

Abstract
Glucose-insulin-potassium (GIK) is a useful adjunct to myocarditis. Besides its essential action in energy metabolism, insulin also exerts an anti-inflammatory effect. This study investigated the effect of insulin on myocardial inflammation in experimental autoimmune myocarditis (EAM) in mice and its potential role in T cell regulation. Mice were divided randomly into a normal control group, a saline-treated EAM group and an insulin-treated EAM group. The histopathological changes of myocardium, α-myosin heavy chain (MyHCα)614–629 antigen-specific autoantibody titre, the serum level of cardiac troponin I (cTnI), mitogen-activated protein kinase (MAPK) family members' activity and content were measured. Furthermore, the phenotype of T lymphocyte subsets in splenocytes was analysed to evaluate the immune status of mice. Insulin reduced serum cTnI of EAM mice on days 14 and 21 (P < 0·05) after immunization, with no changes in blood glucose and autoantibody production. Western blot revealed that extracellular signal-regulated protein kinase (ERK1/2) may be a determining factor in this process. Total ERK1/2 and phospho-ERK1/2 (p-ERK1/2) were both up-regulated in insulin-treated mice after immunization. We also found that insulin treatment promoted T cell recovery without changing the naive-to-memory T-cell ratio; in particular, CD3+ T cells in insulin-treated mice proliferated more vigorously than in control mice (P < 0·05). We report here for the first time that insulin alleviates myocarditis in the EAM model. These data show that insulin has a direct effect on T cell proliferation in EAM. It is possible that GIK or insulin may assist T cell recovery towards normal in myocarditis, especially for diabetic or hyperglycaemic patients.
Keywords: ERK1/2, insulin, myocarditis, proliferation, T cell
Introduction
Myocarditis is an inflammatory disease of the myocardium with a wide range of clinical presentations; some of these cases go on to develop fibrosis and dilated cardiomyopathy [1]. The treatment for myocarditis is aimed mainly at the ensuing viral infection and cardiac muscle inflammation, and hormone therapy is used occasionally to reduce the myocardial inflammatory response and myocardial scar formation.
The immune response plays an important role in the pathogenesis of post-infectious myocarditis [2,3]. Impaired central tolerance to α-myosin heavy chain (MyHCα) contributes to the pathogenesis of myocarditis in both mice and humans [4]. Significantly reduced numbers of peripheral T cells were observed in coxsackievirus B3 (CVB3) and cardiac MyHCα-induced myocarditis [5]. T cell infiltration leads to opposing results in myocarditis: both the clearance of virus-infected myocytes (which is beneficial) and cell-mediated myocardial injury or necrosis (which is detrimental) [6].
Glucose-insulin-potassium (GIK) ingredients, which are used as energy, include adenosine triphosphate (ATP), co-enzyme A, potassium, insulin and glucose; they provide energy for the myocardium and promoting cardiac metabolism, thus speeding the repair process. GIK was first used in cardiology and cardiac surgery 40 years ago; although it has remained a controversial and doubtful cardioprotective intervention, it is still a commonly used adjunct to myocarditis. Insulin, probably the most important component of GIK, also exerts an anti-inflammatory effect [7]. The specific role of insulin in myocarditis, however, is still unknown.
Additionally, mitogen-activated protein kinases (MAPKs) may be common mediators in the pathological remodelling processes of cardiovascular diseases (such as hyperglycaemia, hypertension, cardiac hypertrophy, ageing and obesity) [8]. It has been demonstrated that extracellular signal-regulated protein kinase 1/2 (ERK1/2) may exert a strong cardioprotective effect under different circumstances via multiple downstream targets. The two classes of drugs used commonly to treat cardiac-related diseases, Ca2+ blocker and β-adrenergic receptor blockers, have been reported to be mediated in part through ERK1/2 activity [9]. The other two important members of the MAPK family, p-38 MAPK and c-Jun N-terminal kinase (JNK), also belong to stress-activated protein kinase (SAPKs). They are activated by the ischaemic and inflammatory stresses associated with myocardial injury and have been implicated in promoting myocardial damage and remodelling [10]. However, it is unclear whether insulin is involved in the regulation of the MAPK pathway in myocarditis.
In this study, using haematoxylin and eosin (H&E) staining, we observed that insulin may decrease the heart injury biomarker cardiac troponin I (cTnI) levels after stimulation by MyHC-α614–629 peptides. Furthermore, ERK1/2 kinase may play a role in this protective process. More specifically, we addressed the effects of insulin on the T cell population in this experimental autoimmune myocarditis (EAM) model.
Materials and methods
Mice, immunization and treatment protocols
Male BALB/c mice (8–10 weeks old, weighing 20–24 g) were obtained from our university laboratory animal centre. A murine heart muscle-specific peptide derived from MyHCα614–629 was used as an antigen and was commercially synthesized and purified by high performance liquid chromatography (acetyl-SLKLMATLFSTYASAD-OH [11], >95% purity, MeiLian Biochemical Service and Products, Tianjin, China). The peptide was dissolved in 0·01 M phosphate-buffered saline (PBS) and emulsified with an equal volume of complete Freund's adjuvant (CFA; Sigma-Aldrich, St Louis, MO, USA). Mice were immunized subcutaneously (s.c.) with the myocardiogenic peptide MyHCα614–629 100 μg/0·2 ml on days 0 and 7. Mice were divided randomly into a normal control group, an insulin-treated EAM group and a saline-treated EAM group. For insulin treatment, mice were injected s.c. with 2·5 U/kg body weight of insulin on alternate days for a period of 21 days. Control EAM mice received saline only. Novolin 30R (100 U/ml; Novo Nordisk, Bagsværd, Denmark) was the insulin used in the mice. All experiments using mice were approved by the Fourth Military Medical University Animal Experimental Committee.
H&E staining and histological assessment
Immediately after the mice were killed, their hearts were fixed in 10% paraffin-embedded formalin and stained with H&E. Myocarditis was scored on a semiquantitative scale using grades from 0 to 4 (0, no inflammatory infiltrates; 1, small foci of inflammatory cells between myocytes or inflammatory cells surrounding individual myocytes; 2, a large foci of 100 inflammatory cells or involving 30 myocytes; 3, 10% of a myocardial cross-section involved; and 4, 30% of a myocardial cross-section involved).
Cytokine detection
Sera were collected and assayed using enzyme-linked immunosorbent assay (ELISA) kits. Mouse cTnI ELISA kits were purchased from Yanji Biotech (Shanghai, China). Absorbance was measured at 450 nm using a Bio-Rad (Hercules, CA, USA) microplate reader.
Antibodies to cardiac myosin
Serum levels of MyHCα-reactive antibodies were determined on day 21 using microtitre plates coated with 0·1 g/l of MyHCα614–629 incubated with horseradish peroxidase (HRP)-conjugated goat anti-mouse immunoglobulin (Ig)G antibody (Dako, Glostrup, Denmark). Adjusted optical density (OD) was calculated as follows: adjusted OD = mean OD of a sample – mean OD of a negative control.
Quantitative real-time polymerase chain reaction (qPCR)
Total RNA was extracted from the hearts using Trizol reagent (Invitrogen, Grand Island, NY, USA). First-strand cDNA was synthesized in a volume of 20 μl using 1 μg of total RNA and the PrimeScript® RT reagent (Takara Bio Inc., Shiga, Japan; Code: DRR036A). PCR was performed with a BioRad system and SYBR® Premix Ex Taq™ II Perfect real-time kit (Takara Code: DRR081A), according to the manufacturer's instructions. Mouse interleukin (IL)-1β and tumour necrosis factor (TNF)-α primers were synthesized as the following sequences (Sangon Biotech, Shanghai, China): IL-1β forward: 5′-GGGCTGGACTGTTTCTAATGC-3′, reverse: 5′-ATGGTTTCTTGTGACCCTGAG-3′; TNF-α forward: 5′-TTCACTGGAGCCTCGAATGTC-3′, reverse: 5′-CAGGGAAGAATCTGGAAAGGT-3′; and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (forward): 5′-GGTGAAGGTCGGTGTGAACG-3′, reverse: 5′-CTCGCTCCTGGAAGATGGTG-3′.
The relative amount of specific mRNA was normalized to mouse GAPDH. All PCR reactions were run in triplicate and were performed with 40 cycles. Quantitative real-time PCR analysis was carried out using the 2-ΔΔCt method.
Western blot analysis
The heart cell lysates were electrophoresed on 12% sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to nitrocellulose membranes (Millipore, Billerica, MA, USA). The membranes were blocked with 5% (w/v) bovine skimmed milk in PBS and incubated with specific antibodies (Cell Signaling Technology, Irvine, CA, USA) at 4°C overnight. MAPK activation was examined with specific antibodies as follows: phospho-ERK1/2 (Thr202/Tyr204), phospho-p38 MAPK (Thr180/Tyr182), phospho-SAPK/JNK (Thr183/Tyr185), ERK1/2, p38 MAPK and SAPK/JNK. β-actin was used as a reference gene to analyse the protein quantitatively. The membranes were then washed and incubated with HRP-labelled goat anti-rabbit IgG secondary antibody (Dako) at room temperature for 1 h. After washing, enhanced chemiluminescence (ECL) reagent (Roche, Indianapolis, IN, USA) was applied according to the manufacturer's instructions, and the membranes were exposed to the Chemi Doc XRS imaging system (Bio-Rad).
Flow cytometric analysis
Splenocytes were collected from mice at 21 days after inducing EAM. Splenocytes were extracted into a single cell suspension in PBS, and red blood cells were lysed by incubation in Ack lysis buffer (Beyotime, Shanghai, China). Cells were washed and FcγR II/III blocked with α-CD16/32 antibodies. Surface markers were stained with the appropriate fluorochrome-labelled antibodies: CD3-phycoerythrin (PE)-cyanin 5 (Cy5), CD4-PE, CD8-fluorescein isothiocyanate (FITC), CD4-FITC, CD44-PE, CD62L-allophycocyanin (APC) and appropriate isotype controls. All the antibodies were purchased from Becton-Dickinson (Franklin Lakes, NJ, USA). The cells were examined using a flow cytometer [fluorescence activated cell sorter (FACS)Calibur; Becton-Dickinson] and the results were analysed using WinMDI software version 2·9.
Statistical analysis
All data are expressed as mean ± standard error of the mean (s.e.m.). Normally distributed data were analysed using Student's t-test; otherwise, the Mann–Whitney U-test was used. For histological assessment, the average score was analysed with a non-parametric test [12]. All the statistical tests were performed with GraphPad Prism software version 4·0 (GraphPad Software, San Diego, CA, USA).
Results
Insulin treatment ameliorates cardiac injury in the EAM mouse model
In this study, all the mice survived past day 21 after injection. First, we found that immunization with MyHCα614–629 could induce severe inflammatory myocarditis in our mouse model. As assessed by H&E histological staining, the severity of myocarditis in insulin-treated mice was decreased compared to saline-treated groups on day 21 (Fig. 1a). The ratio of heart weight to body weight (HW : BW) of the saline-treated mice was significantly greater than that of the normal mice (P < 0·05) (Fig. 1b), but no significant differences in HW : BW were observed between insulin-treated mice and the normal mice. To avoid too large a drop in blood glucose, we chose the dose of 2·5 U/kg body weight of insulin on an alternate day. The results showed that there were no significant differences in blood glucose between each of the groups (Fig. 1c).
Fig. 1.
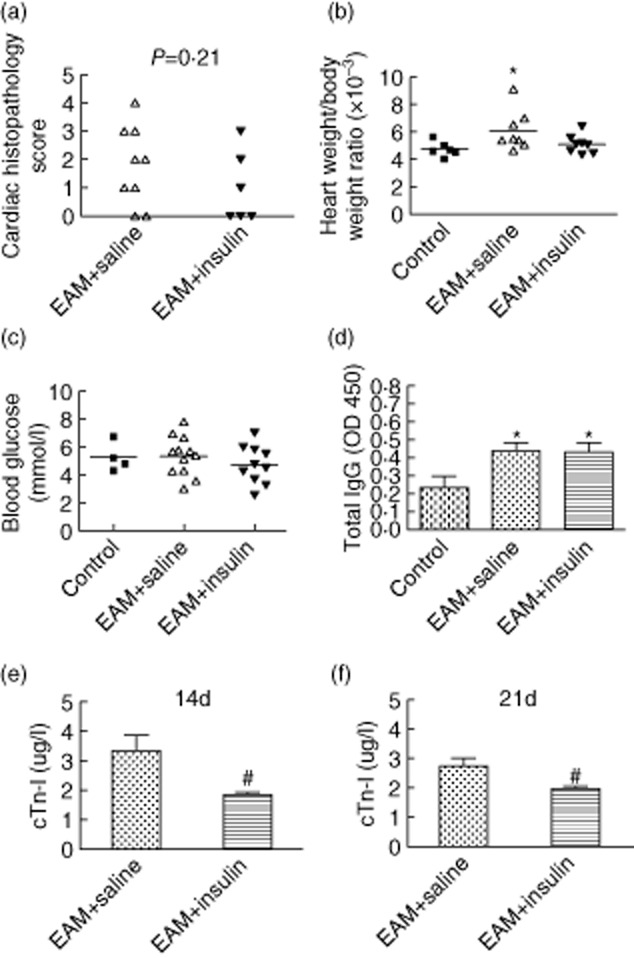
Insulin-treated mice were protected from autoimmune myocarditis. (a) Histopathological score of hearts at day 21 after immunization. There was a tendency for insulin to decrease the myocarditis score (P = 0·21). (b) Relative heart weight of saline-treated (open triangle) and insulin-treated (filled triangle) mice at day 21 of experimental autoimmune myocarditis (EAM). Comparison between saline-treated mice and normal control mice (filled square), *P < 0·05. The Mann–Whitney U-test was used for non-parametric test including the evaluation of severity scores and heart weight : body weight ratios. (c) Blood glucose at day 21 of EAM. There were no differences between each group. (d) The level of α-myosin heavy chain (MyHCα)614–629 peptide-reactive serum antibodies were measured by enzyme-linked immunosorbent assay (ELISA) at day 21 of EAM. The serum was diluted 1 : 2000 (n = 7 in each group). (e,f) The serum cardiac troponin I (cTnI) level was assessed by ELISA on days 14 and 21. Comparison between the saline- and insulin-treated groups indicated less cTnI in serum from EAM mice treated with insulin, #P < 0·05 (n = 10 in each group).
To test the antigen-specific autoantibody response, MyHCα614–629 reactive serum antibodies were measured by ELISA at day 21 of EAM. Peptide-immunized mice developed significantly higher levels of MyHCα614–629-reactive total IgG compared to mock-immunized control mice (P < 0·05), and there was no significant difference between the insulin and saline treatment groups in autoantibody production (Fig. 1d). Meanwhile, we found that insulin significantly lowered the serum cTnI levels of EAM mice on day 14 (P < 0·05) (Fig. 1e) and on day 21 (P < 0·05) (Fig. 1f) compared with that of the saline group.
Insulin did not decrease inflammatory cytokines significantly in heart
BALB/c mice developed severe myocarditis which peaked at day 21, as described previously [13]. Marked inflammation in the myocardium was observed in the saline-treated group as a prominent feature of this myocarditis model, characterized by extensive mononuclear and polymorphonuclear cell infiltration. In contrast, the inflamed area in the 2·5 U/kg insulin group was smaller than that in the saline-treated group (Fig. 2a–c). IL-1β and TNF-α are the key inflammatory cytokines in the pathogenesis of myocarditis; blockade of TNF-α (or IL-β) ameliorates myocarditis in mice [13]. Thus, we analysed these two cytokines in the heart by qPCR at day 21 of EAM. We found that IL-1β and TNF-α tended to be lower in hearts from insulin-treated mice at day 21 of EAM compared to saline-treated controls, but with no significant differences between each group (Fig. 2d).
Fig. 2.
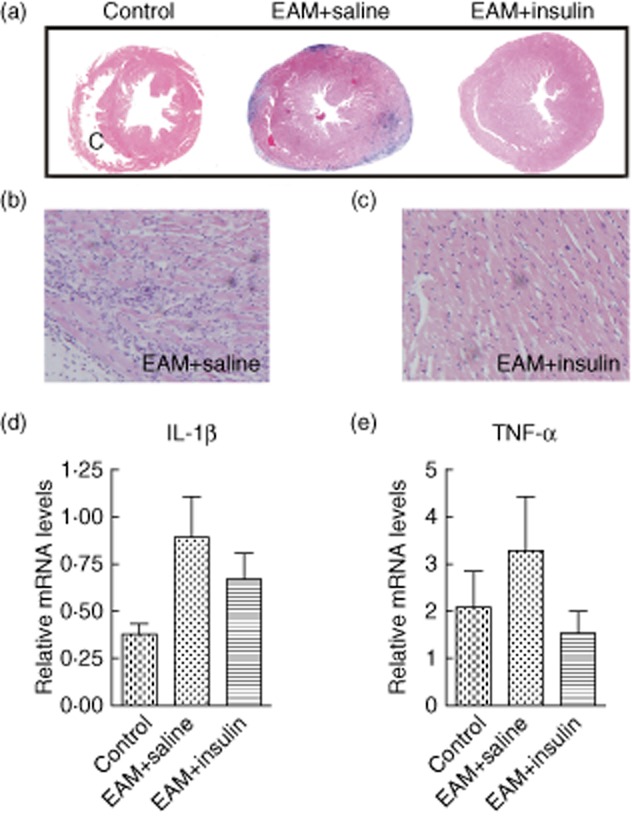
Inflammatory infiltrates were present in the control group but there were fewer infiltrates in the insulin-treated hearts at day 21 after immunization compared to control; (a) ×5 original magnification, representative histopathology of α-myosin heavy chain α (MyHCα)614–629 induced experimental autoimmune myocarditis (EAM) at day 21 from saline-treated (middle), insulin-treated (right) mice and normal mouse heart (left); (b,c) ×20 original magnification. (d,e) Quantitative polymerase chain reaction (PCR) detected the proinflammatory cytokines interleukin (IL)-1β and tumour necrosis factor (TNF)-α in the heart. There was more IL-1β and TNF-α in the saline-treated EAM group than in the normal control group. However, levels of IL-1β and TNF-α were lower in hearts from insulin-treated mice at day 21 after EAM compared to saline-treated controls (P > 0·05) (n = 6 in each group).
ERK1/2 content and phosphorylation were up-regulated in the hearts of insulin-treated EAM mice
We next examined whether insulin increased MAPK activity in hearts of EAM mice. The ERK1/2 : β-actin ratio was increased significantly in EAM mice after insulin treatment, compared with those treated with saline (P < 0·05) (Fig. 3a,b). The amount of p-ERK1/2 also increased; however, the ratio of p-ERK to ERK1/2 did not change after insulin treatment. In addition, the JNK and p38 MAPK pathway in EAM mice did not differ in the presence or absence of insulin (Fig. 3c).
Fig. 3.
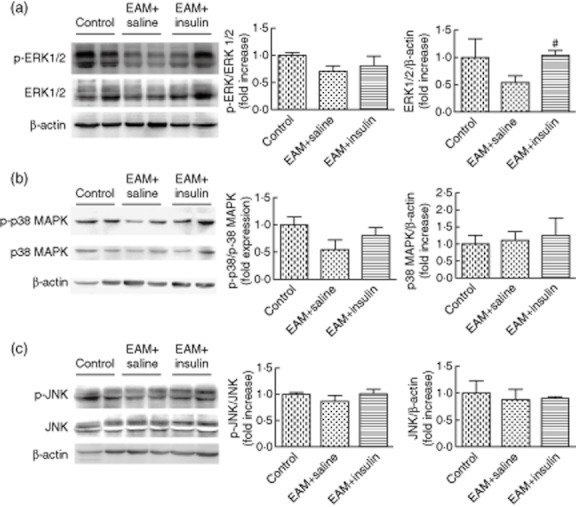
Representative immunoblots using antibodies against (a) phosphorylated extracellular signal-regulated protein kinase (p-ERK) and total ERK1/2; (b) phosphorylated p38 (p-p38) mitogen-activated protein kinase (MAPK) and p38 MAPK; (c) phosphorylated c-Jun N-terminal kinase (p-JNK) and total JNK to probe proteins extracted from hearts. Blots of proteins extracted from hearts of normal mice are shown for comparison. The graphs also show quantification of (a) p-ERK/total ERK and total ERK /β-actin; (b) p-p38/total p38 MAPK and total p38 MAPK/β-actin; (c) p-JNK/total JNK and total JNK /β-actin. Comparison between saline-treated mice and insulin-treated mice, #P < 0·05 (n = 7 in each group).
Insulin treatment could promote T cell recovery in EAM without changing the ratio of memory : naive T cells
EAM in mice has been shown to be predominantly T cell-driven, and MyHCα strikingly reduced the number of peripheral T cells. Therefore, we further explored the possibility that insulin has an effect on T cells in myocarditis. The CD3 antigen is a marker for T cells and a crucial molecule in T cell signal transduction. In this study, we assessed the splenic CD3+ T cell populations in the mice by flow cytometric analysis on day 21 after inducing EAM. In the saline-treated group, the percentage of CD3+ T cells was decreased significantly compared to the mock-immunized group (P < 0·01), while there was a significant increase in the insulin-treated group compared to the saline-treated group (P < 0·05). To investigate further the subpopulation of the CD3+ T cells, double staining with antibodies against CD3 and CD4 or CD8 were performed. No significant differences were observed, but an increased tendency was noted in the relative proportions of CD4+ or CD8+ T cells of insulin-treated mice compared to saline-treated EAM mice at day 21 (Fig. 4). We also set up a late treatment group to examine whether insulin could reverse the established EAM. In this group, insulin was injected subcutaneously at a dose of 2·5 U/kg body weight from days 14 to 21 after immunization, but there was no significant difference between insulin- and saline-treated groups in the T cell subpopulations (data not shown).
Fig. 4.
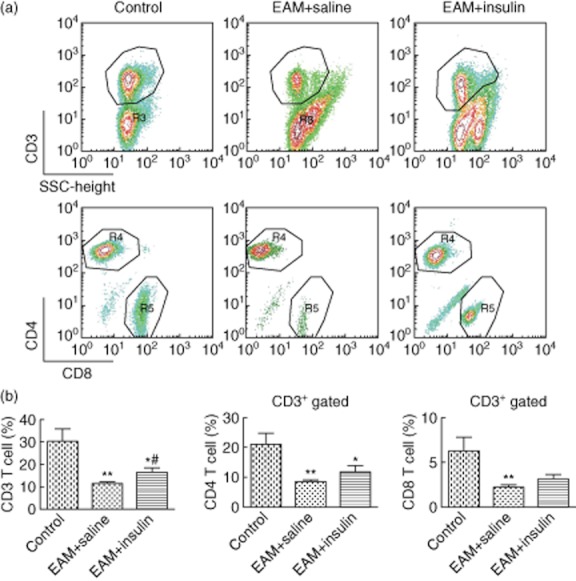
Flow cytometry results of splenic mononuclear cells after induction of experimental autoimmune myocarditis (EAM)by injection with complete Freund's adjuvant (CFA)/α-myosin heavy chain (MyHCα) peptide. Splenic CD3+ T cells from insulin-treated mice proliferated more vigorously than did those from saline-treated mice. Data are expressed as mean ± standard error of the mean (n = 6–8 in each group). (a) Representative flow cytometric analysis of CD3+ T cell proportion and CD3+CD4+ or CD3+CD8+ subpopulations at 21 days after inducing EAM in splenocytes. (b) Comparison between EAM mice and normal mice, *P < 0·05; **P < 0·01. Comparison between saline-treated mice and insulin-treated mice, #P < 0·05.
Naive and memory T cells come from CD4+ T cell precursors, and the balance between these two subpopulations in the peripheral blood and lymphoid tissue is crucial for maintaining the homeostasis of the immune response. To determine whether insulin caused changes in the memory : naive T cell ratio, we examined the proportions of CD44+CD62L– (memory T cells) and CD44–CD62L+ (naive T cells) of splenocytes on day 21 of EAM. A significant increase in the proportions of CD44+CD62L– memory T cells was observed 21 days after immunization compared to that of the normal control group (P < 0·01). In contrast, a significant decrease in the amount of CD44–CD62L+ naive T cells was observed compared to the mock-immunized group (P < 0·01). We did not observe any differences in the proportions of memory : naive T cell ratios at this time-point between the insulin-treated and saline-treated groups (Table 1). Thus, these data indicate that splenic CD3+ T cells from the insulin-treated group were increased proportionally compared to the saline-treated group without changing the memory : naive T cell ratio in EAM mice.
Table 1.
Splenic naive T cell : memory T cell ratio analysis in each group of mice at 21 day after immunization
| Naive T cells (CD44lowCD62L+) (%) | Memory T cell (CD44highCD62L-) (%) | Naive/memory T cell ratio | |
|---|---|---|---|
| Control | 41·2 ± 11·9 | 18·4 ± 3·8 | 2·4 ± 1·15 |
| EAM + saline | 8·3 ± 2·2** | 74·4 ± 6·9** | 0·1 ± 0·06** |
| EAM + insulin | 6·8 ± 2·7** | 74·8 ± 9·5** | 0·1 ± 0·05** |
Data reported as mean ± standard error of the mean, **P < 0·01. EAM: experimental autoimmune myocarditis.
Discussion
EAM is a mouse model of post-infectious cardiomyopathy, and reflects mechanisms of inflammatory cardiomyopathy in humans. Using this model, we demonstrated the possibility that insulin, as an important ingredient of GIK, may alleviate heart inflammation, improve T cell subset recovery and activate the endogenous protective signalling of ERK1/2 after stimulation by MyHCα614–629 peptides.
Many studies suggest an independent role for insulin in heart diseases and other inflammatory diseases. For example, rigorous insulin treatment reduced the number of deaths from multiple-organ failure with sepsis, regardless of whether or not there was a history of diabetes or hyperglycaemia [14]. Ellger et al. reported that deaths in a burn-injury rabbit model were significantly lower in the two normoglycaemic groups independent of insulin levels, and these researchers suggested that the benefits of intensive insulin therapy may require maintenance of normoglycaemia [15]. Also, Vlasselaers et al. demonstrated recently a significant effect of GIK in combination with tight glycaemic control on the release of C-reactive protein and the proinflammatory cytokines IL-6 and IL-8 in neonates undergoing congenital heart defect surgery [16]. Some complex anti-inflammatory effects of insulin have been described, which are mediated via down-regulation of proinflammatory cytokines and reactive oxygen species through inhibition of nuclear factor (NF)-κB [17], down-regulation of acute phase proteins [18] and, conversely, via increasing endothelial nitric oxide synthase [19] and anti-inflammatory cytokine secretion [20]. These actions contribute to the comprehensive anti-inflammatory effect of insulin, but its role in autoimmune heart disease is not completely understood.
In this study, we found that the severity of myocarditis in insulin-treated mice on day 21 of EAM, as assessed by histology, was decreased compared to the severity of myocarditis in saline-treated groups. Heart weight of the saline-treated mice was increased significantly after MyHCα614–629 immunization compared with that of the normal mice (P < 0·05), which demonstrated severe cardiomyopathy after CFA/MyHCα614–629 peptide immunization. However, no significant differences were observed in the heart weight in insulin-treated mice compared with normal mice, suggesting that insulin treatment did not improve dilated cardiomyopathy in EAM. Moreover, there was no significant difference between the insulin and saline treatment groups in autoantibody production. This suggests that immunization with MyHCα614–629 peptides increased the autoreactive B cell proliferation and autoantibody production, but that insulin did not promote these reactions. Thus, insulin may reduce the severity of chronic myocarditis without interfering with blood glucose.
Although insulin tended to decrease heart severity score and also the level of the inflammatory cytokines IL-1β and TNF-α, these differences were not statistically significant. These data demonstrate that, to a certain extent, the ability of insulin to regulate inflammation is limited; insulin should be combined with other drugs to control myocarditis in a clinical setting. Previous studies have indicated that cTnI is a protein associated with the myocyte contractile apparatus, which will be released into the serum when myocytes are injured and can serve as a marker of myocyte injury [21]. cTnI is highly sensitive and specific for myocarditis in mice 17–21 days after MyHCα immunization [22]. We found that insulin treatment lowered serum cTnI levels of EAM mice significantly on days 14 (P < 0·05) and 21 (P < 0·05) compared with the saline-treated group.
In recent years, the properties of insulin as a signal for activation of survival pathways as a suppressor of myocardial apoptosis in the setting of myocardial ischaemia/reperfusion injury and as a modulator of inflammation has come into focus [23]. Previous research has shown that insulin protects the myocardium via the phosphatidylinositol-3-kinase (PI3K)–Akt endothelial nitric oxide synthase signalling pathway, and the resulting nitric oxide generated via this signalling pathway protects the myocardium. However, we did not observe activation of Akt in the hearts of our EAM mouse model. While MAPKs are another well-studied family of proteins that play an integral role in cardiac development, physiological adaptation and pathological manifestation, ERK1/2 has also been shown to compensate for loss of Akt activity in the post-infarct myocardium and promote cardioprotection in response to erythropoietin [24]. Conversely, JNK and p38 MAPK can contribute to adverse myocardial remodelling and development of heart failure [8]. In this study, Western blot revealed that total ERK1/2 and p-ERK1/2 were each up-regulated significantly in insulin-treated mice after immunization (P < 0·05), which would explain why the p-ERK : ERK1/2 ratio did not change significantly after insulin treatment. However, JNK and p38 MAPK did not change after insulin treatment in EAM mice, indicating that ERK1/2 may be the determining factor that is modulated by insulin in this EAM model.
In addition, because EAM is a T cell-mediated mouse model of post-viral cardiomyopathy [11], we explored further the possibility that insulin had a direct effect on T cells in myocarditis. We found that insulin treatment could promote T cell recovery; specifically, total CD3+ T cells from insulin-treated mice proliferated more vigorously than those of saline-treated mice (P < 0·05). Another probable mechanism of improvement in T cell function is the anti-apoptotic effect of insulin. In-vivo administration of insulin decreased post-ischaemic myocardial apoptotic death [25]. The results of this study support that insulin may play a role in the pathogenesis of EAM through modulating T cell function. The mechanism that accounts for this response, however, may be due partially to the activation of ERK pathway in the CD3+ T cells.
In this work, flow cytometric analysis of splenic cells at day 21 demonstrated that the CD3+ T cell population was elevated significantly with early insulin treatment after MyHCα614–629 antigen immunization and, to some degree, the severity of myocarditis was reduced in the presence of insulin. The results of this research demonstrate that T cells of mice with autoimmune myocarditis were influenced by insulin. We report here for the first time that insulin has a direct effect on T cell proliferation in EAM, suggesting that insulin is involved in the complicated endocrine–immune network, but the effect of insulin on the activation of T cells in vivo needs to be investigated further. Clinically, our findings suggest that with the appropriate administration and balance of GIK or insulin, T cells can return to normal levels more quickly in EAM, especially for diabetic or hyperglycaemic patients. Further research is needed to elucidate the specific mechanism by which insulin acts on immune cells in physiological and pathological conditions.
Acknowledgments
Supported by the State Key Program of National Natural Science Foundation of China (no. 81030005).
Disclosures
The authors have no financial conflicts of interest.
References
- 1.Valaperti A, Marty RR, Kania G, et al. CD11b+ monocytes abrogate Th17 CD4+ T cell-mediated experimental autoimmune myocarditis. J Immunol. 2008;180:2686–2695. doi: 10.4049/jimmunol.180.4.2686. [DOI] [PubMed] [Google Scholar]
- 2.Bachmaier K, Neu N, de la Maza LM, et al. Chlamydia infections and heart disease linked through antigenic mimicry. Science. 1999;283:1335–1339. doi: 10.1126/science.283.5406.1335. [DOI] [PubMed] [Google Scholar]
- 3.Caforio AL, Mahon NJ, McKenna WJ. Cardiac autoantibodies to myosin and other heart-specific autoantigens in myocarditis and dilated cardiomyopathy. Autoimmunity. 2001;34:199–204. doi: 10.3109/08916930109007385. [DOI] [PubMed] [Google Scholar]
- 4.Lv H, Havari E, Pinto S, et al. Impaired thymic tolerance to alpha-myosin directs autoimmunity to the heart in mice and humans. J Clin Invest. 2011;121:1561–1573. doi: 10.1172/JCI44583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kishimoto C, Kuribayashi K, Fukuma K, et al. Immunologic identification of lymphocyte subsets in experimental murine myocarditis with encephalomyocarditis virus. Different kinetics of lymphocyte subsets between the heart and the peripheral blood, and significance of Thy 1.2+ (pan T) and Lyt 1+, 23+ (immature T) subsets in the development of myocarditis. Circ Res. 1987;61:715–725. doi: 10.1161/01.res.61.5.715. [DOI] [PubMed] [Google Scholar]
- 6.Yajima T, Knowlton KU. Viral myocarditis: from the perspective of the virus. Circulation. 2009;119:2615–2624. doi: 10.1161/CIRCULATIONAHA.108.766022. [DOI] [PubMed] [Google Scholar]
- 7.Vlasselaers D. Glucose-insulin-potassium: much more than enriched myocardial fuel. Circulation. 2011;123:129–130. doi: 10.1161/CIRCULATIONAHA.110.002709. [DOI] [PubMed] [Google Scholar]
- 8.Rose BA, Force T, Wang Y. Mitogen-activated protein kinase signaling in the heart: angels versus demons in a heart-breaking tale. Physiol Rev. 2010;90:1507–1546. doi: 10.1152/physrev.00054.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kovacs K, Hanto K, Bognar Z, et al. Prevalent role of Akt and ERK activation in cardioprotective effect of Ca(2+) channel- and beta-adrenergic receptor blockers. Mol Cell Biochem. 2009;321:155–164. doi: 10.1007/s11010-008-9929-8. [DOI] [PubMed] [Google Scholar]
- 10.Turner NA. Therapeutic regulation of cardiac fibroblast function: targeting stress-activated protein kinase pathways. Future Cardiol. 2011;7:673–691. doi: 10.2217/fca.11.41. [DOI] [PubMed] [Google Scholar]
- 11.Marty RR, Dirnhofer S, Mauermann N, et al. MyD88 signaling controls autoimmune myocarditis induction. Circulation. 2006;113:258–265. doi: 10.1161/CIRCULATIONAHA.105.564294. [DOI] [PubMed] [Google Scholar]
- 12.Eriksson U, Ricci R, Hunziker L, et al. Dendritic cell-induced autoimmune heart failure requires cooperation between adaptive and innate immunity. Nat Med. 2003;9:1484–1490. doi: 10.1038/nm960. [DOI] [PubMed] [Google Scholar]
- 13.Cihakova D, Rose NR. Pathogenesis of myocarditis and dilated cardiomyopathy. Adv Immunol. 2008;99:95–114. doi: 10.1016/S0065-2776(08)00604-4. [DOI] [PubMed] [Google Scholar]
- 14.Cariou A, Vinsonneau C, Dhainaut JF. Adjunctive therapies in sepsis: an evidence-based review. Crit Care Med. 2004;32:S562–570. doi: 10.1097/01.ccm.0000142910.01076.a5. [DOI] [PubMed] [Google Scholar]
- 15.Ellger B, Debaveye Y, Vanhorebeek I, et al. Survival benefits of intensive insulin therapy in critical illness: impact of maintaining normoglycemia versus glycemia-independent actions of insulin. Diabetes. 2006;55:1096–1105. doi: 10.2337/diabetes.55.04.06.db05-1434. [DOI] [PubMed] [Google Scholar]
- 16.Vlasselaers D, Mesotten D, Langouche L, et al. Tight glycemic control protects the myocardium and reduces inflammation in neonatal heart surgery. Ann Thorac Surg. 2010;90:22–29. doi: 10.1016/j.athoracsur.2010.03.093. [DOI] [PubMed] [Google Scholar]
- 17.Dandona P, Aljada A, Mohanty P, et al. Insulin inhibits intranuclear nuclear factor kappaB and stimulates IkappaB in mononuclear cells in obese subjects: evidence for an anti-inflammatory effect? J Clin Endocrinol Metab. 2001;86:3257–3265. doi: 10.1210/jcem.86.7.7623. [DOI] [PubMed] [Google Scholar]
- 18.Hansen TK, Thiel S, Wouters PJ, et al. Intensive insulin therapy exerts antiinflammatory effects in critically ill patients and counteracts the adverse effect of low mannose-binding lectin levels. J Clin Endocrinol Metab. 2003;88:1082–1088. doi: 10.1210/jc.2002-021478. [DOI] [PubMed] [Google Scholar]
- 19.Hartell NA, Archer HE, Bailey CJ. Insulin-stimulated endothelial nitric oxide release is calcium independent and mediated via protein kinase B. Biochem Pharmacol. 2005;69:781–790. doi: 10.1016/j.bcp.2004.11.022. [DOI] [PubMed] [Google Scholar]
- 20.Jeschke MG, Klein D, Herndon DN. Insulin treatment improves the systemic inflammatory reaction to severe trauma. Ann Surg. 2004;239:553–560. doi: 10.1097/01.sla.0000118569.10289.ad. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sato Y, Kita T, Takatsu Y, et al. Biochemical markers of myocyte injury in heart failure. Heart. 2004;90:1110–1113. doi: 10.1136/hrt.2003.023895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ou L, Li W, Liu Y, et al. Animal models of cardiac disease and stem cell therapy. Open Cardiovasc Med J. 2010;4:231–239. doi: 10.2174/1874192401004010231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sack MN, Yellon DM. Insulin therapy as an adjunct to reperfusion after acute coronary ischemia: a proposed direct myocardial cell survival effect independent of metabolic modulation. J Am Coll Cardiol. 2003;41:1404–1407. doi: 10.1016/s0735-1097(03)00164-5. [DOI] [PubMed] [Google Scholar]
- 24.Miki T, Miura T, Tanno M, et al. Impairment of cardioprotective PI3K-Akt signaling by post-infarct ventricular remodeling is compensated by an ERK-mediated pathway. Basic Res Cardiol. 2007;102:163–170. doi: 10.1007/s00395-006-0622-3. [DOI] [PubMed] [Google Scholar]
- 25.Gao F, Gao E, Yue TL, et al. Nitric oxide mediates the antiapoptotic effect of insulin in myocardial ischemia-reperfusion: the roles of PI3-kinase, Akt, and endothelial nitric oxide synthase phosphorylation. Circulation. 2002;105:1497–1502. doi: 10.1161/01.cir.0000012529.00367.0f. [DOI] [PubMed] [Google Scholar]