebhardt BM, Hill JM, Kaufman HE. Reactivation of herpes virus after lamellar keratoplasty. Japanese journal of ophthalmology. 1999;43:257–261. doi: 10.1016/s0021-5155(99)00023-4. [DOI] [PubMed] [Google Scholar]
- Zhu J, Koelle DM, Cao J, Vazquez J, Huang ML, Hladik F, Wald A, Corey L. Virus-specific CD8+ T cells accumulate near sensory nerve endings in genital skin during subclinical HSV-2 reactivation. J Exp Med. 2007;204:595–603. doi: 10.1084/jem.20061792. [DOI] [PMC free article] [PubMed] [Google Scholar]