

Abstract
Mucosal administration of an antigen eliciting bystander suppression at the site of inflammation results in effective antigen-specific immunotherapy for autoimmune diseases. Heat shock proteins are bystander antigens that are effective in peptide-specific immunotherapy in both experimental and human autoimmune disease. The efficacy of preventive peptide immunotherapy is increased by enhancing peptide-specific immune responses with proinflammatory agents. Combining peptide-specific immunotherapy with general suppression of inflammation may improve its therapeutic effect.
Keywords: arthritis, heat shock protein, mucosal, peptide, therapy
Introduction
How to restore the immune balance in a deranged immune system that attacks self tissues in autoimmune diseases is a continuing focus of research. The current treatment of autoimmunity still depends mainly on conventional life-long general immune suppression. Although a step forward has been made in clinical efficacy by the introduction of biologics that block proinflammatory cytokines, inflammation revives as soon as therapy is discontinued. Moreover, the considerable immune suppression evoked by cytokine blockade has been associated with severe side effects such as serious opportunistic infections, and even malignancies such as lymphoma [1–6].
A more specific approach could overcome drawbacks of non-specific immune suppressive therapy. By specific targeting of autoaggressive T cells in autoimmunity, side effects can be reduced. Indeed, such antigen-specific immunotherapy has been shown to be effective in multiple animal models of autoimmunity without severe side effects (reviewed by [7]). Translation of these findings to human therapies showed promising results, but efficacy has been less than expected (Table 1). To improve the effect of antigen-specific therapy in clinical autoimmune diseases, three issues – choice of antigen, route of administration and peptide immunogenicity – need to be addressed.
Table 1.
Major clinical trials of peptide immunotherapy in autoimmune disease
| Trial name | Design | Type of therapy | Peptide | Route | Patient group | Immunomodulatory effects | Clinical efficacy | Future | Reference | |
|---|---|---|---|---|---|---|---|---|---|---|
| Diabetes | Phase I | Parenteral peptide | Pro-insulin peptide C19-A3 | i.d. | Long-standing DM type I | Increase of IL-10 producing peptide-specific T cells | None measured | Thrower, Clin Exp Immunol 2009 | ||
| Phase I | Parenteral peptide | Human insulin B chain peptide | i.m. | New-onset DM type I | Insulin-specific B and T cell responses | None measured | Orban, Autoimmunity 2010 | |||
| Phase I | Parenteral peptide | NBI-6024 (altered peptide of insulin B epitope B9-23) | s.c. | New-onset DM type I | Shift from Th1 to Th2 immunoregulatory phenotype | None measured | Alleva, Scand J Immunol 2006 | |||
| DIAMYD | Phase II | Parenteral peptide with adjuvant | GAD65-Alum | s.c. | New-onset DM type I | Increased percentage of total Treg and GAD65-specific Treg cells, increased secretion IL-5, IL-10, IL-13 | Preservation of residual insulin secretion | Ongoing phase II NCT00529399 | Ludvigsson, N Engl J Med 2008; Hjorth, Clin Immunol 2011 | |
| MS | PreCISe | Phase I/II | Parenteral peptide | Copaxone (Glatiramer-acetaat, peptide mixture) | s.c. | Early MS | None measured | Risk reduction for developing clinically definite MS, delayed onset, decreased relapse rate | Comi, Lancet 2009; Cohen, Neurology 2007 | |
| Phase II | Parenteral peptide | Altered peptide of MBP83-99 | s.c. | MS | Increased disease activity | Bielekova & Kappos, Nat Med 2000 | ||||
| SLE | Phase II | Parenteral peptide | Spliceosomal peptide p140 | s.c. | SLE | Anti-ds DNA antibody levels reduced | Muller, Arthritis Rheum 2008 | |||
| Uveitis | Open therapeutic trial, no control group | Oral peptide | B27PD (self-HLA peptide) | p.o. | Long-lasting therapy refractive uveitis | None measured | Discontinuation of steroids possible because of reduced intraocular inflammation | Ongoing phase I/II NCT01195948 | Thureau, Immunol Lett 1999, ANYAS 2004 |
MBP: myelin basic protein; i.d.: intradermal; i.m.: intramuscular; s.c.: subcutaneous; p.o.: per os; MS: multiple sclerosis; SLE: systemic lupus erythematosus; DM: diabetes mellitus; HLA: human leucocyte antigen; IL: interleukin.
The choice of antigen in animal models is facilitated by the fact that the disease-inducing antigen is known. The identification of such an antigen in human autoimmunity is more challenging, as the disease-inducing antigen in many autoimmune diseases remains unknown. In addition, it is doubtful whether this single disease-inducing antigen really exists. Therefore new targets for antigen-specific therapy are needed.
The second issue concerns the route of antigen administration. In the majority of clinical trials the antigen is administered by injection, while a more effective option to restore immune tolerance would be the administration of antigen in a tolerogenic environment, such as the gut or nasal mucosa [7,8]. This tolerogenic presentation of the antigen converts the antigen-specific proinflammatory immune response to an antigen-specific regulatory response (reviewed by [9]).
When peptides are administered via the mucosal route the third issue may be a limited immunogenicity, indicating a need for enhancement of peptide recognition [10,11].
When these issues can be addressed, mucosal antigen-specific immunotherapy can be an interesting alternative to generalized immune-suppressive therapy with subsequent unwanted side effects. Results of clinical trials of peptide immunotherapy are promising, but efficiency needs to be enhanced. In this review, we consider ways to improve future antigen-specific immunotherapy with a special focus on heat shock proteins (HSPs).
Heat shock proteins: candidates for immunotherapy
Bystander antigens
At time of diagnosis, human autoimmune diseases are already characterized by a secondary non-specific inflammatory process in which multiple antigens are targets of the immune system (a process known as epitope spreading). The antigens that are specifically up-regulated at the site of inflammation and are immunologically dominant (bystander antigens) are candidates for antigen-specific immunotherapy. Induced tolerogenic immune responses to such bystander antigens could lead to a local down-regulation of the ongoing immune response (bystander suppression).
Heat shock proteins as bystander antigens
HSPs, which are highly conserved intracellular molecular chaperones that are important for cell survival under stressful conditions, fulfil both the above-mentioned criteria for bystander antigens (reviewed in [12]).
First, HSPs are up-regulated upon cell stress and are therefore present exclusively at sites of inflammation. HSPs are indeed abundantly present in muscle cells of juvenile dermatomyositis (JDM) patients [13], in synovial fluid and synovial tissue of juvenile idiopathic arthritis (JIA) [14] and RA patients [15] and in inflamed bowel of Crohn's disease patients [16]. There is also supporting evidence that HSPs are secreted from stressed cells; for example, in blood, free HSP60 is found during various inflammatory conditions [17].
Secondly, in autoimmune disease, HSP-specific responses are immunologically dominant [12,18,19]. For example, HSP60 peptide-specific T cell clones play a significant role in the perpetuation of Crohn's disease and tissue-specific T cell clones from diabetic children recognize human HSP60 as an autoantigen [16,20,21]. Humoral responses to HSPs have also been observed in autoimmune diseases, as antibody responses to multiple HSP families were detected in sera from RA and JIA patients [22,23].
Altogether, HSPs seem to be suitable candidates for the induction of bystander suppression by antigen-specific immunotherapy.
Immunoregulatory properties of HSPs
HSPs are known for their strong evolutionary conservation, resulting in a high level of homology between bacterial and mammalian HSPs [24]. Theoretically, this high homology in combination with their up-regulation during stress and immunodominancy could be dangerous, putting the host at risk for autoimmunity through antigenic mimicry [25]. However, T and B cell responses to self-HSP are present in healthy individuals (and even in cord blood) without widespread inflammation or autoimmunity [26–28]. This could be due to the presence of regulatory immune responses. HSP-specific immune responses have been suggested to have a driving force in the generation of regulatory action via innate and adaptive pathways [12,26,29].
Innate effects of HSP
The innate immune system was thought originally to recognize only pathogen-associated molecular patterns (PAMPs) via their pathogen recognition receptors (PRR), also known as the ‘infectious non-self model’[30]. Matzinger proposed rather that the innate immune system responds to endogenous danger signals (danger-associated molecular patterns, DAMPs), released by damaged or stressed cells with the tissue playing an important role in determining the quality of the immune response [31,32]. As HSPs are up-regulated and excreted during stress, these proteins have long been implicated in triggering innate immune responses. There has been a debate as to whether these innate effects of HSP (reviewed in [17]) could have been the result of contamination by other Toll-like receptor (TLR) agonists (reviewed in [33]). Although the use of contaminated HSP has been a problem in several studies, considerable evidence now exists to support the innate effects of HSP. Properly controlled research revealed that self-HSP60 (and not microbial HSP60) has a direct, lipopolysaccharide (LPS)-independent innate effect on T cells mediated through TLR-2 and on monocytes and macrophages through TLR-4 [34].
Adaptive effects of HSP
The induction of regulation by HSPs has also been described via adaptive immune responses. Presumably as a result of stimulation by homologous HSPs from commensal bacteria in the gut, self-HSP-specific T and B cell responses are present in healthy individuals [18]. To contain these autoreactive T cells that escaped central tolerance safely, peripheral tolerance mechanisms are needed. It has been shown that self-HSP reactive T cells evoke regulatory immune responses. Data from animal models indicate that cross-reactive immunoregulatory T cell responses to self-HSP play a role in disease protection [29,35–37]. In line with these findings, the presence of self-HSP60-specific T cell responses in JIA patients correlates with a benign disease course [38–40]. Self-HSP-specific T cell responses have also been reported to be immunoregulatory in various other autoimmune diseases, such as RA [41] and JDM [13], by the production of anti-inflammatory cytokines such as interleukin (IL)-10, IL-4 and transforming growth factor (TGF)-β[38,42]. A recent study revealed that self-HSP60 could directly induce highly suppressive forkhead box protein 3 (FoxP3+) Treg in vitro [43]. Finally, low concentrations of human HSP60 or p277 (a synthetic human HSP60-derived peptide) have been shown to be able to enhance the regulatory function of CD25+ Treg from human peripheral blood mononuclear cells (PBMC) [19,44].
In conclusion, HSPs are bystander antigens that can elicit regulatory responses in human autoimmune disease, and are therefore interesting targets for antigen-specific immunotherapy.
Peptide immunotherapy
The development of antigen-specific immunotherapy with proteins has been hampered by side effects such as mast cell activation or cytotoxic T cell responses [45–47]. Peptide immunotherapy can be an attractive alternative, as it increases specificity and thereby reduces side effects. Moreover, synthetic peptides are free of microbial products.
Peptide selection
In human disease, selection of appropriate peptides for immunotherapy is a major challenge. The selection process is helped by focusing on desirable characteristics of the peptide.
First, the peptide should be recognized by the human immune system and thus be able to bind disease-associated human leucocyte antigen (HLA) molecules. For this purpose, multiple prediction models of peptide binding to HLA have been shown to be helpful [48–51]. Secondly, the peptide should mimic the naturally processed epitope, as altered peptides may behave unpredictably [52,53]. To fulfil this criterion, selection can be based on elution studies of HLA-peptide complexes. Thirdly, as self-cross-reactive responses have been shown to be important for the disease-protective effect of peptides [29], a peptide needs to have high homology to self and still be immunogenic.
HSP-peptide immunotherapy
HSP-peptides prevent autoimmune disease in multiple experimental animal models (Table 2). However, most peptides used in these models were not selected primarily on their binding capacity of disease-associated human HLA molecules, a feature desired for translation of the experimental results into humans.
Table 2.
Protective heat shock protein (HSP) peptide treatment in experimental models of autoimmunity
| Model | Route | Adjuvant | Regimen | Peptides | HSP | References | |
|---|---|---|---|---|---|---|---|
| Arthritis | DIA | i.d. | IFA | p | Mixture of 120–134 and 213–277 | Self | Moudgil, J Immunol 2005 |
| PIA | i.p. | None | p and t | 261–271 | Non-self | Thompson, J Immunol 1998; Francis, Immunology 2000 | |
| AA and CPIA | i.d. | DDA | p | 256–270 | Non-self | Anderton, J Exp Med 1995 | |
| AA and AIA | i.n. | None | p | 176–190 | Non-self | Prakken, Proc Natl Acad Sci 1997 | |
| AA | i.d. | IFA | p | 180–188 | Non-self | Golden, Agents Actions 1991 | |
| AA | i.n. | None | t | 180–188 | Non-self | Roord, PLoS ONE 2006 | |
| AA | i.d. | IFA | p | 234–252 | Non-self | Tanaka, J Immunol 1999 | |
| AA | i.n. | None | p | 111–125 | Non-self | Wendling, J Immunol 2000 | |
| AA | i.p. | None | p | 61–80 (mHSP65), 31–46, 37–52 (self)HSP60 | Both | Ulmansky, J Immunol 2002 | |
| AA | i.n. | None | p | 254–268 | Non-self | Zonneveld-Huijssoon, Ann Rheum Dis 2011 | |
| AA | s.c. | DDA | p | Mixture of 417–431, 441–455, 465–479, 513–527, 521–535 (BCTD) | Non-self | Moudgil, J Exp Med 1997; Durai, J Immunol 2004 | |
| AA | s.c. | IFA | p | 177–191 | Non-self | Durai, J Rheumatol 2007 | |
| DM | NOD | s.c. | IFA | t | 437–460 (p277) | Self | Cohen, Lancet 1994 |
| NOD | i.p. | IFA | p and t | 437–460 (p277) | Self | Elias, Diabetes 1995, 1997; Ablamunits, J Autoimmun 1998; Tian, J Immunol 1998; Elia, Proc Natl Acad Sci 1991 | |
| STZ | i.p. | Mineral oil | t | 437–460 (p277) | Self | Elias, Diabetes 1996 | |
| NOD | s.c. | IFA | p | 166–185 (p12) | Self | Elias, J Autoimmun 1997 | |
| BB-DP | p.o. | None | p | Peptide analogue of p277 (Diapep277) | Self | Brugman, Diabetologia 2004 | |
| Sjögren | SS | s.c. | IFA | p | 437–460 | Self | Delaleu, Arthritis Rheum 2008 |
Adapted from [12] and [22]. AA: adjuvant arthritis; DIA: dimethyl dioctadecyl ammoniumbromide-induced arthritis; PIA: pristine-induced arthritis; AIA: avridine-induced arthritis; CPIA: CP20961-induced arthritis; STZ: STZ toxin-induced diabetes; BB-DP: BioBreeding-Diabetes Prone rat; SS: spontaneous Sjögren syndrome; i.d.: intradermal; i.p.: intraperitoneal; i.n.: intranasal; s.c.: subcutaneous; p.o.: per os, oral; IFA: incomplete Freund's adjuvant; DDA: dimethyl dioctadecyl ammoniumbromide; p: preventive regimen; t: therapeutic regimen.
In two recent studies, HSP60-derived HLA-binding peptides were tested in an experimental arthritis model [54,55]. In one of the two studies, the identified human HSP60 epitope was modified artificially to increase the HLA binding affinity and to skew towards a regulatory T cell response [54]. Intradermal administration of this altered peptide suppressed experimental arthritis (AA) in vivo by the induction of regulatory T cells (Treg) and increased Treg frequency in ex-vivo assays with PBMC from RA patients in contrast to the native peptide [54]. However, as mentioned previously, native peptides that mimic the naturally processed epitope are preferred for safe antigen-specific immunotherapy, and intradermal administration is not the optimal route for tolerance induction.
The other study performed by our group used a native HLA-binding T cell epitope of mycobacterial HSP60 that evoked a tolerogenic immune response in PBMCs of arthritis patients [41,42]. Nasal administration of this peptide was effective in experimental arthritis and induced a CD4+ T cell population with reduced tumour necrosis factor (TNF)-α production at the site of inflammation. This induced T cell population also expressed FoxP3 and had potent suppressive capacity which, upon transfer, protected against arthritis [55].
These specific experimental results have not yet been translated into clinical trials. So far, clinical trials have been performed with two other interesting HSP-epitopes (Table 3).
Table 3.
Clinical trials of peptide immunotherapy with heat shock protein (HSP) peptides in autoimmune diseases
| Trial name | Design | Type of therapy | Peptide | Route | Patient group | Immunomodulatory effects | Clinical efficacy† | Reference | |
|---|---|---|---|---|---|---|---|---|---|
| Diabetes | DiaPep277 | Phase II | Parenteral peptide | p277 (HSP60 437–460) | s.c. | New-onset T1D | Increased IL-10 production by T cells is associated with preservation of C-peptide up to 12–18 months | Lower need for exogenous insulin | Huurma, Clin Exp Immunol 2008 |
| RA | Phase II | Oral peptide | DnaJP1 peptide (HSP40) | Oral | RA | Immune diviation from TNF-α to IL-10, peptide-induced FoxP3 expression on CD25bright cells | ACR20 and ACR50 score reduced | Koffeman, Arthritis Rheum 2009 |
ACR 20, ACR50, measurements of improvement in rheumatoid arthritis. T1D: type 1 diabetes; RA: rheumatoid arthritis; IL: interleukin; TNF: tumour necrosis factor.
DnaJP1
The first clinical trial with an HSP-peptide in human arthritis was performed with dnaJP1. DnaJP1 is a peptide derived from Escherichia coli HSP40, containing a sequence of five amino acids found in the majority of HLA-DR alleles linked with RA (‘shared epitope’). In a Phase I trial, patients with early active RA received oral dnaJP1 during 6 months. After treatment, in-vitro responses to dnaJP1 changed from proinflammatory to anti-inflammatory, with increased IL-10 production and augmented FoxP3 expression in Treg cells [56].
In a following Phase II trial, patients with active RA with proven immunological reactivity to dnaJP1 received the same mucosal dnaJ treatment. Clinical improvement was achieved at multiple time-points and was accompanied again by anti-inflammatory in-vitro responses to dnaJP1 with reduced production of TNF-α and a trend towards increased production of IL-10 [57].
Diapep277
A vaccination strategy based on HSP60 as a diabetes autoantigen was performed with p277 (DiaPep277), a 24-amino-acid peptide of mouse HSP60 (437–460) that has been shown to have preventive and therapeutic effects in experimental diabetes [58,59].
Multiple Phase II trials have been performed with subcutaneous p277 [60–63]. In adults newly diagnosed with type 1 diabetes (T1D), residual C-peptide levels (reflecting the amount of insulin production) could be preserved [63,64]. A recent immunological study revealed that the preservation of C-peptide was associated with peptide specific tolerance [61]. Phase II trials are currently ongoing [65].
Good candidates for immunotherapy have now been proved to be effective in multiple animal models of autoimmune diseases, and translation into humans has provided encouraging results; the next step will be to improve therapeutic efficiency in human autoimmune disease. Enhancement of peptide immunogenicity when delivered via the mucosal route and combination of peptide therapy with immune modulating agents could be interesting options.
Improving antigen-specific therapy
Enhancing peptide immunogenicity
As regulation of effector T cells is an active process, immune activation is needed for optimal control [32]. A peptide signal delivered via the tolerogenic mucosal route may be too small to induce the desired immune deviation due to the intrinsic weakness of the peptide signal alone, triggering only the adaptive immune system [10,11]. In a healthy immune system, activation of the innate immune system leads to better presentation of the peptide and thereby enhances peptide-triggered adaptive immune responses. The combination of enhancing both adaptive and innate immunity may therefore be an attractive option for the enhancement of mucosal immunotherapy in autoimmune disease.
Adjuvant
Combination of an adjuvant triggering innate immunity with a T cell-epitope triggering adaptive immunity could also enhance peptide immunogenicity. Although adjuvants have been used in non-mucosal vaccination strategies in autoimmunity, the concept of enhancing mucosal vaccination with an adjuvant for preventive peptide therapy in autoimmune disease is somewhat new (Fig. 1). Prerequisites for such an innate triggering mucosal adjuvant would be applicability at mucosal sites, activation of antigen-presenting cells (APCs) and skewing towards Treg cell responses. Co-administration of such an adjuvant would result in more efficient antigen presentation by mucosal APCs and enlarge the beneficial effect of mucosal peptide treatment.
Fig. 1.
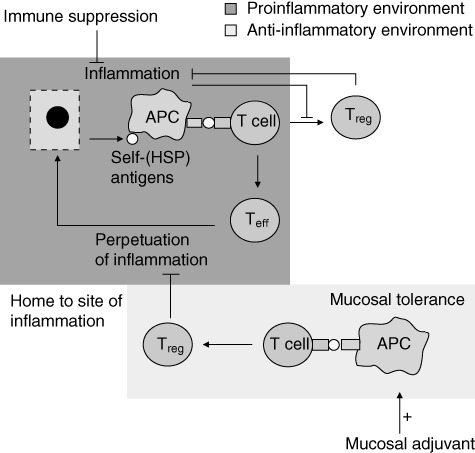
The dual role of heat shock proteins (HSPs), inducing proinflammatory effector T cells (Teff) or tolerogenic/regulatory T cells (Treg), is influenced by the inflammation status of the tissues. At the site of inflammation, self-HSP antigens are released from damaged cells. These antigens are presented by activated antigen-presenting cells (APC) to T cells. In a proinflammatory environment this results in predominantly Teff cells, contributing to perpetuation of inflammation. T cells induced via the mucosal route are directed by the anti-inflammatory environment towards a predominantly tolerogenic response (Treg) (mucosal tolerance induction). The mucosally induced antigen-specific T cells consist of multiple kinds of regulatory cells and which are thought to migrate to the site of inflammation as their cognate antigen (e.g. HSP) is expressed there. At the site of inflammation, these antigen-specific Tregs skew the proinflammatory T cell response towards an anti-inflammatory phenotype by cytokine release-like interleukin (IL)-10 or cell–cell contact. Mucosal adjuvant can enhance peptide presentation by APCs at the site of tolerance induction, enlarging the pool of Treg formed after mucosal tolerance induction. The inflammatory environment hampers the development of (self-HSP-specific) T cells. Generalized immune suppressive therapy reduces inflammation, creating a more favourable environment for the development of Treg. Combination therapy of antigen-specific mucosal tolerance induction with immune suppressive therapy could therefore enhance efficacy of peptide specific immunotherapy.
In clinical diabetes, alum as an adjuvant for subcutaneous antigen-specific immunotherapy led to preservation of residual insulin secretion in adults and children with early-onset T1D [66–69]. The increased effectiveness of alum-adjuvanted peptide immunotherapy has been shown to depend on the activation of innate immunity by DNA released from dying cells [70]. However, alum as an adjuvant could not restore euglycaemia in T1D patients and was not tested mucosally, indicating a need to explore other adjuvants.
An adjuvant with promising results in non-obese diabetic (NOD) mice is the non-toxic B subunit of the cholera enterotoxin (CTB). Oral administration of islet autoantigens linked to CTB improved significantly suppression of hyperglycaemia and pancreatic inflammation [71,72]. CTB has been shown more recently to induce enhanced antigen capture by dendritic cells and migration of the dendritic cells towards the site of antigen administration (the Peyer's patches) [73,74]. Another agent that can be considered an innate-activating adjuvant for mucosal peptide therapy is HSP itself. HSP can up-regulate adaptive immune responses by stimulating innate receptors such as TLR-2 and TLR-4 [34]. Indeed, HSP60 enhanced immunogenicity of CMV peptide vaccines [75] and increased the efficacy of p277 therapy in diabetic mice [76]. HSP60 seems to function as the body's natural adjuvant as a result of its ability to activate both the innate and adaptive responses [19].
Adjuvants of particular interest that have been administered mucosally are cytosine–guanine–oligodeoxynucleotides (CpG-ODN), which consist of a nucleotide sequence common in bacterial DNA. CpG-ODN stimulate TLR-9 on antigen-presenting cells and have been used successfully as nasal vaccine adjuvant in anthrax (AVA) vaccination in mice [77–79]. Data from our group indicate that CpG-ODN enhance antigen-specific immunotherapy in an experimental arthritis model (ARD, in press).
In conclusion, enhancing immunogenicity of a peptide in a preventive regimen seems very efficient in improving peptide-specific immunotherapy. However, caution should be taken in the addition of proinflammatory agents to a peptide in a therapeutic setting as it could, in theory, lead to over-activation of an already deranged immune system.
Combination therapy with general immune modulators
Although boosting the antigen-specific immune response seems effective in a preventive regimen, the ongoing widespread inflammation present in established autoimmune disease probably hampers antigen-specific immune modulation in a therapeutic setting. Short-term non-specific dampening of inflammation before administration of antigen could create an environment in which the antigen-specific response can be detected and modulated, thereby improving therapeutic efficacy of antigen-specific immunotherapy (Fig. 1).
In addition to the reduction of inflammatory background ‘noise’, dampening inflammation is crucial for adequate functioning of Treg (reviewed by [80]). For example, a chronic inflammatory environment causes local dysfunction of Treg or converts them into proinflammatory T helper type 17 (Th17) cells [81–86]. In line with these observations, generalized immune suppression by TNF-α blockade [87,88] or immune modulation by anti-CD3 [89–91] favours the development of Treg cells. However, it is conceivable that due to non-antigen-specific immune therapies only a small number of induced Treg will be specific for antigens expressed in the target autoimmune organ. Combining generalized immune suppression with antigen-specific peptide therapy could therefore expand antigen-specific Treg that are able to migrate to the tissue where their cognate antigen is expressed: the site of inflammation.
Some successful combination therapy strategies in autoimmune diseases have been reported in the literature. For example, combination therapy of anti-CD3 with disease-related peptides has been shown to be effective in experimental models of new-onset diabetes. The combined approach was more efficient than peptide or anti-CD3 alone and induced antigen-specific Treg that could transfer protection [92,93]. Combined anti-CD3 therapy with disease-related peptides has not been tested so far in human T1D, but perhaps such a combined approach could improve recent results of anti-CD3 monotherapy in human T1D [91].
Another proven effective strategy in experimental models is the combination of antigen-specific immunotherapy with TNF-α blockade. In the rat adjuvant arthritis model, low-dose anti-TNF-α (Etanercept) combined with nasally administered HSP60 peptide induced clinical control in a therapeutic setting to a larger extent than peptide treatment alone. The clinical response was accompanied by an increase in peptide-specific FoxP3-expressing T cells to a degree comparable to full-dose Etanercept. Finally, the combination treatment induced more peptide-specific IL-10 production than did Etanercept alone [94].
An interesting finding regarding combination therapy of a peptide with immune modulation in humans has been reported in the earlier-described dnaJP1 clinical trial in RA. Post-hoc analysis revealed that the best clinical results were obtained in a subgroup of patients taking hydroxychloroquine (HCQ), an immune modulating agent [95]. In addition to the earlier-mentioned combination strategies in animal models, this finding indicates the potential therapeutic efficacy of combination treatment in humans as well [57].
In summary, boosting the peptide-specific immune response, on one hand, and short-term dampening of the ongoing systemic inflammation, on the other hand, could improve the therapeutic efficiency of antigen-specific immunotherapy. Combination therapy shows promising results in experimental autoimmunity, but evidence is limited as yet for human autoimmune disease. Enlarging the efficacy of antigen-specific therapy is worth exploring, while the possibility of lowering the dose of immune-suppressive medication reduces side effects associated with life-long drug administration.
Conclusion
In this review, we have discussed strategies to improve the clinical outcome of antigen-specific immunotherapy in human autoimmune disease. Three major issues concerning the choice of antigen, route of administration and peptide immunogenicity were dealt with.
Some issues remain to further optimize antigen-specific immunotherapy for human autoimmune disease. In this regard, dosing is important for mucosal tolerance induction to be effective [8] and dose-finding studies are needed to improve therapeutic results further.
Furthermore, the selection of patients in clinical trials of peptide-specific immunotherapy is crucial. Some prerequisites for treatment response have been identified in experimental and human studies. First, genetic factors play a role as the availability of beneficial antigen-specific T cells varies between different genetic backgrounds in mice models of diabetes and correlates with treatment outcome [93]. Secondly, a high representation of tolerogenic and anergic immune pathways at baseline is associated with clinical responsiveness to peptide immunotherapy [57]. Thirdly, the presence and quality of peptide-specific responses before start of treatment play a role in the eventual efficacy of peptide immunotherapy [57,61]. Selecting patients on these criteria could be of help in further optimization of antigen-specific immunotherapy.
In conclusion, peptide immunotherapy with bystander antigens such as HSPs shows promising results in experimental models, and the first positive results from clinical trials are currently emerging. New approaches aiming for enhanced peptide recognition in a controlled immune environment by the use of adjuvant and/or combining peptide treatment with short-term immune suppressive medication may hold promise for a successful future for peptide-specific immunotherapy in autoimmune diseases.
Disclosure
The authors have no conflicts of interest to declare.
References
- 1.Dekker L, Armbrust W, Rademaker CM, Prakken B, Kuis W, Wulffraat NM. Safety of anti-TNFalpha therapy in children with juvenile idiopathic arthritis. Clin Exp Rheumatol. 2004;22:252–8. [PubMed] [Google Scholar]
- 2.Maini RN, Taylor PC. Anti-cytokine therapy for rheumatoid arthritis. Annu Rev Med. 2000;51:207–29. doi: 10.1146/annurev.med.51.1.207. [DOI] [PubMed] [Google Scholar]
- 3.Keane J, Gershon S, Wise RP, et al. Tuberculosis associated with infliximab, a tumor necrosis factor alpha-neutralizing agent. N Engl J Med. 2001;345:1098–104. doi: 10.1056/NEJMoa011110. [DOI] [PubMed] [Google Scholar]
- 4.Bloom BJ. Development of diabetes mellitus during etanercept therapy in a child with systemic-onset juvenile rheumatoid arthritis. Arthritis Rheum. 2000;43:2606–8. doi: 10.1002/1529-0131(200011)43:11<2606::AID-ANR31>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 5.Lepore L, Marchetti F, Facchini S, Leone V, Ventura A. Drug-induced systemic lupus erythematosus associated with etanercept therapy in a child with juvenile idiopathic arthritis. Clin Exp Rheumatol. 2003;21:276–7. [PubMed] [Google Scholar]
- 6.Food and Drug Administration Drug Safety Communication. Safety review update on reports of hepatosplenic T-cell lymphoma in adolescents and young adults receiving tumor necrosis factor (TNF) blockers, azathioprine and/or mercaptopurine. Silver Spring, MD: US Food and Drug Administration; 2011. [Google Scholar]
- 7.Sabatos-Peyton CA, Verhagen J, Wraith DC. Antigen-specific immunotherapy of autoimmune and allergic diseases. Curr Opin Immunol. 2010;22:609–15. doi: 10.1016/j.coi.2010.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Faria AM, Weiner HL. Oral tolerance. Immunol Rev. 2005;206:232–59. doi: 10.1111/j.0105-2896.2005.00280.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wraith DC. Therapeutic peptide vaccines for treatment of autoimmune diseases. Immunol Lett. 2009;122:134–6. doi: 10.1016/j.imlet.2008.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Decrausaz L, Domingos-Pereira S, Duc M, et al. Parenteral is more efficient than mucosal immunization to induce regression of human papillomavirus-associated genital tumors. Int J Cancer. 2011;129:762–72. doi: 10.1002/ijc.25973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bouvet JP, Decroix N, Pamonsinlapatham P. Stimulation of local antibody production: parenteral or mucosal vaccination? Trends Immunol. 2002;23:209–13. doi: 10.1016/s1471-4906(02)02186-5. [DOI] [PubMed] [Google Scholar]
- 12.van Eden W, van der ZR, Prakken B. Heat-shock proteins induce T-cell regulation of chronic inflammation. Nat Rev Immunol. 2005;5:318–30. doi: 10.1038/nri1593. [DOI] [PubMed] [Google Scholar]
- 13.Elst EF, Klein M, de JW, et al. Hsp60 in inflamed muscle tissue is the target of regulatory autoreactive T cells in patients with juvenile dermatomyositis. Arthritis Rheum. 2008;58:547–55. doi: 10.1002/art.23202. [DOI] [PubMed] [Google Scholar]
- 14.Boog CJ, de Graeff-Meeder ER, Lucassen MA, et al. Two monoclonal antibodies generated against human hsp60 show reactivity with synovial membranes of patients with juvenile chronic arthritis. J Exp Med. 1992;175:1805–10. doi: 10.1084/jem.175.6.1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huang QQ, Sobkoviak R, Jockheck-Clark AR, et al. Heat shock protein 96 is elevated in rheumatoid arthritis and activates macrophages primarily via TLR2 signaling. J Immunol. 2009;182:4965–73. doi: 10.4049/jimmunol.0801563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Puga Yung GL, Fidler M, Albani E, et al. Heat shock protein-derived T-cell epitopes contribute to autoimmune inflammation in pediatric Crohn's disease. PLoS ONE. 2009;4:e7714. doi: 10.1371/journal.pone.0007714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Henderson B, Pockley AG. Molecular chaperones and protein-folding catalysts as intercellular signaling regulators in immunity and inflammation. J Leukoc Biol. 2010;88:445–62. doi: 10.1189/jlb.1209779. [DOI] [PubMed] [Google Scholar]
- 18.Cohen IR. Autoimmunity to chaperonins in the pathogenesis of arthritis and diabetes. Annu Rev Immunol. 1991;9:567–89. doi: 10.1146/annurev.iy.09.040191.003031. [DOI] [PubMed] [Google Scholar]
- 19.Quintana FJ, Cohen IR. The HSP60 immune system network. Trends Immunol. 2011;32:89–95. doi: 10.1016/j.it.2010.11.001. [DOI] [PubMed] [Google Scholar]
- 20.Sobel DO, Creswell K. Characterization of anti-islet cytotoxic human T-cell clones from patients with type 1 diabetes mellitus. Autoimmunity. 2006;39:323–32. doi: 10.1080/08916930600720753. [DOI] [PubMed] [Google Scholar]
- 21.Jiang H, Canfield SM, Gallagher MP, et al. HLA-E-restricted regulatory CD8(+) T cells are involved in development and control of human autoimmune type 1 diabetes. J Clin Invest. 2010;120:3641–50. doi: 10.1172/JCI43522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang MN, Yu H, Moudgil KD. The involvement of heat-shock proteins in the pathogenesis of autoimmune arthritis: a critical appraisal. Semin Arthritis Rheum. 2010;40:164–75. doi: 10.1016/j.semarthrit.2009.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wu CT, Ou LS, Yeh KW, Lee WI, Huang JL. Serum heat shock protein 60 can predict remission of flare-up in juvenile idiopathic arthritis. Clin Rheumatol. 2011;30:959–65. doi: 10.1007/s10067-011-1709-2. [DOI] [PubMed] [Google Scholar]
- 24.Karlin S, Brocchieri L. Heat shock protein 60 sequence comparisons: duplications, lateral transfer, and mitochondrial evolution. Proc Natl Acad Sci USA. 2000;97:11348–53. doi: 10.1073/pnas.97.21.11348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.van WF, Prakken B. Heat shock proteins: darwinistic immune modulation on dangerous grounds. J Leukoc Biol. 2010;88:431–4. doi: 10.1189/jlb.0410236. [DOI] [PubMed] [Google Scholar]
- 26.Cohen IR. Peptide therapy for Type I diabetes: the immunological homunculus and the rationale for vaccination. Diabetologia. 2002;45:1468–74. doi: 10.1007/s00125-002-0937-z. [DOI] [PubMed] [Google Scholar]
- 27.Fischer HP, Sharrock CE, Panayi GS. High frequency of cord blood lymphocytes against mycobacterial 65-kDa heat-shock protein. Eur J Immunol. 1992;22:1667–9. doi: 10.1002/eji.1830220651. [DOI] [PubMed] [Google Scholar]
- 28.Aalberse JA, Kapitein B, de RS, et al. Cord blood CD4+ T cells respond to self heat shock protein 60 (HSP60) PLoS ONE. 2011;6:e24119. doi: 10.1371/journal.pone.0024119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Anderton SM, van der ZR, Prakken B, Noordzij A, van Eden W. Activation of T cells recognizing self 60-kD heat shock protein can protect against experimental arthritis. J Exp Med. 1995;181:943–52. doi: 10.1084/jem.181.3.943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Janeway CA, Jr, Dianzani U, Portoles P, et al. Cross-linking and conformational change in T-cell receptors: role in activation and in repertoire selection. Cold Spring Harb Symp Quant Biol. 1989;54:657–66. doi: 10.1101/sqb.1989.054.01.077. [DOI] [PubMed] [Google Scholar]
- 31.Matzinger P. The danger model: a renewed sense of self. Science. 2002;296:301–5. doi: 10.1126/science.1071059. [DOI] [PubMed] [Google Scholar]
- 32.Matzinger P, Kamala T. Tissue-based class control: the other side of tolerance. Nat Rev Immunol. 2011;11:221–30. doi: 10.1038/nri2940. [DOI] [PubMed] [Google Scholar]
- 33.Tsan MF. Heat shock proteins and high mobility group box 1 protein lack cytokine function. J Leukoc Biol. 2011;89:847–53. doi: 10.1189/jlb.0810471. [DOI] [PubMed] [Google Scholar]
- 34.Zanin-Zhorov A, Nussbaum G, Franitza S, Cohen IR, Lider O. T cells respond to heat shock protein 60 via TLR2: activation of adhesion and inhibition of chemokine receptors. FASEB J. 2003;17:1567–9. doi: 10.1096/fj.02-1139fje. [DOI] [PubMed] [Google Scholar]
- 35.Durai M, Kim HR, Moudgil KD. The regulatory C-terminal determinants within mycobacterial heat shock protein 65 are cryptic and cross-reactive with the dominant self homologs: implications for the pathogenesis of autoimmune arthritis. J Immunol. 2004;173:181–8. doi: 10.4049/jimmunol.173.1.181. [DOI] [PubMed] [Google Scholar]
- 36.Durai M, Kim HR, Bala KK, Moudgil KD. T cells against the pathogenic and protective epitopes of heat-shock protein 65 are crossreactive and display functional similarity: novel aspect of regulation of autoimmune arthritis. J Rheumatol. 2007;34:2134–43. [PubMed] [Google Scholar]
- 37.Wendling U, Paul L, van der ZR, Prakken B, Singh M, van Eden W. A conserved mycobacterial heat shock protein (hsp) 70 sequence prevents adjuvant arthritis upon nasal administration and induces IL-10-producing T cells that cross-react with the mammalian self-hsp70 homologue. J Immunol. 2000;164:2711–7. doi: 10.4049/jimmunol.164.5.2711. [DOI] [PubMed] [Google Scholar]
- 38.Prakken AB, van Eden W, Rijkers GT, et al. Autoreactivity to human heat-shock protein 60 predicts disease remission in oligoarticular juvenile rheumatoid arthritis. Arthritis Rheum. 1996;39:1826–32. doi: 10.1002/art.1780391108. [DOI] [PubMed] [Google Scholar]
- 39.Albani S, Ravelli A, Massa M, et al. Immune responses to the Escherichia coli dnaJ heat shock protein in juvenile rheumatoid arthritis and their correlation with disease activity. J Pediatr. 1994;124:561–5. doi: 10.1016/s0022-3476(05)83134-8. [DOI] [PubMed] [Google Scholar]
- 40.de Graeff-Meeder ER, van der ZR, Rijkers GT, et al. Recognition of human 60 kD heat shock protein by mononuclear cells from patients with juvenile chronic arthritis. Lancet. 1991;337:1368–72. doi: 10.1016/0140-6736(91)93057-g. [DOI] [PubMed] [Google Scholar]
- 41.de JH, Lafeber FF, de JW, et al. PAN-DR-binding Hsp60 self epitopes induce an interleukin-10-mediated immune response in rheumatoid arthritis. Arthritis Rheum. 2009;60:1966–76. doi: 10.1002/art.24656. [DOI] [PubMed] [Google Scholar]
- 42.Kamphuis S, Kuis W, de Jager W, et al. Tolerogenic immune responses to novel T-cell epitopes from heat-shock protein 60 in juvenile idiopathic arthritis. Lancet. 2005;366:50–6. doi: 10.1016/S0140-6736(05)66827-4. [DOI] [PubMed] [Google Scholar]
- 43.De Kleer I, Vercoulen Y, Klein M, et al. CD30 discriminates heat shock protein 60-induced FOXP3+ CD4+ T cells with a regulatory phenotype. J Immunol. 2010;185:2071–9. doi: 10.4049/jimmunol.0901901. [DOI] [PubMed] [Google Scholar]
- 44.Zanin-Zhorov A, Cahalon L, Tal G, Margalit R, Lider O, Cohen IR. Heat shock protein 60 enhances CD4 CD25 regulatory T cell function via innate TLR2 signaling. J Clin Invest. 2006;116:2022–32. doi: 10.1172/JCI28423. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 45.Genain CP, Abel K, Belmar N, et al. Late complications of immune deviation therapy in a nonhuman primate. Science. 1996;274:2054–7. doi: 10.1126/science.274.5295.2054. [DOI] [PubMed] [Google Scholar]
- 46.Blanas E, Carbone FR, Allison J, Miller JF, Heath WR. Induction of autoimmune diabetes by oral administration of autoantigen. Science. 1996;274:1707–9. doi: 10.1126/science.274.5293.1707. [DOI] [PubMed] [Google Scholar]
- 47.Hanninen A, Braakhuis A, Heath WR, Harrison LC. Mucosal antigen primes diabetogenic cytotoxic T-lymphocytes regardless of dose or delivery route. Diabetes. 2001;50:771–5. doi: 10.2337/diabetes.50.4.771. [DOI] [PubMed] [Google Scholar]
- 48.Sette A. Tools of the trade in vaccine design. Science. 2000;290:2074b–5b. doi: 10.1126/science.290.5499.2074b. [DOI] [PubMed] [Google Scholar]
- 49.Singh H, Raghava GP. ProPred: prediction of HLA-DR binding sites. Bioinformatics. 2001;17:1236–7. doi: 10.1093/bioinformatics/17.12.1236. [DOI] [PubMed] [Google Scholar]
- 50.Bian H, Hammer J. Discovery of promiscuous HLA-II-restricted T cell epitopes with TEPITOPE. Methods. 2004;34:468–75. doi: 10.1016/j.ymeth.2004.06.002. [DOI] [PubMed] [Google Scholar]
- 51.Mohanapriya A, Lulu S, Kayathri R, Kangueane P. Class II HLA-peptide binding prediction using structural principles. Hum Immunol. 2009;70:159–69. doi: 10.1016/j.humimm.2008.12.012. [DOI] [PubMed] [Google Scholar]
- 52.Kappos L, Comi G, Panitch H, et al. Induction of a non-encephalitogenic type 2 T helper-cell autoimmune response in multiple sclerosis after administration of an altered peptide ligand in a placebo-controlled, randomized phase II trial. The Altered Peptide Ligand in Relapsing MS Study Group. Nat Med. 2000;6:1176–82. doi: 10.1038/80525. [DOI] [PubMed] [Google Scholar]
- 53.Bielekova B, Goodwin B, Richert N, et al. Encephalitogenic potential of the myelin basic protein peptide (amino acids 83-99) in multiple sclerosis: results of a phase II clinical trial with an altered peptide ligand. Nat Med. 2000;6:1167–75. doi: 10.1038/80516. [DOI] [PubMed] [Google Scholar]
- 54.Dominguez MC, Lorenzo N, Barbera A, et al. An altered peptide ligand corresponding to a novel epitope from heat-shock protein 60 induces regulatory T cells and suppresses pathogenic response in an animal model of adjuvant-induced arthritis. Autoimmunity. 2011;44:471–82. doi: 10.3109/08916934.2010.550590. [DOI] [PubMed] [Google Scholar]
- 55.Zonneveld-Huijssoon E, Roord ST, de JW, et al. Bystander suppression of experimental arthritis by nasal administration of a heat shock protein peptide. Ann Rheum Dis. 2011;70:2199–206. doi: 10.1136/ard.2010.136994. [DOI] [PubMed] [Google Scholar]
- 56.Prakken BJ, Samodal R, Le TD, et al. Epitope-specific immunotherapy induces immune deviation of proinflammatory T cells in rheumatoid arthritis. Proc Natl Acad Sci USA. 2004;101:4228–33. doi: 10.1073/pnas.0400061101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Koffeman EC, Genovese M, Amox D, et al. Epitope-specific immunotherapy of rheumatoid arthritis: clinical responsiveness occurs with immune deviation and relies on the expression of a cluster of molecules associated with T cell tolerance in a double-blind, placebo-controlled, pilot phase II trial. Arthritis Rheum. 2009;60:3207–16. doi: 10.1002/art.24916. [DOI] [PubMed] [Google Scholar]
- 58.Birk OS, Elias D, Weiss AS, et al. NOD mouse diabetes: the ubiquitous mouse hsp60 is a beta-cell target antigen of autoimmune T cells. J Autoimmun. 1996;9:159–66. doi: 10.1006/jaut.1996.0019. [DOI] [PubMed] [Google Scholar]
- 59.Elias D, Cohen IR. Peptide therapy for diabetes in NOD mice. Lancet. 1994;343:704–6. doi: 10.1016/s0140-6736(94)91582-2. [DOI] [PubMed] [Google Scholar]
- 60.Raz I, Elias D, Avron A, Tamir M, Metzger M, Cohen IR. Beta-cell function in new-onset type 1 diabetes and immunomodulation with a heat-shock protein peptide (DiaPep277): a randomised, double-blind, phase II trial. Lancet. 2001;358:1749–53. doi: 10.1016/S0140-6736(01)06801-5. [DOI] [PubMed] [Google Scholar]
- 61.Huurman VA, van der Meide PE, Duinkerken G, et al. Immunological efficacy of heat shock protein 60 peptide DiaPep277 therapy in clinical type I diabetes. Clin Exp Immunol. 2008;152:488–97. doi: 10.1111/j.1365-2249.2008.03656.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Raz I, Avron A, Tamir M, et al. Treatment of new-onset type 1 diabetes with peptide DiaPep277 is safe and associated with preserved beta-cell function: extension of a randomized, double-blind, phase II trial. Diabetes Metab Res Rev. 2007;23:292–8. doi: 10.1002/dmrr.712. [DOI] [PubMed] [Google Scholar]
- 63.Lazar L, Ofan R, Weintrob N, et al. Heat-shock protein peptide DiaPep277 treatment in children with newly diagnosed type 1 diabetes: a randomised, double-blind phase II study. Diabetes Metab Res Rev. 2007;23:286–91. doi: 10.1002/dmrr.711. [DOI] [PubMed] [Google Scholar]
- 64.Huurman VA, Decochez K, Mathieu C, Cohen IR, Roep BO. Therapy with the hsp60 peptide DiaPep277 in C-peptide positive type 1 diabetes patients. Diabetes Metab Res Rev. 2007;23:269–75. doi: 10.1002/dmrr.691. [DOI] [PubMed] [Google Scholar]
- 65.Eldor R, Kassem S, Raz I. Immune modulation in type 1 diabetes mellitus using DiaPep277: a short review and update of recent clinical trial results. Diabetes Metab Res Rev. 2009;25:316–20. doi: 10.1002/dmrr.942. [DOI] [PubMed] [Google Scholar]
- 66.Ludvigsson J, Hjorth M, Cheramy M, et al. Extended evaluation of the safety and efficacy of GAD treatment of children and adolescents with recent-onset type 1 diabetes: a randomised controlled trial. Diabetologia. 2011;54:634–40. doi: 10.1007/s00125-010-1988-1. [DOI] [PubMed] [Google Scholar]
- 67.Ludvigsson J, Faresjo M, Hjorth M, et al. GAD treatment and insulin secretion in recent-onset type 1 diabetes. N Engl J Med. 2008;359:1909–20. doi: 10.1056/NEJMoa0804328. [DOI] [PubMed] [Google Scholar]
- 68.Agardh CD, Cilio CM, Lethagen A, et al. Clinical evidence for the safety of GAD65 immunomodulation in adult-onset autoimmune diabetes. J Diabetes Complications. 2005;19:238–46. doi: 10.1016/j.jdiacomp.2004.12.003. [DOI] [PubMed] [Google Scholar]
- 69.Agardh CD, Lynch KF, Palmer M, Link K, Lernmark A. GAD65 vaccination: 5 years of follow-up in a randomised dose-escalating study in adult-onset autoimmune diabetes. Diabetologia. 2009;52:1363–8. doi: 10.1007/s00125-009-1371-2. [DOI] [PubMed] [Google Scholar]
- 70.Marichal T, Ohata K, Bedoret D, et al. DNA released from dying host cells mediates aluminum adjuvant activity. Nat Med. 2011;17:996–1002. doi: 10.1038/nm.2403. [DOI] [PubMed] [Google Scholar]
- 71.Arakawa T, Yu J, Langridge WH. Food plant-delivered cholera toxin B subunit for vaccination and immunotolerization. Adv Exp Med Biol. 1999;464:161–78. doi: 10.1007/978-1-4615-4729-7_13. [DOI] [PubMed] [Google Scholar]
- 72.Arakawa T, Yu J, Chong DK, Hough J, Engen PC, Langridge WH. A plant-based cholera toxin B subunit-insulin fusion protein protects against the development of autoimmune diabetes. Nat Biotechnol. 1998;16:934–8. doi: 10.1038/nbt1098-934. [DOI] [PubMed] [Google Scholar]
- 73.Anosova NG, Chabot S, Shreedhar V, Borawski JA, Dickinson BL, Neutra MR. Cholera toxin, E. coli heat-labile toxin, and non-toxic derivatives induce dendritic cell migration into the follicle-associated epithelium of Peyer's patches. Mucosal Immunol. 2008;1:59–67. doi: 10.1038/mi.2007.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Shreedhar VK, Kelsall BL, Neutra MR. Cholera toxin induces migration of dendritic cells from the subepithelial dome region to T- and B-cell areas of Peyer's patches. Infect Immun. 2003;71:504–9. doi: 10.1128/IAI.71.1.504-509.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Rouvio O, Dvorkin T, mir-Kroll H, et al. Self HSP60 peptide serves as an immunogenic carrier for a CTL epitope against persistence of murine cytomegalovirus in the salivary gland. Vaccine. 2005;23:3508–18. doi: 10.1016/j.vaccine.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 76.Liang J, Aihua Z, Yu W, Yong L, Jingjing L. HSP65 serves as an immunogenic carrier for a diabetogenic peptide P277 inducing anti-inflammatory immune response in NOD mice by nasal administration. Vaccine. 2010;28:3312–7. doi: 10.1016/j.vaccine.2010.02.100. [DOI] [PubMed] [Google Scholar]
- 77.Krieg AM. CpG motifs: the active ingredient in bacterial extracts? Nat Med. 2003;9:831–5. doi: 10.1038/nm0703-831. [DOI] [PubMed] [Google Scholar]
- 78.Klinman D, Shirota H, Tross D, Sato T, Klaschik S. Synthetic oligonucleotides as modulators of inflammation. J Leukoc Biol. 2008;84:958–64. doi: 10.1189/jlb.1107775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Klinman DM, Currie D, Lee G, Grippe V, Merkel T. Systemic but not mucosal immunity induced by AVA prevents inhalational anthrax. Microbes Infect. 2007;9:1478–83. doi: 10.1016/j.micinf.2007.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wehrens EJ, van WF, Roord ST, Albani S, Prakken BJ. Treating arthr by immunomodulation: is there a role for regulatory T cells? Rheumatology (Oxf) 2010;49:1632–44. doi: 10.1093/rheumatology/keq130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wehrens EJ, Mijnheer G, Duurland CL, et al. Functional human regulatory T cells fail to control autoimmune inflammation due to PKB/c-akt hyperactivation in effector cells. Blood. 2011;118:3538–48. doi: 10.1182/blood-2010-12-328187. [DOI] [PubMed] [Google Scholar]
- 82.Ruprecht CR, Gattorno M, Ferlito F, et al. Coexpression of CD25 and CD27 identifies FoxP3+ regulatory T cells in inflamed synovia. J Exp Med. 2005;201:1793–803. doi: 10.1084/jem.20050085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Pasare C, Medzhitov R. Toll pathway-dependent blockade of CD4+CD25+ T cell-mediated suppression by dendritic cells. Science. 2003;299:1033–6. doi: 10.1126/science.1078231. [DOI] [PubMed] [Google Scholar]
- 84.Koenen HJ, Smeets RL, Vink PM, van RE, Boots AM, Joosten I. Human CD25highFoxp3pos regulatory T cells differentiate into IL-17-producing cells. Blood. 2008;112:2340–52. doi: 10.1182/blood-2008-01-133967. [DOI] [PubMed] [Google Scholar]
- 85.Radhakrishnan S, Cabrera R, Schenk EL, et al. Reprogrammed FoxP3+ T regulatory cells become IL-17+ antigen-specific autoimmune effectors in vitro and in vivo. J Immunol. 2008;181:3137–47. doi: 10.4049/jimmunol.181.5.3137. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 86.Valencia X, Stephens G, Goldbach-Mansky R, Wilson M, Shevach EM, Lipsky PE. TNF downmodulates the function of human CD4+CD25hi T-regulatory cells. Blood. 2006;108:253–61. doi: 10.1182/blood-2005-11-4567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ricciardelli I, Lindley KJ, Londei M, Quaratino S. Anti tumour necrosis-alpha therapy increases the number of FOXP3 regulatory T cells in children affected by Crohn's disease. Immunology. 2008;125:178–83. doi: 10.1111/j.1365-2567.2008.02839.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ehrenstein MR, Evans JG, Singh A, et al. Compromised function of regulatory T cells in rheumatoid arthritis and reversal by anti-TNFalpha therapy. J Exp Med. 2004;200:277–85. doi: 10.1084/jem.20040165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Boettler T, von HM. Immunotherapy of type 1 diabetes – how to rationally prioritize combination therapies in T1D. Int Immunopharmacol. 2010;10:1491–5. doi: 10.1016/j.intimp.2010.07.008. [DOI] [PubMed] [Google Scholar]
- 90.Penaranda C, Tang Q, Bluestone JA. Anti-CD3 therapy promotes tolerance by selectively depleting pathogenic cells while preserving regulatory T cells. J Immunol. 2011;187:2015–22. doi: 10.4049/jimmunol.1100713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sherry N, Hagopian W, Ludvigsson J, et al. Teplizumab for treatment of type 1 diabetes (Protege study): 1-year results from a randomised, placebo-controlled trial. Lancet. 2011;378:487–97. doi: 10.1016/S0140-6736(11)60931-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Bresson D, Togher L, Rodrigo E, et al. Anti-CD3 and nasal proinsulin combination therapy enhances remission from recent-onset autoimmune diabetes by inducing Tregs. J Clin Invest. 2006;116:1371–81. doi: 10.1172/JCI27191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Bresson D, Fradkin M, Manenkova Y, Rottembourg D, von HM. Genetic-induced variations in the GAD65 T-cell repertoire governs efficacy of anti-CD3/GAD65 combination therapy in new-onset type 1 diabetes. Mol Ther. 2010;18:307–16. doi: 10.1038/mt.2009.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Roord ST, Zonneveld-Huijssoon E, Le T, et al. Modulation of T cell function by combination of epitope specific and low dose anticytokine therapy controls autoimmune arthritis. PLoS ONE. 2006;1:e87. doi: 10.1371/journal.pone.0000087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Fox RI, Kang HI. Mechanism of action of antimalarial drugs: inhibition of antigen processing and presentation. Lupus. 1993;2(Suppl 1):S9–12. [PubMed] [Google Scholar]