

Abstract
It is now well accepted that sex hormones have immunoregulatory activity and may prevent exacerbations in multiple sclerosis during pregnancy. Our previous studies demonstrated that oestrogen (17β-oestradiol; E2) protection against experimental autoimmune encephalomyelitis (EAE) is mediated mainly through oestrogen receptor-α (ERα) and the membrane receptor G-protein-coupled receptor 30 (GPR30) and is abrogated in the absence of B cells and the co-inhibitory receptor, Programmed Death-1 (PD-1). To critically evaluate the cell source of the E2 and PD-1 co-inhibitory pathways in EAE regulation, we assessed the requirement for ERs on transferred B cells and downstream effects on expression of PD-1/PD-ligand on CD4+ Foxp3+ regulatory T (Treg) cells in B-cell-replenished, E2-treated B-cell-deficient (μMT−/−) mice with EAE. The results clearly demonstrated involvement of ERα and GPR30 on transferred B cells that mediated the protective E2 treatment effect on EAE and further showed an E2-mediated B-cell-dependent up-regulation of PD-1 on CD4+ Foxp3+ Treg cells. These findings identify regulatory B-cell populations as key players in potentiating Treg-cell activity during E2-mediated protection against EAE.
Keywords: experimental autoimmune encephalomyelitis, multiple sclerosis, oestrogen and receptors, programmed death-1/programmed death ligand, regulatory B cells
Introduction
The increased incidence of multiple sclerosis (MS) in females has prompted extensive evaluation of possible gender-related mechanisms of disease induction and therapeutic strategies. One major conclusion from these studies is that increased levels of endogenous or exogenous sex hormones, including oestrogen (E2), oestriol (E3) and progesterone, can reduce lesion formation and severity of MS.1–7 Our laboratory has focused on the ability of E2 to induce protection against onset and severity of experimental autoimmune encephalomyelitis (EAE), an animal model of MS.8,9 Collectively, our findings implicate regulatory B (Breg) cells,10 oestrogen receptor-α (ERα)11 and G-protein-coupled receptor 30 (GPR30)12 and the programmed death-1 (PD-1)/programmed death ligand (PD-L) co-inhibitory pathway13–16 as critical factors contributing to the E2 protective mechanism. However, the cellular interactions that prevent or reduce EAE severity during E2 treatment are not yet fully understood.
Our previous work demonstrated that B cells are indispensable for E2-mediated protection against EAE.10 The purpose of the current study was to establish the sufficiency of B cells and their oestrogen receptors to restore E2-mediated protection against EAE and to evaluate downstream effects on infiltration of inflammatory cells into the central nervous system (CNS) and cellular expression of other critical regulatory molecules, including the PD-1 receptor and its ligands, PD-L1 and PD-L2. We therefore expanded upon our previous studies 10 that involved isolation and transfer of B cells from naive wild-type (WT) mice into recipient B-cell-deficient μMT−/− mice, in which signs of EAE disease were delayed but not reduced by E2 treatment. Specifically, we transferred B cells from myelin oligodendrocyte glycoprotein (35–55) (MOG35–55)/complete Freund's adjuvant (CFA) immunized donor mice that were reported to have increased levels of Breg-cell subtypes and enhanced treatment effects on EAE.17,18 The recipient μMT−/− mice were E2-preconditioned a week before the B-cell transfers. Our results demonstrated clearly that passively transferred B cells from MOG35–55/CFA-immunized WT donors, expressing ERα or GPR30, could restore E2-mediated protection in B-cell-deficient μMT−/− mice, which resulted in significantly reduced infiltration of T cells, monocytes and dendritic cells into the brain, reduced CNS pathology and enhanced downstream induction of PD-1 by CD4+ Foxp3+ regulatory T (Treg) cells.
Materials and methods
Animals
Wild-type female C57BL/6 mice were purchased from Harlan Laboratories (Livermore, CA). The μMT−/− mice containing no B cells because of targeted disruption of the membrane exon of the immunoglobulin µ-chain gene were originally obtained from Jackson Laboratories (Bar Harbor, ME) and were bred at the Animal Resource Facility at the Portland Veteran Affairs Medical Center. The ERα−/− mice on a C57BL/6 background, also bred in-house, were originally obtained from the Korach Laboratory (National Institutes of Health, Research Triangle Park, NC) and GPR30−/− mice were bred using in-house colonies. The generation of ERα−/− 19 and GPR30−/− mice (also on a C57BL/6 background)12 has been previously described. Female B6.129-Esr2tm1Unc (Esr-2KO, Esr2−/−, ERβ−/−) and ERα/ERβ−/− (Double-knockout) mice were a gift from Stephanie Murphy, PhD (Department of Anesthesiology and Perioperative Medicine, Oregon Health & Science University, Portland, OR, USA). All mice (on a C57BL/6 background) were used at 7–8 weeks of age and were housed in the Animal Resource Facility at the Portland Veterans Affairs Medical Center in accordance with institutional guidelines. The study was conducted in accordance with National Institutes of Health guidelines for the use of experimental animals, and the protocols were approved by the Institutional Animal Care and Use Committee.
Donor mice
Female WT, ERα−/−, ERβ−/−, ERα/β−/− and GPR30−/− mice that served as donors of B cells were immunized with 200 μg MOG35–55 peptide (PolyPeptide Laboratories, San Diego, CA) in 200 μg CFA (H37Ra, Difco). Pertussis toxin was not given to ensure the retention of MOG-primed cells in the spleens.
Adoptive transfer of B cells
Spleens from MOG-immunized WT, ERα−/−, ERβ−/−, ERα/β−/− and GPR30−/− mice were harvested on day 15 post-immunization (p.i.) and processed for B-cell isolation. Splenic CD19+ B cells were purified using paramagnetic bead-conjugated antibodies from the CD19 cell isolation kit and subsequently separated by AutoMACS (Miltenyi Biotec, Auburn, CA). The positive fraction of the cells separated in this way were CD19+ B cells with a purity of ≥ 95%. Approximately 10 × 106 purified splenic B cells from the donor mice were transferred intravenously into μMT−/− mice, on the same day as EAE induction.
Hormone treatment and induction of EAE in the recipient μMT−/− mice
Female μMT−/− mice (recipients) were implanted with 2·5 mg/60-day release E2 pellets (Innovative Research of America, Sarasota, FL) or sham-treated (control) 1 week before B-cell transfer and immunization with 200 μg MOG35–55 peptide (PolyPeptide Laboratories, San Diego, CA) in 200 μg CFA (H37Ra, Difco). The recipient mice received pertussis toxin (Ptx, List Biologicals, Campbell, CA) on the day of immunization (75 ng) and 2 days later (200 ng). All recipient mice were monitored daily for clinical signs of disease and scored using the following scale: 0 = no signs; 1 = limp tail or mild hind limb weakness; 2 = moderate hind limb weakness or mild ataxia; 3 = moderately severe hind limb weakness; 4 = severe hind limb weakness or mild forelimb weakness or moderate ataxia; 5 = paraplegia with no more than moderate forelimb weakness; and 6 = paraplegia with severe forelimb weakness or severe ataxia or moribund condition.
Histopathology
Intact spinal columns removed from recipient mice at the end of the study (i.e. day 20–28 p.i.) were fixed in 10% formalin. Dissected spinal cords were embedded in paraffin before sectioning. Sections were stained with haematoxylin & eosin to assess inflammatory lesions. Transverse sections were stained with a modified eriochrome cyanine protocol to assess the sparing of the white and grey matter (demyelination).20 Slides were analysed by light microscopy.
Flow cytometry
Spleens and brains from control and E2-treated μMT−/− mice were processed for lymphocyte isolation. Cells were stained with a combination of the following antibodies obtained from BD Bioscience (San Diego, CA): CD4 (L3T4), CD19 (1D3), CD1d (1B1), CD5 (53-7.3), PD-L1 (MIH5), PD-L2 (Ty25), CD11b (M1/70), CD11c (HL-3), CD45 (30-F11). The intracellular staining of Foxp3 (MF23) and PD-1 (J43) was completed following overnight incubation in fixation/permeabilization buffer (eBiosciences, San Diego, CA). Dead cells were gated out using propidium iodide discrimination. Cells were gated on CD19 to determine expression of the CD1dhigh CD5+ and PD-L1 populations.
Intracellular interleukin-10 (IL-10) expression was also visualized by immunofluorescence staining and analysed by flow cytometry, as described in ref. 17. Briefly, isolated splenocytes were resuspended (2 × 106 cells/ml) in complete medium [RPMI-1640 medium containing 10% fetal calf serum, 200 μg/ml penicillin, 200 U/ml streptomycin, 4 mm l-glutamine and 5 × 10−5 m 2-mercaptoethanol (all from Life Technologies, Carlsbad, CA)] with lipopolysaccharide (10 μg/ml, Escherichia coli serotype 0111:B4; Sigma-Aldrich, St Louis, MO), PMA (50 ng/ml; Sigma-Aldrich), ionomycin (500 ng/ml; Sigma-Aldrich), and Brefeldin A (1 μl/ml of medium; BD Biosciences) for 4 hr, in 24-well flat-bottom plates. For IL-10 detection, FcRs were blocked with mouse FcR monoclonal antibody (2.4G2; BD Biosciences) before surface staining. Stained cells were fixed and permeabilized using a Cytofix/Cytoperm kit (BD Biosciences), according to the manufacturer's instructions, and stained with allophycocyanin-conjugated mouse anti-IL-10 monoclonal antibody (JES5-16E3). Data were collected with CellQuest (BD Biosciences, San Jose, CA) and FCS Express (De Novo Software, Los Angeles, CA) software on a FACSCalibur (BD Biosciences). Absolute numbers were calculated from live-gated cells.
Statistical analysis
Data are reported using graphpad prism (v 4·0, San Diego, CA) and expressed as the mean ± SEM. Statistical significance for the disease course between recipient μMT−/− control and E2-implanted mice was calculated using one-way analysis of variance with multiple comparison post-test (Bonferroni). The Student's t-test and the Kruskal–Wallis test (non-parametric analysis of variance) with Dunn's multiple comparison of means post-test were used for analysis of the cumulative disease index (CDI). Student's t-test was used to compare flow cytometry data between the recipient μMT−/− control and E2-implanted mice. P-values ≤ 0·05 were considered significant.
Results
B cells from mMOG35–55-immunized WT mice are sufficient to restore E2-mediated protection in B-cell-deficient mice
Our previous work demonstrates that B cells are indispensable for E2-mediated protection against EAE.10 As demonstrated in ref. 10, our preliminary studies involved isolating B cells from naive WT mice and adoptively transferring into recipient μMT−/− mice that were either sham-treated or E2-implanted a day after the transfers and MOG-immunized 7 days later. We demonstrated that the E2-implanted μMT−/− mice that were B-cell recipients were significantly protected from EAE compared with the sham-treated recipients. However, this E2-mediated protection, upon B-cell transfers from naive WT mice, was short-lived and the recipient mice lost protection from day 21 onwards. Previous studies by others indicated an enhanced ability of antigen-primed B cells to mediate protection against EAE.17,18 Hence, as an extension to our previous studies, we evaluated whether B cells from mMOG35–55-immunized WT mice were sufficient to restore E2-mediated protection in E2-preconditioned B-cell-deficient μMT−/− mice when transferred on the same day as immunization with mMOG35–55/CFA/Ptx rather than 7 days before immunization, as done earlier. Briefly, 10 million B cells were transferred via tail vein injections into recipient μMT−/− mice that were either sham-treated or implanted with 2·5 mg per 60-day release E2 pellets 7 days before the B-cell transfer and immunization protocol. The transferred B cells could be detected in the blood, inguinal lymph nodes and spleens until at least 15 days p.i. Disease course in the recipient μMT−/− mice was followed by monitoring changes in clinical EAE scores for 26–30 days p.i. to determine the sufficiency of B cells in E2-implanted mice. Subsequently, the same protocol was followed for all the adoptive transfer experiments.
The control μMT−/− recipients of MOG-specific B cells from WT donors demonstrated an onset of EAE at day 11 p.i. and exhibited a steady increase in the disease scores with the peak of the disease at day 15 p.i. (Fig. 1a). However, the E2-implanted μMT−/− recipients of MOG-specific WT B cells were significantly protected from EAE until day 24 p.i. with a loss of protection, here onwards, as compared with the control WT B-cell-recipient counterparts. These results were as per our expectation because the group of μMT−/− mice implanted with E2 pellets was anticipated to mimic a WT scenario. That is, replenishment with WT B cells would enable E2 protection against EAE compared with vehicle-treated μMT−/− mice which, despite receiving B cells, would not be protected from EAE (similar to the WT sham-treated mice). As demonstrated earlier, the E2-implanted μMT−/− mice with no cell transfers exhibited a delay in the onset of EAE (day 15 p.i.), but completely lost E2-mediated protection by day 18, with their disease scores being comparable to the respective sham-treated counterparts by day 19. The CDI and the peak of the disease of E2-implanted μMT−/− recipients of WT B cells were also significantly lower than those of control recipients. Spinal cord sections were assessed for the extent of inflammation (haematoxylin & eosin staining) and demyelination (eriochrome staining) in the CNS of the control and E2-implanted μMT−/− recipient mice. We demonstrated earlier10 that spinal cord sections of E2-implanted μMT−/− mice (receiving no B cells) demonstrated massive leucocyte infiltration with several foci of inflammation along with severe demyelination similar to control μMT−/− mice. Here, the control μMT−/− recipient mice demonstrated leucocyte infiltration along with demyelination. However, the spinal cord sections from the E2-implanted μMT−/− recipient mice showed no obvious signs of inflammation and demyelination and appeared healthy (day 15 p.i.) (Fig. 1b), mimicking a phenomenon demonstrated by the WT E2-treated mice.8 Hence, the above results confirm that B cells are sufficient to restore E2-mediated protection in the recipient μMT−/− mice and the protection is longer and more pronounced than that demonstrated by the E2-implanted μMT−/− mice with no cell transfers.
Figure 1.
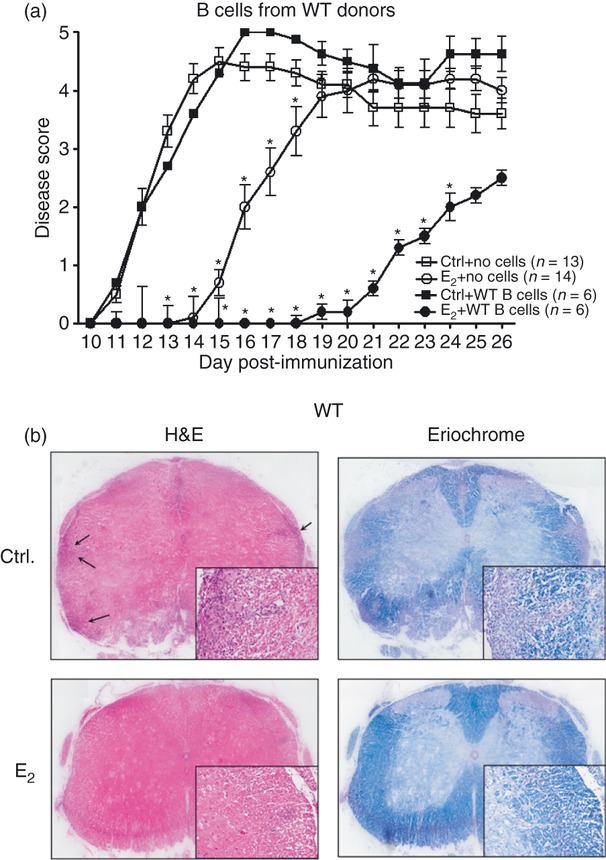
B cells are sufficient to restore 17β-oestradiol (E2) -mediated protection in the B-cell-deficient mice. Female wild-type (WT) mice that served as donors of B cells were immunized with mouse myelin oligodendrocyte glycoprotein (35–55) (mMOG35–55) peptide for 15 days in complete Freund's adjuvant (CFA) before collection of splenocytes and enrichment of B cells by MACS. Seven- to 8-week-old μMT−/− mice that served as recipients of B cells were either sham-treated (control) or implanted with E2 pellets 1 week before transfer of 10 million B cells and immunization with MOG35–55 peptide in CFA/pertussis toxin (Ptx). Recipient mice were monitored for signs of clinical experimental autoimmune encephalomyelitis over 26 days. Data presented are (a) mean daily disease scores from two independent experiments with three mice/group/experiment. *P ≤ 0·05, compared with the respective control mice (i.e. controls with no B cells and controls receiving WT B cells but no E2) (one-way analysis of variance with Bonferroni post hoc test). (b) Histopathological evaluation of spinal cords of WT B-cell-recipient μMT−/− (sham and E2-implanted) mice. Spinal cords from each group, collected on day 15 post-immunization, were fixed in PFA and embedded in paraffin. Transverse sections, 10 μm thick, from different regions of the spinal cord from each of the groups were stained with haematoxylin & eosin to enumerate infiltrating leucocytes and with Eriochrome cyanine to visualize the extent of demyelination. Arrows denote foci of inflammation; magnification was 50 × and 200 × (inset). Sections are representative of two experiments (n = 3/group/experiment).
Presence of ERα in B cells is indispensable for E2-mediated protection against EAE
As B cells are known to express ERs, it was crucial to determine the contribution of these ERs to the protective function of B cells in E2-related protection against EAE. To characterize the ERs involved in E2-mediated protection, our laboratory has demonstrated earlier that the immunomodulatory effects of E2 in EAE are dependent on ERα and not ERβ signalling.11 Also based on our studies on the recently discovered membrane ER, GPR30,12 it is likely that both GPR30 and ERα participate in E2-mediated protection in an additive manner. In our recent studies,10 we demonstrated that direct treatment of B cells with E2 significantly reduced proliferation of MOG35–55-specific T cells, suggesting a requirement for ERα on B cells. It was therefore important to ascertain the role of ERs on B cells as critical players responsible for mediating E2-directed protection against EAE. We first assessed the role of ERα in B cells as a contributor to E2-mediated protection in the recipient μMT−/− mice. B cells were obtained from MOG-immunized ERα−/− (ERKO) donors, day 14 p.i., and transferred into sham-treated or E2-implanted recipient μMT−/− mice. The E2-implanted μMT−/− recipients of B cells from ERα−/− mice lost protection on about the same day (day 16 p.i.) as the E2-implanted μMT−/− mice with no cell transfers (Fig. 2a). They also demonstrated a significantly earlier onset compared with the E2-implanted μMT−/− mouse recipients of WT B cells (Table 1). The CDI and peak of the disease for these E2-implanted recipients of MOG-specific ERα−/− B cells was also significantly higher compared with the E2-implanted recipients of WT B cells and similar to the E2-implanted μMT−/− mice with no cell transfers (Table 1). The spinal cord sections from the E2-implanted ERα−/− B-cell-recipient mice demonstrated equivalent leucocyte infiltration along with demyelination compared with the control recipients (Fig. 2b). These data indicate a critical role of ERα on the B cells in mediating the protective effects of E2.
Figure 2.

17β-Oestradiol (E2) -mediated protection against experimental autoimmune encephalomyelitis (EAE) is mediated through oestrogen receptor-α (ERα) and G-protein-coupled receptor 30 (GPR30) on B cells. Female ER knockout mice (i.e. ERα−/−, ERβ−/−, GPR30−/− or ERα/β−/− mice) that served as donors of B cells were immunized for 15 days with mouse myelin oligodendrocyte glycoprotein (35–55) (mMOG35–55) peptide in complete Freund's adjuvant (CFA) before collection of splenic B cells for transfer. Seven- to 8-week-old μMT−/− mice that served as recipients of B cells were either sham-treated (control) or implanted with E2 pellets 1 week before B-cell transfer and immunization with MOG35–55 peptide in CFA/pertussis toxin (Ptx). Recipient mice were monitored for signs of clinical EAE for 26 days. Data presented are (a) mean daily EAE disease scores from two to four independent experiments in sham versus E2-treated B-cell-deficient mice receiving 10 million B cells from mMOG35–55-immunized ERα−/−, ERβ−/−, GPR30−/− and ERα/β−/− mice with three or four mice/group/experiment. *P ≤ 0·05, compared with the respective control mice (i.e. sham-treated versus E2-implanted μMT−/− mice receiving no cells and sham-treated versus E2-implanted μMT−/− B-cell recipients) (one-way analysis of variance with Bonferroni post hoc test). Histopathological evaluation of spinal cords from (b) ERα−/−, (c) ERβ−/−, (d) GPR30−/− and (e) ERα/β−/− B-cell-recipient μMT−/− mice (sham-treated and E2-implanted, day 26 post-immunization). Spinal cords from each group were fixed in PFA and embedded in paraffin. Transverse sections, 10 μm thick, from different regions of the spinal cord from each of the groups were stained with haematoxylin & eosin to enumerate infiltrating leucocytes and with Eriochrome cyanine to visualize the extent of demyelination. Arrows denote foci of inflammation; magnification was 50 × and 200 × (inset). Sections are representative of two experiments (n = 3–4/group/experiment).
Table 1.
Clinical course of EAE in B-cell-recipient mMT−/− mice*
| µMT−/− recipient mice | Incidence | Onset | Peak | CDI | Mortality |
|---|---|---|---|---|---|
| No cells Ctrl | 13/13 | 11·6 ± 0·5 | 4·6 ± 0·1 | 43·8 ± 13·7 | 1 |
| No cells E2 | 13/14 | 16·4 ± 12·21 | 4·7 ± 0·7 | 20·8 ± 10·91 | 2 |
| + WT B cells Ctrl | 6/6 | 12·2 ± 1·6 | 4·6 ± 0·9 | 53·7 ± 7·5 | 1 |
| + WT B cells E2 | 4/6 | 20·6 ± 1·51 | 2·6 ± 2·4 | 11·3 ± 10·41 | 0 |
| + ER-α−/− B cells Ctrl | 11/11 | 10·2 ± 3·0 | 4·7 ± 0·3 | 57·5 ± 15·7 | 0 |
| + ER-α−/− B cells E2 | 13/13 | 16·3 ± 2·92 | 4·5 ± 0·32 | 30·3 ± 11·42 | 0 |
| + ER-β−/− B cells Ctrl | 8/8 | 11·6 ± 1·8 | 4·5 ± 0·4 | 53·3 ± 24·8 | 0 |
| + ER-β−/− B cells E2 | 5/6 | 20·2 ± 2·41 | 2·8 ± 1·91,3 | 15·8 ± 14·81,3 | 0 |
| + GPR30−/− B cells Ctrl | 8/8 | 10·5 ± 0·7 | 5·0 ± 0·2 | 48·8 ± 14·4 | 0 |
| + GPR30−/− B cells E2 | 6/8 | 16·7 ± 2·42 | 3·3 ± 1·91 | 23·6 ± 25·71 | 1 |
| + ER-α/β−/− B cells Ctrl | 6/6 | 10·5 ± 0·6 | 5·0 ± 0 | 58·8 ± 16·6 | 0 |
| + ER-α/β−/− B cells E2 | 6/6 | 16·8 ± 1·01 | 3·9 ± 1·6 | 20·2 ± 24·41 | 2 |
Statistical evaluation of EAE disease course for recipient mMT−/− Control and E2-implanted mice, including the Cumulative Disease Index (CDI), through day 26 post-immunization for the 2–4 experiments (n = 3–4 mice/group/experiment). The daily disease scores from pooled data and corresponding CDI data from each of the independent experiments (mean ± SEM) are presented. Additional information for the experiments is shown in Figs. 1 and 2.
CDI, cumulative disease index; Ctrl, control; E2, 17β-oestradiol; ER, oestrogen receptor; GPR30, G-protein-coupled receptor 30.
Significant (P < 0·05) as compared with the respective Control group.
Significant (P < 0·05) as compared with the E2-implanted wild-type B-cell-recipient group.
Significant (P < 0·05) as compared with the E2-implanted ERα−/− B-cell-recipient group.
Upon transferring B cells from MOG-immunized ERβ−/− (BERKO) donors, the E2-implanted μMT−/− recipient mice had a significantly late onset compared with not only the sham-treated ERβ−/− B-cell recipients (Fig. 2a) but also the E2-implanted ERα−/− B-cell recipients (Table 1). The E2-implanted μMT−/− recipients of B cells from ERβ−/− mice lost protection at day 24 p.i., about the same day as the E2-implanted recipients of B cells from WT donors. The day of disease onset and the disease peak were comparable with those of the E2-implanted WT B-cell-recipient μMT−/− mice (Table 1). Spinal cord sections from E2-implanted ERβ−/− B-cell-recipient μMT−/− mice demonstrated fewer inflammatory foci and a lesser extent of demyelination (Fig. 2c). These data demonstrate the dispensable role of ERβ in B cells for E2-mediated protection in the recipient μMT−/− mice, with observed protection potentially the result of residual functional ERα and GPR30 E2 receptors.
Our studies in GPR30−/− mice12 demonstrated that the mER, GPR30, is sufficient, yet not exclusively responsible, for the full E2-mediated protection against EAE. The same study led us to conclude that that ERα and GPR30 work together additively to achieve optimal protection, and each receptor may adequately compensate the functional loss of the other. To verify the requirement of GPR30 on the B cells in E2-mediated protection, we transferred B cells from MOG-immunized GPR30−/− donors into sham-treated or E2-implanted μMT−/− mice on the day of induction of EAE in the recipient mice. Upon transferring B cells from MOG-immunized GPR30−/− donors, as expected, the control recipient μMT−/− mice had an early onset (day 10·5 p.i.); however, the E2-implanted recipients had an onset of disease around day 16–17 p.i. (Fig. 2a). This trend of an early disease onset was similar to the E2-implanted recipients of ERα−/− B cells. However, the CDI and the peak of the disease in the E2-implanted GPR30−/− B-cell-recipient μMT−/− mice were lower than those of the E2-implanted ERα−/− B-cell-recipient μMT−/− mice. The E2-implanted recipients of GPR30−/− B cells lost protection from day 19 p.i. compared with the sham-treated recipients. Spinal cord sections from E2-implanted GPR30−/− B-cell-recipient μMT−/− mice also demonstrated massive leucocyte infiltration with several foci of inflammation (indicated by arrows, Fig. 2d) along with severe demyelination similar to control recipient μMT−/− mice. These data indicate a partial role of GPR30 in B cells for E2-mediated protection in recipient μMT−/− mice.
When B cells from MOG-immunized ERα/β−/− (ERKO/BERKO) donors (which still express GPR30) were transferred into control and E2-implanted μMT−/− mice, the E2-implanted recipients had an onset similar to that of the E2-implanted ERα−/− and GPR30−/− B-cell recipients (Fig. 2a). The E2-implanted recipients lost protection from day 19 p.i. onwards compared with the sham-treated recipients. The CDI and peak of disease in the E2-implanted recipients of ERα/β−/− B cells were similar to those of the E2-implanted ERα−/− and GPR30−/− B-cell recipients. The spinal cord sections of the E2-implanted ERα/β−/− also demonstrated leucocyte infiltration with several foci of inflammation and demyelination (Fig. 2e). Hence, these results confirm that GPR30 and ERα act in unison in E2-mediated protection.
Donor B cells from mMOG35–55-primed WT and different ER-deficient mice have similar regulatory markers
With the difference in the protection rendered by the B cells from WT and various ER receptor-deficient mice, we sought to determine whether the donor B cells attain a distinct phenotype in the presence of MOG35–55 peptide, which causes them to elicit different levels of protection in the recipient μMT−/− mice. B cells from MOG-immunized WT, ERα−/−, ERβ−/− and GPR30−/− donors were isolated on day 14 p.i. and characterized via flow cytometry for the presence of various regulatory markers. Our results10 demonstrated that the requirement for B cells in E2-mediated protection against EAE involved direct E2 effects on Breg cells mediated through ERα and the PD-1/PD-L1 negative co-stimulatory pathway, indicating that B cells may mediate immunoregulation via these markers. Therefore, we assessed the donor B cells from different strains of mice for the percentage of PD-L1 and Breg cells (CD1dhigh CD5+) and for IL-10 production. As demonstrated in Fig. 3, the per cent expressions of each of these regulatory markers by the B cells from different strains were not significantly distinct from each other. However, as the donor B cells from different ER-deficient mice mediate different outcomes of protection against EAE, it implies that the donor B cells acquire regulatory properties in an E2-rich milieu, which was provided in the E2-implanted μMT−/− recipients.
Figure 3.

Donor B cells from mouse myelin oligodendrocyte glycoprotein (35–55) (mMOG35–55) -primed wild-type (WT) and oestrogen receptor (ER) -deficient mice have similar regulatory markers. Mononuclear cells were isolated from the spleens of mMOG35–55-immunized donor mice of different ER-deficient strains: i.e. WT, ERα−/−, ERβ−/−, and G-protein-coupled receptor 30 (GPR30−/−) mice. On day 14 post-immunization, splenocytes were assessed for the expression of programmed death ligand 1 (PD-L1) on CD19+ B cells, percentage of CD1dhigh CD5+ B cells and interleukin-10 (IL-10) -producing B cells after lipopolysaccharide + PMA-ionomycin stimulation for 4 hr within the indicated gates among total CD19+ B cells. Data represent PD-L1 expression, CD1dhigh CD5+ expression and IL-10 expression on gated CD19+ cells of total gated and live/fixed cells. Similar results were obtained when B cells from the donor mice were characterized in at least two independent experiments with (n = 2/strain/experiment).
Less infiltration of immune cells in CNS of E2-implanted μMT−/− recipients of WT B cells
As the E2-implanted μMT−/− recipients of WT B cells demonstrate protection from EAE compared with not only the sham-treated recipients but also the no transfer E2-implanted μMT−/− mice, we characterized the immune cell trafficking into the CNS in these mice. Leucocytes isolated from brains of sham-treated (control) and E2-implanted μMT−/− recipient of WT B cells were stained for several cell surface markers. Brains of E2-implanted μMT−/− recipients not only demonstrated lower numbers of total leucocytes compared with control μMT−/− recipient mice (data not shown), but also had a significant decrease in percentages of CD4+ T cells (Fig. 4). Brains of E2-implanted μMT−/− recipients also demonstrated significant decrease in percentages of infiltrating macrophages (CD11b+ CD45high) and dendritic cells (CD11c+ CD45+), which participate in disease pathogenesis through effects on blood–brain barrier permeability, antigen presentation and immune regulation21 (Fig. 4). Hence, replenishment of B cells redeems the immunosuppressive and protective effects of E2 in the μMT−/− recipient mice, as demonstrated by not only significantly less demyelination and fewer inflammatory foci (histology) but also less infiltration of immune cells into the CNS.
Figure 4.
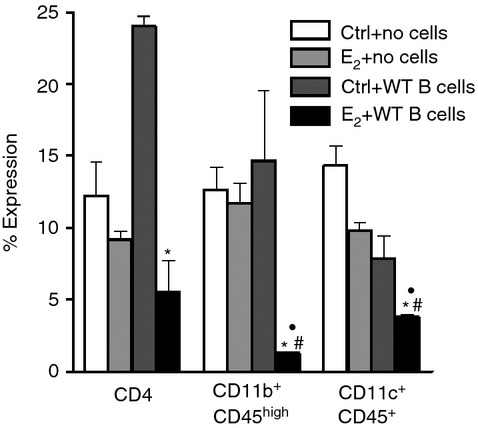
Less infiltration of immune cells in central nervous system of 17β-oestradiol (E2) -implanted μMT−/− recipients of wild-type (WT) B cells. On day 15 post-immunization with mouse myelin oligodendrocyte glycoprotein (35–55) /complete Freunds' adjuvant/pertussis toxin, mononuclear cells isolated from brains of sham-treated versus E2-implanted μMT−/− mice that received no cells or 10 million WT B cells were analysed by FACS for percentages of CD4+ T cells, CD11b+ CD45high macrophages and CD11c+ CD45+ dendritic cells. Frequencies of CD4+ T cells, macrophages and dendritic cells were determined in individual brains and data indicate the percentages of total gated live leucocytes (n = 6). Data are pooled from two independent experiments (mean ± SEM). *P ≤ 0·05 compared with sham control μMT−/− mice that received WT B cells; #P ≤ 0·05 compared with the control μMT−/− mice that did not receive cell transfers and • P ≤ 0·05 compared with E2-implanted μMT−/− mice that did not receive cell transfers (Student's t-test).
The E2-implanted μMT−/− recipients of WT B cells have significantly increased percentages of PD-L1-expressing cells, PD-1 expression in Treg cells and CD1dhigh CD5+ expression by the transferred B cells
Next, we assessed the induction of immune responses in the periphery (spleens) because it is known that after induction of immune responses in the periphery, pathogenic T cells migrate to the CNS to initiate an inflammatory process responsible for clinical signs of EAE.22 To evaluate MOG-specific responses generated in spleens, mononuclear cells from sham-treated and E2-implanted μMT−/− recipient mice were stained for the negative co-inhibitory markers, PD-L1 and PD-L2, and the expression of PD-1 on CD4+ Foxp3+ Treg cells. First, we also ascertained that the transferred B cells still survived in the recipients (Fig. 5a). Moreover, the per cent expression of total PD-L1 in the spleens of E2-implanted μMT−/− B-cell-recipient mice was significantly higher than in the sham-treated recipients and the no-cell-transfer mice (Fig. 5b). There was no difference in the PD-L2 expression in the two groups of recipient mice (data not shown). Since E2 induces an increase in the CD4+ Foxp3+ (Treg) cell population,14,23 we evaluated the percentage of Treg cells in the B-cell-transferred recipient mice. There was a trend towards an increased percentage of the CD4+ Foxp3+ population in the E2-implanted μMT−/− B-cell-recipient mice but the increase was not statistically significant compared with the control μMT−/− B-cell-recipient mice, as demonstrated in Fig. 5(c). Our laboratory has earlier demonstrated the critical role of PD-1, a co-inhibitory receptor, in E2-mediated protection against EAE.13,14,24 Hence, we assessed the expression of PD-1 in the Treg-cell population (PD-1+ Foxp3+ CD4+ cells) in the recipient mice. The E2-implanted recipients of MOG-specific B cells indeed had significantly higher expression of PD-1 compared with the control recipients (Fig. 5d). The Breg-cell (CD1dhigh CD5+) percentage among the transferred and surviving B cells was also calculated to quantify any differences between the sham-treated and E2-implanted μMT−/− recipient mice. As indicated in Fig. 5(e), the percentage of Breg cells in the E2-implanted recipients of WT B cells was significantly higher than that in the control recipients. These data demonstrate that B cells in an E2-rich milieu attain regulatory properties and also that the transferred B cells induce immunoregulatory properties in other immune cells in the B-cell-deficient recipients.
Figure 5.

17β-Oestradiol (E2) -implanted μMT−/− recipients of wild-type (WT) B cells have significantly increased percentages of splenic programmed death ligand 1 (PD-L1) -expressing cells, PD-1+ CD4+ Foxp3+ regulatory T (Treg) cells and CD1dhi CD5+ B cells. On day 15 post-immunization with mouse myelin oligodendrocyte glycoprotein (35–55) /complete Freunds' adjuvant/pertussis toxin, mononuclear cells isolated from spleens of control and E2-implanted μMT−/− mice that received WT B cells and their no cell transfer counterparts were analysed for the presence of (a) B cells (CD19+ cells), (b) total PD-L1-expressing cells, (c) total Foxp3+ CD4+ (Treg) cells, (d) PD-1+ CD4+ Foxp3+ Treg cells and (e) CD1dhi CD5+ CD19+ B cells. Frequencies of B cells, total PD-L1-expressing cells, Treg cells, PD-1-expressing cells in the Treg population and regulatory B-cell subset (CD1dhi CD5+ CD19+ cells) were determined in individual spleens and data indicate the percentages of total gated live leucocytes (n = 6). Data are pooled from two independent experiments (mean ± SEM). *P ≤ 0·05 compared with sham control μMT−/− mice that received WT B cells; # P ≤ 0·05 compared with the control μMT−/− mice that did not receive cell transfers and • P ≤ 0·05 compared with E2-implanted μMT−/− mice that did not receive cell transfers (Student's t-test).
Discussion
The results presented above demonstrate the requirement for expression of ERα or GPR30 by transferred B cells from MOG35–55-immunized donors to restore responsiveness to E2 therapy in μMT−/− mice developing EAE. Furthermore, the data confirm E2-dependent B-cell involvement in inhibiting infiltration of CD4+ T cells, monocytes and dendritic cells into the CNS and up-regulating PD-1 on CD4+ Foxp3+ Treg cells and PD-L1-expressing cells in recipient mice. These findings are important because they define the identity of immune cells involved in regulatory interactions that lead to protection against clinical and histological signs of EAE after treatment with E2.
The model system used in the current study used transfer of B cells from MOG35–55-immunized donors into sham or E2-pretreated B-cell-deficient μMT−/− mice followed by EAE induction to establish the sufficiency of B cells in E2 protection. This study design represents a departure from our previous protocols to take advantage of the reported increase in regulatory activity of using antigen-specific B cells for the transfers17,18 and places the B cells into a pre-existing E2-enhanced environment upon EAE induction. Although E2 treatment of the B-cell-deficient μMT−/− mice does not inhibit EAE disease severity, it does delay onset of clinical signs by ∼ 5 days, producing a later baseline (day 16) for comparison with B-cell-transferred recipients. This delay may be a result of the effects of E2 on other cell types, which could include Treg cells,21 myeloid cells and CNS cells.25 To support this reasoning, studies demonstrate the link between B cells and CD4+ Foxp3+ Treg-cell homeostasis, wherein the adoptive transfer of naive WT splenic B cells into μMT−/− mice resulted in a significant increase in the numbers of Treg cells by day 10 post-transfer.26 Likewise, in our study the E2-implanted μMT−/− mice, with no cell transfers, demonstrated a higher percentage of Foxp3+ Treg cells than the no cell-transfer control μMT−/− mice (Fig 5c), though this increase was not significant. Also, the E2-implanted μMT−/− recipient mice transferred with WT MOG-specific B cells demonstrated a further increase in the Treg-cell percentages though it was not statistically significant. Another study indicates the crucial role for Treg cells in inhibiting late-phase EAE disease.27 Our previous study also demonstrated reduced IL-17 production by the splenocytes of the E2-implanted μMT−/− mice, suggesting a relatively less inflammatory environment in an E2-rich milieu, even in the absence of B cells.10 Therefore, we postulate that the delayed onset of disease in the μMT−/− mice, with no cell transfers, may be a result of the effects of E2 on other cell types, such as Treg cells. The transfer of MOG-specific B cells from WT mice further delayed the onset of EAE by an additional 4 days (∼ day 20) and inhibited both peak and cumulative disease severity scores.
The transfer of B cells from various ER knockout mice provided new and critical information regarding the direct effects of E2 on the transferred regulatory B cells. Restoration of μMT−/− mice with B cells lacking ERα did not produce any clinical benefit, indicating a pivotal role for B-cell-expressed ERα. In contrast, restoration with B cells lacking ERβ but sufficient in ERα produced a similar effect to WT B cells. Interestingly, restoration with B cells lacking both ERα and ERβ or with B cells lacking the membrane GPR30 ER had a partial effect on EAE severity, implicating a role for GPR30 in the E2 protective mechanism, albeit secondary to that of ERα. These findings put into perspective the crucial importance of ERα and GPR30 on B cells for E2-dependent protection against EAE.
Our study further defined the downstream effects of transferred MOG-reactive B cells on E2-dependent immunoregulatory mechanisms involving Treg cells, Breg cells and the PD-1/PD-L co-inhibitory pathway. The transferred WT B cells (i) strongly inhibited infiltration of CD4+ T cells, monocytes/macrophages and dendritic cells into the CNS of recipient mice in the presence of E2, similar to the pattern observed in E2-treated WT mice; (ii) strongly increased expression of PD-1 from 11% to 28% in CD4+ Foxp3+ Treg cells in the presence of E2; (iii) enhanced expression of PD-L1 but not PD-L2 on lymphocytes in both control and B-cell-transferred mice in the presence of E2; and (iv) produced an expanded population of CD1dhigh CD5+ regulatory B cells (increased from 9% to 13% of CD19+ B cells) in the E2-conditioned recipient mice. Although the above E2-mediated effects were mediated by only 10 million B cells, it is possible that higher numbers of transferred MOG-reactive B cells might provide even more potent and longer-lasting protection against EAE.
These results suggest interactions between PD-L1 present on both the regulatory B-cell10,16 and T-cell subsets (Fig. 5b) may activate and up-regulate expression of PD-1 on CD4+ Foxp3+ Treg cells (Fig. 5d) in the presence of E2. Most of the protective effects of oestrogen appear to be mediated through ERα expressed by B cells, but as we have recently published,12 the crucial up-regulation of PD-1 that potentiates suppressive activity of CD4+ Foxp3+ Treg cells may involve mainly GPR30. This scenario is both plausible and consistent with the idea that Breg cells (i.e. IL-10-producing B cells including the CD1dhigh CD5+ regulatory B10 cells) have a protective role during EAE initiation, whereas Breg-potentiated Treg cells have a therapeutic role in the later stages after onset of clinical disease.17
Acknowledgments
The authors thank Lisa Miller and Mary Chase for technical assistance, Danielle Galipeau for assistance in histology images, Zefora Alderman for assistance in preparing the figures and Eva Niehaus for assistance with manuscript preparation. This work was supported by National Institutes of Health grant NS045445 and National Multiple Sclerosis Society grant RG3405-C-6. This material is based upon work supported in part by the Department of Veterans Affairs, Veterans Health Administration, Office of Research and Development, Biomedical Laboratory Research and Development. The contents do not represent the views of the Department of Veterans Affairs or the US Government.
Disclosure
The authors have no conflict of interest to declare.
References
- 1.Abramsky O. Pregnancy and multiple sclerosis. Ann Neurol. 1994;36(Suppl):S38–41. doi: 10.1002/ana.410360712. [DOI] [PubMed] [Google Scholar]
- 2.Bernardi S, Grasso MG, Bertollini R, Orzi F, Fieschi C. The influence of pregnancy on relapses in multiple sclerosis: a cohort study. Acta Neurol Scnd. 1991;84:403–6. doi: 10.1111/j.1600-0404.1991.tb04977.x. [DOI] [PubMed] [Google Scholar]
- 3.Confavreux C, Huchinson M, Hours MM, Corinovis-Tourniaire P, Moreau T. Rate of pregnancy-related relapse in multiple sclerosis. N Engl J Med. 1998;339:285–91. doi: 10.1056/NEJM199807303390501. [DOI] [PubMed] [Google Scholar]
- 4.Damek DM, Shuster EA. Pregnancy and multiple sclerosis. Mayo Clin Proc. 1997;72:977–89. doi: 10.1016/S0025-6196(11)63371-5. [DOI] [PubMed] [Google Scholar]
- 5.Sicotte NL, Liva SM, Klutch R, Pfieffer P, Bouvier S, Odesa S, Wu TCJ, Voskuhl RR. Treatment of multiple sclerosis with pregnancy hormone estriol. Ann Neurol. 2002;52:421–8. doi: 10.1002/ana.10301. [DOI] [PubMed] [Google Scholar]
- 6.Soldan SS, Alvarez RetuertoAI, Sicotte NL, Voskuhl RR. Immune modulation in multiple sclerosis patients treated with the pregnancy hormone estriol. J Immunol. 2003;171:6267–74. doi: 10.4049/jimmunol.171.11.6267. [DOI] [PubMed] [Google Scholar]
- 7.Kipp M, Amor S, Krauth R, Beyer C. Multiple sclerosis: neuroprotective alliance of estrogen-progesterone and gender. Front Neuroendocrinol. 2012;33:1–16. doi: 10.1016/j.yfrne.2012.01.001. [DOI] [PubMed] [Google Scholar]
- 8.Bebo BF, Fyfe-Johnson A, Adlard K, Beam AG, Vandenbark AA, Offner H. Low-dose estrogen therapy ameliorates experimental autoimmune encephalomyelitis in two different inbred mouse strains. J Immunol. 2001;166:2080–9. doi: 10.4049/jimmunol.166.3.2080. [DOI] [PubMed] [Google Scholar]
- 9.Ito A, Bebo BF, Jr, Matejuk A, Zamora A, Silverman M, Fyfe-Johnson A, Offner H. Estrogen treatment down-regulates TNF-α production and reduces the severity of experimental autoimmune encephalomyelitis in cytokine knockout mice. J Immunol. 2001;167:542–52. doi: 10.4049/jimmunol.167.1.542. [DOI] [PubMed] [Google Scholar]
- 10.Bodhankar S, Wang C, Vandenbark AA, Offner H. Estrogen-induced protection against experimental autoimmune encephalomyelitis is abrogated in the absence of B cells. Eur J Immunol. 2011;41:1165–75. doi: 10.1002/eji.201040992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Polanczyk M, Zamora A, Subramanian S, et al. The protective effect of 17β-estradiol on experimental autoimmune encephalomyelitis is mediated through estrogen receptor-α. Am J Pathol. 2003;163:1599–605. doi: 10.1016/s0002-9440(10)63516-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang C, Dehghani B, Magrisso IJ, et al. GPR30 contributes to estrogen-induced thymic atrophy. Mol Endocrinol. 2008;22:636–48. doi: 10.1210/me.2007-0359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Polanczyk MJ, Hopke C, Vandenbark AA, Offner H. Estrogen-mediated immunomodulation involves reduced activation of effector T cells, potentiation of Treg cells, and enhanced expression of the PD-1 costimulatory pathway. J Neurosci Res. 2006;84:370–8. doi: 10.1002/jnr.20881. [DOI] [PubMed] [Google Scholar]
- 14.Polanczyk MJ, Hopke C, Vandenbark AA, Offner H. Treg suppressive activity involves estrogen-dependent expression of programmed death-1 (PD-1) Int Immunol. 2007;19:337–43. doi: 10.1093/intimm/dxl151. [DOI] [PubMed] [Google Scholar]
- 15.Wang C, Li Y, Proctor TM, Vandenbark AA, Offner H. Down-modulation of programmed death 1 alters regulatory T cells and promotes experimental autoimmune encephalomyelitis. J Neurosci Res. 2010;88:7–15. doi: 10.1002/jnr.22181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Carter LL, Leach MW, Azoitei ML, et al. PD-1/PD-L1, but not PD-1/PD-L2, interactions regulate the severity of experimental autoimmune encephalomyelitis. J Neuroimmunol. 2007;182:124–34. doi: 10.1016/j.jneuroim.2006.10.006. [DOI] [PubMed] [Google Scholar]
- 17.Matsushita T, Horikawa M, Iwata Y, Tedder TF. Regulatory B cells (B10 cells) and regulatory T cells have independent roles in controlling experimental autoimmune encephalomyelitis initiation and late-phase immunopathogenesis. J Immunol. 2010;185:2240–52. doi: 10.4049/jimmunol.1001307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Su Y, Zhang AH, Li X, Owusu-Boaitey N, Skupsky J, Scott DW. B cells “transduced” with TAT-fusion proteins can induce tolerance and protect mice from diabetes and EAE. Clin Immunol. 2011;140:260–7. doi: 10.1016/j.clim.2011.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu H, Loo KK, Palaszynski K, Ashouri J, Lubahn DB, Voskuhl RR. Estrogen receptor α mediates estrogen's immune protection in autoimmune disease. J Immunol. 2003;171:6936–40. doi: 10.4049/jimmunol.171.12.6936. [DOI] [PubMed] [Google Scholar]
- 20.Rabchevsky AG, Fugaccia I, Sullivan PG, Scheff SW. Cyclosporin A treatment following spinal cord injury to the rat: behavioral effects and stereological assessment of tissue sparing. J Neurotrauma. 2001;18:513–22. doi: 10.1089/089771501300227314. [DOI] [PubMed] [Google Scholar]
- 21.Polanczyk MJ, Carson BD, Subramanian S, Afentoulis M, Vandenbark AA, Ziegler SF, Offner H. Estrogen drives expansion of the CD4+ CD25+ regulatory T cell compartment. J Immunol. 2004;173:2227–30. doi: 10.4049/jimmunol.173.4.2227. [DOI] [PubMed] [Google Scholar]
- 22.Zamvil SS, Steinman L. The T lymphocyte in experimental allergic encephalomyelitis. Annu Rev Immunol. 1990;8:579–621. doi: 10.1146/annurev.iy.08.040190.003051. [DOI] [PubMed] [Google Scholar]
- 23.Polanczyk MJ, Hopke C, Huan J, Vandenbark AA, Offner H. Enhanced FoxP3 expression and Treg cell function in pregnant and estrogen-treated mice. J Neuroimmunol. 2005;170:85–92. doi: 10.1016/j.jneuroim.2005.08.023. [DOI] [PubMed] [Google Scholar]
- 24.Wang C, Dehghani B, Li Y, Kaler LJ, Vandenbark AA, Offner H. Oestrogen modulates experimental autoimmune encephalomyelitis and interleukin-17 production via programmed death 1. Immunology. 2009;126:329–35. doi: 10.1111/j.1365-2567.2008.03051.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Polanczyk MJ, Jones RE, Subramanian S, et al. T lymphocytes do not directly mediate the protective effect of estrogen on experimental autoimmune encephalomyelitis. Am J Pathol. 2004;165:2069–77. doi: 10.1016/S0002-9440(10)63257-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ray A, Basu S, Williams CB, Salzman NH, Dittel BN. A novel IL-10-independent regulatory role for B cells in suppressing autoimmunity by maintenance of regulatory T cells via GITR ligand. J Immunol. 2012;188:3188–98. doi: 10.4049/jimmunol.1103354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Matsushita T, Horikawa M, Iwata Y, Tedder TF. Regulatory B cells (B10 cells) and regulatory T cells have independent roles in controlling experimental autoimmune encephalomyelitis initiation and late-phase immunopathogenesis. J Immunol. 2010;185:2240–52. doi: 10.4049/jimmunol.1001307. [DOI] [PMC free article] [PubMed] [Google Scholar]