

Abstract
The optimum use of allogeneic blood and marrow transplantation (BMT) as a curative therapy for hematological malignancies lies in the successful separation of mature donor T cells that are host-reactive and induce graft-versus-host disease (GVHD) from those that are tumor-reactive and mediate graft-versus-leukemia (GVL) effects. To study whether this separation was possible in an MHC-matched murine BMT model (B10.BR→CBA) with a CBA-derived myeloid leukemia line, MMC6, we used TCR Vβ CDR3-size spectratype analysis to first show that the Vβ13 family was highly skewed in the B10.BR anti-MMC6 CD8+ T cell response but not in the alloresponse against recipient cells alone. Transplantation of CD8+Vβ13+ T cells at the dose equivalent of their constituency in 1×107 CD8+ T cells, a dose that had been shown to mediate lethal GVHD in recipient mice, induced a slight GVL response with no concomitant GVHD. Increasing doses of CD8+Vβ13+ T cells led to more significant GVL responses, but also increased GVHD symptoms and associated mortality. Subsequent spectratype analysis of GVHD target tissues revealed involvement of gut-infiltrating CD8+Vβ13+ T cells accounting for the observed in vivo effects. When BMT recipients were given MMC6 presensitized CD8+Vβ13+ T cells, they displayed a significant GVL response with minimal GVHD. Spectratype analysis of tumor-presensitized, gut-infiltrating CD8+Vβ13+ T cells showed preferential usage of tumor-reactive CDR3-size lengths, and these cells expressed increased effector memory phenotype (CD44+CD62L-/lo). Thus, Vβ spectratyping can identify T cells involved in anti-host and anti-tumor reactivity and tumor-presensitization can aid in the separation of GVHD and GVL responses.
Keywords: graft-versus-host disease, graft-versus-leukemia, spectratype analysis, bone marrow transplantation
INTRODUCTION
Immunotherapy in the form of blood and marrow transplantation (BMT) for the treatment of hematological malignancies has been hampered by the inability to successfully separate alloreactive graft-versus-host disease (GVHD) from graft-versus-leukemia (GVL) responses. Both of these events are mediated by mature T cells present in the donor inoculum (1-6). Approaches, such as T cell depletion or alteration of T cell subset composition have been implemented with limited success, due to delayed hematopoietic or immune reconstitution, graft failure, and increased susceptibility to opportunistic infections (1-5). In addition, because these approaches rely on the depletion of broad cellular populations with overlapping alloreactive and anti-tumor specificities, the incidence of tumor relapse has also increased (6-9).
Our work has focused on identifying alloreactive (capable of mediating GVHD) and GVL-reactive cells by analyzing the T cell receptor (TCR) Vβ chain utilization of these responses using complementarity-determining region 3 (CDR3)-size spectratype analysis. This method allows for the separation of allo- and tumor-reactive T cells without the need to define specific reactive antigens. As previously reported, spectratype analysis has led to the identification of donor alloreactive CD8+ and CD4+ Vβ families in the C57Bl/6 (B6)→CXBE and B6→BALB.B MHC-matched, minor histocompatibility antigen (miHA)-mismatched murine models of BMT (10-12). In addition, we have shown a significant GVL effect with minimal GVHD in the haploidentical B6→[B6×DBA/2]F1 BMT murine model, when recipients were transplanted with spectratype-identified Vβ families that were uniquely anti-tumor reactive (13).
In the current study, we used CDR3-size spectratype analysis to determine the Vβ families involved in the GVL response against MMC6, a CBA-derived myeloid leukemia cell line, in the B10.BR→CBA MHC-matched (H2k), miHA-disparate BMT model. Previous studies have shown that this is a CD8+ T cell-mediated/CD4+ T cell independent GVHD model (3). We have now extended these findings to demonstrate significant anti-MMC6 reactivity in the CD8+Vβ13+ T cell family, which was not detected in the B10.BR anti-CBA alloresponse to antigens of the peripheral lymphoid compartment, but was found to be reactive in the gut of CBA recipients at a different and unique CDR3-size length (i.e., non-overlapping with the anti-MMC6 response). Presensitization of donor B10.BR CD8+Vβ13+ T cells to MMC6 tumor induced a significant GVL effect that co-occurred only minimally with the incidence of lethal GVHD. Spectratype analysis of these tumor-presensitized, CD8+Vβ13+ T cells infiltrating the gut showed preferential usage of tumor-reactive CDR3-size lengths. Furthermore, tumor-presensitized CD8+Vβ13+ donor T cells had an increased percentage of effector memory cells (i.e., CD44+CD62L- phenotype). In summary, we demonstrate here that the GVL potential of tumor-reactive Vβ families identified using spectratype analysis can be enhanced using tumor presensitization. Furthermore, we show that this approach minimizes alloresponse to tissue-specific antigens which in the clinical scenario would be hard to identify a priori. Lastly, we also found evidence to suggest that effector memory T cells play an important role in favoring anti-tumor immunity with diminished GVHD in this model.
MATERIALS & METHODS
Mice
Male B10.BR-H-2k H2-T18a/SgSnJJrep (B10.BR) and CBA/J (CBA) mice were purchased from the Jackson Laboratory (Bar Harbor, ME), housed in a sterile environment in microisolator cages, and given autoclaved food and acidified water (pH 2.5) ad libitum. Both donor and recipient mice were used between the ages of 8 and 12 weeks. All protocols used in this study were approved by Hackensack University Medical Center’s Institutional Animal Care and Use Committee.
Cell line and media
MMC6, a c-myc retrovirus-transformed myeloid leukemia line of CBA origin, was generated in our laboratory according to previously established methods (14), and grown in complete RPMI 1640 media (Mediatech, Herndon, VA) containing 10% FBS (Hyclone, Logan, UT), 2mM L-glutamine, penicillin (50 IU/mL)/streptomycin (50 μg) (Mediatech) and 2-ME at 37°C in 7% CO2.
Preparation of cells for GVHD experiments
Anti-Thy-1.2 depleted bone marrow (ATBM) cells were prepared by flushing bone marrow from the femurs of donor mice, followed by incubation with J1j (anti-Thy-1.2) mAb (1:100) and guinea pig C’ (1:5) at 37°C. T cell-enriched donor cell populations were prepared from pooled lymph nodes and RBC-lysed splenocyte suspensions. B cells and CD4+ T cells were removed by subsequent incubations with the B cell-specific mAb, J11d.2 (1:500) and anti-CD4 mAb (RL172; 1:100), respectively, in the presence of C’ at 37°C. Vβ-enriched T cell populations were obtained by magnetic bead separation using the AutoMacs system (Miltenyi Biotec, Auburn, CA), using anti-Vβ13 FITC-conjugated mAb (clone MR12-3; Pharmingen, San Jose, CA) at a concentration of 0.5ug/ml and anti-FITC mAb-conjugated microbeads. The positive fraction was typically greater than 90% FITC-positive as determined by flow cytometry. In some experiments, donor mice were presensitized with an i.p. injection of 1×107 irradiated (40 Gy) MMC6 tumor cells 17-21 d prior to BMT experiments.
GVHD and GVL experiments
Recipient mice were exposed to lethal irradiation (11 Gy; split dose, 4 h apart) using a 137Ce source (Gammacell 40 Exactor; MDS Nordion), and after another 2 h, mice were injected i.v. with ATBM (2×106) cells alone or in combination with different donor T cell populations and quantities (as specified in figure legends). On d 1 post-BMT, recipient mice were challenged with an i.p injection of 5×103 MMC6 cells, when specified. Mice were checked daily for morbidity and mortality and were weighed twice weekly until the termination of the experiments (approximately 100 d post-BMT). Experiments were repeated at least twice with 3-5 mice per group. Median survival times (MST) were calculated and the non-parametric Wilcoxon test was used for statistical analysis of survival curves. A p value ≤0.05 was considered statistically significant.
Preparation of RNA and complimentary DNA for CDR3-size spectratyping
B10.BR CD8+ T cells were transplanted into irradiated CBA mice in the absence of ATBM cells. After 8-10 d, spleens from 5 recipient mice were pooled and homogenized in Ultraspec reagent (Biotecx Laboratories, Houston, TX). Total RNA extraction, generation of cDNA, and RT-PCR were performed as previously described (15). Control CD8+ T cell populations were obtained from the spleens of naïve B10.BR mice. For anti-MMC6 spectratype analysis, B10.BR mice were presensitized with 1×107 MMC6 tumor cells (40 Gy) by i.p. injection followed by a footpad boost with 1×106 MMC6 cells (40 Gy) 3 wk later. One wk after the last presensitization, CD8+ T cells were isolated from the popliteal, inguinal, and mesenteric draining lymph nodes and put in Ultraspec. Alternatively, harvested CD8+ T cells were stimulated in vitro with MMC6 tumor cells (100 Gy) at a 1:2 ratio for 4 d. Semi-nested PCR was performed by using a panel of Vβ sense oligoprimers (IDT Technologies) and two Cβ antisense oligoprimers, with the second Cβ being fluorescently labeled (PE Applied Biosystems). The fluorescently-labeled PCR products were run on an ABI 3130 capillary gel system at the Molecular Resource Facility of the University of Medicine and Dentistry of New Jersey (Newark, NJ). Analyses were performed using GeneMapper software (version 3.7) from PE Applied Biosystems.
Quantitation of CDR3-size usage
Experiments were repeated a minimum of thrice with 3-5 mice in each group. The mean of the areas under the corresponding peaks was calculated for each Vβ histogram. A band (i.e., CDR3-size length) in the anti-CBA or the anti-MMC6 spectratype analysis was considered skewed if the mean area under the peak was greater than 5x the SD above the mean area of the corresponding control peak from naïve B10.BR CD8+ T cells.
Isolation of tissue-infiltrating mononuclear cells
CBA mice were exposed to lethal irradiation (11 Gy, split dose, 4 h apart) and injected i.v. with 2×107 naïve or MMC6 presensitized B10.BR CD8+ T cells along with 5×105 ATBM cells. After 10-13 d, mice were euthanized by CO2 inhalation and perfused by intracardiac injection with PBS. GVHD-target tissues (i.e., spleen, liver, and distal ileum of the intestine) were harvested and tissue-infiltrating mononuclear cells were isolated as previously described (15). Preparation of RNA and cDNA for CDR3-size spectratyping was performed as described above.
Histopathology
At the termination of some experiments, surviving mice were sacrificed and GVHD target organs including skin (ear and tongue), gut and liver were harvested for histopathology. Organs were placed in Tissue-Tek OCT compound (Sakura Finetek, Torrance, CA), snap-frozen in liquid nitrogen, and stored at -80°C. Sections (6um thick) were created from frozen tissue using a Leica CM 1850 cryostat (Leica Microsystems, Buffalo Grove, IL). Histological staining was performed using H&E dyes (Sigma-Aldrich).
Flow cytometry
For flow cytometric analysis, cells were stained in FACS buffer (1×PBS containing 1% BSA and 0.02% sodium azide) with the following fluorochrome-conjugated mAb (Pharmigen, San Jose, CA) diluted 1:100 in FACS buffer: anti-Vβ13 FITC (clone MR12-3), anti-CD62L PE (clone MEL-14), anti-α4β7 PE (clone DATK72), anti-CD8 PE Cy5 (clone 53-6.7). Biotinylated anti-CD44 (clone IM7) was also used, followed by secondary staining with streptavidin-PE-Cy7. A total of 100,000 events were acquired and analyzed using a Beckman Coulter FC500 flow cytometer (Beckman Coulter, Miami, FL). Staining was performed on at least 3-5 separate groups of 3 mice and statistical significance between groups was determined using Student’s t test. A p value ≤0.05 was considered statistically significant.
RESULTS
CD8+ T cells mediate GVL in the B10.BR→CBA model of BMT
Previous experiments have indicated that in the B10.BR→CBA murine model of BMT, CD8+ T cells are responsible for mediating severe lethal GVHD, and can do so in the absence of CD4+ T cells (3). Based on this finding, we set out to determine the role of B10.BR donor CD8+ T cells in the GVL response to a c-myc retrovirus-transformed myeloid leukemia cell line of CBA origin, MMC6. CBA mice were exposed to lethal irradiation (11 Gy, split dose) and transplanted with 2×106 B10.BR ATBM cells alone or in combination with 1×107 CD8+ T cells. On day 1, recipient mice were challenged i.p with 5×103 MMC6 tumor cells. In the absence of CD8+ T cells, all tumor challenged mice succumbed with a MST of 26 days (Fig. 1). Mice that were not tumor challenged, but received B10.BR CD8+ T cells developed GVHD and displayed the typical signs of diarrhea, weight loss, hunched posture, and ruffled fur (11% survival, MST = 37.5 days). Likewise, tumor challenged mice that also received CD8+ T cells succumbed mainly to GVHD (only 3 of 17 mice had visible tumor at time of death; 11% survival) but experienced a significant GVL response compared to tumor challenged mice that received ATBM cells alone (MST = 46 days, p < 0.01) (Fig. 1).
FIGURE 1. B10.BR CD8+ T cells induce both lethal GVHD and GVL effects in MMC6-challenged CBA recipients.
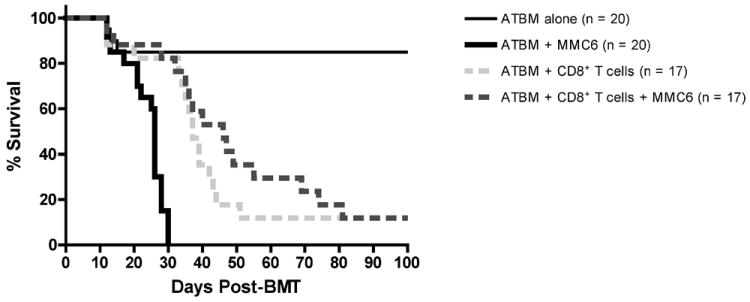
CBA mice were exposed to lethal irradiation (11 Gy, split dose) and transplanted with 2×106 B10.BR ATBM cells with or without 1×107 CD8+ T cells. The next day, mice were challenged i.p. with 5×103 MMC6 tumor cells. Results were combined from 4 separate experiments, each with 3-5 mice per group. Statistical significance between survival curves was determined using the non-parametric Wilcoxon test. ATBM + MMC6 versus ATBM + CD8+ T cells + MMC6, p < 0.01; ATBM + CD8+ T cells versus ATBM + CD8+ T cells + MMC6, p > 0.2.
TCR Vβ CDR3-size spectratype analysis of the B10.BR anti-CBA and B10.BR anti-MMC6 responses
After determining the GVL potential of B10.BR CD8+ T cells against the MMC6 tumor cells, we next sought to differentiate alloreactive from tumor-specific reactive CD8+ T cells using TCR Vβ CD3-size spectratype analysis. For the B10.BR anti-CBA analysis, CBA mice were exposed to lethal irradiation (11 Gy, split dose, 4 h apart) and transplanted with 1-2×107 B10.BR CD8+ T cells in the absence of ATBM cells. Potential reactivity against hematopoietically-derived miHA was assessed by spectratyping the splenocytes of recipient mice harvested 10-13 d post-transplant. The results revealed 12 alloreactive Vβ families: 3, 5, 7, 8.2, 8.3, 9, 11, 12, 14, 15, 18 and 20 (Table I). For the B10.BR anti-MMC6 analysis, responses were quantified both in vivo and in vitro. For in vivo analysis, B10.BR mice were presensitized with irradiated (40 Gy) MMC6 cells via i.p. and footpad injections, given 3 wk apart. Draining lymph nodes were harvested from mice 1 wk later and CD8+ T cells were selected for spectratype analysis. For in vitro analysis of anti-tumor responses, CD8+ T cells were harvested from tumor-presensitized B10.BR mice and further stimulated with MMC6 tumor cells for 4 d in vitro. The results of both in vivo and in vitro analyses revealed that Vβ families 1, 6, 7, 8.2, 13, and 15 were skewed (indicating clonal or oligoclonal T cell expansion) in the anti-tumor response, and that Vβ1, 6, and 13 were unique to the MMC6-response, for neither of these families were found to be reactive against hematopoietically-derived CBA miHA (Table I). Vβ1 was excluded as a candidate for future in vivo GVL reactivity studies due to the lack of a commercially available Vβ1 antibody. Since Vβ6 was skewed at only one CDR3-size length (138), we decided to focus solely on the Vβ13 family due to the strength of its anti-tumor response, which involved three CDR3-size lengths (160, 163, 166). A representative spectratype of the CD8+Vβ13+ T cell family (Fig. 2) demonstrated the skewing of peaks in the anti-MMC6 response (Fig. 2C), whereas no alloreactivity was detected by lymphocytes from transplanted CBA mice (Fig. 2B).
Table I.
Comparison of the skewed CDR3-size lengths of B10.BR CD8+ T cells from the spleens of CBA recipients or MMC6 tumor presensitized B10.BR micea
| Vβ | α-CBA | α-MMC6 |
|---|---|---|
| 1 | — | 186, 189 |
| 2 | — | — |
| 3 | 149 | — |
| 4 | — | — |
| 5 | 172 | — |
| 6 | — | 138 |
| 7 | 176 | 167, 176 |
| 8.1 | — | — |
| 8.2 | 134 | 153 |
| 8.3 | 155, 170 | — |
| 9 | 147 | — |
| 10 | — | — |
| 11 | 157 | — |
| 12 | 201 | — |
| 13 | — | 160, 163, 166 |
| 14 | 160 | — |
| 15 | 182 | 182 |
| 16 | — | — |
| 18 | 225 | — |
| 20 | 196 | — |
For the anti-CBA response, lethally irradiated CBA recipient mice were injected with 1 × 107 B10.BR CD8+ T cells and spleens were harvested after 10 days. For the anti-MMC6 response, B10.BR mice were injected i.p. with irradiated MMC6 cells followed by footpad boost 3 weeks later. One week later, CD8+ T cells were isolated from inguinal, mesenteric, and popliteal draining lymph nodes. CDR3-size spectratype analysis was performed as described in Materials and Methods. Skewing of bands was defined as an average peak area greater than the average control area plus 5×SD. —, No skewing; CDR3 lengths are shown for skewed Vβ families. Results were averaged from 5 separate experiments.
FIGURE 2. Representative spectratype histograms in the B10.BR anti-CBA and anti-MMC6 CD8+Vβ13+ T cell responses in lymphoid tissue.
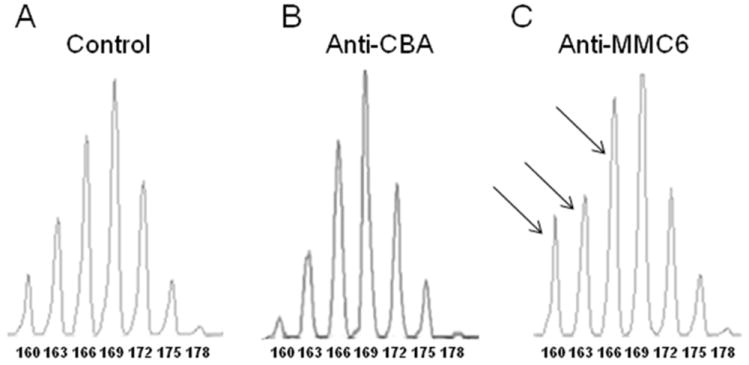
Spectratype analyses were performed on CD8+ T cell populations as described in Materials and Methods. (A) CD8+ splenic T cells harvested from naïve B10.BR mice acted as the control group. (B) For alloreactive responses (anti-CBA), CBA mice were exposed to lethal irradiation (11 Gy, split dose) and transplanted with 1-2×107 B10.BR CD8+ T cells. Splenocytes were harvested at 10-13 d post-BMT. (C) For the anti-tumor response (anti-MMC6), B10.BR mice were presensitized (i.p.) with 1×107 MMC6 tumor cells (40 Gy), followed by a second injection of 1×106 MMC6 cells (40 Gy) in the footpad 17-21 d later. After 1 wk, draining lymph nodes were harvested and CD8+ T cell isolated. Skewing was defined as a peak area greater than the mean of the control plus 5×SD. CDR3 lengths are shown under each peak. Skewed peaks are marked with an arrow.
GVHD- and GVL-inducing potential of B10.BR CD8+Vβ13+ T cells in CBA recipient mice
The GVL potential of the B10.BR CD8+Vβ13+ T cell family was further evaluated in CBA BMT recipients challenged with MMC6 tumor cells. CBA mice were exposed to lethal irradiation (11 Gy, split dose) and transplanted with B10.BR ATBM cells alone or in combination with 1×107 CD8+ T cells, a dose previously shown to induce lethal GVHD (Fig. 1). Another group of mice received the equivalent dose of CD8+Vβ13+ T cells found in 1×107 CD8+ T cells (i.e., 8.34×105; 1x; as determined by flow cytometry). As shown in Fig. 3A, mice receiving a 1x dose of CD8+Vβ13+ T cells displayed 100% survival with no mice developing GVHD. Furthermore, this amount of T cells also provided a significant GVL effect in tumor challenged mice compared to the MMC6 control group, although 100% of mice eventually succumbed to tumor challenge (Fig. 3B) (MST = 32 d vs. MST = 25 d, respectively, p < 0.01).
FIGURE 3. GVHD and GVL potential of B10.BR CD8+Vβ13+ donor T cells in MMC6-challenged CBA recipients.
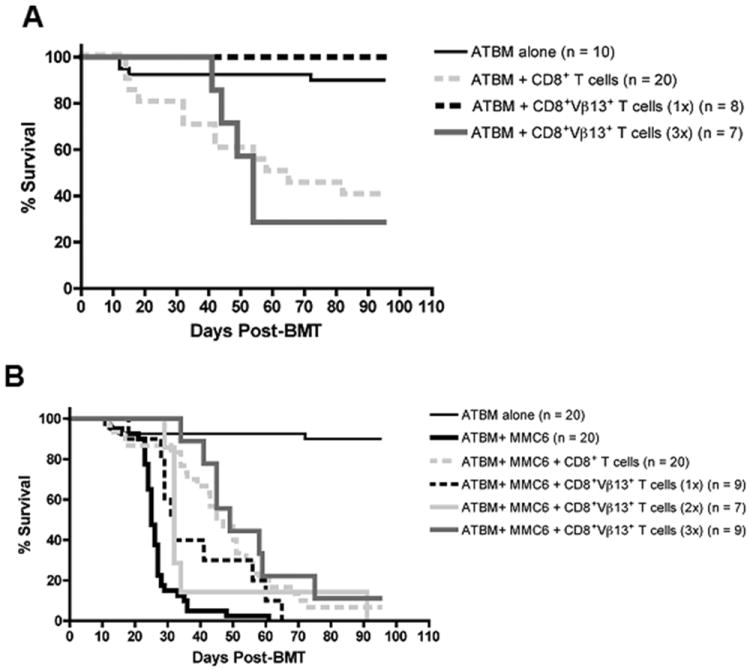
A and B, CBA mice were exposed to lethal irradiation (11 Gy, split dose) and transplanted with 2×106 ATBM cells alone or along with either unseparated CD8+ T cells (1×107) or CD8+Vβ13+ T cells (1x, 8.34×105; 2x, 1.67×106; or 3x, 2.5×106) from B10.BR donor mice. A, Statistical significance between survival curves was determined using the non-parametric Wilcoxon test. ATBM + CD8+ T cells versus ATBM + CD8+Vβ13+ T cells (1x), p < 0.02; ATBM + CD8+ T cells versus ATBM + CD8+Vβ13+ T cells (3x), p = 0.9. B, All groups except ATBM alone were challenged with MMC6 tumor cells (5×103) on d 1 post-BMT. Experiments were repeated twice with 3-5 mice per group. ATBM + MMC6 versus ATBM + MMC6 + CD8+Vβ13+ T cells (1x), p < 0.01; ATBM + MMC6 versus ATBM + MMC6 + CD8+Vβ13+ T cells (2x), p < 0.01; ATBM + MMC6 versus ATBM + MMC6 + CD8+Vβ13+ T cells (3x), p < 0.01; ATBM + MMC6 + CD8+Vβ13+ T cells (1x) versus ATBM + MMC6 + CD8+Vβ13+ T cells (2x), p = 0.59; ATBM + MMC6 + CD8+Vβ13+ T cells (1x) versus ATBM + MMC6 + CD8+Vβ13+ T cells (3x), p = 0.05; ATBM + MMC6 + CD8+Vβ13+ T cells (2x) versus ATBM + MMC6 + CD8+Vβ13+ T cells (3x), p < 0.02.
Based on these results, we increased the dose of B10.BR CD8+Vβ13+ T cells transplanted in order to enhance the observed GVL effect. A 2x dose of CD8+Vβ13+ T cells (1.67×106) did not lead to an increased GVL effect over a 1x dose (Fig. 3B) (MST = 32 d, p = 0.59). In addition, these mice began to display symptoms of GVHD (weight loss, diarrhea; data not shown). Administering a 3x dose of CD8+Vβ13+ T cells (2.5×106) significantly increased the survival rate of tumor challenged mice (Fig. 3B) (MST = 49 d, p < 0.01). However, mice receiving the 3x dose alone (not tumor challenged) experienced a 70% mortality rate, associated with increased symptoms of GVHD (MST = 54 d) (Fig. 3A). These results demonstrated that while a low dose of B10.BR CD8+Vβ13+ T cells was unable to induce GVHD in CBA recipients, higher doses could indeed cause disease. Because the purity of the CD8+Vβ13+ T cell fraction ranged between 90-95%, a separate group of mice was injected with the equivalent amount of contaminating cells present in a 3x dose (i.e., 7.4×104) to ensure that the observed GVHD at the higher dose was not caused by other alloreactive CD8+ T cells in the CD8+Vβ13+ T cell inoculum. These mice displayed no symptoms of GVHD and all survived until the termination of the experiment (data not shown).
The CD8+Vβ13+ T cell family is skewed in GVHD target tissues
Despite the fact that Vβ13 was not found to be skewed in the B10.BR anti-CBA alloresponse against hematopoietically-derived miHA, as determined by the spectratype analysis of splenocytes from CBA recipients of B10.BR CD8+ T cells (Table I; Fig. 2B), CD8+Vβ13+ T cells were indeed capable of causing GVHD when infused at higher dose equivalents, particularly at 3x (Fig. 3A). This unexpected result prompted us to conduct spectratype analysis on infiltrating T cells of GVHD-target tissues (i.e, liver and gut). The analyses revealed skewing of the Vβ13 family in the intestine of CBA recipient mice (Fig. 4A; Table II), although at a different non-overlapping CDR3-size length (169) than those found in the anti-MMC6 response (160,163,166). Furthermore, while there were many overlapping skewed CDR3-size lengths among the Vβ families between the intestines and the liver, the Vβ13 family was not found to be skewed in the liver (Table II).
FIGURE 4. Representative spectratype histograms in the B10.BR anti-CBA and anti-MMC6 CD8+Vβ13+ T cell responses in intestinal tissue.
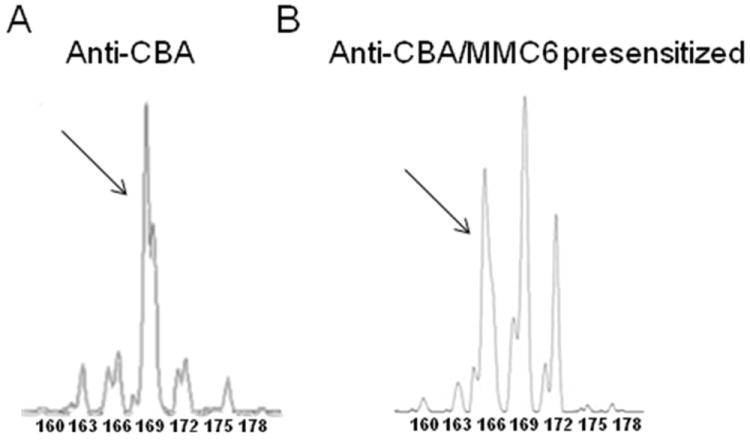
(A) For alloreactive responses (anti-CBA), CBA mice were exposed to lethal irradiation (11 Gy, split dose) and transplanted with 1-2×107 B10.BR CD8+ T cells. Intestinal tissue was harvested at 10-13 d post-BMT. (B) For the anti-tumor response in the intestine, CBA mice were exposed to lethal irradiation (11 Gy, split dose) and transplanted with 2×107 MMC6 presensitized CD8+ T cells and tissue was harvested at 10-13 d post-BMT. Tissues were processed and spectratype analyses were performed. CD8+ splenic T cells harvested from naïve B10.BR mice acted as the control group (shown in Fig. 2A). Skewing was defined as a peak area greater than the mean of the control plus 5×SD. CDR3 lengths are shown under each peak. Skewed peaks are marked with an arrow.
Table II.
Comparison of the skewed CDR3-size lengths of B10.BR CD8+ T cells from the GVHD-target organs of transplanted CBA recipient micea
| Vβ | Spleen | Intestine | Liver |
|---|---|---|---|
| 1 | — | 192 | 192 |
| 2 | — | 151, 163 | 163 |
| 3 | 149 | 149 | — |
| 4 | — | — | — |
| 5 | 172 | — | — |
| 6 | — | 148, 151, 154 | 148 |
| 7 | 176 | 176 | 176 |
| 8.1 | — | 133 | — |
| 8.2 | 134 | 137 | 140 |
| 8.3 | 155, 170 | 164 | 164 |
| 9 | 147 | 147, 150 | 144, 147 |
| 10 | — | 145 | 145 |
| 11 | 157 | 160, 163 | 148, 157 |
| 12 | 201 | 201, 216 | 210 |
| 13 | — | 169 | — |
| 14 | 160 | 160 | 160 |
| 15 | 182 | 170, 176, 182 | 167, 179 |
| 16 | — | — | — |
| 18 | 225 | 219 | 216 |
| 20 | 196 | 199 | 199, 202 |
Lethally irradiated CBA recipients were injected i.v. with 2 × 107 CD8+ B10.BR T cells. 10 days post-BMT, GVHD-target tissue infiltrating CD8+ T cells were isolated and CDR3-size spectratype analysis was performed as described in Materials and Methods. Skewing of bands was defined as an average peak area greater than the average control area plus 5×SD. —, No skewing; CDR3 lengths are shown for skewed Vβ families. Results were averaged from 3 separate groups of 3-5 mice/group.
Tumor-presensitized CD8+Vβ13+ T cells induce a potent GVL effect with minimal GVHD
Previous studies have demonstrated that tumor-presensitized T cells can exert potent GVL responses in the absence of GVHD (14), hence we also tested if in the B10.BR→CBA model, tumor-presensitization could help bias the reactivity of the cells towards an enhanced GVL response with minimal GVHD complication. To this end, donor B10.BR mice were injected i.p. with 1×107 irradiated (40 Gy) MMC6 tumor cells 17-21 days prior to BMT. The results from these experiments (Fig. 5) indicated that tumor-challenged CBA mice receiving a 1x dose of tumor-presensitized B10.BR CD8+Vβ13+ T cells (8.34×105) at time of BMT displayed a potent GVL effect compared to mice that received an equal amount of naïve CD8+Vβ13+ T cells (MST = 77 d vs. 31.5 d, respectively; p = 0.03). Of the nine mice that died in the former group, 4 had bloody ascites upon post-mortem necropsy, indicative of tumor progression, 2 exhibited symptoms of GVHD, 1 displayed both symptoms of GVHD and had evidence of tumor growth, and 2 had an undetermined cause of death. In contrast, only 37.5% of the CBA mice receiving CD8+Vβ13+ T cells in the absence of tumor challenge succumbed to GVHD. The remaining mice, while displaying mild symptoms of GVHD, such as moderate weight loss (70% of initial body weight), achieved 112% of their initial body weight at the time of termination of the experiment (100 d post-BMT) (Fig. 6A). In addition, ear tissue samples were collected at the termination of some experiments to determine the extent of GVHD pathology, and a representative analysis is shown in Fig. 6B. Surviving CBA recipients injected with B10.BR CD8+ T cells [panel (b)] exhibited classical GVHD pathology in the ear as evidenced by a 4-7 fold increase in dermal thickness (double arrow) compared to control mice transplanted with ATBM cells alone [panel (a)] or with B10.BR CD8+Vβ13+ T cells (1x presensitized) [panel (c)]. Furthermore, epidermal hyperplasia (single arrows) was also present in specimens from CBA mice receiving CD8+ T cells, accompanied by papillary folds in the epidermal-dermal border. A magnified image (100X) demonstrates the collagen layer and cellularity found in the dermal layer [panels (d), (e) and (f)]. Similar to the ATBM control group, surviving recipients of CD8+Vβ13+ (1x presensitized) T cells displayed minimal infiltration and collagen deposition (an indicator of tissue damage) [panels (d) & (f)] compared to the CD8+ T cell specimen [panel (e)]. The recovery of the gut epithelium of surviving mice transplanted with CD8+Vβ13+ (1x presensitized) T cells is depicted in Fig. 6C. Specimens from the ATBM control group [panel (a)] had a perfect and healthy epithelial arrangement. Samples from CD8+ T cell injected mice [panel (b)] had an abnormal epithelium suggestive of a severe stress of the small intestine, characterized by disruption of the intestinal villi (arrows). Specimens from the CD8+Vβ13+ (1x presensitized) T cell group [panel (c)] had more defined epithelial crypts (arrows) and overall less damage, which supported the lack of diarrhea and the increase in weight observed in this group at the termination of the experiment.
FIGURE 5. Tumor-presensitized donor B10.BR CD8+Vβ13+ T cells elicit a significant GVL effect with minimal GVHD.
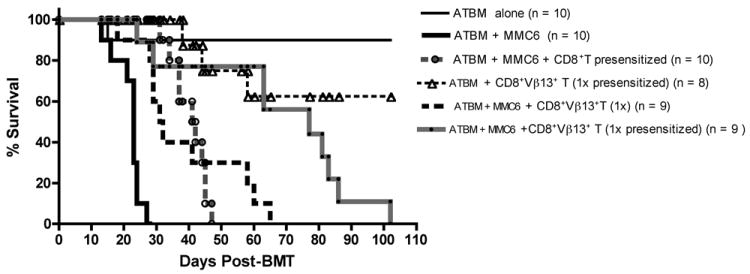
CBA mice were exposed to lethal irradiation (11 Gy, split dose) and transplanted with ATBM cells (2×106) alone or along with either CD8+ T cells (1×107) or CD8+Vβ13+ T cells (1x, 8.34×105 or 3x, 2.5×106) from MMC6 presensitized or naïve donor B10.BR mice. Transplanted mice were challenged with MMC6 tumor cells (5×103) on d 1 post-BMT. Experiments were repeated twice, each with 4-5 mice per group. Statistical significance between survival curves was determined using the non-parametric Wilcoxon test. ATBM + MMC6 versus ATBM + MMC6 + CD8+Vβ13+ T cells (1x presensitized), p <0.01; ATBM + CD8+ T cells + MMC6 versus ATBM + MMC6 + CD8+Vβ13+ T cells (1x presensitized), p < 0.05; ATBM + MMC6 + CD8+Vβ13+ T cells (1x), versus ATBM + MMC6 + CD8+Vβ13+ T cells (1x presensitized), p = 0.03.
FIGURE 6. CBA recipients of tumor-presensitized B10.BR CD8+Vβ13+ T cells exhibit minimal histological evidence of GVHD.
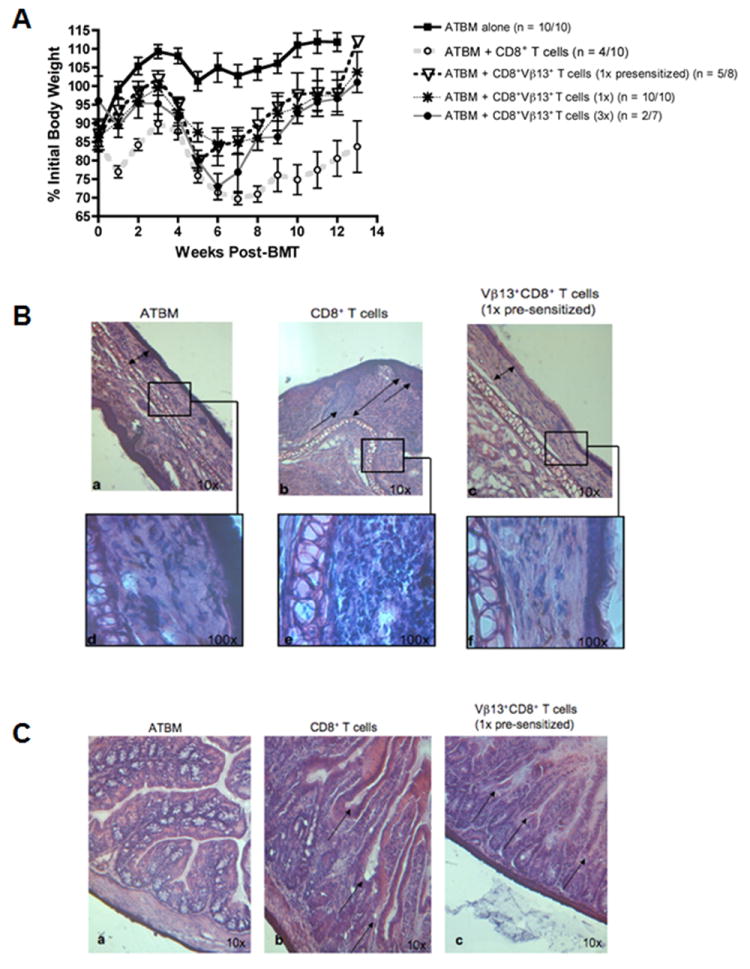
CBA mice were exposed to lethal irradiation (11 Gy, split dose) and transplanted with 2×106 ATBM cells alone or in combination with either CD8+ T cells (1×107) or CD8+Vβ13+ T cells (1x, 8.34×105 or 3x, 2.5×106) from MMC6 presensitized or naïve B10.BR donor mice, as indicated. A, The mean ± SE % initial body weight of surviving mice in each group was derived relative to the mean weight of the group on d 0. n = # mice surviving at termination of experiment/ # mice at initiation of experiment. B, Comparison of the dermis and epidermis of the ear between mice transplanted with ATBM cells alone, CD8+ T cells, or CD8+Vβ13+ T cells (1x presensitized). H&E reveals normal and regular thickness of the epithelium (double arrows) of the ATBM [a] and CD8+Vβ13+ T cells (1x presensitized) specimens [c], while there is severe irregularity of the epidermal-dermal border (single arrows) in the CD8+ T cell sample [b] with evident hyperplasia of the epidermis. Likewise, the collagen layer in the dermis (double arrow) is regular and well arranged in the ATBM and CD8+Vβ13+ T cell sample [a&c] and has low cellularity [d&f]. In contrasts, the dermis in the CD8+ T cell sample exhibits increased thickness and vast cellularity (double arrow) [e]. C, comparison of the epithelium of the small intestine between mice transplanted with ATBM cells alone, CD8+ T cells, or CD8+Vβ13+ T cells (1x presensitized). H&E reveals the integrity of the epithelium in the ATBM specimen [a] while there is loss of the architecture of the villi and crypts in the CD8+ T cell sample (arrows) [b] compared to the more defined epithelial structure observed in the CD8+Vβ13+ T cell (1x presensitized) sample (arrows) [c].
The CD8+Vβ13+ skewing pattern is altered in GVHD target tissue in the presence of tumor antigens
Since GVHD appeared to be alleviated in CBA recipients of tumor-presensitized B10.BR CD8+Vβ13 T cells, spectratype analysis was performed on gut-infiltrating CD8+ T cells to determine what CDR3-lengths were exhibiting skewing. As mentioned earlier, in the absence of tumor antigens, skewing of Vβ13 at one CDR3-size length (169) was observed in the intestine (Fig. 4A). However, when recipient mice were transplanted with MMC6-presensitized CD8+ T cells, Vβ13 skewing was only detected at CDR3-size length 166 (Fig. 4B) which corresponded with that length found skewed in the anti-tumor response in the lymphoid compartment. Table III lists the skewed Vβ13 CDR3-size lengths in the alloreactive and anti-tumor response in the hematopoietic compartment compared to the naïve and tumor-presensitized alloreactive response in the hematopoietic compartment and the intestine.
Table III.
Comparison of skewed Vβ13 CDR3-size lengths in the anti-CBA and anti-MMC6 response in the spleen versus the intestines or recipient mice.
| 160 | 163 | 166 | 169 | 172 | 175 | 178 | |
|---|---|---|---|---|---|---|---|
| α-CBA, spleen | - | - | - | - | - | - | - |
| α-MMC6, spleen | + | + | + | - | - | - | - |
| α-CBA, intestine | - | - | - | + | - | - | - |
| α-CBA/MMC6 pre-sensitized, spleena | + | - | + | - | - | - | - |
| α-CBA/MMC6 pre-sensitized, intestinea | - | - | + | - | - | - | - |
Lethally irradiated CBA recipients were injected i.v. with 2 × 107 MMC6 pre-sensitized CD8+ B10.BR T cells. 10 days post-BMT, GVHD-target tissue infiltrating CD8+ T cells were isolated and CDR3-size spectratype analysis was performed as described in Materials and Methods. Skewing of bands was defined as an average peak area greater than the average control area plus 5×SD. —, No skewing; +, skewed CDR3-size length. Results were averaged from 3 separate groups of 3-5 mice/group.
Increased effector memory T cell phenotype of tumor-presensitized CD8+Vβ13+ T cells
In order to elucidate the mechanism underlying the observed differences between the GVHD/GVL potential of MMC6-presensitized and naïve B10.BR CD8+Vβ13+ T cells, we conducted flow cytometric analysis to determine the phenotype of the injected donor T cells. Although there did not appear to be an increase in the overall expression of CD44 in the CD8+Vβ13+ T cell population from MMC6-presensitized mice as compared to naïve mice, differences were observed in expression levels of the lymphoid homing receptor L-selectin (CD62L), a marker that differentiates central memory T cells (TCM; CD44+CD62L+) from effector memory T cells (TEM; CD44+CD62Llo/-) (16). The overall results (a representative sample is displayed in Fig. 7), showed a dramatic decrease in CD62L expression on CD8+Vβ13+ T cells that had been MMC6-presensitized as compared to naïve cells (mean CD62L+ of 32.06 ±16.2% vs. 60.8±19.5%, respectively, n = 5, p = 0.02), corresponding to a TEM phenotype. Given the presence of gut-associated GVHD in mice transplanted with CD8+Vβ13+ T cells, we also looked at the expression of α4β7 integrin, an adhesion molecule involved in lymphocyte homing in the gut, in cells from the mesenteric lymph nodes of tumor-presensitized mice (17). After gating on the CD44+ memory cells in the CD8+Vβ13+ T population, we observed a slight, but significant increase in the expression of α4β7 as compared to this cell population from naïve mice (mean α4β7+ of 8.2 ±5.8% vs. 3.8 ±4.4%, respectively, n = 4, p = 0.03) (a representative sample is displayed in Fig. 8).
FIGURE 7. Memory (CD44+) CD8+Vβ13+ T cells from the spleens and lymph nodes of tumor pre-sensitized B10.BR mice exhibit decreased CD62L expression as compared to naïve mice.
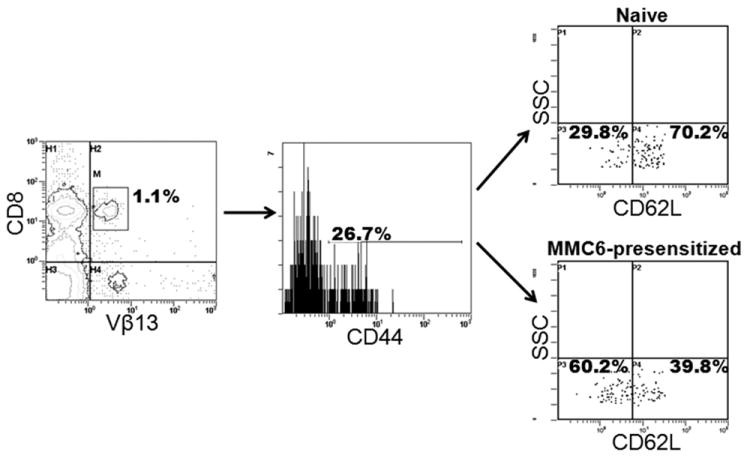
B10.BR mice were injected i.p. with 1×107 MMC6 cells (40Gy). Three weeks later, cell suspensions from spleens and lymph nodes were stained for flow cytometric analysis. Cells were first gated on CD8+ Vβ13+ cells (left panel) followed by CD44 expression (middle panel). The percent expression of CD62L on naïve cells is shown in the top right panel compared to the MMC6 presensitized cells in the bottom right panel. One representative experiment is shown.
FIGURE 8. Tumor-presensitized memory (CD44+) CD8+Vβ13+ T cells from the mesenteric lymph nodes of B10.BR mice upregulate the expression of α4β7 integrin.
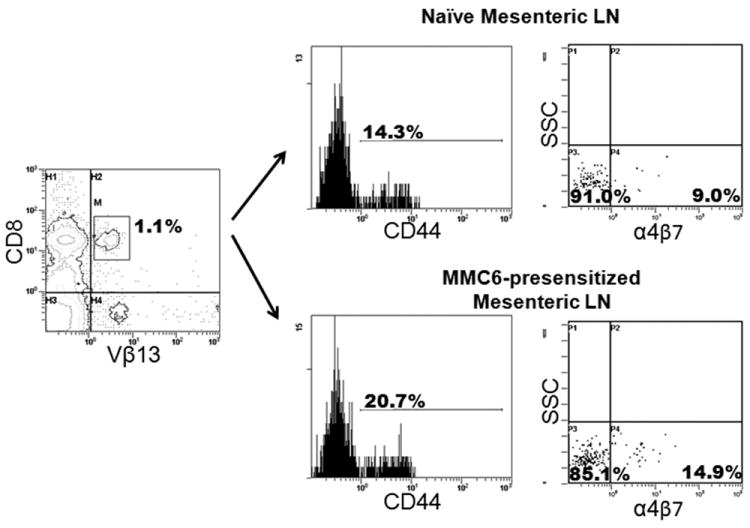
B10.BR mice were injected i.p. with 1×107 MMC6 cells (40 Gy). Mesenteric lymph nodes were harvested 17-21 d later for flow cytometric analysis. Cells were first gated on CD8+Vβ13+ cells (left panel) followed by expression of CD44 (middle panels). The percent expression of α4β7 on naïve cells is shown in the top right panel compared to the MMC6-presensitized cells in the bottom right panel. One representative experiment is shown.
DISCUSSION
It has long been observed that following BMT, patients who develop clinical GVHD have a reduced incidence of leukemic relapse (6, 18, 19). This can be directly correlated to the presence of mature T cells in the donor inoculum which are known to contribute to both GVHD and GVL effects. Many methods have been proposed in order to reduce the incidence of GVHD while shifting the balance towards the favorable GVL potentials of BMT, yet there is much to be done on the road to a complication-free implementation of this immunotherapy for the treatment of hematological malignancies. For example, approaches attempting to deplete the entire compartment or major subsets of T cells in the donor inoculum have not only led to an increase in the incidence of leukemic relapse, but also resulted in an increase in graft failure and opportunistic infections (20, 21). In recent years, we and others have demonstrated that CDR3-size spectratyping of the TCR Vβ chain can be utilized to differentiate alloreactive from uniquely tumor-reactive T cells, opening the door for potential tailoring of the donor inoculum to recipient’s malignancy (13, 22, 23). In other words, donor T cells could be selected for their specific tumor-reactivity, or depleted if confirmed to be uniquely alloreactive, in order to impart maximum benefit with reduction/elimination of deleterious side effects.
In the current study, we mimic the common scenario of minimal residual disease following an MHC-matched transplant for acute myeloid leukemia, using the well established MHC-matched/miHA-disparate B10.BR→CBA BMT model (1) and the MMC6 recipient-derived myeloid leukemia cell line, to demonstrate that spectratype analysis can be successfully used to identify unique donor anti-tumor responses. Only six Vβ families were skewed in the CD8+ T cell anti-MMC6 response; of these, three were also skewed in the alloresponse against CBA splenocytes, while three remained unique to the anti-tumor response. For our in vivo studies, we decided to focus solely on the CD8+Vβ13+ T cells because it was the only family composed of three skewed CDR3-size lengths and the anti-Vβ mAb was commercially available for separation. Interestingly, the spectratype analysis revealed that 12 Vβ families were skewed in the B10.BR anti-CBA alloresponse, whereas only 6 were found to be participating in the B10.BR anti-MMC6 response (Table I). Since the tumor is of recipient CBA origin, it would follow that MMC6 cells might express many of the alloantigens along with some tumor-specific antigens and therefore would have been expected to elicit a response from the same number, if not more Vβ families. One possible explanation is that the tumor cells express a monocytic-myeloid phenotype, and some of the alloreactive Class I-restricted antigens may not be expressed in cells of this particular stage or subtype. Alternatively, tumor cells may alter certain self-protein pathways, thereby ultimately reducing the presence of some protein degradation products which in turn account for less miHA being presented. In addition, tumor cells often develop mechanisms to evade the immune response including the ability to down-regulate the expression of Class I molecules and some immunogenic antigens (24, 25), albeit MMC6 tumor-specific antigens are still being recognized.
The in vivo experiments confirmed that B10.BR CD8+Vβ13+ T cells have the ability to mediate a GVL response against MMC6 tumor challenge in CBA recipients (Fig. 3B), and that tumor-free survival is further extended when given at higher (2x and 3x) dosages. That notwithstanding, increased numbers of donor CD8+Vβ13+ T cells also led to acute GVHD, characterized particularly by gut-related symptoms and increased recipient mortality (Fig. 3A). Since the spectratype data against hematopoietically-derived host miHA antigens, found in the spleen of recipient mice, did not reveal alloreactivity of the Vβ13 family, we conducted further spectratype analysis on the tissue-infiltrating T cells in other target organs, as had been previously examined in another BMT model (15). We found that the B10.BR CD8+Vβ13+ family was indeed skewed in the intestines of CBA recipients of naïve B10.BR CD8+ donor T cells at CDR3-size length 169, albeit a length distinct from those found expanded in the anti-tumor response (160, 163, and 166; Table III). Since the B10.BR CD8+Vβ13+ T cell tissue alloresponse did not originate in the hematopoietic compartment, the observed results may suggest the presence of a tissue-specific cryptic epitope, inducing the peak 169 expansion, that emerged only upon sufficient tissue damage caused by infiltration of a large number of naïve T cells (3x). It has been postulated that T cells which react to a specific immunodominant epitope may induce an inflammatory response at the site of antigen encounter that leads to the generation of tissue debris that is later processed and cross-presented by host antigen presenting cells. The response is thus amplified due to the recruitment of other T cells reacting to the subdominant and/or cryptic epitopes (26). Furthermore, only one CDR3-size length, 166, was found to be expanded in the intestine of recipients inoculated with MMC6-presensitized B10.BR CD8+ T cells while the tissue-specific alloresponse associated with peak 169 was actually lost (or potentially mitigated) in the CD8+Vβ13+ gut-infiltrating T cells, possibly because there was not enough tissue injury mediated to release the cryptic epitope recognized by that subset of T cells. The expansion of the 166 peak, associated with a tumor-specific response in the lymphoid compartment, concomitant with the disappearance of the alloreactive 169 peak, suggests immunodominance of this anti-tumor response. However because peak 166 was also found skewed in the spleen of mice receiving tumor-presensitized cells we cannot rule out the possibility that the detected tissue skewing is related to non-specific entry of the already expanded and recently activated cells into the tissue as either bystanders or drawn in by the inflammatory conditions following irradiation exposure. It is also possible that the tumor-presensitized CD8+Vβ13+ T cells were cross-reactive in their recognition of MMC6 tumor antigens and miHA epitopes expressed at low levels in the intestinal epithelium of CBA mice.
Presensitization to tumor prior to harvesting and transplantation of donor T cells, potentiated the GVL effects of the CD8+Vβ13+ T cell family in our model, while circumventing the unwanted GVHD observed at higher cell doses. This strong GVL response was characterized by a statistically significant increase in the mean survival time of tumor-challenged mice receiving a 1x dose of presensitized CD8+Vβ13+ T cell compared to mice injected with an equal dose of naïve cells (MST = 77 vs. MST = 31.5 days; p = 0.03, Fig. 5). In addition, the incidence of lethal GVHD was also reduced in mice that received 1x presensitized T cell fractions compared to those receiving a 3x dose of naïve T cells (62.5% survival vs. 29% survival, respectively). In the absence of tumor burden, 37.5 % of mice receiving 1x presensitized T cells succumbed to GVHD, whereas the surviving mice fully recovered from mild symptoms of the disease by the termination of the experiment (Fig. 6A, B, C). These results suggested that presensitizing donor cells to host tumor prior to transplant can be an advantageous therapeutic approach to increase the GVL response while minimizing acute, lethal GVHD related to the transplantation of large numbers of donor T cells.
We subsequently examined the IFN-γ response of B10.BR CD8+Vβ13+ cells to either alloantigen or tumor, by ELISpot assay. While there was an overall higher IFN-γ response in the tumor-presensitized cells, there was no significant difference between the alloresponse and the anti-tumor response (data not shown). Again, these results correlate with our in vivo observation that tumor-presensitized CD8+Vβ13+ T cells did mediate some GVHD-related pathology.
The role memory T cells play in the development of GVHD and GVL responses is actively under investigation. Memory T cells can be divided into two types, central memory T cells (TCM) which express CD44, along with CD62L, a cell adhesion molecule important in lymph node homing, and effector memory T cells (TEM) which lack CD62L. It is believed that TEM, which have enhanced abilities to induce effector mechanisms such as IFNγ production and perforin release, function as sentinels in peripheral tissues armed for immediate protection, while TCM home to the lymph nodes where they are able to generate a second wave of effector cells (16, 27, 28). Flow cytometric analysis was used to determine any phenotypic changes that might correlate with the enhanced capacity of tumor-presensitized B10.BR CD8+Vβ13+ T cells to eradicate tumor. The results revealed that there was a higher percentage of cells with TEM phenotype (i.e., CD44+CD62L-) in the CD8+Vβ13+ T cell population in the spleen and lymph nodes of presensitized animals compared to naïve cells (67.7 ±15.9% vs. 39.0 ±19.2%, CD62L-, respectively, p=0.02, Fig. 7). Furthermore, presensitized CD8+Vβ13+ T cells of the mesenteric lymph nodes also expressed a higher percentage of the heterodimic integrin α4β7, which is present in activated T cells and facilitates their infiltration in the gut epithelium (8.2 ±5.8% vs. 3.8 ±4.4%, respectively, p=0.03, Fig. 8), corroborating the low-grade intestinal pathology observed in animals transplanted with tumor-presensitized T cells.
Several studies have found that TEM and TCM to previously encountered environmental antigens failed to mediate GVHD in either MHC-matched or MHC-mismatched models of BMT (29-31). This inactivity was hypothesized to be due to a lack of cross-reactivity between environmental antigens and host alloantigens. On the other hand, studies examining the effects of alloantigen primed memory T cells have shown TCM to be highly GVH reactive, with TEM unable to mediate GVHD (32-35). This is not due to differences in the capacity of these populations to home to secondary lymphoid tissue as initially postulated (36, 37), but rather due to an abortive response within target tissues (31, 35). In regard to their GVL potential, several investigators have demonstrated the anti-tumor effects of CD62L- T cells (29, 38, 39). Of most interest, Chen et al found that CD62L- T cells lacked a response to alloantigens, and tumor-presensitized CD62L- T cells were able to inhibit tumor growth while remaining unable to induce GVHD in third-party recipients (29). Likewise, Yang et al have also shown that shedding of CD62L is necessary for T cells to acquire lytic activity required for tumor eradication (40). Others however, have found that in vitro generated CD8+ TCM are more adept at eradicating tumor in vivo than other T cell populations, including naïve T cells and TEM. (36, 41). Yet as previously stated, the TCM population has been shown to also play a predominate roll in GVHD. Ultimately, the immunotherapeutic use of BMT comes down to determining the population that can mediate antitumor responses with minimal GVHD. Results from our model using spectratype identified, tumor presensitized CD8+ T cells suggest that TEM are the cells responsible for mediating the observed GVL effects while keeping GVHD responses to a minimum. Further experiments using purified memory T cell populations in the current model will help to confirm this hypothesis.
Here, we have expanded the current knowledge regarding the GVL/GVHD potential of antigen-experienced cells by transplanting an anti-tumor reactive Vβ T cell population, as determined by TCR Vβ spectratyping, into tumor-challenged mice in a BMT setting. Since we found superior GVL capability of the donor CD8+Vβ13+T cells after presensitization with host tumor, and the phenotype of these cells indicated an increase in the TEM (CD62L-CD8+Vβ13+) population, it stands to reason that these cells are most likely responsible for the conferred anti-tumor effects. That notwithstanding, we also noted an increase in the incidence of mild GVHD in recipients of tumor-presensitized CD8+Vβ13+ T cells compared to mice that received an equal number of naïve CD8+Vβ13+ T cells, potentially due to cross-reactivity between some tumor and tissue alloantigens. Further experiments are currently being conducted in our laboratory to elucidate the GVL/GVHD role of the CD62L-CD8+Vβ13+ T cell population in the B10.BR→CBA BMT model.
In order to translate our findings into the clinical setting, ex vivo priming of donor T cells against recipient tumor antigens would be the most likely scenario. This procedure has been successfully performed in several studies, particularly in the form of dendritic cell-based tumor vaccines (42, 43). However, in the case of a known tumor antigen, it is also possible to presensitize a normal donor, as described by Kwak et al. and others (44, 45). In addition, tumor cell purging of ex vivo expanded hematopoietic stem cells has been successfully implemented in autologous transplant for both liquid and solid tumors (46, 47).
It would also be of interest to determine if other methods of activation, aside from or concomitant with tumor presensitization, can lead to increased GVL potential of the CD8+Vβ13+ T cell population while keeping the GVHD response to a minimum. An array of cytokines and costimulatory ligands have been shown to successfully activate, expand, or promote the survival of T cells, as well as increase the memory T cell population which we have shown to be important in this model (48-51). Klebanoff et al have examined an array of γc cytokines, including IL-2, IL-7, IL-15, and IL-21, and showed that all were capable of augmenting in vivo antitumor responses of adoptively transferred CD8+ T cells (41). Treatment with IL-2 in conjunction with costimulation with anti-CD3 mAb has been used to activate CD62Llo T cells from tumor-draining lymph nodes to increase antitumor responses (52). In addition, IL-2/anti-IL2 antibody complexes have been shown to not only enhance CD8+ T cell activation, but also to increase the number of antigen-specific effector/memory CD8+ T cells in a viral vaccine model (53). A variety of cell-intrinsic modulators of memory formation (CIMMs) have been reported in the literature (54). These small molecules target key metabolic pathways such as the mTOR, PI3K, and Wnt/β-catenin signaling pathways that can augment the development of CD8+ memory T cells. For instance, investigators have demonstrated that the immunosuppressive drug rapamycin, which affects the intacellular kinase mTOR, can enhance memory T cell responses in vaccinated mice (55).
Acknowledgments
In the process of preparing this manuscript, Dr. Friedman battled and succumbed to ALS. Her impetus and constant dedication to her research will continue to be a source of inspiration for all of us who had the privilege of working with her.
Footnotes
This work was supported in part by the National Institute of Health grants RO1 HL075622 (to T.M.F.) and RO1 HL055593 (to R.K.).
Abbreviations used in this paper: BMT, blood and marrow transplant; GVHD, graft-versus-host disease; GVL, graft-versus-leukemia; CDR3, complementarity-determining region 3; miHA, minor histocompatibility antigen; ATBM, anti-T cell-depleted bone marrow; LN, lymph nodes; MST, median survival time.
Disclosures
The authors have no financial conflict of interest.
References
- 1.Korngold R, Sprent J. Lethal graft-versus-host disease after bone marrow transplantation across minor histocompatibility barriers in mice. Prevention by removing mature T cells from marrow. J Exp Med. 1978;148:1687–1698. doi: 10.1084/jem.148.6.1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Korngold R, Sprent J. H-2 restriction of T cells causing lethal graft-versus-host disease across minor histocompatibility barriers in mice. Transplant Proc. 1981;13:1217–1219. [PubMed] [Google Scholar]
- 3.Korngold R, Sprent J. Variable capacity of L3T4+ T cells to cause lethal graft-versus-host disease across minor histocompatibility barriers in mice. J Exp Med. 1987;165:1552–1564. doi: 10.1084/jem.165.6.1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hamilton BL, Bevan MJ, Parkman R. Anti-recipient cytotoxic T lymphocyte precursors are present in the spleens of mice with acute graft versus host disease due to minor histocompatibility antigens. J Immunol. 1981;126:621–625. [PubMed] [Google Scholar]
- 5.Fowler DH. Shared biology of GVHD and GVT effects: potential methods of separation. Crit Rev Oncol Hematol. 2006;57:225–244. doi: 10.1016/j.critrevonc.2005.07.001. [DOI] [PubMed] [Google Scholar]
- 6.Horowitz MM, Gale RP, Sondel PM, Goldman JM, Kersey J, Kolb HJ, Rimm AA, Ringden O, Rozman C, Speck B, et al. Graft-versus-leukemia reactions after bone marrow transplantation. Blood. 1990;75:555–562. [PubMed] [Google Scholar]
- 7.Marmont AM, Horowitz MM, Gale RP, Sobocinski K, Ash RC, van Bekkum DW, Champlin RE, Dicke KA, Goldman JM, Good RA, et al. T-cell depletion of HLA-identical transplants in leukemia. Blood. 1991;78:2120–2130. [PubMed] [Google Scholar]
- 8.Pavletic SZ, Kumar S, Mohty M, de Lima M, Foran JM, Pasquini M, Zhang MJ, Giralt S, Bishop MR, Weisdorf D. NCI First International Workshop on the Biology, Prevention, and Treatment of Relapse after Allogeneic Hematopoietic Stem Cell Transplantation: report from the Committee on the Epidemiology and Natural History of Relapse following Allogeneic Cell Transplantation. Biol Blood Marrow Transplant. 2010;16:871–890. doi: 10.1016/j.bbmt.2010.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Miller JS, Warren EH, van den Brink MR, Ritz J, Shlomchik WD, Murphy WJ, Barrett AJ, Kolb HJ, Giralt S, Bishop MR, Blazar BR, Falkenburg JH. NCI First International Workshop on The Biology, Prevention, and Treatment of Relapse After Allogeneic Hematopoietic Stem Cell Transplantation: Report from the Committee on the Biology Underlying Recurrence of Malignant Disease following Allogeneic HSCT: Graft-versus-Tumor/Leukemia Reaction. Biol Blood Marrow Transplant. 2010;16:565–586. doi: 10.1016/j.bbmt.2010.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Friedman TM, Gilbert M, Briggs C, Korngold R. Repertoire analysis of CD8+ T cell responses to minor histocompatibility antigens involved in graft-versus-host disease. J Immunol. 1998;161:41–48. [PubMed] [Google Scholar]
- 11.Jones SC, Friedman TM, Murphy GF, Korngold R. Specific donor Vbeta-associated CD4 T-cell responses correlate with severe acute graft-versus-host disease directed to multiple minor histocompatibility antigens. Biol Blood Marrow Transplant. 2004;10:91–105. doi: 10.1016/j.bbmt.2003.10.002. [DOI] [PubMed] [Google Scholar]
- 12.Friedman TM, Statton D, Jones SC, Berger MA, Murphy GF, Korngold R. Vbeta spectratype analysis reveals heterogeneity of CD4+ T-cell responses to minor histocompatibility antigens involved in graft-versus-host disease: correlations with epithelial tissue infiltrate. Biol Blood Marrow Transplant. 2001;7:2–13. doi: 10.1053/bbmt.2001.v7.pm11215694. [DOI] [PubMed] [Google Scholar]
- 13.Patterson AE, Korngold R. Infusion of select leukemia-reactive TCR Vbeta+ T cells provides graft-versus-leukemia responses with minimization of graft-versus-host disease following murine hematopoietic stem cell transplantation. Biol Blood Marrow Transplant. 2001;7:187–196. doi: 10.1053/bbmt.2001.v7.pm11349805. [DOI] [PubMed] [Google Scholar]
- 14.Korngold R, Leighton C, Manser T. Graft-versus-myeloid leukemia responses following syngeneic and allogeneic bone marrow transplantation. Transplantation. 1994;58:278–287. [PubMed] [Google Scholar]
- 15.Zilberberg J, McElhaugh D, Gichuru LN, Korngold R, Friedman TM. Inter-strain tissue-infiltrating T cell responses to minor histocompatibility antigens involved in graft-versus-host disease as determined by Vbeta spectratype analysis. J Immunol. 2008;180:5352–5359. doi: 10.4049/jimmunol.180.8.5352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sallusto F, Geginat J, Lanzavecchia A. Central memory and effector memory T cell subsets: function, generation, and maintenance. Annu Rev Immunol. 2004;22:745–763. doi: 10.1146/annurev.immunol.22.012703.104702. [DOI] [PubMed] [Google Scholar]
- 17.Petrovic A, Alpdogan O, Willis LM, Eng JM, Greenberg AS, Kappel BJ, Liu C, Murphy GJ, Heller G, van den Brink MR. LPAM (alpha 4 beta 7 integrin) is an important homing integrin on alloreactive T cells in the development of intestinal graft-versus-host disease. Blood. 2004;103:1542–1547. doi: 10.1182/blood-2003-03-0957. [DOI] [PubMed] [Google Scholar]
- 18.Passweg JR, Tiberghien P, Cahn JY, Vowels MR, Camitta BM, Gale RP, Herzig RH, Hoelzer D, Horowitz MM, Ifrah N, Klein JP, Marks DI, Ramsay NK, Rowlings PA, Weisdorf DJ, Zhang MJ, Barrett AJ. Graft-versus-leukemia effects in T lineage and B lineage acute lymphoblastic leukemia. Bone Marrow Transplant. 1998;21:153–158. doi: 10.1038/sj.bmt.1701064. [DOI] [PubMed] [Google Scholar]
- 19.Kolb HJ, Schattenberg A, Goldman JM, Hertenstein B, Jacobsen N, Arcese W, Ljungman P, Ferrant A, Verdonck L, Niederwieser D, van Rhee F, Mittermueller J, de Witte T, Holler E, Ansari H. Graft-versus-leukemia effect of donor lymphocyte transfusions in marrow grafted patients. Blood. 1995;86:2041–2050. [PubMed] [Google Scholar]
- 20.Apperley JF, Jones L, Hale G, Waldmann H, Hows J, Rombos Y, Tsatalas C, Marcus RE, Goolden AW, Gordon-Smith EC, et al. Bone marrow transplantation for patients with chronic myeloid leukaemia: T-cell depletion with Campath-1 reduces the incidence of graft-versus-host disease but may increase the risk of leukaemic relapse. Bone Marrow Transplant. 1986;1:53–66. [PubMed] [Google Scholar]
- 21.Small TN, Papadopoulos EB, Boulad F, Black P, Castro-Malaspina H, Childs BH, Collins N, Gillio A, George D, Jakubowski A, Heller G, Fazzari M, Kernan N, MacKinnon S, Szabolcs P, Young JW, O’Reilly RJ. Comparison of immune reconstitution after unrelated and related T-cell-depleted bone marrow transplantation: effect of patient age and donor leukocyte infusions. Blood. 1999;93:467–480. [PubMed] [Google Scholar]
- 22.Kurokawa T, Fischer K, Bertz H, Hoegerle S, Finke J, Mackensen A. In vitro and in vivo characterization of graft-versus-tumor responses in melanoma patients after allogeneic peripheral blood stem cell transplantation. Int J Cancer. 2002;101:52–60. doi: 10.1002/ijc.10555. [DOI] [PubMed] [Google Scholar]
- 23.Friedman TM, Goldgirsh K, Berger SA, Zilberberg J, Filicko-O’Hara J, Flomenberg N, Donato M, Rowley SD, Korngold R. Overlap between in vitro donor antihost and in vivo posttransplantation TCR Vbeta use: a new paradigm for designer allogeneic blood and marrow transplantation. Blood. 2008;112:3517–3525. doi: 10.1182/blood-2008-03-145391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Higgins JP, Bernstein MB, Hodge JW. Enhancing immune responses to tumor-associated antigens. Cancer Biol Ther. 2009;8:1440–1449. doi: 10.4161/cbt.8.15.9133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Leen AM, Rooney CM, Foster AE. Improving T cell therapy for cancer. Annu Rev Immunol. 2007;25:243–265. doi: 10.1146/annurev.immunol.25.022106.141527. [DOI] [PubMed] [Google Scholar]
- 26.el-Shami K, Tirosh B, Bar-Haim E, Carmon L, Vadai E, Fridkin M, Feldman M, Eisenbach L. MHC class I-restricted epitope spreading in the context of tumor rejection following vaccination with a single immunodominant CTL epitope. Eur J Immunol. 1999;29:3295–3301. doi: 10.1002/(SICI)1521-4141(199910)29:10<3295::AID-IMMU3295>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 27.Sallusto F, Lenig D, Forster R, Lipp M, Lanzavecchia A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature. 1999;401:708–712. doi: 10.1038/44385. [DOI] [PubMed] [Google Scholar]
- 28.Unsoeld H, Pircher H. Complex memory T-cell phenotypes revealed by coexpression of CD62L and CCR7. J Virol. 2005;79:4510–4513. doi: 10.1128/JVI.79.7.4510-4513.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen BJ, Cui X, Sempowski GD, Liu C, Chao NJ. Transfer of allogeneic CD62L- memory T cells without graft-versus-host disease. Blood. 2004;103:1534–1541. doi: 10.1182/blood-2003-08-2987. [DOI] [PubMed] [Google Scholar]
- 30.Anderson BE, McNiff J, Yan J, Doyle H, Mamula M, Shlomchik MJ, Shlomchik WD. Memory CD4+ T cells do not induce graft-versus-host disease. J Clin Invest. 2003;112:101–108. doi: 10.1172/JCI17601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen BJ, Deoliveira D, Cui X, Le NT, Son J, Whitesides JF, Chao NJ. Inability of memory T cells to induce graft-versus-host disease is a result of an abortive alloresponse. Blood. 2007;109:3115–3123. doi: 10.1182/blood-2006-04-016410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang Y, Joe G, Hexner E, Zhu J, Emerson SG. Host-reactive CD8+ memory stem cells in graft-versus-host disease. Nat Med. 2005;11:1299–1305. doi: 10.1038/nm1326. [DOI] [PubMed] [Google Scholar]
- 33.Zhang Y, Joe G, Hexner E, Zhu J, Emerson SG. Alloreactive memory T cells are responsible for the persistence of graft-versus-host disease. J Immunol. 2005;174:3051–3058. doi: 10.4049/jimmunol.174.5.3051. [DOI] [PubMed] [Google Scholar]
- 34.Zhang Y, Joe G, Zhu J, Carroll R, Levine B, Hexner E, June C, Emerson SG. Dendritic cell-activated CD44hiCD8+ T cells are defective in mediating acute graft-versus-host disease but retain graft-versus-leukemia activity. Blood. 2004;103:3970–3978. doi: 10.1182/blood-2003-09-3135. [DOI] [PubMed] [Google Scholar]
- 35.Juchem KW, Anderson BE, Zhang C, McNiff JM, Demetris AJ, Farber DL, Caton AJ, Shlomchik WD, Shlomchik MJ. A repertoire-independent and cell-intrinsic defect in murine GVHD induction by effector memory T cells. Blood. 2011;118:6209–6219. doi: 10.1182/blood-2011-01-330035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Klebanoff CA, Gattinoni L, Torabi-Parizi P, Kerstann K, Cardones AR, Finkelstein SE, Palmer DC, Antony PA, Hwang ST, Rosenberg SA, Waldmann TA, Restifo NP. Central memory self/tumor-reactive CD8+ T cells confer superior antitumor immunity compared with effector memory T cells. Proc Natl Acad Sci U S A. 2005;102:9571–9576. doi: 10.1073/pnas.0503726102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dutt S, Tseng D, Ermann J, George TI, Liu YP, Davis CR, Fathman CG, Strober S. Naive and memory T cells induce different types of graft-versus-host disease. J Immunol. 2007;179:6547–6554. doi: 10.4049/jimmunol.179.10.6547. [DOI] [PubMed] [Google Scholar]
- 38.Kagamu H, Shu S. Purification of L-selectin(low) cells promotes the generation of highly potent CD4 antitumor effector T lymphocytes. J Immunol. 1998;160:3444–3452. [PubMed] [Google Scholar]
- 39.Kagamu H, Touhalisky JE, Plautz GE, Krauss JC, Shu S. Isolation based on L-selectin expression of immune effector T cells derived from tumor-draining lymph nodes. Cancer Res. 1996;56:4338–4342. [PubMed] [Google Scholar]
- 40.Yang S, Liu F, Wang QJ, Rosenberg SA, Morgan RA. The Shedding of CD62L (L-Selectin) Regulates the Acquisition of Lytic Activity in Human Tumor Reactive T Lymphocytes. PLoS ONE. 2011;6:e22560. doi: 10.1371/journal.pone.0022560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Klebanoff CA, Gattinoni L, Palmer DC, Muranski P, Ji Y, Hinrichs CS, Borman ZA, Kerkar SP, Scott CD, Finkelstein SE, Rosenberg SA, Restifo NP. Determinants of Successful CD8+ T-Cell Adoptive Immunotherapy for Large Established Tumors in Mice. Clinical Cancer Research. 2011;17:5343–5352. doi: 10.1158/1078-0432.CCR-11-0503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Figdor CG, I, de Vries J, Lesterhuis WJ, Melief CJ. Dendritic cell immunotherapy: mapping the way. Nat Med. 2004;10:475–480. doi: 10.1038/nm1039. [DOI] [PubMed] [Google Scholar]
- 43.Montagna D, Maccario R, Locatelli F, Rosti V, Yang Y, Farness P, Moretta A, Comoli P, Montini E, Vitiello A. Ex vivo priming for long-term maintenance of antileukemia human cytotoxic T cells suggests a general procedure for adoptive immunotherapy. Blood. 2001;98:3359–3366. doi: 10.1182/blood.v98.12.3359. [DOI] [PubMed] [Google Scholar]
- 44.Dang Y, Knutson KL, Goodell V, dela Rosa C, Salazar LG, Higgins D, Childs J, Disis ML. Tumor antigen-specific T-cell expansion is greatly facilitated by in vivo priming. Clin Cancer Res. 2007;13:1883–1891. doi: 10.1158/1078-0432.CCR-06-2083. [DOI] [PubMed] [Google Scholar]
- 45.Kwak LW, Taub DD, Duffey PL, Bensinger WI, Bryant EM, Reynolds CW, Longo DL. Transfer of myeloma idiotype-specific immunity from an actively immunised marrow donor. Lancet. 1995;345:1016–1020. doi: 10.1016/s0140-6736(95)90757-2. [DOI] [PubMed] [Google Scholar]
- 46.Cremer FW, Kiel K, Sucker C, Wacker J, Atzberger A, Haas R, Goldschmidt H, Moos M. A rationale for positive selection of peripheral blood stem cells in multiple myeloma: highly purified CD34+ cell fractions of leukapheresis products do not contain malignant cells. Leukemia. 1997;11(Suppl 5):S41–46. [PubMed] [Google Scholar]
- 47.Leone F, Cavalloni G, Gunetti M, Pignochino Y, Piacibello W, Aglietta M. Tumor cell purging by ex vivo expansion of hemopoietic stem cells from breast cancer patients combined with targeting ErbB receptors. Biol Blood Marrow Transplant. 2006;12:68–74. doi: 10.1016/j.bbmt.2005.06.003. [DOI] [PubMed] [Google Scholar]
- 48.Ward RC, Kaufman HL. Targeting Costimulatory Pathways for Tumor Immunotherapy. International Reviews of Immunology. 2007;26:161–196. doi: 10.1080/08830180701365941. [DOI] [PubMed] [Google Scholar]
- 49.Westwood JA, Berry LJ, Wang LXJ, Duong CPM, Pegram HJ, Darcy PK, Kershaw MH. Enhancing adoptive immunotherapy of cancer. Expert Opinion on Biological Therapy. 10:531–545. doi: 10.1517/14712591003610622. [DOI] [PubMed] [Google Scholar]
- 50.Gattinoni L, Klebanoff CA, Restifo NP. Paths to stemness: building the ultimate antitumour T cell. Nat Rev Cancer. 2012;12:671–684. doi: 10.1038/nrc3322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Overwijk WW, Schluns KS. Functions of gammaC cytokines in immune homeostasis: Current and potential clinical applications. Clinical Immunology. 2009;132:153–165. doi: 10.1016/j.clim.2009.03.512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang L-X, Chen B-G, Plautz GE. Adoptive Immunotherapy of Advanced Tumors with CD62 L-Selectinlow Tumor-Sensitized T Lymphocytes Following Ex Vivo Hyperexpansion. The Journal of Immunology. 2002;169:3314–3320. doi: 10.4049/jimmunol.169.6.3314. [DOI] [PubMed] [Google Scholar]
- 53.Mostbock S, Lutsiak MEC, Milenic DE, Baidoo K, Schlom J, Sabzevari H. IL-2/Anti-IL-2 Antibody Complex Enhances Vaccine-Mediated Antigen-Specific CD8+ T Cell Responses and Increases the Ratio of Effector/Memory CD8+ T Cells to Regulatory T Cells. The Journal of Immunology. 2008;180:5118–5129. doi: 10.4049/jimmunol.180.7.5118. [DOI] [PubMed] [Google Scholar]
- 54.Gattinoni L, Klebanoff CA, Restifo NP. Pharmacologic Induction of CD8+ T Cell Memory: Better Living Through Chemistry. Science Translational Medicine. 2009;1:11ps12. doi: 10.1126/scitranslmed.3000302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Araki K, Turner AP, Shaffer VO, Gangappa S, Keller SA, Bachmann MF, Larsen CP, Ahmed R. mTOR regulates memory CD8 T-cell differentiation. Nature. 2009;460:108–112. doi: 10.1038/nature08155. [DOI] [PMC free article] [PubMed] [Google Scholar]