

Abstract
This study evaluates the correlation between natural killer (NK) cell function and human immunodeficiency virus (HIV)-1 disease progression in 133 untreated HIV-1 positive Chinese subjects, including 41 former plasma donors (FPDs) and 92 men who have sex with men, and 35 HIV-negative controls. Flow cytometry was used to determine the abundance of NK cell subsets, the expression levels of receptor species, human leucocyte antigen (HLA) genotyping and the antibody-dependent cell-mediated cytotoxicity (ADCC) responses of NK cells. We observed a decreased expression of CD56dimCD16+ NK cell subsets and an increased expression of CD56−CD16+ with HIV-1 infection. As well, the expression of activating and inhibitory receptors increased significantly in NK cells, but CD16 receptor levels and the NKG2A/NKG2C ratio were down-regulated with HIV-1 infection. ADCC responses were higher in elite controllers than in all other groups, and were correlated inversely with HIV-1 viral load but correlated positively with CD4 count only in FPDs. Furthermore, individuals infected for < 1 year have lower ADCC responses than those infected for > 1 year. We also observed a negative association between ADCC responses and viral load in those who carry the HLA-A*30/B*13/Cw*06 haplotype. The positive correlation between CD16 expression and ADCC responses and a negative correlation trend between CD158a and ADCC responses were also observed (P = 0·058). Our results showed that the ADCC response is associated with patients' disease status, receptor expression levels, infection time and specific HLA alleles, which indicates that ADCC may offer protective effects against HIV-1 infection.
Keywords: antibody-dependent cell-mediated cytotoxicity, human immunodeficiency virus type 1, human leucocyte antigen class I, natural killer cells, receptors
Introduction
The human immunodeficiency virus (HIV) directly targets and devastates the host's immune system; it is therefore paramount to examine ways in which the host may mount effective defences against HIV attacks. Natural killer (NK) cells are critically involved in several pathways that combat HIV infection, such as through secreting degranulated perforin and granzyme B to kill target cells; the Fas–FasL pathway to induce lysis of infected cells; production of cytokines to regulate immunity; and antibody-dependent cell-mediated cytotoxicity (ADCC) to lysis infected cells [1]. NK cells are classified into three subsets, CD3negCD16posCD56dim, CD3negCD56bright and CD3negCD16posCD56neg [2]. The ability of NK cells to mediate protection against viral infection depends not only on the relative abundance of each subset, but also on the composition of receptors on the NK cell surface: inhibitory NK receptors (iNKRs) and activating receptors [3]. During infections, the binding of NK cell receptors to ligands on the target cell can cause it to overcome inhibitory signals and become activated, culminating in the killing of the target cell [4]. As the effector cells of ADCC, NK cells can also be activated by the binding of its receptors (such as CD16) to the Fc portion of antibodies attached to the surface of target cells [1].
As described previously by Zhang et al., both the proportion of total NK cells and the cytotoxic subset are down-regulated during HIV-1 infection, but the expression of NKG2A and NKG2C on the NK cell surface is increased [5]. However, the exact composition and status of NK cell receptors during infection is still under debate. Some studies have indicated that the expression of activating receptors are decreased and inhibiting receptors are increased during progressive HIV infection [6], while others showed that inhibitory receptor levels are not elevated significantly [7]. In addition, NK cell receptors could bind specific human leucocyte antigen class I (HLA-1) molecules; the studies have shown that particular HLA alleles appear to be associated with stemming the progression of HIV-1 infection [8]. Jia et al. found that specific HLA alleles may be associated with restricted viral load in Chinese HIV-1 patients, and this may be due to Gag-specific cytotoxic T lymphocyte (CTL) responses; however, the study was focused solely on adaptive immune responses [9]. Recently, research found that NK cells are protective against HIV disease progression through combinations of killer cell immunoglobulin-like receptors (KIR) and their HLA class I ligands [10]. Parsons et al. reported that the KIR3DL1/HLA-Bw4 combinations have higher responsiveness on NK cells expressing the 3DL1 receptor, which mediate protection through lysis of HIV-infected cells [11]. Tiemessen et al. demonstrated that possession of one HLA-C1 allele and more KIR2DL/S genes was associated with increased magnitude of NK cell responses and significantly decreased HIV viral load [12].
Recently, a study by Gomez-Roman et al. demonstrated that antibody responses could inhibit HIV-1 replication, due at least partly to ADCC [13]. Indeed, there is still debate regarding the exact role of ADCC in HIV-1 infection. Some research has indicated that HIV replication is correlated negatively with ADCC response [14,15], but others have demonstrated a lack of correlation [16]. The inconsistencies in outcome may be due in part to differences in experimental measures (e.g. rapid fluorometric ADCC assay, intracellular 51chromium assay or intracellular cytokine staining (ICS) ADCC assay [17]).
Thus, there is currently no firm consensus on the function of NK cells during HIV-1 infection. At the same time, HIV-1 infection is impacted by multiple factors, including the viral genetic diversity, host–pathogen interactions and host immunological reactions. This study therefore sought to evaluate the correlation between ADCC and HIV-1 disease progression through examining the distribution of NK cell subpopulations and measuring receptor expression and specific ADCC responses in a chronically infected cohort of treatment-naive former plasma donors (FPDs). The infection time of this cohort was highly uniform (17–19 years), and all individuals were infected by a single closely related HIV-1 strain via unregulated commercial plasma donation practices in the mid-1990s. This cohort included 10 elite controllers, who maintained HIV viral load below detection for almost 10 years without therapy; the mechanism through which this occurs is not well understood, but is presumed to be associated with ADCC [18]. Furthermore, we examined the correlation between ADCC responses and HIV-1 viral load in FPDs with or without the HLA-1 alleles of interest. Finally, to determine whether the duration of infection would influence ADCC responses, we also recruited and analysed a cohort of HIV-1-positive men who have sex with men (MSM) infected for different lengths of time (3 months–5 years).
Methods
Subjects
We recruited 133 HIV-1-infected participants and 35 healthy HIV-negative controls. Infection was defined by HIV-1-specific antibody tests with enzyme-linked immunosorbent assay (anti-HIV ELISA kit; Wantai, Beijing, China) and confirmed by Western blot (anti-HIV-1 Western blot kit; Aoya, China). All HIV-positive individuals from Fuyang City (Anhui Province, China) and Beijing City were naive of therapy. The protocols used were approved by the Institutional Review Board of the National Center for AIDS/Sexually Transmitted Disease Control and Prevention, Chinese Center for Disease Control and Prevention. Subject recruitment was approved by an institutional human ethics committee. All subjects provided written informed consent before sample collection.
FPD
This cohort included 41 chronically infected HIV individuals from local clinics in Anhui Province of China, infected between 1992 and 1995 [19]. The subjects were divided into three groups according to their clinical states: elite controllers, non-controllers with high CD4+ count (CD4+high) and non-controllers with low CD4+ count (CD4+low) (Table 1).
Table 1.
Characteristics of the study participants
| Cohort | FPD | MSM | Healthy | ||||
|---|---|---|---|---|---|---|---|
| Group | CD4+low * | CD4+high * | EC* | < 1 year | 1–3 years | > 3 years | Healthy |
| No. | 10 | 21 | 10 | 45 | 33 | 14 | 35 |
| Age (years) | 41·9 ± 5·0 | 48·1 ± 7·2 | 46·5 ± 7·1 | 28·0 ± 7·1 | 31·9 ± 8·6 | 34·0 ± 7·1 | 30·1 ± 5·4 |
| Gender (female/male) | 5/5 | 8/13 | 8/2 | 0/45 | 0/33 | 0/14 | 18/17 |
| CD4+ count (cells/μl) | 276 ± 49 | 566 ± 186 | 613 ± 133 | 384 ± 173 | 363 ± 112 | 352 ± 258 | n.a. |
| Viral load (copies RNA/ml) | 12 657 ± 110 144 | 11 058 ± 289 153 | <LDL† | 35 611 ± 44320 | 22 227 ± 47376 | 25 996 ± 33 140 | n.a. |
| Infection time (years) | 15·8 ± 1·3 | 16·4 ± 0·6 | 16·4 ± 0·3 | 0·5 ± 0·2 | 2·0 ± 0·7 | 5·2 ± 2·4 | n.a. |
| HLA-A*30/B*13/Cw*06 no. (freq)‡ | 4 (40%) | 8 (38%) | 5 (50%) | n.a. | n.a. | n.a. | n.a. |
All data displayed as mean ± standard deviation (SD).
The former plasma donors (FPD) cohort is divided by CD4+ cell count and by viral load: CD4+low: non-controllers with viral load > 10 000 copies RNA/ml, CD4 decline > 50 cells per year, and CD4+ cells count < 350 cells/μl; CD4+high: non-controllers with viral load > 10 000 copies RNA/ml, CD4 decline > 50 cells per year, and CD4+ cells count > 350 cells/μl; EC: elite controllers who keep viral load < LDL over 10 years.
LDL: low detection limit = 40 copies RNA/ml.
Numbers and frequency of subjects who carried HLA-A*30/B*13/Cw*06 in three groups. HLA: human leucocyte antigen; MSM: men who have sex with men; n.a.: not available.
MSM
This cohort included 92 HIV-1 positive subjects. Subjects were divided into three groups according to the length of their infected time: < 1 year, 1–3 years and > 3 years (Table 1).
HIV-1 specific peptides
We used a 95% purified viral envelope peptide pool containing 20-mers (overlapping by 10 amino acids) from HIV-1 strain CN54 (Bio-scientific Co., Shanghai, China). Each peptide stock used was 1 mg/ml dissolved in RPMI-1640 with 10% dimethyl sulphoxide (DMSO), then diluted in RPMI-1640 to a working concentration of 1 μg/ml.
Sample processing
Blood samples were collected in sodium-heparin (anti-coagulated) and sent to the laboratory within 8 h; 100 μl of fresh whole blood was used for CD4+ T cell counting and 200 μl for the ADCC ICS assay. DNA was extracted from 300 μl whole blood using a commercial kit (Qiagen, Valencia, CA, USA). The plasma was separated and stored at −70°C for viral load assay. We also used peripheral blood mononuclear cells (PBMC) for the NK receptor phenotype assays, which were isolated by density-gradient centrifugation on Ficoll-Hypaque (Pharmacia, Stockholm, Sweden).
ADCC ICS assay
Whole blood samples were incubated at 37°C for 5 h with envelope (env) peptide pool. Brefeldin-A 5 μg/ml (Sigma, St Louis, MO, USA), GolgiStop 0·3 μg/ml and CD107a-allophycocyanin (APC) (BD Biosciences, Franklin Lakes, NJ, USA) were added according to protocol. Negative (DMSO 1 μg/ml; Sigma) and positive controls [phorbol-12-myristate-13-acetate (PMA) 1 μg/ml and ionomycin 1 μg/ml] were also included for each sample. Samples were surface-stained with live/dead dye Amcyan (Invitrogen, Carlsbad, CA, USA), CD14-Pacific Blue, CD3-peridinin chlorophyll (PerCP) and CD56-phycoerythrin (PE), then lysed, permeabilized and intracellularly stained with interferon (IFN)-γ-Alexa Fluor700 (BD). Data were acquired on Aria I and analysis was performed on FlowJo software (Tree Star Inc., Ashland, OR, USA).
NK phenotype assay
PBMC were surface-stained with monoclonal flow antibodies: live/dead dye Amcyan (Invitrogen), CD14-Pacific Blue, CD3-PE-Texas Red (Beckman Coulter, Brea, CA, USA), CD16-APC cyanin 7 (cy7), CD56-PEcy7, CD158a-fluorescein isothiocyanate (FITC), NKP46-APC (BD), NKB1-Alexa Fluor 700 (Biolegend, San Diego, CA, USA), NKG2A-PE and NKG2C- PerCP (R&D Systems, Minneapolis, MN, USA).
All sample data were acquired with fluorescence activated cell sorter (FACS) Aria I (BD); 3 × 105 lymphocytes were collected per sample. Data were analysed with FlowJo software (Tree Star Inc.).
CD4+ T cell count and viral load assay
Absolute CD4+ T cell count was obtained by a fluorescence activated cell sorter (FACS) Calibur TruCount tubes (BD) and Tritest reagents CD3-FITC/CD4-PE/CD45-PerCP (BD). Results were analysed by MultiSETTM software (BD). Plasma HIV viral load was also measured by fluorescent real-time polymerase chain reaction (PCR) (Cobas Amplicor; Roche, Basle, Switzerland) and analysed using Amplicor ultrasensitive assay (Roche). The RNA detection threshold was 40 copies/ml.
HLA class I typing
HLA genotyping was performed using sequence-specific PCR primers with a commercial typing kit [HLA-ABC sequence-specific priming (SSP) Morgan™ Kits, Texas Biosystems, Inc., TX, USA], according to the manufacturer's protocol.
Statistical analysis
Data analysis and figure production were performed using SigmaPlot version 10·0, SigmaStat version 3·5 (SPSS Inc., Chicago, IL, USA), GraphPad Prism version 5 (GraphPad Software Inc., San Diego, CA, USA) and Microsoft Excel 2007 (Microsoft Corp.). As our data did not pass normality tests, multiple comparison among groups was performed using the Dunn's t-test (Kruskal–Wallis one-way analysis of variance on ranks). When there were significant differences among groups (P < 0·05), Mann–Whitney U-tests were adopted to compare differences between groups, using a significance threshold of P < 0·05. Spearman's non-parametric rank test was used in the regression analysis of non-normal data, and Pearson's two-parametric correlation analysis was performed for normal data. In all analyses, we used a viral load value of 39 RNA copies/ml for samples with viral load below the limit of detection (40 RNA copies/ml). In order to investigate the interaction between HLA and ADCC, we standardized our continuous variables (ADCC, HLA) by subtracting the mean and dividing by the standard deviation, and fitted them into the generalized linear model (sas version 8·2).
Results
NK subsets changed in frequency during HIV-1 infection
Compared to healthy individuals, total NK cell count (Fig. 1c) in elite controllers showed no significant reduction, while NK cell counts for the non-controller groups were decreased significantly. In contrast, in all HIV-infected FPDs, the proportion of the CD16posCD56dim subset relative to total NK cells was decreased significantly (P < 0·001) compared to healthy controls. As well, the abundance of the CD16posCD56dim in the CD4+low group was reduced significantly compared with the other infected groups and with controls (Fig. 1d). Expression of the CD56bright subset was decreased in all HIV-positive groups compared to healthy controls (Fig. 1e). Moreover, the proportions of the CD16posCD56neg NK subset relative to total NK cells were higher in the HIV-positive groups than in healthy controls (P < 0·001), and was lower in elite controllers compared to the CD4+low group (Fig. 1f). Overall, we observed that the CD16posCD56dim subset of NK cells decreased and the CD16posCD56neg subset increased during HIV-1 infection, which is in line with previous research [20]. We also found that the prevalence of CD16posCD56neg NK cells relative to functional subsets was aggravated with decreasing CD4+ T cell count.
Fig. 1.
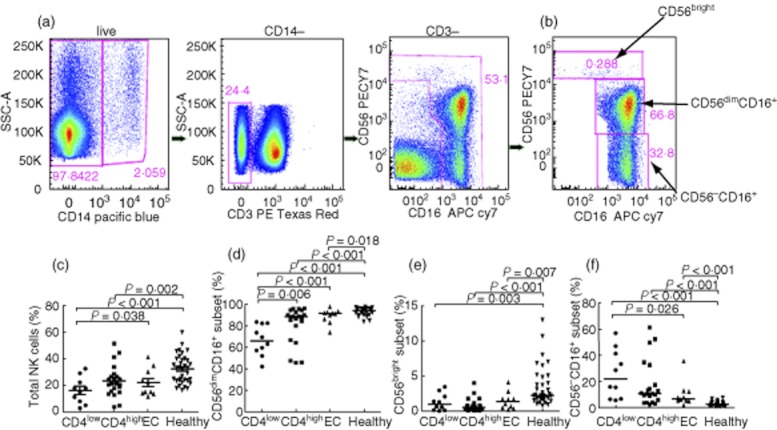
Comparison of natural killer (NK) cell subsets in the peripheral blood of human immunodeficiency virus(HIV)-infected patients and healthy controls. (a,b) Gating of NK cells subsets in representative subjects is shown. NK cells from chronic viraemic HIV-1-positive individuals [CD14negCD3neg peripheral blood mononuclear cells (PBMC) gate] were classified into three subsets: CD16posCD56dim, CD56bright and CD16posCD56neg. (c–f) The expression levels of total NK cells and the CD16posCD56dim, CD56bright and CD16posCD56neg subsets, respectively, in former plasma donors (FPDs) with different groups and in healthy controls.
NK receptor expression is altered during HIV-1 infection
Activating receptors
In chronically infected FPDs, the expression of the activating receptor NKG2C was significantly higher in total NK cells compared to healthy controls (P < 0·05); NKG2C levels were also higher in CD4+low non-controllers compared to elite controllers (P < 0·05; Fig. 2b). For the receptor NKP46, expression in total NK cells was higher in both non-controller groups compared to healthy controls (P < 0·001), and also higher in CD4+low non-controllers compared to elite controllers (P < 0·05) (Fig. 2c). Further, the expression of receptor CD16 in total NK cells in non-controllers was lower than in healthy controls, and significantly lower in CD4+low non-controllers compared to all other groups (Fig. 2d).
Fig. 2.
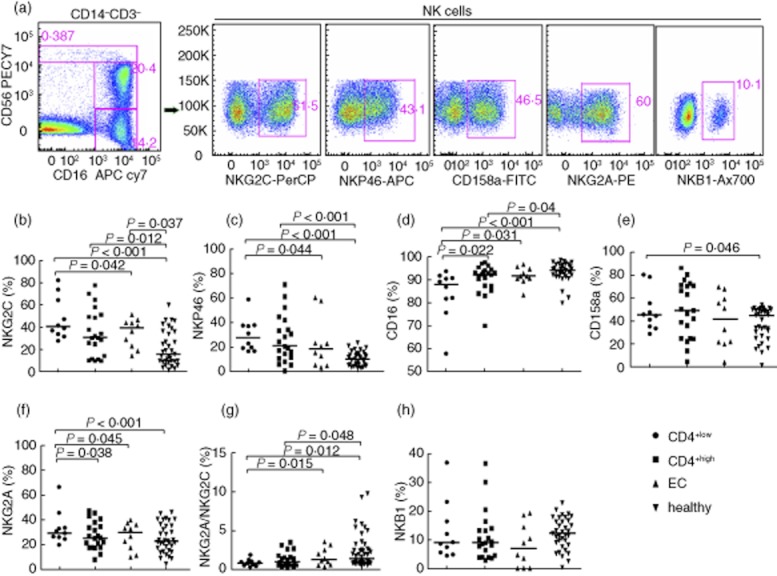
Natural killer (NK) cell receptor species were determined through flow cytometry using peripheral blood mononuclear cells (PBMC) from former plasma donors (FPDs). (a) Gating of NK cells (CD14negCD3neg CD56+/−CD16+/− lymphocyte gate) and NK cell receptors in representative subjects is shown. (b,c) NK activating receptors NKG2C and NKP46 were elevated in the total NK cells during human immunodeficiency virus(HIV)infection, and were highest in CD4+low non-controllers. (d) Expression of activating receptor CD16 was lower in total NK cells during HIV infection. (e,f) Inhibitory receptors CD158a and NKG2A were also elevated during HIV infection. (g) The ratio of NKG2A/NKG2C was lower in non-controllers compared to healthy controls. (h) Inhibitory receptor NKB1 was not different among groups; (b–h) marked with median expression levels.
Inhibitory receptors
The expression of receptor CD158a was higher in CD4+low non-controllers compared to healthy controls in total NK cells (Fig. 2e). The expression of inhibitory receptor NKG2A in total NK cells was increased significantly in the CD4+low non-controllers compared with the other three groups (Fig. 2f).
Interestingly, the ratio of NKG2A receptors (inhibitory) to NKG2C receptors (activating) on NK cells was lower in CD4+low non-controllers compared to either healthy individuals or elite controllers, whose ratio was usually above 1; the ratio in CD4+high non-controllers was also lower compared to healthy individuals (Fig. 2g). However, there was no difference of NKB1 among groups, but a reductive trend in infected individuals (Fig. 2h). In addition, we did not find a significant association between the altered expression of NK cells receptors during HIV-1 infection with either absolute CD4+ T cell count or HIV-1 viral load (data not shown).
We also analysed the expression level of receptors in each NK subset (Supplementary Fig. S1), and found the expression of all receptors in CD16posCD56dim were similar to those in total NK cells. There was no significant difference among different subgroups in the CD56bright subset. In the CD16posCD56neg subset, however, we observed that NKP46 expression was lower in the HIV-positive groups compared to healthy controls. In contrast, the expression of NKG2A was higher in the HIV-positive groups compared to healthy controls.
NK cells induced ADCC in chronically HIV-1-infected individuals
HIV-1-specific NK cell-induced ADCC response was characterized by CD107a or IFN-γ expression in NK cells (Fig. 3a). In response to stimulation by HIV-1 viral envelope peptides, CD107a or IFN-γ expression was significantly lower in non-controllers compared to elite controllers. A higher trend of ADCC responses was observed in the CD4high group compared with the CD4low group, although their difference was not statistically significant (Fig. 3b).
Fig. 3.
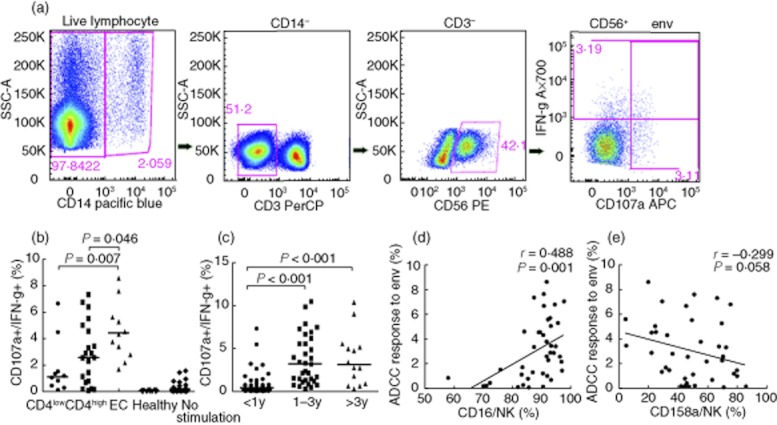
Human immunodeficiency virus(HIV)-1-specific antibody-dependent cell-mediated cytotoxicity (ADCC) in former plasma donors (FPDs) with different disease progressions. (a) ADCC response was identified by intracellular expression of CD107a or interferon (IFN)-γ in natural killer (NK) cells (gated CD14−CD3−CD56+) using flow cytometry. (b) In response to stimulation by HIV-1 viral envelope peptides, CD107a and IFN-γ expression was significantly lower in non-controllers compared to elite controllers in FPDs. (c) Among men who have sex with men (MSM), CD107a and IFN-γ expression was lower in individuals infected for < 1 year compared to those at 1–3 years or > 3 years of infection. (d) ADCC responses were correlated positively with CD16 expression levels in FPDs. (e) There was a general inverse trend between ADCC responses and CD158a expression in FPDs.
ADCC is correlated with NK cell receptor expression in FPDs
We analysed the correlation between ADCC responses and NK receptor expression, and found that only the expression of CD16 was associated positively with ADCC (Fig. 3d). In addition, we observed an inverse trend between ADCC responses and CD158a expression, although no statistically significant correlation was observed (P = 0·058) (Fig. 3e). No significant correlation between ADCC and NKB1 was observed (data not shown).
NK-induced ADCC is increased in individuals infected for longer than 1 year
We enrolled a separate cohort of HIV-1-infected MSM to examine the association between the duration of infected time and ADCC responses. Interestingly, the ADCC response was significantly lower in individuals infected for < 1 year compared to those at 1–3 years and > 3 years (average 5 years) of infection (P < 0·001). There was no significant difference between those infected for 1–3 years and those at > 3 years (Fig. 3c). Therefore, it is likely that the in-vivo maturation time of ADCC-inducing antibodies measures at about 1 year of HIV-1 infection, which requires further research to validate.
The correlation between ADCC response and disease progression is influenced by infected time
There was no significant association between ADCC responses and viral load or CD4+ cell count in the MSM cohort (Fig. 4a,b), which is infected with HIV-1 for a mean time of fewer than 5 years. However, in the long-term-infected FPD cohort, ADCC responses were correlated inversely with viral load (Fig. 4c), but correlated positively with CD4+ cell count (Fig. 4d).
Fig. 4.
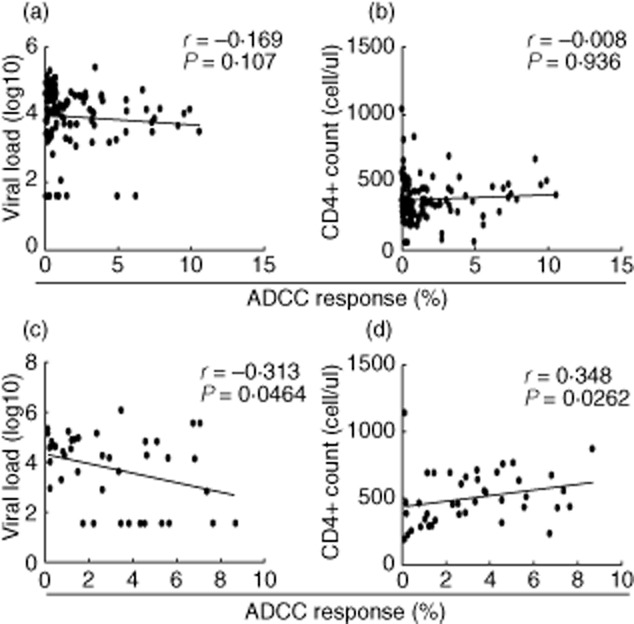
The correlation between human immunodeficiency virus(HIV)-1-specific antibody-dependent cell-mediated cytotoxicity (ADCC) and disease progression in cohorts with different infected time. (a,b) In men who have sex with men (MSM), ADCC responses had no correlation with viral load or CD4+ cell count. (c,d) Among former plasma donors (FPDs), ADCC responses were correlated inversely with viral load, but correlated positively with CD4+ cell count.
HLA alleles associated with ADCC responses
To determine the relationship between HLA-1 alleles and HIV-1 viral control, we assessed genotype distribution of the HLA class I allele in the FPD cohort. We found 11 variants for HLA-A, 16 for HLA-B and 10 for HLA-C; however, no statistically significant difference was detected in the distribution of these alleles among the elite controller and non-controller groups (details of HLA variants in Supplementary Table S1). Conversely, the presence of either the HLA-A*30, B*13 or Cw*06 alleles, as well as the presence of any or more than one of the three variants (HLA-A*30/B*13/Cw*06), were observed to have lower viral loads compared to non-carriers (P < 0·05) (Fig. 5a). Although the generalized linear model (R2 > 0·05) did not find a statistically significant difference between the ADCC responses of those with the HLA-A*30, B*13, Cw*06 alleles and those without (data not shown), there was a general trend of higher ADCC responses in those carrying one of the three variants (Fig. 5b). Furthermore, we divided FPDs into two groups according to whether or not they were carrying either the HLA-A*30, B*13 or Cw*06 alleles, and assessed if correlations between ADCC response and disease progression markers – such as viral load and CD4+ cell count – were affected by allele status. In addition, the negative correlation between ADCC response and viral load was found only in those with the HLA-A*30/B*13/Cw*06 alleles combination rather than those without (P < 0·05; Fig. 5c,d). Similarly, the positive correlation between ADCC response and CD4+ count was also observed only in those with the HLA-A*30/B*13/Cw*06 alleles of interest (Fig. 5e,f).
Fig. 5.
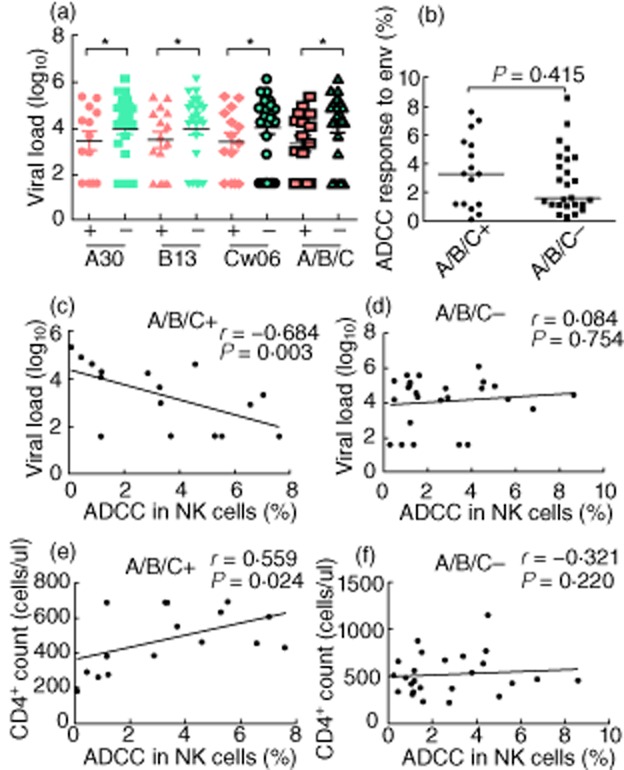
The correlation between human leucocyte antigen (HLA) class I alleles and human immunodeficiency virus (HIV) viral replication. (a) The viral loads in individuals with the HLA-A*30, B*13 or Cw*06 alleles (red), as well as those with any or more than one of the three variants, were significantly lower than those without these alleles (green) (P < 0·05). (b) Antibody-dependent cell-mediated cytotoxicity (ADCC) responses were not significantly different in individuals with any or more than one of the three variants, compared to those without these alleles. (c,d) Viral load is correlated negatively with ADCC response in patients with the HLA-A*30/B*13/Cw*06 alleles, but no correlation between viral load and ADCC responses in patients without these alleles. (e,f) Similarly, CD4+ cell count was correlated positively with ADCC response in patients with the HLA-A*30/B*13/Cw*06 alleles; no correlation was observed between viral load and ADCC responses in patients without these alleles.
Discussion
ADCC, as an essential means of NK cell activity against infection, is at least partly responsible for controlling HIV-1 viral replication. Indeed, some studies have shown that ADCC responses were associated with slower progression of HIV infection [1], although debate persists about this effect and its mechanisms. Our study demonstrated that ADCC responses are associated negatively with the clinical progression of HIV-1 infection and correlated positively with infected time and specific HLA alleles (HLA-A*30/B*13/Cw*06) in long-term survivors in a uniformly infected FPD cohort. This result indicates that ADCC may offer protective effects against HIV-1 infection. However, using an ADCC ICS assay, we did not observe similar correlations between ADCC and CD4 count or viral load in the MSM cohort. The differences in results may be due to differences in the homogeneity of infection time in each cohort. The FPD cohort was infected through a uniform route and time-period, while the MSM consisted of individuals infected for shorter and more varied periods of time. Furthermore, a study by Chung et al. using similar methodology had reported no correlation between ADCC response to env viral peptide pool and CD4 count or viral load in a patient cohort with diverse infection time and disease status [21]. This agrees with our results in the MSM cohort. Our study therefore constitutes the first detailed analysis of the ADCC response to HIV-1 in patient cohorts at different stages of disease progression with different infection times, using an ICS assay. It is also the first study in Chinese HIV-1-infected individuals of the effects of infection time and HLA haplotypes on ADCC responses induced by NK cells.
Our study first assessed the distribution of NK cell subpopulations in our cohort of elite controllers and non-controllers. Consistent with previous findings, we observed that the clinical progression of HIV is correlated with changes in the distribution of NK cell subpopulations, namely a decrease in CD16posCD56dim subsets and an increase in CD16posCD56neg subsets.
We next assessed the expression levels of several key activating and inhibitory NK receptors. In line with previous findings [5], we found the frequencies of activating receptors (including NKG2C) and inhibitory receptors (including NKG2A) both increased in chronic HIV-infected individuals. However, we also found that the ratio of NKG2A to NKG2C receptors decreased with advanced clinical progression of disease. Therefore, increased activation of NK cells (through modulated expression of NKG2A compared to NKG2C) may be a marker for indentifying the clinical course of HIV-1 infection.
Consistent with evidence from Ballan et al. [22], we observed the inhibitory killer immunoglobulin-like receptor CD158a (KIR2DL1) to be up-regulated in HIV-positive individuals. However, the expression of NKB1 (KIR3DL1) was not significantly different among the groups. There was a negative trend between CD158a expression and ADCC responses (P = 0·058), indicating that KIR2DL1 may inhibit ADCC. No statistically significant difference was found in correlation between ADCC responses and CD158a or NKB1 expression, which may be due to HIV-1 escape mutations, and failure to recognize some viral sequences by ADCC antibodies during disease progression. Ward et al. also found that the combination of HLA-C with CD158a, CD158b and CD159a receptors can inhibit ADCC induced by NK cells [23]. Meanwhile, we also found increased expression of the activating receptor NKP46 in patients with deteriorating clinical status. Conversely, De Maria et al. reported a correlation between decreased NK cell cytotoxic function and reduced expression of natural cytotoxicity receptors (NKp46, NKp30 and NKp44) in viraemic HIV-1 individuals [24]. The discrepancy in outcome may be attributed to the differences in the cohorts studied (e.g. the infective stages of HIV-1-positive subjects). The combined evidence suggests that the pathological effects of HIV-1 infection may include the disabling of NK cells through altering NK receptor expression, which inhibits cytotoxicity and protects infected CD4+ cells from destruction by NK cells.
Subsequently, we assessed the expression of the activating receptor CD16, an FcIII receptor implicated in protective immune responses against HIV-1 infection, through inducing ADCC and antibody-dependent cell-mediated viral inhibition (ADCVI) [25,26]. We observed a reduced expression of CD16 in non-controllers, which points to a link between attenuated ADCC response and deteriorated clinical progression of HIV infection in long-term survivors. We also observed a positive association between CD16 expression and ADCC responses. We therefore investigated the ADCC responses in patients at different stages of HIV-1 infection; to our knowledge, this was the first study of its kind among Chinese cohorts with this method. Our results showed that, along with the decrease of CD4+ counts and increase of viral loads, ADCC responses decreased in HIV-1-infected individuals with more severe clinical progression. ADCC antibodies usually exist at relative high titres in HIV-1-infected individuals and are correlated inversely with viral replication; functional antibodies are especially high in elite controllers [27]. Furthermore, HIV-1 controllers were known to have lower neutralizing antibodies but similar levels of binding antibodies compared with viraemic patients [27,28]. Thus, we hypothesized that ADCC responses play a predominant role in restricting HIV replication in elite controllers. Interestingly, Chung et al. found that ADCC antibodies play an important role in forcing HIV-1 escape mutations, such that more and more ADCC antibodies failed to recognize viral sequences during disease progression [29]. Some research has shown that rapid progressors had significantly lower titres of antibodies capable of mediating ADCC compared to those of non-rapid progressors [15,30]. This may explain our finding that ADCC responses were relatively high in CD4+high non-controllers, even as their viral loads differed widely.
In characterizing ADCC responses in HIV patients at different stages of disease, using a cohort of Chinese MSM, we also examined individuals who had been infected for different lengths of time. We found that individuals in the early days of their infection (< 1 year) had lower responses than those at > 1 year infection. This may be explained by the fact that HIV-specific ADCC responds to conformational viral epitopes presented in early HIV infection, and that responses broaden to include linear epitopes over time [31]. Therefore, we posit that the infection time duration of 1 year may be the turning-point for NK-induced ADCC to broaden its response specificity to include linear epitopes. Because ADCC responses may be induced by monocytes, which respond very weakly to the env peptide pool (data not shown), we consider the ADCC responses in the CD14−CD3−CD56+ population were induced mainly by NK cells.
Furthermore, we also found that ADCC's modulation of HIV replication may be associated closely with HLA-A*30/B*13/Cw*06 alleles, which is one of the main three-loci haplotypes in the Chinese Han population [32]. Unlike most research on HLA and HIV-1 infection, which examined T cell responses, our study focused on ADCC responses induced by NK cells. HLA-B*13, a HLA-Bw4 allele, was reported previously to be linked with successful control of disease progression [33,34]. HLA-Bw4 is the KIR3DL1 ligand, and NK cells can mediate ADCC against HIV-infected target cells through the interaction between KIR3DL1 and HLA-Bw4. The interaction induces NK cells to produce more cytokine and degranulation against target cells [11]. In addition, HLA-A*30 and Cw*06 were reported to be associated with lower viral loads [9]. HLA-Cw*06 is an HLA-C group 1 (C1) allotype, which plays an important role in reducing HIV viral load and increased HIV peptide-specific NK cell responses [11]. In contrast, the ligand for KIR2DL1/S1 is HLA-C2, which is not found to have any protective function against HIV infection. Higher expression of KIR2DL1 and lower NK functions are therefore observed in patients with more severe clinical status.
Furthermore, although ADCC responses were not statistically significantly higher in individuals with any or more than one of the HLA-A*30/B*13/Cw*06 alleles, none the less there was a general trend of higher ADCC responses compared to those without the alleles. We also demonstrated an amplified correlation between ADCC response and disease progression markers (i.e. CD4+ count and viral load) in patients carrying these alleles. While some studies have shown that HLA-B*27 and B*57 and HLA-C1 were associated with the control of clinical progression in HIV infection [12,35], our findings did not observe this association. This may be due to differences in both the genetic composition of the virus (i.e. different HIV-1 strains) and in the subjects (i.e. different ethnicity, etc.). In our peptide-specific ADCC assay, externally delivered peptides bound to HLA-1 molecules on antigen-presenting cells, and these complexes were recognized by specific KIR receptors on NK cells, ultimately inducing NK cell activation. The triggering of ADCC induced by NK cells was through the CD16 receptor on NK cells. Recently, peptide antagonism has been indicated as the reason of NK cell activation [36].
In conclusion, our study found a association between NK cell-induced ADCC responses and the clinical progression of HIV-1 infection. We hypothesize a model in which ADCC responses are low within 1 year of infection due to specificity to conformational viral epitopes, and become elevated with longer infection times as antibodies also respond to linear epitopes. Conversely, as the clinical progression of disease advances, both the distribution of functional NK cell populations and the composition of NK cell surface receptors become upset. Thus, NK cells fail to induce normal ADCC responses, which further aggravate HIV-1 disease progression. Meanwhile, ADCC responses may be relatively high in some individuals (such as CD4+high non-controllers) but none the less fail to reduce the HIV-1 viral load, which may be due to escape mutations in the viral genome as infection progresses.
In addition, we found that HIV-1-infected individuals with the HLA-A*30/B*13/Cw*06 haplotype exhibiting the ADCC response were capable of controlling viral replication effectively. While the exact mechanism of this effect is not well understood, it may point to potential methods for disrupting disease progression, with applicability in the development of HIV vaccines or gene therapy.
Acknowledgments
This project was funded by the National Mega Projects on Key Infectious Disease Control (2008ZX10001-010, 2012ZX10001-008) of China Ministry of Science and Technology, National Nature Science Foundation of China (81020108030), Key Project of State Key Laboratory for Infectious Disease Prevention and Control (2008SKLID101, 2011SKLID207). We thank Stephen De Rosa (University of Washington/Fred Hutchinson Cancer Research Center) and Jenny Huai Chen Hsi for critical editing of the manuscript.
Disclosure
None.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Figure S1. natural killer (NK) cell receptor expressions were determined in each NK subset from FPDs. (A,B) NK activating receptors NKG2C and NKP46 were elevated in the CD16posCD56dim NK subset during human immunodeficiency virus (HIV) infection. (C,D) Inhibitory receptors NKG2A and CD158a were also elevated in the CD16posCD56dim NK subset of HIV-positive individuals. No significant difference was found among different subgroups in the CD56bright subset except CD158a expression. In CD16posCD56neg subset, however, we observed lower expression of NKP46 in HIV-positive groups compared to healthy controls. In contrast, the expression of NKG2A was higher in HIV-positive groups compared to healthy controls, and the level of CD158a in CD4low group was also higher than healthy controls.
Table S1. Detailed information of human leucocyte antigen (HLA) alleles in 41 human immunodeficiency virus (HIV)-1 infected former plasma donors (FPDs).
References
- 1.Chung A, Rollman E, Johansson S, Kent SJ, Stratov I. The utility of ADCC responses in HIV infection. Curr HIV Res. 2008;6:515–519. doi: 10.2174/157016208786501472. [DOI] [PubMed] [Google Scholar]
- 2.Mavilio D, Lombardo G, Benjamin J, et al. Characterization of CD56−/CD16+ natural killer (NK) cells: a highly dysfunctional NK subset expanded in HIV-infected viremic individuals. Proc Natl Acad Sci USA. 2005;102:2886–2891. doi: 10.1073/pnas.0409872102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Moretta A, Bottino C, Vitale M, et al. Activating receptors and coreceptors involved in human natural killer cell-mediated cytolysis. Annu Rev Immunol. 2001;19:197–223. doi: 10.1146/annurev.immunol.19.1.197. [DOI] [PubMed] [Google Scholar]
- 4.Lanier LL. NK cell recognition. Annu Rev Immunol. 2005;23:225–274. doi: 10.1146/annurev.immunol.23.021704.115526. [DOI] [PubMed] [Google Scholar]
- 5.Zhang R, Xu J, Hong K, et al. Increased NKG2A found in cytotoxic natural killer subset in HIV-1 patients with advanced clinical status. AIDS. 2007;21(Suppl. 8):S9–17. doi: 10.1097/01.aids.0000304691.32014.19. [DOI] [PubMed] [Google Scholar]
- 6.Trinchieri G. Biology of natural killer cells. Adv Immunol. 1989;47:187–376. doi: 10.1016/S0065-2776(08)60664-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mavilio D, Lombardo G, Kinter A, et al. Characterization of the defective interaction between a subset of natural killer cells and dendritic cells in HIV-1 infection. J Exp Med. 2006;203:2339–2350. doi: 10.1084/jem.20060894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Martin MP, Qi Y, Gao X, et al. Innate partnership of HLA-B and KIR3DL1 subtypes against HIV-1. Nat Genet. 2007;39:733–740. doi: 10.1038/ng2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jia M, Hong K, Chen J, et al. Preferential CTL targeting of Gag is associated with relative viral control in long-term surviving HIV-1 infected former plasma donors from China. Cell Res. 2012;22:903–914. doi: 10.1038/cr.2012.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Boulet S, Kleyman M, Kim JY, et al. A combined genotype of KIR3DL1 high expressing alleles and HLA-B*57 is associated with a reduced risk of HIV infection. AIDS. 2008;22:1487–1491. doi: 10.1097/QAD.0b013e3282ffde7e. [DOI] [PubMed] [Google Scholar]
- 11.Parsons MS, Wren L, Isitman G, et al. HIV infection abrogates the functional advantage of natural killer cells educated through KIR3DL1/HLA-Bw4 interactions to mediate anti-HIV antibody-dependent cellular cytotoxicity. J Virol. 2012;86:4488–4495. doi: 10.1128/JVI.06112-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tiemessen CT, Paximadis M, Minevich G, et al. Natural killer cell responses to HIV-1 peptides are associated with more activating KIR genes and HLA-C genes of the C1 allotype. J Acquir Immune Defic Syndr. 2011;57:181–189. doi: 10.1097/QAI.0b013e3182174a76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gomez-Roman VR, Patterson LJ, Venzon D, et al. Vaccine-elicited antibodies mediate antibody-dependent cellular cytotoxicity correlated with significantly reduced acute viremia in rhesus macaques challenged with SIVmac251. J Immunol. 2005;174:2185–2189. doi: 10.4049/jimmunol.174.4.2185. [DOI] [PubMed] [Google Scholar]
- 14.Forthal DN, Gilbert PB, Landucci G, Phan T. Recombinant gp120 vaccine-induced antibodies inhibit clinical strains of HIV-1 in the presence of Fc receptor-bearing effector cells and correlate inversely with HIV infection rate. J Immunol. 2007;178:6596–6603. doi: 10.4049/jimmunol.178.10.6596. [DOI] [PubMed] [Google Scholar]
- 15.Johansson SE, Rollman E, Chung AW, et al. NK cell function and antibodies mediating ADCC in HIV-1-infected viremic and controller patients. Viral Immunol. 2011;24:359–368. doi: 10.1089/vim.2011.0025. [DOI] [PubMed] [Google Scholar]
- 16.Dalgleish A, Sinclair A, Steel M, Beatson D, Ludlam C, Habeshaw J. Failure of ADCC to predict HIV-associated disease progression or outcome in a haemophiliac cohort. Clin Exp Immunol. 1990;81:5–10. doi: 10.1111/j.1365-2249.1990.tb05283.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stratov I, Chung A, Kent SJ. Robust N. K cell-mediated human immunodeficiency virus (HIV)-specific antibody-dependent responses in HIV-infected subjects. J Virol. 2008;82:5450–5459. doi: 10.1128/JVI.01952-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Deeks SG, Walker BD. Human immunodeficiency virus controllers: mechanisms of durable virus control in the absence of antiretroviral therapy. Immunity. 2007;27:406–416. doi: 10.1016/j.immuni.2007.08.010. [DOI] [PubMed] [Google Scholar]
- 19.Li D, Chen J, Jia M, et al. Loss of balance between T helper type 17 and regulatory T cells in chronic human immunodeficiency virus infection. Clin Exp Immunol. 2011;165:363–371. doi: 10.1111/j.1365-2249.2011.04435.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bonaparte MI, Barker E. Inability of natural killer cells to destroy autologous HIV-infected T lymphocytes. AIDS. 2003;17:487–494. doi: 10.1097/00002030-200303070-00003. [DOI] [PubMed] [Google Scholar]
- 21.Chung AW, Navis M, Isitman G, et al. Activation of NK cells by ADCC antibodies and HIV disease progression. J Acquir Immune Defic Syndr. 2011;58:127–131. doi: 10.1097/QAI.0b013e31822c62b9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ballan WM, Vu BA, Long BR, et al. Natural killer cells in perinatally HIV-1-infected children exhibit less degranulation compared to HIV-1-exposed uninfected children and their expression of KIR2DL3, NKG2C, and NKp46 correlates with disease severity. J Immunol. 2007;179:3362–3370. doi: 10.4049/jimmunol.179.5.3362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ward JP, Bonaparte MI, Barker E. HLA-C and HLA-E reduce antibody-dependent natural killer cell-mediated cytotoxicity of HIV-infected primary T cell blasts. AIDS. 2004;18:1769–1779. doi: 10.1097/00002030-200409030-00005. [DOI] [PubMed] [Google Scholar]
- 24.De Maria A, Fogli M, Costa P, et al. The impaired NK cell cytolytic function in viremic HIV-1 infection is associated with a reduced surface expression of natural cytotoxicity receptors (NKp46, NKp30 and NKp44) Eur J Immunol. 2003;33:2410–2418. doi: 10.1002/eji.200324141. [DOI] [PubMed] [Google Scholar]
- 25.Florese RH, Demberg T, Xiao P, et al. Contribution of nonneutralizing vaccine-elicited antibody activities to improved protective efficacy in rhesus macaques immunized with Tat/Env compared with multigenic vaccines. J Immunol. 2009;182:3718–3727. doi: 10.4049/jimmunol.0803115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hessell AJ, Hangartner L, Hunter M, et al. Fc receptor but not complement binding is important in antibody protection against HIV. Nature. 2007;449:101–104. doi: 10.1038/nature06106. [DOI] [PubMed] [Google Scholar]
- 27.Lambotte O, Ferrari G, Moog C, et al. Heterogeneous neutralizing antibody and antibody-dependent cell cytotoxicity responses in HIV-1 elite controllers. AIDS. 2009;23:897–906. doi: 10.1097/QAD.0b013e328329f97d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pereyra F, Addo MM, Kaufmann DE, et al. Genetic and immunologic heterogeneity among persons who control HIV infection in the absence of therapy. J Infect Dis. 2008;197:563–571. doi: 10.1086/526786. [DOI] [PubMed] [Google Scholar]
- 29.Chung AW, Isitman G, Navis M, et al. Immune escape from HIV-specific antibody-dependent cellular cytotoxicity (ADCC) pressure. Proc Natl Acad Sci USA. 2011;108:7505–7510. doi: 10.1073/pnas.1016048108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Baum LL, Cassutt KJ, Knigge K, et al. HIV-1 gp120-specific antibody-dependent cell-mediated cytotoxicity correlates with rate of disease progression. J Immunol. 1996;157:2168–2173. [PubMed] [Google Scholar]
- 31.Chung AW, Navis M, Isitman G, et al. Activation of NK cells by ADCC responses during early HIV infection. Viral Immunol. 2011;24:171–175. doi: 10.1089/vim.2010.0108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yuan F, Sun YY, Luo Y, et al. [Haplotype and linkage analysis of HLA-I classical genes in Chinese Han population] Zhongguo Shi Yan Xue Ye Xue Za Zhi. 2007;15:1084–1089. [PubMed] [Google Scholar]
- 33.Harrer EG, Bergmann S, Eismann K, et al. A conserved HLA B13-restricted cytotoxic T lymphocyte epitope in Nef is a dominant epitope in HLA B13-positive HIV-1-infected patients. AIDS. 2005;19:734–735. doi: 10.1097/01.aids.0000166099.36638.56. [DOI] [PubMed] [Google Scholar]
- 34.Honeyborne I, Prendergast A, Pereyra F, et al. Control of human immunodeficiency virus type 1 is associated with HLA-B*13 and targeting of multiple gag-specific CD8+ T-cell epitopes. J Virol. 2007;81:3667–3672. doi: 10.1128/JVI.02689-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Carrington M, O'Brien SJ. The influence of HLA genotype on AIDS. Annu Rev Med. 2003;54:535–551. doi: 10.1146/annurev.med.54.101601.152346. [DOI] [PubMed] [Google Scholar]
- 36.Fadda L, Borhis G, Ahmed P, et al. Peptide antagonism as a mechanism for NK cell activation. Proc Natl Acad Sci USA. 2010;107:10160–10165. doi: 10.1073/pnas.0913745107. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.