

Abstract
The gram-negative obligate intracellular bacterium Anaplasma phagocytophilum is the causative agent of human granulocytic anaplasmosis (HGA), an emerging tick-borne infectious disease occuring worldwide. HGA is generally self-limiting, however, the underlying mechanisms, particularly the innate immune pathways that mediate the immune clearance of A. phagocytophilum, are less understood. We herein report an unexpected role for Receptor interacting protein-2 (Rip2), the adaptor protein for the Nod-Like Receptors (NLRs), Nod1/Nod2, in the host immune response against A. phagocytophilum infection. Although A. phagocytophilum genome is reported to lack the genes encoding the known ligands of Nod1 and Nod2, its infection up-regulated the transcription of Rip2 in human primary neutrophils. Our results revealed that Rip2 deficient mice had significantly higher bacterial load than wild type controls throughout the infection period. In addition, the Rip2 deficient mice took strikingly longer duration to clear A. phagocytophilum infection. Detailed analysis identified that interferon gamma (IFNγ) and interleukin -18 (IL-18) but not interleukin -12 (IL-12), macrophage inflammatory protein-2 (MIP-2), and KC response were diminished in A. phagocytophilum-challenged Rip2 deficient mice. Together, these results revealed that Rip2 play important roles in the immune control of A. phagocytophilum, and may contribute to our understanding of the host response to Rickettsiales.
Keywords: Rip2, Anaplasma phagocytophilum, NLR, IFNγ
Introduction
The tick-borne obligate intracellular gram negative bacterium A. phagocytophilum primarily colonizes within human neutrophils, and causes the illness Human Granulocytic Anaplasmosis (HGA) (Carlyon & Fikrig, 2003). The infected individuals may develop a range of symptoms from mild to severe, and include fever, malaise, headache, septic shock–like syndromes, hemorrhage, coagulopathy, respiratory distress syndrome, acute abdominal syndromes, acute renal failure, heart failure and myocarditis, opportunistic infections, and neuropathies (Bakken & Dumler, 2000). Thrombocytopenia and leucopenia are common in A. phagocytophilum infected individuals. The infection can become fatal in aged and immune-compromised subjects.
Although A. phagocytophilum is among the most prevalent tick-borne pathogens in the USA (von Loewenich, et al., 2004), how this bacterium interacts with mammalian immune system is still unclear. Generally, the A. phagocytophilum infection is self-limiting in immuno-competent subjects (Dumler, et al., 2005). The laboratory model for A. phagocytophilum infection is mouse, which often clears infection within 10–12 days (Sun, et al., 1997, Hodzic, et al., 1998, Borjesson & Barthold, 2002). Both T and B cells have been shown to play important roles in the control and clearance of A. phagocytophilum (Telford, et al., 1996, Bunnell, et al., 1999, von Loewenich, et al., 2004). A key role for CD4+ T cells and T-helper 1 (Th1) response is widely established in the immune response to the infection of A. phagocytophilum (Pedra, et al., 2007, Birkner, et al., 2008). It has been shown that IFNγ, IL- 12, and IL-18 play important roles in the early clearance of A. phagocytophilum (Akkoyunlu & Fikrig, 2000, Pedra, et al., 2007, Pedra, et al., 2007). Immune effector components such as iNOS and phagocyte NADPH oxidase were dispensable for the in vivo clearance of A. phagocytophilum infection (Banerjee, et al., 2000). Although some of the critical players of adaptive response required for controlling A. phagocytophilum are known, our understanding of the innate immune components involved in the recognition and clearance of this bacterium is rather incomplete. Interestingly, prominent anti-bacterial innate immune detection system TLR2, TLR4, and their adaptor MyD88 are shown to play no role in the immune response to A. phagocytophilum infection (von Loewenich, et al., 2004). A recent study demonstrated that signaling through the Nod Like Receptor (NLR) family member IPAF (NLRC4), its adaptor ASC, and Caspase-1 is critical for the control of A. phagocytophilum infection during the early phase of infection (Pedra, et al., 2007). However, mice deficient in NLRC4, ASC, and Caspase-1 were still able to clear A. phagocytophilum infection by day 6, indicating there are additional pathogen detection and clearance systems involved in the innate and adaptive immune response to A. phagocytophilum infection. Thus the mechanisms involved in the immune detection and clearance of A. phagocytophilum remains unresolved and warrants more studies.
We previously reported that A. phagocytophilum infection up regulates Rip2 (Sukumaran, et al., 2005), the adaptor molecule of the cytoplasmic pattern recognition receptor Nod1 and 2, (Magalhaes, et al., 2011) in immune cells. Nod1 and 2 belong to NLR family of proteins, and are known to detect bacterial membrane derived peptidoglycan components iE-DAP and muramyl dipeptide (MDP), respectively (Inohara & Nunez, 2003, Franchi, et al., 2009). Following peptidoglycan detection, Nod1/Nod2 recruit and associate with the adaptor protein Rip2, triggering proinflammatory signaling pathways via NF-κB and the mitogen-activated protein (MAP) kinases p38, JNK, and ERK (Kobayashi, et al., 2002). Activation of Nod1 and Nod2 also triggers proinflammatory responses, leading to the induction of cytokine and chemokines. Interestingly A. phagocytophilum genome does not encode genes for the synthesis of iE-DAP and MDP (Dunning Hotopp, et al., 2006). Therefore it is intriguing that Rip2 is activated during A. phagocytophilum infection. We hypothesized that Rip2 may play a role in the immune response to A. phagocytophilum, through a potentially unknown mechanism. In this study, we investigated whether Rip2 play any role in A. phagocytophilum infection clearance, and demonstrate that Rip2 plays an unexpected role in controlling the A. phagocytophilum infection.
Methods
A. phagocytophilum propagation
The A. phagocytophilum HZ strain (Prof. Yasuko Rikihisa, Ohio State University) was used in this study (Sukumaran, et al., 2005). The A. phagocytophilum was propagated in the promyelocytic cell line (HL-60) (ATCC, 240 CCL) (Sukumaran, et al., 2005). A previously reported method was used to propagate and purify A. phagocytophilum (Carlyon, et al., 2004). Briefly, equal volume of A. phagocytophilum-infected and uninfected HL-60 cells were mixed and diluted 1/5 with RPMI medium with 10% fetal bovine serum. For the purification of cell-free A. phagocytophilum, maximally infected cells were pelleted by centrifugation for 10 min at 400 × g, lysed first by six passage through a 25-gauge needle, followed by another six passages through a 27-gauge needle. After lysis, the cell debris was removed by centrifugation at 1200rpm for 5 min, and the supernatant containing host cell free bacterium was used for the in vitro (human neutrophils) and ex vivo (mouse splenocytes) infection studies.
Infection of neutrophils with A. phagocytophilum in vitro
The infection of neutrophils was done as reported earlier (Sukumaran, et al., 2005). Briefly, suspensions of freshly prepared A. phagocytophilum (isolated from 5 × 106 heavily infected HL-60 cells) were added to neutrophils (106 neutrophils/ml) maintained in the Ultra Low Attachment six-well plates (Corning Inc., N.Y.), and incubated at 37°C in 5% CO2 for 4 h. The percentage of infected neutrophils was confirmed by immunofluorescence microscopy, and the number of A. phagocytophilum isolates per neutrophil was calculated as previously described (Carlyon, et al., 2004). The ratio of bacteria to neutrophils was ~5:1. At various time points post infection, the cells were recovered by centrifugation at 210 × g, RNA was isolated using RNAeasy kit (Qiagen) and cDNA was made using iSCRIPT kit (Biorad). The Rip2 levels were determined by SYBR Green dye based Quantitative real-time PCR (qRT-PCR), with primers detecting Rip2 (GenBank accession AF064824), and normalized with human β actin gene. Rip2 was amplified using the following primers: Forward: 5′-CCATCCCGTACCACAAGCTC-3′ and Reverse: 5′-GCAGGATGCGGAATCTCAAT-3′. The primers used for amplifying human β actin gene were: Forward: 5′-TGATATCGCCGCGCTCGTCGTC -3′ and Reverse: 5′-GCCGATCCACACGGAGTACT-3′.
Infection of mice and quantification of A. phagocytophilum in the peripheral blood
Wild type C57BL/6, mice were purchased from Jackson Laboratory (Bar Harbor, ME). Rip2 null mice (backcrossed to C57BL/6 for at least 10 generations) have been previously described (Kobayashi, et al., 2002, Kobayashi, et al., 2005). For the in vivo infection, 4 to 12 weeks age, sex matched, and mice maintained under specific pathogen-free conditions (such as maintenance in barrier-filtered cages, given standard laboratory diet, and sterile water ad libitum throughout the study) were used. The experiments were performed according to the guidelines of the Yale Universityinstitutional animal care and research committee.
For the in vivo infection studies, 100μl of blood from A. phagocytophilum HZ strain infected rag1-null mice (25% neutrophil infection) was used as the inoculum to infect immunocompetent C57BL/6 or Rip2−/− mice, by intra peritoneal injection. To determine the infection level in rag1-null mice, Giemsa staining of the blood cells was performed, followed by quantification of the percentage of granulocytes containing morulae (A. phagocytophilum aggregates) (Borjesson & Barthold, 2002).
For the quantification of the A. phagocytophilum level in the peripheral blood, 100ul of peripheral blood from mice was incubated twice with 900 μl of erythrocyte lysis buffer (Sigma-Aldrich) at room temperature for 20 min, followed by DNA was extraction using DNeasy Tissue Kit (Qiagen). The bacterial levels were determined by SYBR Green dye based qRT-PCR, with primers detecting A. phagocytophilum 16S rRNA (GenBank accession M73224), and normalized with mouse actin gene (GenBank accession X03672) as described before (Pedra, et al., 2007, Pedra, et al., 2007). Mouse actin was amplified using the following primers: Forward: 5′-AGAGGGAAATCGTGCGTGAC-3′ and Reverse: 5′-CAATAGTGATGACCTGGCCGT -3′. The primers used to amplify the 16S rRNA of A. phagocytophilum were: Forward:5′-CCATTTCTAGTGGCTATCCCATACTAC -3′ and Reverse: 5′-TCGAACGGATTATTCTTTATAGCTTG -3′
Measurement of cytokines
The protein standards and antibody pairs for IL-12p40/p70, IL-2, MIP-2, KC and IFNγ were purchased from BD Pharmingen, and IL-18 from MBL. Retro- orbital bleeding was performed at the desired time points from wild type and Rip2−/− mice, and the ELISA experiments for cytokine measurement were performed as described previously (Sukumaran, et al., 2005).
A. phagocytophilum restimulation assays
For the ex vivo splenocyte restimulation assays, the spleens were first removed from euthanized mice 6 days post-infection with A. phagocytophilum. The splenocytes (106 cells) were prepared following RBC lysis, and restimulated with live purified A. phagocytophilum (108 bacteria) in vitro for 18 h and the culture supernatant was used to perform ELISA for cytokine measurements. A. phagocytophilum was propagated and purified as described in the methods section. Uninfected spleen cells were used as negative controls.
Statistical analysis
We performed statistical analysis by using unpaired Student’s t test. A. phagocytophilum genome copy numbers present in the peripheral blood of wild-type and gene-deficient mice were compared, and values of p ≤ 0.05 were considered statistically significant.
Results
A. phagocytophilum infection up-regulates Rip2 expression
In the present study, we first investigated the potential induction of Rip2 by A. phagocytophilum in human primary neutrophils. For this, we infected human primary neutrophils with A. phagocytophilum, and quantified the levels of Rip2 using quantitative real-time PCR (qRT-PCR). As shown in Figure 1, there was a four-fold increase (p<0.05) in Rip2 transcripts at four hour post infection with A. phagocytophilum. This observation was consistent with our earlier study. We further determined the time course of the transcription of Rip2. It was observed that, as shown in Figure 1, up-regulation of Rip2 transcription starts as early as 2 h post infection, and continued to increase over a period of 4 h, and reaches a plateau thereafter. In conclusion, A. phagocytophilum infection results in the up regulation of transcription of Rip2.
Figure 1.
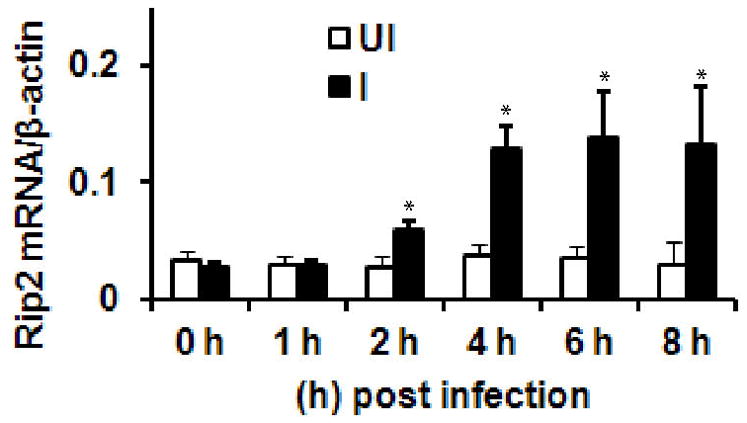
Rip2 transcription is induced during A. phagocytophilum infection. RNAs from 1h, 2h, 4h, 6h and 8h infected (I) and uninfected (UI) neutrophils were used to perform qRT-PCR. The results are expressed as number of copies of Rip2 mRNA/human β-actin. The results are expressed as mean ±SD from three independent experiments. The statistical significance was calculated using student’s t-test and *denotes p<0.05.
Rip2 is important for the immune control of A. phagocytophilum infection in vivo
Since we observed an increase in the transcription of Rip2 in A. phagocytophilum infected cells, and Rip2 has a known role in controlling intracellular bacterial infections, we addressed the important question whether Rip2 has any functional role in the immune defense and clearance of A. phagocytophilum infection in vivo. For this, we infected Rip2−/− mice with A. phagocytophilum intra-peritoneally, and monitored bacterial load and infection clearance kinetics in the blood using qRT-PCR detecting A. phagocytophilum 16S rRNA. Previous studies have shown that in the C57BL/6 murine model of A. phagocytophilum infection, bacterial load increases from day 2 onwards, reaching peak at day 3–4, followed by infection decline and clearance by day 10–12 (von Loewenich, et al., 2004). Strikingly, genetic deletion of Rip2 noticeably altered A. phagocytophilum infection pattern in the mouse model. As shown in Figure 2, Rip2−/− mice showed an increased A. phagocytophilum burden, compared to gene sufficient controls. In addition, there was a prominent delay in the kinetics of clearance of A. phagocytophilum in the blood of Rip2 −/− animals, compared to wild type controls (Fig. 2). In wild type C57BL/6 mice, A. phagocytophilum burden peaked at day 3, and then rapidly declined and there was complete clearance by day 12. In contrast, even though there was comparable bacterial load in Rip2−/− and wild-type mice at day 3, the A. phagocytophilum load was consistently higher in Rip2−/− mice than in wild type controls during the entire course of infection following day 3. Contrast to that in wild type, in Rip2−/−mice, the A. phagocytophilum burden progressively increased with a peak bacterial load reaching at day 7 post-infection, with 8 fold (p<0.05) more bacterial load than in wild type mice. While wild type animals cleared infection completely by day 10–12, Rip2−/− mice had a very prolonged infection, and the bacteria could be detected even at day 20.
Figure 2.
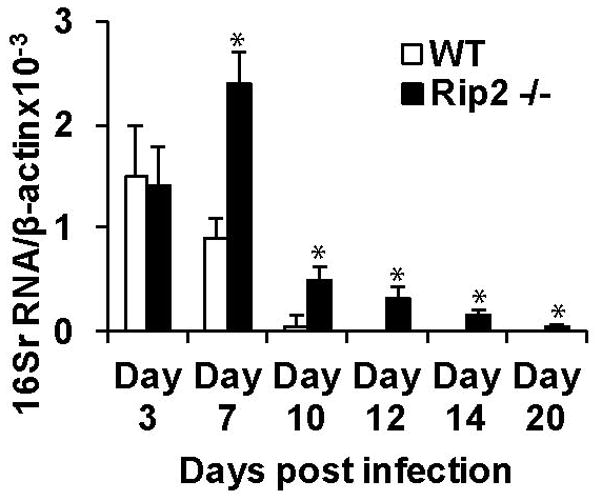
Rip2 deficiency caused increased load and delayed clearance of A. phagocytophilum in peripheral blood of mice. Rip2−/− and WT (n=15) mice were infected with A. phagocytophilum, and the bacterial load was determined in the peripheral blood (by measuring the levels of bacterial 16S rRNA normalized to mouse β actin) at the indicated time points using qRT-PCR analysis as detailed in methods section. The experiment was repeated 3 times and the result from a representative experiment is shown. The results are expressed as median ±SD. The statistical significance was calculated using student’s t-test and *denotes p<0.05.
In conclusion, Rip2−/− mice showed a significantly higher bacterial load at all time points following day 3 and a prominent delay of infection clearance extending up to day 20. These data identify an unexpected, but important role for Rip2 in the immune clearance of A. phagocytophilum infection.
Rip2 is dispensable for the MIP-2 and KC production in response to A. phagocytophilum infection
We next sought to find out the impaired immune and inflammatory mechanisms that resulted in the increased susceptibility of Rip2 −/− mice to A. phagocytophilum infection. For this, we first investigated whether Rip2 plays any role in the signature inflammatory host response typically manifested during A. phagocytophilum infection, by analyzing the levels of major chemokines and cytokines associated with A. phagocytophilum infection (Akkoyunlu & Fikrig, 2000, Akkoyunlu, et al., 2001, Pedra, et al., 2007, Pedra, et al., 2007) in the knockout mouse model. One of the key products of human and mouse neutrophils are chemokines. Therefore, we first determined if Rip2 contributes to the IL-8 chemokine response during A. phagocytophilum infection by measuring the levels of murine IL-8 homologues MIP-2 and KC (Scorpio, et al., 2004). We infected Rip2−/− mice with A. phagocytophilum intra-peritoneally and measured the levels of MIP-2 and KC, in the mice sera. The results showed that both MIP-2 and KC levels were unaltered in A. phagocytophilum infected Rip2 −/− mice (Figure 3A and 3B) compared to wild type controls at all time points tested. These results indicate that Rip2 is not involved in the chemokine response during A. phagocytophilum challenge.
Figure 3.
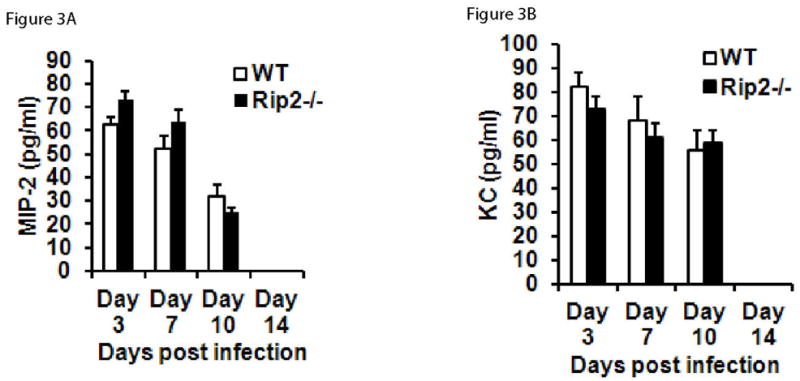
Rip2 is not involved in the MIP-2 and KC response to A. phagocytophilum infection. Sera from wild type (n =5) or Rip2−/− (n = 5) mice were harvested at days 3, 7,10 and 14 post infection, pooled, and the levels of MIP-2 (Figure 3A), and KC (Figure 3B) were measured using ELISA. The results represent mean±SD of samples from three independent experiments. The statistical significance was calculated using student’s t-test and *denotes p<0.05.
Rip2 is required for IL-18 but not IL-12 production during A. phagocytophilum challenge
Earlier studies have shown that the pro-inflammatory cytokines such as IL-12 and IL-18 were important components in the immunity against A. phagocytophilum infection by inducing optimal production of IFN-γ and Th1 immune responses (Pedra, et al., 2007, Pedra, et al., 2007). To determine whether Rip2 plays any role in the cytokine response, we studied the serum levels of major NF-κB -dependent cytokine IL-12, and inflammasome-dependent cytokine IL-18, in Rip2-deficient mice following A. phagocytophilum challenge. As shown in Figure 4A, there was no significant difference in the levels of IL-12 in Rip2−/− animals. However, A. phagocytophilum infected Rip2−/− mice showed a modest but significant reduction (1.78 fold, p<0.05) in IL-18 levels at day 10 (Figure 4B). This evidence indicates that Rip2 plays a differential role in the cytokine response to A. phagocytophilum challenge.
Figure 4.
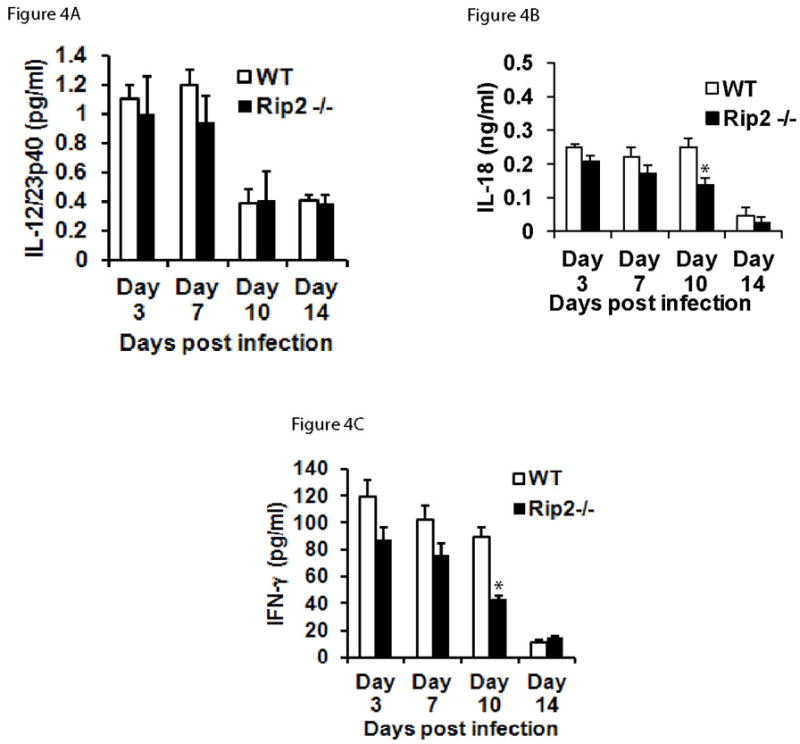
Rip2 deficiency resulted in altered immune response to A. phagocytophilum infection. Sera from wild type (n =6) or Rip2−/− (n = 6) mice were harvested at days 3, 7,10 and 14 post infection, pooled, and the levels of IL-12 (Figure 4A), IL-18 (Figure 4B) and IFNγ (Figure 4C), were measured using ELISA. The results are expressed as mean±SD of samples from three independent experiments. The statistical significance was calculated using student’s t-test and *denotes p<0.05.
Rip2 deficiency impairs IFNγ production during A. phagocytophilum infection
Earlier studies have shown that mice deficient in either the production or signaling of IFNγ had a prominent delay in the immune clearance of A. phagocytophilum (Akkoyunlu & Fikrig, 2000, Dumler, et al., 2000, Lepidi, et al., 2000, Martin, et al., 2000, Wang, et al., 2004, Pedra, et al., 2007, Pedra, et al., 2007, Birkner, et al., 2008, Pedra, et al., 2008). Moreover, the level of IL-18 (also known as interferon-gamma inducing factor) was reduced in the serum of Rip2 deficient mice during A. phagocytophilum infection. Therefore, we next analyzed whether Rip2 impacts on the IFNγ production during A. phagocytophilum infection. For this, Rip2−/− animals were infected with A. phagocytophilum and the IFNγ response was measured. As shown in Figure 4C, Rip2−/− mice displayed significantly reduced IFNγ levels in the sera at day 10 post infection. The IFNγ levels in Rip2−/− animals were 2.1 fold (p<0.05) less than that in the infected wild type mice, indicating a potential role for Rip2 in the IFNγ response to A. phagocytophilum.
To further confirm the role of Rip2−/− in the cytokine response to A. phagocytophilum infection, we performed ex vivo experiments. Since Th1-type immune response is important in the immune response to A. phagocytophilum infection, we isolated murine splenocytes from infected Rip2 deficient mice, re-stimulated with A. phagocytophilum ex vivo and measured the levels of IFNγ and also IL-2. The results of these experiments identified a reduction in the production of both IFNγ (Figure 5A) and IL-2 (Figure 5B) by the splenocytes of Rip2−/− mice in response to A. phagocytophilum infection. When splenocytes from A. phagocytophilum infected wild type and Rip2−/− mice were restimulated, the IFNγ response in Rip2−/− splenocytes was 16.8 fold less than (p<0.05) that in wild type animals. We also observed a 3.4 fold reduction (p<0.05) in the IL-2 response in Rip2−/− splenocytes when restimulated with A. phagocytophilum. These results collectively identify the notable defect in Th1-like response, in Rip2-deficient mice in response to A. phagocytophilum infection, indicating that Rip2 likely acts as a contributor to the immune response against this bacterium.
Figure 5.
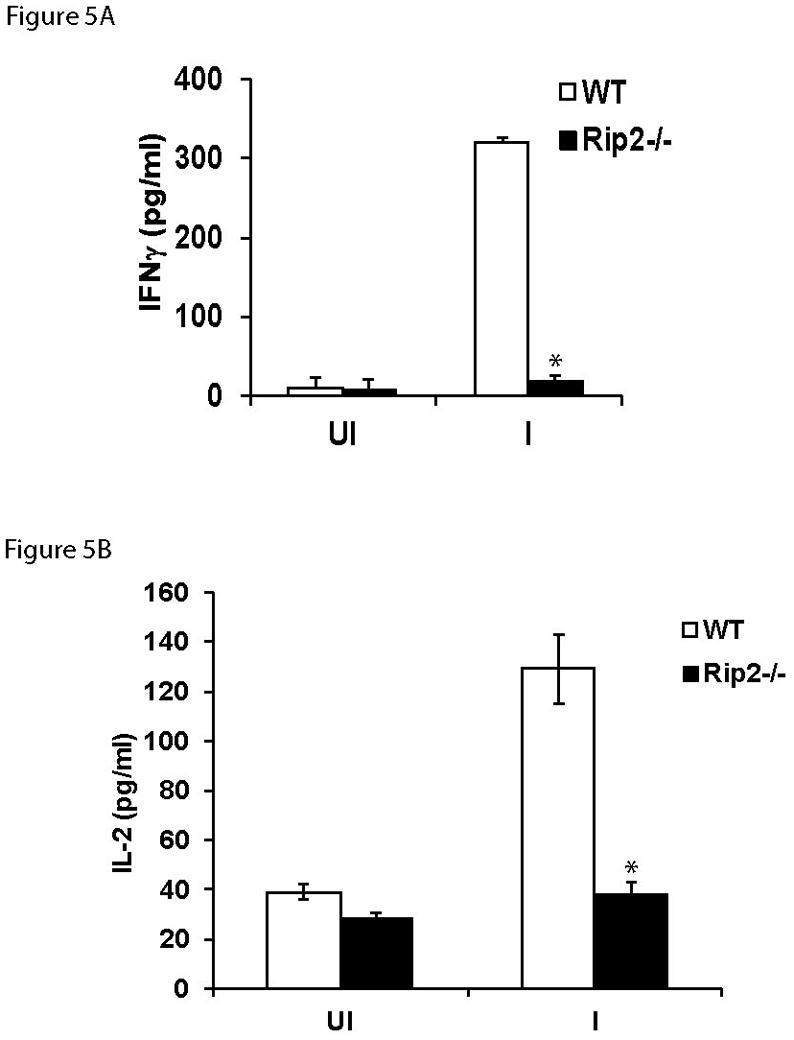
Splenocytes from A. phagocytophilum-infected Rip2−/− mice have reduced IFNγ and IL-2 secretion during restimulation with A. phagocytophilum. Splenocytes collected from wild type (n =5) or Rip2−/− (n = 5) mice were stimulated with live A. phagocytophilum for 18 h, and IFNγ (Figure 5A) and IL-2 (Figure 5B) levels were measured using ELISA. The results are expressed as mean±SD of five mice from each group. The experiment was repeated twice with similar results. The statistical significance was calculated using student’s t-test and *denotes p<0.05. UI = uninfected samples and I = A. phagocytophilum-infected samples.
Discussion
Although mammals often successfully clear the infection by A. phagocytophilum, the underlying innate immune mechanisms, particularly the pattern recognition receptors (PRR) and innate immune pathways that initiate its clearance, are still a mystery. A previously published gene microarray based study from our group analyzing transcriptional response of human neutrophils to A. phagocytophilum indicated that the transcription of Rip2 is up regulated during infection (Sukumaran, et al., 2005). Following up on this earlier observation, in this study we identified that Rip2, the key adaptor molecule mediating signaling from the cytoplasmic pattern recognition receptors Nod1/2, plays an important role in the control of A. phagocytophilum infection.
Rip2 has been previously shown to play an essential role in the immunity against various intracellular pathogens including Listeria monocytogenes (Chin, et al., 2002), Legionella pneumophila (Archer, et al., 2010), Chlamydophila pneumonia (Shimada, et al., 2009), Mycobacteirum tuberculosis (Shimada, et al., 2009) and extracellular pathogens such as E. coli (Balamayooran, et al., 2011).
Compared to wild type mice, Rip2−/− mice were significantly more susceptible to A. phagocytophilum infection and had a markedly higher bacterial burden at all time points following day 3 post-infection. The deficiency of Rip2 also resulted in delayed clearance of A. phagocytophilum from the blood in the mouse model. While wild type mice took 10–12 days to clear A. phagocytophilum infection, Rip2−/− mice took ~20 days to clear infection. Over all the role of Rip2 became prominent during the early-mid phase of the immune response to A. phagocytophilum infection. The fact that the Rip2 knockout mice were still able to clear the infection suggests that there may be other un-identified pathways that may be important in the clearance of A. phagocytophilum.
A major inflammatory chemokine that is heavily induced during A. phagocytophilum infection is IL-8 in humans (Akkoyunlu, et al., 2001) and MIP-2 and KC (murine IL-8 homologues) in mice (Scorpio, et al., 2004). These chemokines are suggested to be important in A. phagocytophilum pathogenesis by inducing trafficking of neutrophils to the sites of infection via binding to receptors in neutrophils. Signaling through Rip2 can activate NF-κB target genes such as IL-8 (Buchholz & Stephens, 2006, Buchholz & Stephens, 2008). Furthermore, Rip2 has been previously shown to play a crucial role in neutrophil recruitment in vivo (Magalhaes, et al., 2011). But interestingly, our study found that the levels of the chemokines (MIP-2 and KC) were un-affected in A. phagocytophilum-infected Rip2−/− mice hinting the possibility that these chemokines may not play any role in the immune defect manifested by Rip2−/− mice.
IFNγ, another inflammatory cytokine, has been shown to play a very critical role in early host defense to A. phagocytophilum infection (Akkoyunlu & Fikrig, 2000, Dumler, et al., 2000, Lepidi, et al., 2000, Martin, et al., 2000, Wang, et al., 2004, Pedra, et al., 2007, Pedra, et al., 2007, Birkner, et al., 2008, Pedra, et al., 2008). Our study observed a defect in the IFN-γ and IL-18 levels in Rip2 deficient mice during A. phagocytophilum infection. Both in vivo and ex vivo data showed that the levels of IFNγ, a major inflammatorycytokine was significantly lower in Rip2−/− mice. These results are consistent with the previous reports that IFN-γ plays a major role in the immune pathology and early clearance of A. phagocytophilum infection (Akkoyunlu & Fikrig, 2000), even though it is dispensable for the complete elimination of the pathogen (Birkner, et al., 2008). It is also known that an adaptive CD4+ T cell mediated response is critical for the complete clearance of A. phagocytophilum infection (Birkner, et al., 2008). Whether Rip2 plays any role in the CD4+ T cell response against A. phagocytophilum infection needs to be addressed in the future studies. Also, the identity of the specific cell type in which the IFNγ production is impaired during A. phagocytophilum infection of the Rip2−/− mouse remains to be determined. Since previous reports have shown the importance of natural killer (NK) cells, NKT cells (Choi, et al., 2007) and CD4+T cells (Birkner, et al., 2008) in the IFNγ production and host defense to A. phagocytophilum infection, we speculate that Rip2 pathway could contribute to the IFNγ production by these cell types. Although IL-12 secretion was previously shown to be dependent on Rip2 (Kobayashi, et al., 2002), interestingly, there was no difference in IL-12 in Rip2−/− mouse when challenged with A. phagocytophilum. But the levels of the inflammasome dependent cytokine, IL-18, was less in infected Rip2−/− animals. It is also intriguing that even though the bacterial load in Rip2−/− mice became significantly different from controls on day 7 onwards, the difference in the levels of IFNγ and IL-18 in Rip2−/− mice became more prominent only at day 10 post-infection. This hints to the possibility that Rip2 may also have a role independent of IFNγ and IL-18. Future studies are required to delineate the mechanism.
Since Rip2 is not a PRR but mediates the innate immune response by acting as an adaptor for the innate immune receptors Nod1 and Nod2, it will be important to identify the receptor that detects A. phagocytophilum and signals through Rip2. The role for Rip2 in the immune response to A. phagocytophilum is rather unexpected, given the fact that the genome of A. phagocytophilum is reported to lack the genes encoding peptidoglycan, the known ligand for Nod1/2. So it is unlikely that the role of Rip2 in the immune response to A. phagocytophilum infection is linked to mechanisms involving peptidoglycan sensing. This leaves us with a major question as to which mechanism A. phagocytophilum uses to activate Rip2 mediated immune response. Our speculation would be that (1) a non-peptidoglycan microbial component (e.g., virulence factor) or (2) endogenous danger-associated molecular patterns (DAMPs, e.g., High-mobility group box proteins, heparin sulfate, ATP, DNA etc.) (Chen & Nunez, 2010) that are released by damaged host cells during infection could potentially play roles in activating Rip2 during A. phagocytophilum invasion. As a precedence for the first hypothesis, a Salmonella type III secretion system effector was recently identified to activate Nod/Rip2 signaling pathway (Keestra, et al., 2011). A. phagocytophilum is also known to possess a functional type IV secretion system (Rikihisa, et al., 2010). Previous studies also reported that DAMPs can initiate signaling through innate immune receptors such as TLRs (Yanai, et al., 2009) and NLRs (Petrilli, et al., 2007, Benko, et al., 2008). It is also possible that Rip2 may interact or act synergistically with other molecules that have been previously associated with A. phagocytophilum clearance such as the inflammasome NLR proteins Ipaf, Asc or Caspase 1, thereby leading to the delayed immune clearance. Supporting this possibility are previous reports showing the interaction of Rip2 pathway with Asc and caspase1 pathway (Sarkar, et al., 2006, Pan, et al., 2007). In addition, the observation that the production of inflammasome-dependent cytokine IL-18 was less in A. phagocytophilum-infected Rip2 knockout mice again hints towards a possibility of cross-talk between Rip2 and inflammasome pathways during A. phagocytophilum infection. Further studies are required to delineate the underlying mechanisms.
In conclusion, our studies revealed a previously unanticipated role for Rip2−/− in the immune clearance of A. phagocytophilum. This study thus provides novel insights into the mechanisms underlying immune response to the infection by A. phagocytophilum.
Acknowledgments
The authors would like to thank Debbie Beck for assistance with mice studies. We also thank Prof. Yasuko Rikihisa for providing A. phagocytophilum strain HZ for the studies. This work was supported by grants from the NIH.
Footnotes
Conflict of interest: The authors have no conflict of interest to declare.
References
- Akkoyunlu M, Fikrig E. Gamma interferon dominates the murine cytokine response to the agent of human granulocytic ehrlichiosis and helps to control the degree of early rickettsemia. Infection and immunity. 2000;68:1827–1833. doi: 10.1128/iai.68.4.1827-1833.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akkoyunlu M, Malawista SE, Anguita J, Fikrig E. Exploitation of interleukin-8-induced neutrophil chemotaxis by the agent of human granulocytic ehrlichiosis. Infection and immunity. 2001;69:5577–5588. doi: 10.1128/IAI.69.9.5577-5588.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Archer KA, Ader F, Kobayashi KS, Flavell RA, Roy CR. Cooperation between multiple microbial pattern recognition systems is important for host protection against the intracellular pathogen Legionella pneumophila. Infection and immunity. 2010;78:2477–2487. doi: 10.1128/IAI.00243-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakken JS, Dumler JS. Human granulocytic ehrlichiosis. Clinical infectious diseases: an official publication of the Infectious Diseases Society of America. 2000;31:554–560. doi: 10.1086/313948. [DOI] [PubMed] [Google Scholar]
- Balamayooran T, Batra S, Balamayooran G, Cai S, Kobayashi KS, Flavell RA, Jeyaseelan S. Receptor-interacting protein 2 controls pulmonary host defense to Escherichia coli infection via the regulation of interleukin-17A. Infection and immunity. 2011;79:4588–4599. doi: 10.1128/IAI.05641-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee R, Anguita J, Fikrig E. Granulocytic ehrlichiosis in mice deficient in phagocyte oxidase or inducible nitric oxide synthase. Infection and immunity. 2000;68:4361–4362. doi: 10.1128/iai.68.7.4361-4362.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benko S, Philpott DJ, Girardin SE. The microbial and danger signals that activate Nod-like receptors. Cytokine. 2008;43:368–373. doi: 10.1016/j.cyto.2008.07.013. [DOI] [PubMed] [Google Scholar]
- Birkner K, Steiner B, Rinkler C, Kern Y, Aichele P, Bogdan C, von Loewenich FD. The elimination of Anaplasma phagocytophilum requires CD4+ T cells, but is independent of Th1 cytokines and a wide spectrum of effector mechanisms. European journal of immunology. 2008;38:3395–3410. doi: 10.1002/eji.200838615. [DOI] [PubMed] [Google Scholar]
- Borjesson DL, Barthold SW. The mouse as a model for investigation of human granulocytic ehrlichiosis: current knowledge and future directions. Comparative medicine. 2002;52:403–413. [PubMed] [Google Scholar]
- Buchholz KR, Stephens RS. Activation of the host cell proinflammatory interleukin- 8 response by Chlamydia trachomatis. Cellular microbiology. 2006;8:1768–1779. doi: 10.1111/j.1462-5822.2006.00747.x. [DOI] [PubMed] [Google Scholar]
- Buchholz KR, Stephens RS. The cytosolic pattern recognition receptor NOD1 induces inflammatory interleukin-8 during Chlamydia trachomatis infection. Infection and immunity. 2008;76:3150–3155. doi: 10.1128/IAI.00104-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunnell JE, Trigiani ER, Srinivas SR, Dumler JS. Development and distribution of pathologic lesions are related to immune status and tissue deposition of human granulocytic ehrlichiosis agent-infected cells in a murine model system. The Journal of infectious diseases. 1999;180:546–550. doi: 10.1086/314902. [DOI] [PubMed] [Google Scholar]
- Carlyon JA, Fikrig E. Invasion and survival strategies of Anaplasma phagocytophilum. Cellular microbiology. 2003;5:743–754. doi: 10.1046/j.1462-5822.2003.00323.x. [DOI] [PubMed] [Google Scholar]
- Carlyon JA, Abdel-Latif D, Pypaert M, Lacy P, Fikrig E. Anaplasma phagocytophilum utilizes multiple host evasion mechanisms to thwart NADPH oxidase-mediated killing during neutrophil infection. Infection and immunity. 2004;72:4772–4783. doi: 10.1128/IAI.72.8.4772-4783.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen GY, Nunez G. Sterile inflammation: sensing and reacting to damage. Nature reviews Immunology. 2010;10:826–837. doi: 10.1038/nri2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin AI, Dempsey PW, Bruhn K, Miller JF, Xu Y, Cheng G. Involvement of receptor-interacting protein 2 in innate and adaptive immune responses. Nature. 2002;416:190–194. doi: 10.1038/416190a. [DOI] [PubMed] [Google Scholar]
- Choi KS, Webb T, Oelke M, Scorpio DG, Dumler JS. Differential innate immune cell activation and proinflammatory response in Anaplasma phagocytophilum infection. Infection and immunity. 2007;75:3124–3130. doi: 10.1128/IAI.00098-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumler JS, Trigiani ER, Bakken JS, Aguero-Rosenfeld ME, Wormser GP. Serum cytokine responses during acute human granulocytic ehrlichiosis. Clinical and diagnostic laboratory immunology. 2000;7:6–8. doi: 10.1128/cdli.7.1.6-8.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumler JS, Choi KS, Garcia-Garcia JC, et al. Human granulocytic anaplasmosis and Anaplasma phagocytophilum. Emerging infectious diseases. 2005;11:1828–1834. doi: 10.3201/eid1112.050898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunning Hotopp JC, Lin M, Madupu R, et al. Comparative genomics of emerging human ehrlichiosis agents. PLoS genetics. 2006;2:e21. doi: 10.1371/journal.pgen.0020021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franchi L, Warner N, Viani K, Nunez G. Function of Nod-like receptors in microbial recognition and host defense. Immunological reviews. 2009;227:106–128. doi: 10.1111/j.1600-065X.2008.00734.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodzic E, Ijdo JW, Feng S, et al. Granulocytic ehrlichiosis in the laboratory mouse. The Journal of infectious diseases. 1998;177:737–745. doi: 10.1086/514236. [DOI] [PubMed] [Google Scholar]
- Inohara N, Nunez G. NODs: intracellular proteins involved in inflammation and apoptosis. Nature reviews Immunology. 2003;3:371–382. doi: 10.1038/nri1086. [DOI] [PubMed] [Google Scholar]
- Keestra AM, Winter MG, Klein-Douwel D, et al. A Salmonella virulence factor activates the NOD1/NOD2 signaling pathway. mBio. 2011:2. doi: 10.1128/mBio.00266-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi K, Inohara N, Hernandez LD, et al. RICK/Rip2/CARDIAK mediates signalling for receptors of the innate and adaptive immune systems. Nature. 2002;416:194–199. doi: 10.1038/416194a. [DOI] [PubMed] [Google Scholar]
- Kobayashi KS, Chamaillard M, Ogura Y, Henegariu O, Inohara N, Nunez G, Flavell RA. Nod2-dependent regulation of innate and adaptive immunity in the intestinal tract. Science. 2005;307:731–734. doi: 10.1126/science.1104911. [DOI] [PubMed] [Google Scholar]
- Lepidi H, Bunnell JE, Martin ME, Madigan JE, Stuen S, Dumler JS. Comparative pathology, and immunohistology associated with clinical illness after Ehrlichia phagocytophila-group infections. The American journal of tropical medicine and hygiene. 2000;62:29–37. doi: 10.4269/ajtmh.2000.62.29. [DOI] [PubMed] [Google Scholar]
- Magalhaes JG, Lee J, Geddes K, Rubino S, Philpott DJ, Girardin SE. Essential role of Rip2 in the modulation of innate and adaptive immunity triggered by Nod1 and Nod2 ligands. European journal of immunology. 2011;41:1445–1455. doi: 10.1002/eji.201040827. [DOI] [PubMed] [Google Scholar]
- Martin ME, Bunnell JE, Dumler JS. Pathology, immunohistology, and cytokine responses in early phases of human granulocytic ehrlichiosis in a murine model. The Journal of infectious diseases. 2000;181:374–378. doi: 10.1086/315206. [DOI] [PubMed] [Google Scholar]
- Pan Q, Mathison J, Fearns C, et al. MDP-induced interleukin-1beta processing requires Nod2 and CIAS1/NALP3. Journal of leukocyte biology. 2007;82:177–183. doi: 10.1189/jlb.1006627. [DOI] [PubMed] [Google Scholar]
- Pedra JH, Sutterwala FS, Sukumaran B, et al. ASC/PYCARD and caspase-1 regulate the IL-18/IFN-gamma axis during Anaplasma phagocytophilum infection. Journal of immunology. 2007;179:4783–4791. doi: 10.4049/jimmunol.179.7.4783. [DOI] [PubMed] [Google Scholar]
- Pedra JH, Tao J, Sutterwala FS, et al. IL-12/23p40-dependent clearance of Anaplasma phagocytophilum in the murine model of human anaplasmosis. FEMS immunology and medical microbiology. 2007;50:401–410. doi: 10.1111/j.1574-695X.2007.00270.x. [DOI] [PubMed] [Google Scholar]
- Pedra JH, Mattner J, Tao J, et al. c-Jun NH2-terminal kinase 2 inhibits gamma interferon production during Anaplasma phagocytophilum infection. Infection and immunity. 2008;76:308–316. doi: 10.1128/IAI.00599-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrilli V, Dostert C, Muruve DA, Tschopp J. The inflammasome: a danger sensing complex triggering innate immunity. Current opinion in immunology. 2007;19:615–622. doi: 10.1016/j.coi.2007.09.002. [DOI] [PubMed] [Google Scholar]
- Rikihisa Y, Lin M, Niu H. Type IV secretion in the obligatory intracellular bacterium Anaplasma phagocytophilum. Cellular microbiology. 2010;12:1213–1221. doi: 10.1111/j.1462-5822.2010.01500.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar A, Duncan M, Hart J, Hertlein E, Guttridge DC, Wewers MD. ASC directs NF-kappaB activation by regulating receptor interacting protein-2 (RIP2) caspase-1 interactions. Journal of immunology. 2006;176:4979–4986. doi: 10.4049/jimmunol.176.8.4979. [DOI] [PubMed] [Google Scholar]
- Scorpio DG, Akkoyunlu M, Fikrig E, Dumler JS. CXCR2 blockade influences Anaplasma phagocytophilum propagation but not histopathology in the mouse model of human granulocytic anaplasmosis. Clinical and diagnostic laboratory immunology. 2004;11:963–968. doi: 10.1128/CDLI.11.5.963-968.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada K, Chen S, Dempsey PW, et al. The NOD/RIP2 pathway is essential for host defenses against Chlamydophila pneumoniae lung infection. PLoS pathogens. 2009;5:e1000379. doi: 10.1371/journal.ppat.1000379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sukumaran B, Carlyon JA, Cai JL, Berliner N, Fikrig E. Early transcriptional response of human neutrophils to Anaplasma phagocytophilum infection. Infection and immunity. 2005;73:8089–8099. doi: 10.1128/IAI.73.12.8089-8099.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun W, JWIJ, Telford SR, 3rd, Hodzic E, Zhang Y, Barthold SW, Fikrig E. Immunization against the agent of human granulocytic ehrlichiosis in a murine model. The Journal of clinical investigation. 1997;100:3014–3018. doi: 10.1172/JCI119855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telford SR, 3rd, Dawson JE, Katavolos P, Warner CK, Kolbert CP, Persing DH. Perpetuation of the agent of human granulocytic ehrlichiosis in a deer tick-rodent cycle. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:6209–6214. doi: 10.1073/pnas.93.12.6209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Loewenich FD, Scorpio DG, Reischl U, Dumler JS, Bogdan C. Frontline: control of Anaplasma phagocytophilum, an obligate intracellular pathogen, in the absence of inducible nitric oxide synthase, phagocyte NADPH oxidase, tumor necrosis factor, Toll-like receptor (TLR)2 and TLR4, or the TLR adaptor molecule MyD88. European journal of immunology. 2004;34:1789–1797. doi: 10.1002/eji.200425029. [DOI] [PubMed] [Google Scholar]
- Wang T, Akkoyunlu M, Banerjee R, Fikrig E. Interferon-gamma deficiency reveals that 129Sv mice are inherently more susceptible to Anaplasma phagocytophilum than C57BL/6 mice. FEMS immunology and medical microbiology. 2004;42:299–305. doi: 10.1016/j.femsim.2004.06.001. [DOI] [PubMed] [Google Scholar]
- Yanai H, Ban T, Wang Z, et al. HMGB proteins function as universal sentinels for nucleic-acid-mediated innate immune responses. Nature. 2009;462:99–103. doi: 10.1038/nature08512. [DOI] [PubMed] [Google Scholar]