

Abstract
The molecular mechanisms underpinning central nervous system damage in multiple sclerosis (MS) are complex and it is widely accepted that there is an autoimmune component. Both adaptive and innate immune effector mechanisms are believed to contribute to tissue disease aetiology. HLA-E is a non-classical MHC class Ib molecule that acts as the ligand for the NKG2A inhibitory receptor present on natural killer (NK) and CD8+ cells. Peptide binding and stabilization of HLA-E is often considered to signal infection or cell stress. Here we examine the up-regulation of HLA-E in MS brain tissue. Expression is significantly increased in white matter lesions in the brain of MS patients compared with white matter of neurologically healthy controls. Furthermore, using quantitative immunohistochemistry and confocal microscopy, we show increased HLA-E protein expression in endothelial cells of active MS lesions. Non-inflammatory chronic lesions express significantly less HLA-E protein, comparable to levels found in white matter from controls. Increased HLA-E protein levels were associated with higher scores of inflammation. These results suggest the potential for an effect in central nervous system pathogenesis from HLA-E modulation in stressed tissue. Co-localization with infiltrating CD8+ cells implicates a possible role for HLA-E-restricted regulatory CD8+ cells, as has been proposed in other autoimmune diseases.
Keywords: autoimmunity, major histocompatibility complex, human leucocyte antigen, multiple sclerosis
Multiple sclerosis (MS) is the most common cause of chronic neurological disability among adults aged 20–40 years. The mechanisms that trigger and mediate MS, a presumed autoimmune disease, remain unclear.1–3 Ultimately, the disease process results in areas of central nervous system (CNS) demyelination leading to axon dysfunction, neuronal death and hence neurological disability.
Recent, large-scale genome-wide association studies echo earlier candidate gene analysis in the overwhelming focus they place on genes of the human leucocyte antigen (HLA) complex and, to a lesser extent, multiple, additional, immune system genes.4 The strongest association is seen in Caucasian MS patients with HLA-DRB1*1501, although there are also independent associations in the HLA class I region. Clearly, associations with HLA class I allelic proteins may indicate a role in the pathogenesis of CD8+ T cells and natural killer (NK) cells.5 There is supportive data from both functional and histopathological studies for involvement of both cell types.6–9
The structure of MHC class Ib molecules is similar to that of the classical MHC class I molecules (class Ia) –consisting of a heavy chain with a peptide-binding cleft and a light chain of β2-microglobulin.10 While the class Ia molecules display extreme polymorphism, the class Ib molecules are much less varied in structure and can be considered ‘oligomorphic’.11 The class Ib molecules are believed to be important in innate immunity and immune regulation. They possess more anchor residues than class Ia molecules, placing greater constraints on their peptide-binding ability.12,13 Of the class Ib molecules, HLA-E is the least polymorphic. It has eight alleles, encoding three distinct protein sequences. The HLA-E peptide-binding groove is believed to bind primarily hydrophobic, nonameric peptides derived from the leader sequences of other MHC class I molecules.12 HLA-E acts as a ligand for the CD94/NKG2 family of receptors expressed on NK cells and some subsets of CD8+ T cells.14,15 These receptors are part of the C-type lectin superfamily and are comprised of both activating (2C, 2E and 2H) and inhibitory (2A and 2B) members.16 The best characterized of these receptor–ligand interactions is with CD94/NKG2A, whereby HLA-E expression prevents target cell lysis from CD94/NKG2A+ NK cells.17 Although decreased HLA-E expression provides one potential mechanism for CD94/NKG2A+ lymphocyte activation, it has also been reported that peptides derived from heat-shock protein 60 can bind and stabilize HLA-E, yet completely abrogate CD94/NKG2A binding and alleviate HLA-E-dependent inhibition of NK cells.18 These findings suggest a possible role for HLA-E as a reporter of cellular stress to the innate immune system.
The role of NK cells in MS and in experimental allergic encephalomyelitis (EAE) is of interest, because NK cell subsets may function in both disease exacerbation and regulation.7,19–21 These effects are likely to be seen in the peripheral and central nervous systems. Studies on the function of CD8+ T cells in MS have suggested a role for HLA-E in disease regulation. Regulatory CD8+ T cells capable of lysing myelin-reactive CD4+ T cells have been found in the cerebrospinal fluid of MS patients and appear to be HLA-E restricted.22 Related observations have been made in type I diabetes, where recognition of HLA-E bound to a heat-shock protein 60 peptide was implicated in recognition by regulatory CD8+ cells.23 Therapeutic induction of regulatory CD8+ cells in MS using Copaxone (glatiramer acetate) depends on an HLA-E-dependent interaction with CD4+ cells.24 Furthermore, up-regulation of CD94/NKG2A expression on regulatory CD8+ cells during exacerbation in MS reduces cytolytic activity.22 Paradoxically, EAE can be ameliorated by giving antibody against NKG2A.25 A prerequisite for the further analysis of related mechanisms in clinical MS would presumably be to demonstrate the co-localization of HLA-E expression and CD8 infiltrate in disease tissue: although many studies have focused on the role of classical MHC molecules in MS, little is known about the presence or function of MHC class Ib molecules in this disease. Here we have investigated the expression and localization of HLA-E in the white matter of MS patients and healthy controls.
Materials and methods
Tissue samples
The MS and control brain tissue samples were donated from the UK Multiple Sclerosis Tissue Bank (UKMSTB; Centre for Neuroscience, Imperial College Faculty of Medicine, Hammersmith Campus) and the Human Brain Tissue Bank, Budapest (Department of Anatomy, Semmelweis University, Budapest, Hungary). Fully informed consent and ethical approval were obtained for the collection and study of post-mortem tissue following local guidelines and guidelines published by the BrainNet Europe Brain Bank Consortium.26 All post-mortem MS tissues were obtained via a UK prospective donor scheme with full ethical approval (08/MRE09/31). Neuropathological confirmation of MS was carried out according to the International Classification of Diseases of the Nervous System criteria (http://www.ICDNS.org). Samples were taken from 12 MS patients, 11 women and one man, with ages at death ranging from 34 to 59 years (mean 46·6 years). Two blocks with lesion were available for 10 of 12 the MS cases. All lesions were selected for ongoing disease activity status and were active or chronic active. Ongoing disease activity status was confirmed with presence of lipids phagocytosed by macrophages or phagocytic microglia determined by standard Oil Red O stain, hence the presence of active focal inflammatory demyelination.27 The largest lesion per MS case was chosen for extraction of RNA. The majority of MS cases were secondary progressive and disease duration ranged from 2 to 36 years (mean 16·7 years). The 10 control patients, seven women and three men, were free of any evidence of known neurological disease, and had an average age at death of 47 years (range 26–60 years). Further details of control and MS cases can be found in the Supplementary material, Tables S1 and S2, respectively. Groups were matched for gender and age at death and when compared were not statistically different based on gender (Fisher's exact test; P = 0·2932) or age at death (independent t-test; P = 0·91) and showed similar homogeneity of variances for both groups (F test = 0·33). The MS lesions were identified on serial sections by standard immunostaining for myelin oligodendrocyte glycoprotein (MOG) expression and by Luxol® fast blue (LFB) solution (Sigma-Aldrich Company Ltd, Dorset, UK). Appendix and tonsil inflamed tissue was donated from the Human Biomaterials Resource Centre (Hammersmith Hospitals NHS Trust, Hammersmith Hospital, London). In addition to basic clinical information, clinical milestone and baseline assessment of post-mortem brain was available. All cases at the UKMSTB undergo a thorough neuropathological assessment and are graded on their cortical pathology, axonal damage and lymphocytic infiltrate (degree of inflammation) as described in Reynolds et al.27
RNA extraction and quantitative real-time polymerase chain reaction
RNA was extracted from white matter of MS and control patients. White matter tissues were resected from snap-frozen MS and control brain-tissue blocks and stored at −80°. For MS tissue samples, white matter was taken from within a lesion. Total RNA extraction was performed using the RNeasy lipid tissue mini kit (Qiagen, Crawley, UK). Concentrations and purity of RNA were analysed using a Nanodrop 1000 Spectrophotometer (Thermo Scientific, Wilmington, NC).
Oligonucleotide forward and reverse primers and fluorophore and quencher labelled hydrolysis probes were synthesized by Integrated DNA Technologies (Coralville, IA) and combined in a single tube in a primer : probe ratio of 2 : 1. In all cases, the detection probe for the gene of interest included the non-emissive quenching molecule 6-carboxy-tertamethylrhodamine (TAMRA) and the fluorescent label 6-carboxyfluorescein (6-FAM). The detection probe for the reference gene included the quenching molecule Iowa Black FQ (IBFQ) and the fluorescent label hexachloro fluorescein (HEX). X-prolyl aminopeptidase (aminopeptidase P) 1 (XPNPEP1) and gylceraldehyde 3-phosphate dehydrogenase (GAPDH) were used as reference genes. An assay was designed that would amplify within the coding sequence of HLA-E mRNA but not any other HLA class I sequences and was designed to detect common sequence across all allelic variants. A two-step real-time reverse transcriptase quantitative PCR (RT-qPCR) was performed using the QuantiTect® reverse transcription kit (Qiagen) and Brillant® II QPCR master mix with low 6-carboxy-X-rhodamine (ROX) from Agilent Technologies UK Ltd (Edinburgh, UK). For cDNA synthesis, up to 1 μg total RNA from each sample was reverse transcribed using a QuantiTect® reverse transcription kit with integrated removal of genomic DNA contamination. No-reverse-transcriptase (NoRT) reactions consisted of the same protocol as above but the Quantiscript reverse transcriptase was omitted and replaced with RNAse free-water. Real-time PCR experiments were performed using the Mx3000P™ real-time PCR system with software version 4.10 (Stratagene, La Jolla, CA). For each sample, 20-μl reactions were set up in duplicate and in duplex, with each reaction containing 10 μl 2× master mix, 2 μl 10× primetime assay (1 μl GOI + 1 μl normalizer), 7 μl RNAse-free water and 1 μl template cDNA. PrimeTime™ qPCR assays were purchased from Integrated DNA Technology and are listed in Table 1. Reactions were carried out with the following cycling protocol: 95° for 10 min, then 45 cycles with a three-step programme (95° for 15 s, 50° for 30 s and 72° for 30 s). Fluorescence data collection was performed during the annealing step. Control NoRT reactions to test for contaminating DNA and a negative control containing no cDNA template were introduced in each run. Efficiencies of the primer/probe assays were tested individually and in duplex. Expression levels of target genes were normalized to the levels of the novel XPNPEP1 reference gene and calibrated using a standard curve method for quantification. Some experiments were duplicated using a more commonly used normalizer; GAPDH. The calibrator was generated by creating a pool from all control cDNA samples. Levels of the calibrator represented the baseline (of one) from which all RNA expression values were calculated within an experiment. The standard curve was used to determine relative quantity expression values for each target gene after RT-qPCR analysis of each test specimen. Relative expression values for each target gene are expressed as a ratio of target gene expression level to the reference gene expression level in the same specimen.
Table 1.
Primers and probes used for RT-PCR
| Gene | Forward primer | Reverse primer | Probe |
|---|---|---|---|
| XPNPEP1 | 5′-GTT CCA TCC TTG TAC TGA GCA-3′ | 5′-TTC CCA ACG ATT TCC AGC A-3′ | 5′-/5HEX/CCA TCA TTC ACT ACG CGC CGA TCC /3IABkFQ/-3′ |
| GAPDH | 5′-GGC CAT CCA CAG TCT TCT G-3′ | 5′-CAG CCT CAA GAT CAT CAG CAA-3′ | 5′-/5HEX/ATG ACC ACA GTC CAT GCC ATC ACT /3IABkFQ/-3′ |
| IL-15 | 5′-AGC TGG CAT TCA TGT CTT CA-3′ | 5′-ATT ACA TTC ACC CAG TTG GCT-3′ | 5′-/56-FAM/CCC TGC ACT /ZEN/GAA ACA GCC CAA /3IABkFQ/-3′ |
| NKG2A | 5′-AGC TTC TCA GGA TTT TCA AGG G-3′ | 5′-GAC AGA TAA TTC CCA GGA TCC C-3′ | 5′-/56-FAM/ACT GCA AAG /ZEN/ATT TAC CAT CAG CTC CAG A/3IABkFQ/-3′ |
| HLA-E | 5′-CTT GGA TCT GTG GTC TCT GG-3′ | 5′-AGC TGT CTC AGG CTT TAC AAG-3′ | 5′-/56-FAM/TGG TTG CTG CTG TGA TAT GGA GGA AG/3IABkFQ/-3′ |
XPNPEP1, X-prolyl aminopeptidase (aminopeptidase P) 1; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; IL-15, interleukin-15.
Immunohistochemistry and immunofluorescence
Snap-frozen tissue sections were fixed in 4% paraformaldehyde for 10 min at 4° and sections were then permeabilized and endogenous peroxidase activity was removed by incubation in methanol containing 1% hydrogen peroxide. Primary antibodies (listed in Table 2) were applied and left to incubate overnight at room temperature (RT). After overnight incubation, sections were rinsed (PBS) and biotinylated secondary antibodies were applied for 45 min. Avidin biotin complex (ABC) was applied for 1 hr (ABC, prepared 30 min before application) and slides were then rinsed in PBS. NovaRed™ (Vector®, Burlingame, CA) chromogen was prepared to the manufacturer's instructions and applied for 1–5 min. Sections were counterstained in Mayer's Haemalum for 2–5 s and rinsed for 5 min under running water. Sections were dehydrated and placed in 70% methylated spirit for 1 min, then 90% for 1 min, then 100% for 1 min, and again at 100% for 1 min. Immunofluorescence staining was as described for immunohistochemistry (up to the step of ABC application) except that the secondary antibodies used were fluorescently labelled and used at 1 : 500 (Invitrogen, Paisley, UK). After 45 min in the dark, the sections were rinsed in PBS (three times). Coverslips were applied using fluorescent mounting medium (DAKO, Ely, UK) and DAPI (Invitrogen) and the slides were dried under pressure. Images were ascertained using ImagePro7 software (MediaCybernetics, Inc., Bethesda, MD) and captured with a Nikon Eclipse E1000M microscope/digital camera system. The CD8-positive immunolabelled cells (Mab EP1150Y; see Table 2) were counted by two independent, blinded observers, within the lesion area defined by MOG and LFB staining and given a mean score (vision inspection scale: 0 = no cells; 1 = 0–10 cells; 2 = 10–20 cells; 3 = > 20 cells). Positive immunolabelling was highlighted by setting the grey-level detection limits to threshold using ImageJ software28 and the area of highlighted immunolabelling was obtained as percentage area of the field scanned. Five fields per tissue section were scanned and the mean values were used in subsequent statistical analysis. Fluorescence images were ascertained by fluorescent microscopy using the same Nikon Eclipse E1000M microscope/digital camera system (QImaging, Surrey, BC, Canada) and the Leica TCS STED confocal microscope (Leica Microsystems GmbH, Wetzlar, Germany). Digitized images were processed using Image ProPlus (Media Cybernetics, Rockville, MD) and ImageJ and prepared in Adobe Photoshop.
Table 2.
Primary antibodies used in this study
| Antigen | Dilution | Target | Donor species | Source |
|---|---|---|---|---|
| HLA-E | 1 : 100 | Denatured heavy chain of human HLA-E | Mouse monoclonal | Abcam; clone MEM-E/02 |
| MOG | 1 : 50 | Myelin and oligodendrocytes | Mouse monoclonal | Gift of S. Piddlesden, Cardiff, UK |
| CD3 | 1 : 200 | T cells, NKT cells | Mouse monoclonal | DakoCytomation; clone F7.2.38 |
| CD8 | 1 : 100 | CD8 T cells | Rabbit monoclonal | Epitomics; clone EP1150Y |
| GFAP | 1 : 200 | Astrocytes | Rabbit polyclonal | DakoCytomation |
| vWf | 1 : 1000 | Endothelial cells, platelets and megakaryocytes | Rabbit polyclonal | Abcam |
| Iba1 | 1 : 2500 | Macrophages and microglia | Rabbit polyclonal | Wako Pure Chemical Industries |
| HLA-DP, DQ, DR | 1 : 100 | DP, DQ and DR heterodimers | Mouse monoclonal | DakoCytomation |
| CD68 | 1 : 100 | Monocytes and macrophages | Mouse monoclonal | DakoCytomation |
| IgG1 isotype control | 1 : 100 | No target | Mouse monoclonal | Abcam |
Iba1, Ionized calcium-binding adaptor molecule 1; vWf, von Willebrand factor; MOG, myelin oligodendrocyte glycoprotein; GFAP, glial fibrillary acidic protein; HLA-E, human leucocyte antigen-E.
Statistical analysis
Group difference was established by using paired or independent Student's t-test (one-tailed or two-tailed) unless otherwise stated. Multiple group comparison was assessed using a one-way analysis of variance with Bonferroni's Multiple Comparison Test. Homogeneity of variance was assessed using F-test. Data are shown as mean ± SEM unless stated; differences were taken as significant when P < 0·05.
Results
MS active lesions show up-regulation of HLA-E
We investigated up-regulation of HLA-E mRNA expression in CNS white matter lesions (WML) from patients with secondary progressive MS relative to control tissue. Significantly increased levels (P = 0·0002; two-tailed) of HLA-E mRNA were detected in WML from MS patients (2·96 ± 0·39; n = 12) compared with controls (0·84 ± 0·14; n = 10; Fig. 1).
Figure 1.
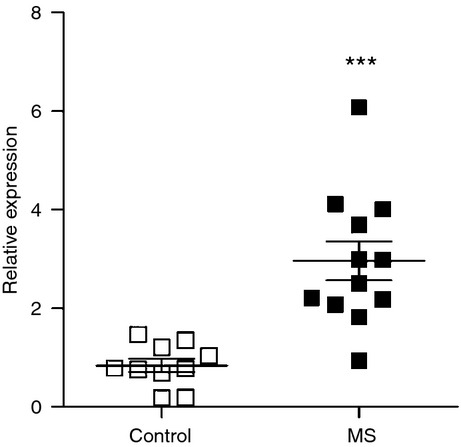
Increased HLA-E mRNA levels in multiple sclerosis (MS) white matter lesions. Snap-frozen tissue from the largest identified lesion from each patient tissue block, established by myelin oligodendrocyte glycoprotein staining, was macrodissected and RNA was extracted as described in the Materials and methods. Increased levels (P = 0.0002; two-tailed) of HLA-E mRNA were detected in white matter lesions from MS patients (2.96 ± 0.39; n = 12) compared with controls (0.84 ± 0.14; n = 10). Data were normalized using X-prolyl aminopeptidase (aminopeptidase P) 1.
We then investigated and quantified HLA-E protein expression in WML by immunohistochemistry and confirmed the increases seen at the mRNA level. Mean expression of HLA-E was determined from two different lesions per case. White matter from non-MS controls showed little or no detectable HLA-E protein expression (Fig. 2a). The HLA-E protein levels (HLA-E-immnolabelling, HLA-E-il) were significantly up-regulated (P = 0·018; one-tailed) in MS tissue (0·767 ± 0·21) compared with controls (0·2541 ± 0·062; Fig. 2b,c). Overall, staining was mostly detected on endothelial cells and on cells that may have been ramified microglia (Fig. 2d). To confirm HLA-E expression on endothelial cells, immunofluorescence co-expression studies were performed (Fig. 3). Expression of HLA-E by endothelial cells was confirmed by co-staining with anti-von Willebrand factor staining (Fig. 3d–f). There was relatively poor co-localization with anti- glial fibrillary acidic protein (GFAP), arguing against HLA-E expression by astrocytes (Fig. 3a–c), though the apparent cytoplasmic staining in a minority of GFAP+ cells will require further investigation.
Figure 2.
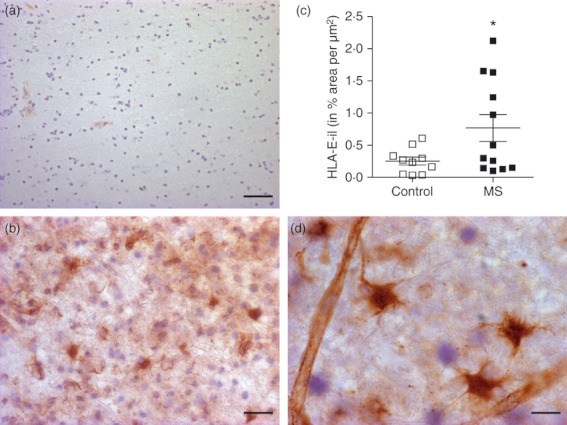
Increased HLA-E protein expression in multiple sclerosis (MS) white matter lesion. Little HLA-E immunolabelling (HLA-il) was detectable in white matter from control tissues (a) compared with MS white matter lesions (b). Staining was mostly detected on endothelial cells and astrocytes (d). (c) Levels were significantly up-regulated (P = 0.018; one-tailed) in MS tissue (0.767 ± 0.21) compared with controls (0.2541 ± 0.062). Scale bar = 30 μm for a; 20 μm for b, 10 μm for d.
Figure 3.
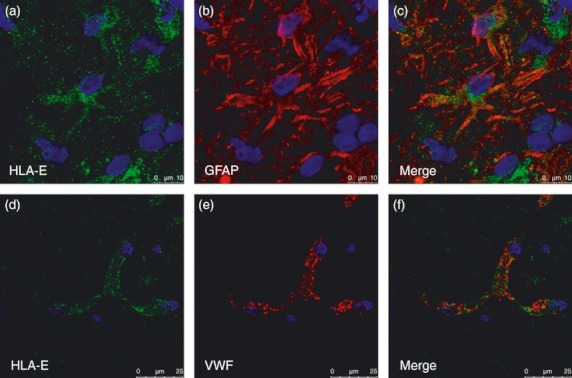
Central nervous system co-staining of HLA-E with glial fibrillary acidic protein (GFAP) and von Willebrand factor antibodies. (a) HLA-E (green) in general does not co-localize with the astrocyte marker GFAP (red) (b). HLA-E (green) (d) was observed on endothelial cells (e) using antibody to the endothelial marker von Willebrand factor. Scale bar = 10 μm.
HLA-E protein expression in MS patients is largely restricted to active lesions and not chronic lesions
The heterogeneous distribution of HLA-E observed in MS lesions warranted further investigation. Multiple sclerosis lesions can be classified according to the extent of demyelination and the abundance of infiltrating immune cells. Consequently, MOG staining was used to assess demyelination and HLA class II and CD3 staining were used to determine the number of infiltrating macrophages and CD3+ T cells respectively (data not shown). Active lesions were defined as those that had a diffusely demarcated edge with patchy demyelination within the core and many infiltrating cells. Chronic active lesions represent the stage between active lesions and chronic lesions, being largely clear of myelin and infiltrating immune cells, except around the lesion edge.27 Among the MS sections, the distinction between the chronic and chronic active lesions was not always apparent, with many of the larger lesions showing characteristics of both. For this reason, the chronic and chronic active lesions were grouped together for analysis. The MS sections that contained active lesion had more HLA-E than sections that contained a chronic/chronic active lesion (Fig. 4a). In addition, HLA-E protein expression was more pronounced in cases that presented more active lesions at the time of death (P = 0·024; Mann–Whitney test; Fig. 4b). Increase in HLA-E protein expression was associated with a higher degree of inflammation (scale 0–5; Spearman r = 0·71; P = 0·0098; Fig. 4c).
Figure 4.
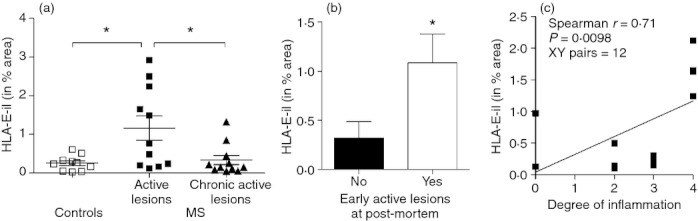
Increased HLA-E protein expression in early active lesions. HLA-E protein expression was assessed in individual lesions divided into active or chronic active lesions (a). There was a significant difference (analysis of variance; P = 0.006) in HLA-E expression in active lesions (1.16 ± 0.31) compared with chronic active lesions (0.33 ± 0.12) and controls (0.25 ± 0.06). HLA-E protein expression was also more pronounced in cases that presented more post-mortem early active lesions based on neuropathological assessment (b; P = 0.024; Mann–Whitney U-test; out of 12 cases, five were negative and seven were positive for presence of early active lesions). Finally (c), when correlated with degree of inflammation scores (0–5) attributed to the 12 multiple sclerosis (MS) cases as described in the Materials and methods, there was a positive correlation with HLA-E protein expression (Spearman r = 0.71; P < 0.01). Higher levels of HLA-E protein expression correlated with higher degrees of inflammation scores suggesting a direct relation between levels of central nervous system inflammation and HLA-E expression. *P < 0.05.
HLA-E up-regulation and infiltration by CD8+ or NK cells
It was of interest to ascertain whether lesions showing up-regulation of HLA-E showed associated infiltrates of NK cells, CD8+ cells, or both. The latter cell-type is of particular interest in this context with respect to the recent discussion of HLA-E-restricted regulatory CD8+ cells in autoimmunity.23 We have recently reported on the paucity of infiltrating NK cells in the lesions analysed from this donor cohort.29 Active lesions showed more extensive CD8+ T-cell infiltrates compared with chronic active lesions (Fig. 5a) and when CD8+ cell activity was correlated with HLA-E expression (Fig. 5b), there was significant positive correlation, suggesting that increased levels of HLA-E correlated with increased number of CD8+ cells (XY = 22 pairs, r2 = 0·2062, P = 0·0337). Immunofluorescent CD8+ cells were detected in the vicinity of HLA-E+ cells, though only rarely were CD8+ cells themselves positive for HLA-E expression (Fig. 5c).
Figure 5.
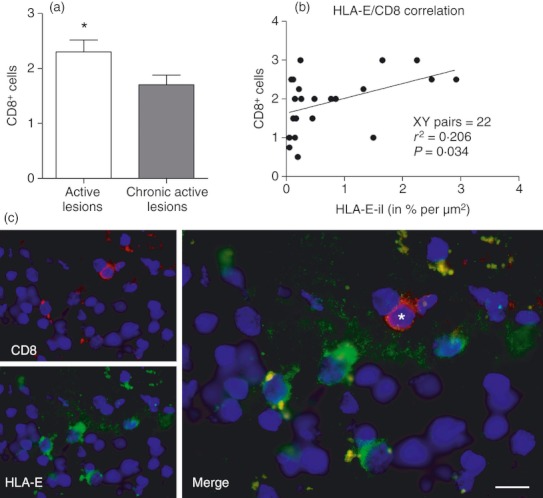
CD8 cells in active lesions co-localize with HLA-E. More CD8+ cells were found in active lesions (a; P = 0.0228; Mann–Whitney U-test). Also, more HLA-E protein expression was detected in lesion tissue enriched with CD8+ cells. There was mild but positive correlation (r2 = 0.2; P = 0.03) between the presence of CD8+ cells and HLA-E expression, suggesting that increased levels of HLA-E correlates with increased infiltration of CD8+ cells (b). Co-localization confirms presence of CD8+ cells (marked by asterisk) near HLA-E+ cells (c). Scale bar = 10 μm.
Lack of difference in NKG2A mRNA expression between white matter lesions of MS patients and normal white matter of controls
NKG2A is believed to be the major receptor for HLA-E.14 It was therefore hypothesized that NKG2A mRNA expression, for example on infiltrating CD8+ cells, may correlate with HLA-E expression and so be significantly greater in MS patients than controls. However, NKG2A expression was not significantly up-regulated in MS lesions (Fig. 6).
Figure 6.
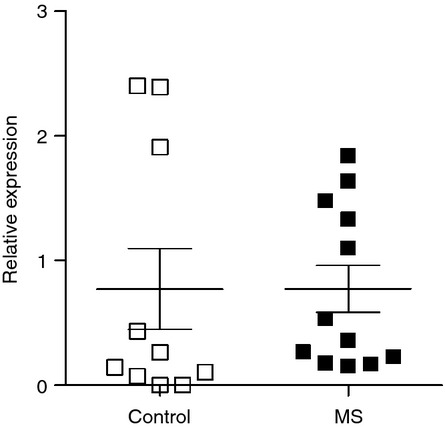
NKG2A mRNA expression. No difference in NKG2A mRNA expression was determined between white matter lesions of multiple sclerosis (MS) patients and white matter of non-MS controls.
Discussion
There is considerable interest in the role of innate immunity, pathogenic or regulatory, in MS, including potential roles of NK cells and the role of killer immunoglobulin-like receptor genotypes in susceptibility.6,7,19,20,29,30 Furthermore, the potential role of NKG2A+ regulatory CD8 cells in MS has been debated.22 Current mainstream models of NK cell surveillance are inadequate to account for an activation signal that may operate in the context of a disease such as MS. That is, NK cells are classically considered to survey down-modulation of HLA class I expression, generally as a consequence of viral subversion or tumorigenesis, or in rare cases may be shown to detect viral peptides bound in HLA class I grooves.31,32 Despite the widespread interest in NK cell actions in a wide range of autoimmune diseases including MS,33 there is a lack of any clear notion as to which ligands might be preferentially expressed by autoimmune target tissue. In the case of MS, although there is considerable literature on the functions of NK cells, we have no model for the changes that may be recognized by these NK cells and account for recognition. There has been little investigation into the expression of MHC class Ib molecules in MS. Here we have taken the first steps in exploring the possibility that this ligand might be supplied by the up-regulation of HLA-E as a consequence of local cellular stress. We have shown an increase in HLA-E expression in the CNS white matter of MS patients both at the level of mRNA production and protein expression.
We have shown HLA-E expression on endothelial cells of both MS patients and controls, but generally more intense in MS patient tissue. In vitro studies previously showed endothelial cell HLA-E up-regulation in response to tumour necrosis factor-α, interferon-γ and interleukin-1β.34 High expression of HLA-E is therefore likely against a backdrop of CNS inflammation and would seem to serve as a marker of endothelial cell activation. In lymphoid organs, HLA-E expression has been observed on T and B cells, and macrophages. In non-lymphoid tissue the assumption has been that HLA-E expression is restricted to endothelial cells.34 Kren et al.,35 analysing normal CNS tissue, found HLA-E expressed on endothelial cells and also, rarely (one case in eight), on microglia; among cases of glioblastoma, tumour-infiltrating microglia/macrophages were strongly positive for HLA-E expression although the staining appeared cytoplasmic rather than cell surface. We also observed few HLA-E+ microglia. Macrophages laden with myelin debris are often observed in MS36,37 and in EAE.38 It is possible that the HLA-E in foamy macrophages may be not of endogenous origin, but may originate from ingested cell debris from surrounding dead cells. The evidence supporting a role for regulatory CD8 cells in control of MS was recently reviewed.39 Although a case has previously been made for a potentially regulatory role of class 1b-positive T cells in the cerebrospinal fluid during neuroinflammatory disease,40 we could here find only a minority of cells in which HLA-E staining in the CNS was accounted for by co-expression in HLA-E+ T cells.
To conclude, we have found that the white matter lesions of MS patients express significantly more HLA-E mRNA and protein than the non-MS white matter. Moreover, we have shown that HLA-E protein expression is largely restricted to active MS lesions. We hypothesize that HLA-E may play a protective role through the activation of HLA-E-restricted regulatory CD8+ T cells. Although the presence of CD8 T cells in CNS tissue from MS patients has been regarded as a marker of immune pathogenesis, it may be that in some cases, CD8 infiltrates should be regarded as markers of regulation, albeit ultimately unsuccessful.
Acknowledgments
We thank the UK Multiple Sclerosis Tissue Bank (Imperial College), Dr Djordje Gveric and Julia Steele for their assistance in the collection of the material used in this study. We also thank Professor Miklos Palkovits, Magdolna Toronyay-Kasztner and the Human Brain Tissue Bank at Semmelweis University, Budapest. This work was funded by the Wellcome Trust, Grant number (ref. WT087999MA). The authors are grateful for support from the NIHR Biomedical Research Centre funding scheme. This study made use of the Department of Medicine, Hammersmith Campus, BRC Imaging and FACS Facility. We thank the donors and their families.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Table S1. The cohort of MS brain donors used.
Table S2. The cohort of control brain donors used.
References
- 1.McFarland HF, Martin R. Multiple sclerosis: a complicated picture of autoimmunity. Nat Immunol. 2007;8:913–9. doi: 10.1038/ni1507. [DOI] [PubMed] [Google Scholar]
- 2.Bhat R, Steinman L. Innate and adaptive autoimmunity directed to the central nervous system. Neuron. 2009;64:123–32. doi: 10.1016/j.neuron.2009.09.015. [DOI] [PubMed] [Google Scholar]
- 3.Amor S, Puentes F, Baker D, van der Valk P. Inflammation in neurodegenerative diseases. Immunology. 2010;129:154–69. doi: 10.1111/j.1365-2567.2009.03225.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sawcer S, Hellenthal G, Pirinen M, et al. Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature. 2011;476:214–9. doi: 10.1038/nature10251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Martin R. HLA class I: friend and foe of multiple sclerosis. Nat Med. 2008;14:1150–1. doi: 10.1038/nm1108-1150. [DOI] [PubMed] [Google Scholar]
- 6.Traugott U, Raine CS. Further lymphocyte characterization in the central nervous system in multiple sclerosis. Ann N Y Acad Sci. 1984;436:163–80. doi: 10.1111/j.1749-6632.1984.tb14788.x. [DOI] [PubMed] [Google Scholar]
- 7.Kastrukoff LF, Lau A, Wee R, Zecchini D, White R, Paty DW. Clinical relapses of multiple sclerosis are associated with ‘novel’ valleys in natural killer cell functional activity. J Neuroimmunol. 2003;145:103–14. doi: 10.1016/j.jneuroim.2003.10.001. [DOI] [PubMed] [Google Scholar]
- 8.Goverman J. Autoimmune T cell responses in the central nervous system. Nat Rev Immunol. 2009;9:393–407. doi: 10.1038/nri2550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Weiss HA, Millward JM, Owens T. CD8+ T cells in inflammatory demyelinating disease. J Neuroimmunol. 2007;191:79–85. doi: 10.1016/j.jneuroim.2007.09.011. [DOI] [PubMed] [Google Scholar]
- 10.O'Callaghan CA, Bell JI. Structure and function of the human MHC class Ib molecules HLA-E, HLA-F and HLA-G. Immunol Rev. 1998;163:129–38. doi: 10.1111/j.1600-065x.1998.tb01192.x. [DOI] [PubMed] [Google Scholar]
- 11.Rodgers JR, Cook RG. MHC class Ib molecules bridge innate and acquired immunity. Nat Rev Immunol. 2005;5:459–71. doi: 10.1038/nri1635. [DOI] [PubMed] [Google Scholar]
- 12.Braud V, Jones EY, McMichael A. The human major histocompatibility complex class Ib molecule HLA-E binds signal sequence-derived peptides with primary anchor residues at positions 2 and 9. Eur J Immunol. 1997;27:1164–9. doi: 10.1002/eji.1830270517. [DOI] [PubMed] [Google Scholar]
- 13.Strong RK, Holmes MA, Li P, Braun L, Lee N, Geraghty DE. HLA-E allelic variants. Correlating differential expression, peptide affinities, crystal structures, and thermal stabilities. J Biol Chem. 2003;278:5082–90. doi: 10.1074/jbc.M208268200. [DOI] [PubMed] [Google Scholar]
- 14.Braud VM, Allan DS, O'Callaghan CA, et al. HLA-E binds to natural killer cell receptors CD94/NKG2A, B and C. Nature. 1998;391:795–9. doi: 10.1038/35869. [DOI] [PubMed] [Google Scholar]
- 15.Arlettaz L, Villard J, de Rham C, Degermann S, Chapuis B, Huard B, Roosnek E. Activating CD94:NKG2C and inhibitory CD94:NKG2A receptors are expressed by distinct subsets of committed CD8+ TCR αβ lymphocytes. Eur J Immunol. 2004;34:3456–64. doi: 10.1002/eji.200425210. [DOI] [PubMed] [Google Scholar]
- 16.Brostjan C, Bellon T, Sobanov Y, Lopez-Botet M, Hofer E. Differential expression of inhibitory and activating CD94/NKG2 receptors on NK cell clones. J Immunol Methods. 2002;264:109–19. doi: 10.1016/s0022-1759(02)00084-4. [DOI] [PubMed] [Google Scholar]
- 17.Lee N, Llano M, Carretero M, Ishitani A, Navarro F, Lopez-Botet M, Geraghty DE. HLA-E is a major ligand for the natural killer inhibitory receptor CD94/NKG2A. Proc Natl Acad Sci U S A. 1998;95:5199–204. doi: 10.1073/pnas.95.9.5199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Michaelsson J, Teixeira de Matos C, Achour A, Lanier LL, Karre K, Soderstrom K. A signal peptide derived from hsp60 binds HLA-E and interferes with CD94/NKG2A recognition. J Exp Med. 2002;196:1403–14. doi: 10.1084/jem.20020797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jiang W, Chai NR, Maric D, Bielekova B. Unexpected role for granzyme K in CD56bright NK cell-mediated immunoregulation of multiple sclerosis. J Immunol. 2011;187:781–90. doi: 10.4049/jimmunol.1100789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hao J, Liu R, Piao W, et al. Central nervous system (CNS)-resident natural killer cells suppress Th17 responses and CNS autoimmune pathology. J Exp Med. 2010;207:1907–21. doi: 10.1084/jem.20092749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lunemann JD, Munz C. Do natural killer cells accelerate or prevent autoimmunity in multiple sclerosis? Brain. 2008;131(Pt 7):1681–3. doi: 10.1093/brain/awn132. [DOI] [PubMed] [Google Scholar]
- 22.Correale J, Villa A. Isolation and characterization of CD8+ regulatory T cells in multiple sclerosis. J Neuroimmunol. 2008;195:121–34. doi: 10.1016/j.jneuroim.2007.12.004. [DOI] [PubMed] [Google Scholar]
- 23.Jiang H, Canfield SM, Gallagher MP, Jiang HH, Jiang Y, Zheng Z, Chess L. HLA-E-restricted regulatory CD8+ T cells are involved in development and control of human autoimmune type 1 diabetes. J Clin Invest. 2010;120:3641–50. doi: 10.1172/JCI43522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tennakoon DK, Mehta RS, Ortega SB, Bhoj V, Racke MK, Karandikar NJ. Therapeutic induction of regulatory, cytotoxic CD8+ T cells in multiple sclerosis. J Immunol. 2006;176:7119–29. doi: 10.4049/jimmunol.176.11.7119. [DOI] [PubMed] [Google Scholar]
- 25.Leavenworth JW, Schellack C, Kim HJ, Lu L, Spee P, Cantor H. Analysis of the cellular mechanism underlying inhibition of EAE after treatment with anti-NKG2A F(ab')2. Proc Natl Acad Sci U S A. 2010;107:2562–7. doi: 10.1073/pnas.0914732107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bell JE, Alafuzoff I, Al-Sarraj S, et al. Management of a twenty-first century brain bank: experience in the BrainNet Europe consortium. Acta Neuropathol. 2008;115:497–507. doi: 10.1007/s00401-008-0360-8. [DOI] [PubMed] [Google Scholar]
- 27.Reynolds R, Roncaroli F, Nicholas R, Radotra B, Gveric D, Howell O. The neuropathological basis of clinical progression in multiple sclerosis. Acta Neuropathol. 2011;122:155–70. doi: 10.1007/s00401-011-0840-0. [DOI] [PubMed] [Google Scholar]
- 28.Abramoff MD, Magalhaes PJ, Ram SJ. Image Processing with ImageJ. Biophotonics Int. 2004;11:36–42. [Google Scholar]
- 29.Durrenberger PF, Ettorre A, Kamel F, et al. Innate immunity in multiple sclerosis white matter lesions: expression of natural cytotoxicity triggering receptor 1 (NCR1) J Neuroinflammation. 2012;9:1. doi: 10.1186/1742-2094-9-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lorentzen AR, Karlsen TH, Olsson M, et al. Killer immunoglobulin-like receptor ligand HLA-Bw4 protects against multiple sclerosis. Ann Neurol. 2009;65:658–66. doi: 10.1002/ana.21695. [DOI] [PubMed] [Google Scholar]
- 31.Blais ME, Dong T, Rowland-Jones S. HLA-C as a mediator of natural killer and T-cell activation: spectator or key player? Immunology. 2011;133:1–7. doi: 10.1111/j.1365-2567.2011.03422.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fadda L, Borhis G, Ahmed P, et al. Peptide antagonism as a mechanism for NK cell activation. Proc Natl Acad Sci U S A. 2010;107:10160–5. doi: 10.1073/pnas.0913745107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schleinitz N, Vely F, Harle JR, Vivier E. Natural killer cells in human autoimmune diseases. Immunology. 2010;131:451–8. doi: 10.1111/j.1365-2567.2010.03360.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Coupel S, Moreau A, Hamidou M, Horejsi V, Soulillou JP, Charreau B. Expression and release of soluble HLA-E is an immunoregulatory feature of endothelial cell activation. Blood. 2007;109:2806–14. doi: 10.1182/blood-2006-06-030213. [DOI] [PubMed] [Google Scholar]
- 35.Kren L, Muckova K, Lzicarova E, et al. Production of immune-modulatory nonclassical molecules HLA-G and HLA-E by tumor infiltrating ameboid microglia/macrophages in glioblastomas: a role in innate immunity? J Neuroimmunol. 2010;220:131–5. doi: 10.1016/j.jneuroim.2010.01.014. [DOI] [PubMed] [Google Scholar]
- 36.Boyle EA, McGeer PL. Cellular immune response in multiple sclerosis plaques. Am J Pathol. 1990;137:575–84. [PMC free article] [PubMed] [Google Scholar]
- 37.Bowen S, Ateh DD, Deinhardt K, et al. The phagocytic capacity of neurones. Eur J Neurosci. 2007;25:2947–55. doi: 10.1111/j.1460-9568.2007.05554.x. [DOI] [PubMed] [Google Scholar]
- 38.Nguyen KB, McCombe PA, Pender MP. Macrophage apoptosis in the central nervous system in experimental autoimmune encephalomyelitis. J Autoimmun. 1994;7:145–52. doi: 10.1006/jaut.1994.1011. [DOI] [PubMed] [Google Scholar]
- 39.Saxena A, Martin-Blondel G, Mars LT, Liblau RS. Role of CD8 T cell subsets in the pathogenesis of multiple sclerosis. FEBS Lett. 2011;585:3758–63. doi: 10.1016/j.febslet.2011.08.047. [DOI] [PubMed] [Google Scholar]
- 40.Wiendl H, Feger U, Mittelbronn M, et al. Expression of the immune-tolerogenic major histocompatibility molecule HLA-G in multiple sclerosis: implications for CNS immunity. Brain. 2005;128(Pt 11):2689–704. doi: 10.1093/brain/awh609. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.