

Abstract
Dietary I− absorption in the gastrointestinal tract is the first step in I− metabolism. Given that I− is an essential constituent of the thyroid hormones, its concentrating mechanism is of significant physiological importance. We recently described the expression of the Na+/I− symporter (NIS) on the apical surface of the intestinal epithelium as a central component of the I− absorption system and reported reduced intestinal NIS expression in response to an I−-rich diet in vivo. Here, we evaluated the mechanism involved in the regulation of NIS expression by I− itself in enterocytes. Excess I− reduced NIS-mediated I− uptake in IEC-6 cells in a dose- and time-dependent fashion, which was correlated with a reduction of NIS expression at the plasma membrane. Perchlorate, a competitive inhibitor of NIS, prevented these effects, indicating that an increase in intracellular I− regulates NIS. Iodide induced rapid intracellular recruitment of plasma membrane NIS molecules and NIS protein degradation. Lower NIS mRNA levels were detected in response to I− treatment, although no transcriptional effect was observed. Interestingly, I− decreased NIS mRNA stability, affecting NIS translation. Heterologous green fluorescent protein-based reporter constructs revealed a significant repressive effect of the I−-targeting NIS mRNA 3′ untranslated region. In conclusion, excess I− downregulates NIS expression in enterocytes by virtue of a complex mechanism. Our data suggest that I− regulates intestinal NIS mRNA expression at the post-transcriptional level as part of an autoregulatory effect of I− on its own metabolism.
Key points
Expression of the Na+/I− symporter (NIS) at the apical surface of the epithelium of the small intestine is key to I− absorption, the first step in I− metabolism.
Intracellular I− at high concentrations in enterocytes decreases its own NIS-mediated uptake by a newly discovered mechanism, downregulating NIS expression at the plasma membrane, increasing NIS protein degradation and decreasing NIS mRNA levels by reducing NIS mRNA stability, involving the NIS 3′-untranslated region.
In conclusion, we have uncovered that I− regulates intestinal NIS expression, and thus its own intestinal absorption, by a complex array of post-transcriptional mechanisms.
Introduction
The Na+/I− symporter (NIS) is an integral plasma membrane glycoprotein that mediates the active transport of I− into the thyroid follicular cells. This process constitutes the crucial first step in the biosynthesis of the only iodine-containing hormones in vertebrates, triiodothyronine (T3) and thyroxine (T4; Carrasco, 1993). Iodide is extremely scarce in the environment and is exclusively supplied to the body through the diet. Given that iodine is an essential component of the thyroid hormones, normal thyroid physiology relies on the proper function of NIS and on an adequate I− intake to prevent I− deficiency disorders (Dohán et al. 2003). Insufficient dietary I−, depending on its severity, causes hypothyroidism and subsequently goiter, stunted growth, retarded psychomotor development and even irreversible mental retardation (Zimmermann, 2009).
NIS also mediates active I− transport in some extrathyroidal tissues, including salivary glands, stomach, small intestine and lactating breast (Tazebay et al. 2000; Wapnir et al. 2003; Altorjay et al. 2007; Nicola et al. 2009a). The presence of NIS in the organism appears to be an adaptive mechanism to compensate for the environmental scarcity of I−. NIS avidly concentrates I− by coupling the inward transport of Na+ down its electrochemical gradient to the translocation of I− against its electrochemical gradient. Iodide transport mediated by NIS is electrogenic; two Na+ ions are transported with each I− (Eskandari et al. 1997). Remarkably, NIS translocates different substrates with different stoichiometries, as NIS-mediated transport of perrhenate or of the environmental pollutant perchlorate is electroneutral (1Na+ with one anion; Dohán et al. 2007; Paroder-Belenitsky et al. 2011).
Iodide absorption in the gastrointestinal tract is the first step in I− metabolism. Given the physiological importance of iodine, the question of where and how dietary I− is absorbed in the gastrointestinal tract has long been of major interest. Studies involving ligation procedures of the gastrointestinal tract suggested that I− might be absorbed in the small intestine (Josefsson et al. 2002). Consistent with this notion, we have recently described functional expression of NIS on the apical surface of the small intestinal epithelium and postulated NIS to be responsible for a major component of I− absorption (Nicola et al. 2009a).
Iodide is a key regulator of NIS expression and function (Carrasco, 1993; Dohán et al. 2003; Bizhanova & Kopp, 2009). High concentrations of I− downregulate thyroid function by inhibiting thyroid hormone biosynthesis, a phenomenon known as the Wolff–Chaikoff effect (Wolff & Chaikoff, 1948). This effect, in turn, is self-limiting, because it is followed hours later by an ‘escape’ from the Wolff–Chaikoff effect, i.e. the downregulation of I− uptake. This leads to lower intracellular I− concentration and thus protects the thyrocytes from the ill effects of I− overload (Braverman & Ingbar, 1963); however, the mechanism of the escape is not fully understood (Wolff et al. 1949; Eng et al. 1999). It has been associated with a decrease in NIS expression, and consequently I− transport as well (Eng et al. 2001). In thyroid cells, the control of NIS mRNA abundance by excess I− has mainly been linked to a transcriptional effect (Uyttersprot et al. 1997; Eng et al. 1999; Spitzweg et al. 1999). Nonetheless, recent evidence suggests that NIS regulation by I− takes place at a post-transcriptional level (Serrano-Nascimento et al. 2010, 2012; Leoni et al. 2011).
We have reported that an I−-enriched diet decreases NIS protein expression in small intestinal enterocytes (Nicola et al. 2009a); however, the mechanism involved in intestinal NIS regulation by I− remains to be elucidated. Here, we investigated this phenomenon in the small intestinal cell line IEC-6. In agreement with our previous observations, the intracellular accumulation of high concentrations of I− decreased NIS-mediated I− uptake in enterocytes by reducing NIS expression at the plasma membrane. Moreover, high concentrations of I− decreased NIS mRNA levels by lowering NIS mRNA stability. Interestingly, we observed that the NIS 3′ untranslated region is involved in I−-induced NIS mRNA decay. In conclusion, our data show that I− regulates intestinal NIS expression at the post-transcriptional level, providing a novel mechanism by which I− exerts an autoregulatory effect on its NIS-mediated intestinal absorption.
Methods
Reagents and antibodies
Bovine insulin, cycloheximide, chloroquine, amiloride and actinomycin D were purchased from Sigma-Aldrich (St Louis, MO, USA). MG132 was obtained from Calbiochem (San Diego, CA, USA). Affinity-purified anti-rat NIS polyclonal antibody was as described by Levy et al. (1997). Polyclonal antibody anti-green fluorescent protein (GFP) was from Clontech (Palo Alto, CA, USA). Monoclonal anti-human E-cadherin and anti-human α-tubulin antibodies were from BD Transduction (San Jose, CA, USA) and Sigma-Aldrich, respectively. Polyclonal anti-mouse α1-Na+–K+-ATPase antibody was from Upstate Biotechnology (Lake Placid, NY, USA). Anti-rabbit Alexa-488 and anti-mouse Alexa-594 secondary antibodies were obtained from Molecular Probes (Eugene, OR, USA). Secondary horseradish peroxidase (HRP)-conjugated antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
Cell culture and transient transfection assays
Intestinal epithelial cell line 6 (IEC-6) and 18 (IEC-18), derived from normal rat small intestinal epithelium, were purchased from American Type Culture Collection (Manassas, VA, USA). Cells were cultured in Dulbecco's modified Eagle's medium (Sigma-Aldrich) adjusted to contain 4.5 g l−1 l-glucose, 3.7 g l−1 sodium bicarbonate and 4 mm l-glutamine, and supplemented with 0.1 U ml−1 bovine insulin and 5% (v/v) heat-inactivated fetal bovine serum (Gibco, Langley, OK, USA).
Transient transfection procedures were performed by using Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA, USA) following the manufacturer's recommendations. Luciferase reporter assay system (Promega, Madison, WI, USA) was employed to evaluate luciferase activity. Transfection efficiency was normalized against the β-galactosidase activity from the cotransfected pCMV-β-Gal vector (Nicola et al. 2011).
Isolation of rat intestinal villus enterocytes
Male Sprague–Dawley rats (six animals per group) were given either distilled water (control group) or 0.05% potassium I−-supplemented water for 12–48 h (Eng et al. 1999; Nicola et al. 2009a). Both groups were subjected to overnight water deprivation before the start of treatment. Male C57BL/6 mice (five animals per group) were subjected to an iodine-deficient diet (TD.95007; Harlan-Teklad, Madison, WI, USA) for 1–4 weeks (Levy et al. 1997). Animals were killed by CO2 asphyxiation at the indicated time points. Animal protocols and procedures were carried out in compliance with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH publication no. 85-23, revised 1996) and local institutional animal care committee guidelines.
Villus tip intestinal epithelial cells were isolated as described (Nicola et al. 2009a). Briefly, small intestine was washed with a physiological solution. To isolate villus epithelial cells, the intestine was filled with a solution containing 1.5 mm KCl, 96 mm NaCl, 27 mm sodium citrate, 8 mm KH2PO4, and 5.6 mm Na2HPO4 (pH 7.3) for 10 min to dissociate cells. Then, the intestine was treated with PBS containing 1.5 mm EDTA and 0.5 mm DTT for 4 min, and released cells were collected.
Iodide uptake
An I− transport assay in IEC-6 cells was carried out as previously described (Nicola et al. 2009a,b). For steady-state uptake experiments, IEC-6 cells were incubated with culture medium or buffered Hanks’ balanced salt solution (HBSS) containing 100 μm I− supplemented with 5 μCi μmol−1 carrier-free Na125I for the indicated period of time in cell culture conditions. For initial-rate I− transport assays, cells were incubated for 2 min with 0.625–160 μm I− (50 μCi μmol−1 carrier-free Na125I), maintaining a constant 140 mm Na+ concentration, and uptake activity was evaluated as above. Experimental data were analysed using the equation: v = (Vmax*[I−])/(Km+[I−]) and fitted by non-linear least squares using Gnuplot software (http://www.gnuplot.info). The 125I− accumulated in the cells was extracted with ice-cold ethanol and quantified in a γ-counter. For standardization, the amount of DNA was determined by the diphenylamine method after trichloroacetic acid precipitation (Fozzatti et al. 2007).
To rule out a reduction of 125I− uptake caused by the remaining free I− from the treatment, cells were incubated with a tracer amount of 125I−, and washout experiments were performed in order to determine the minimal number of washing steps (12 washes, 1 min each) to ensure that no significant (<5%) amount of I− remained in the cells before performing uptake assays.
Iodide efflux
Cells were incubated in culture medium containing 100 μm I− supplemented with 5 μCi μmol−1 carrier-free Na125I for 45 min (control group) or 24 h at 37°C, and I− efflux was measured as described by Weiss et al. (1984). Briefly, the medium was removed and, after washing, it was replaced every 2 min with a volume of fresh HBSS at room temperature. After the last removal of medium, remaining intracellular I− was extracted with ice-cold ethanol. The total radioactivity originally present in the cell (100%) was calculated by adding the counts of the ethanol extraction to the summation of the counts in the collected samples of medium. Results are expressed as a percentage of the remaining intracellular I− as a function of time. Experimental data were analyzed using the one-component exponential decay equation [iI−t= iI−0*e(−k*t)], where iI−t is the amount of intracellular I− at time t; iI−0 is the value at t= 0, and k is the first-order decay constant.
Plasma membrane and whole protein analysis
Cell surface biotinylation and total protein extraction were performed as previously described (Levy et al. 1997; Dohán et al. 2006). Proteins were resolved by SDS-PAGE and electrotransferred to nitrocellulose membranes (Whatman, Clifton, NJ, USA). Membranes were blocked and incubated with 0.4 μg ml−1 affinity-purified anti-rat NIS or 0.5 μg ml−1 anti-GFP antibodies. Equal loading and purification controls were performed with monoclonal antibodies against α-tubulin (0.2 μg ml−1) or E-cadherin (0.2 μg ml−1). HRP-conjugated secondary antibodies were used at 0.1 μg ml−1. Proteins were visualized by the enhanced chemiluminescence Western blot detection system (Pierce, Rockford, IL, USA), as described by the manufacturer. Band intensities were measured densitometrically using ImageJ Software (National Institutes of Health, Bethesda, MD, USA).
Immunofluorescence analysis
IEC-6 cells were seeded onto poly-l-lysine-coated glass coverslips (BD BioCoat, San Jose, CA, USA). After treatment, cells were washed with PBS containing 0.1 mm CaCl2 and 1 mm MgCl2 and fixed with 2% paraformaldehyde for 15 min. Cells were permeabilized with 0.1% bovine serum albumin, 0.1% Triton X-100 in PBS containing 0.1 mm CaCl2 and 1 mm MgCl2 for 20 min, and incubated for 2 h with anti-rat NIS antibody (1 μg ml−1) and anti-α1-Na+–K+-ATPase antibody (1.5 μg ml−1) in permeabilization solution. Cells were washed three times for 10 min with PBS, incubated for 1 h in the dark with anti-rabbit Alexa-488 and anti-mouse Alexa-594 secondary antibodies (4 μg ml−1), and washed three times for 10 min with PBS. Coverslips were mounted onto slides with SlowFade-antifade reagent (Molecular Probes). Cells were visualized in a Bio-Rad Radiance 2000 laser scanning confocal microscope (Bio-Rad Laboratories, Hercules, CA, USA). Quantification of co-localization was performed on randomly selected images (n= 10) using ImageJ software with the co-localization analysis plug-in Manders’ coefficient.
RNA extraction and RT-qPCR
Total RNA purification and cDNA synthesis were performed as previously described (Fozzatti et al. 2007). The qPCR analysis was performed using an ABI Prism 7500 detection system (Applied Biosystems, Foster City, CA, USA) and SYBR green chemistry as previously described (Nicola et al. 2010). Gene-specific primer sets are described in Table 1. For relative quantification of changes in gene expression, the 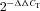 method was used (Livak & Schmittgen, 2001). The housekeeping genes β-actin or 18S ribosomal RNA were used as internal loading controls. For each pair of primers, a dissociation plot resulted in a single peak, indicating that a single PCR product was generated. Specific target amplification was confirmed by automatic sequencing (Macrogen Inc., Seoul, South Korea). All primers were from Sigma-Genosys (Houston, TX, USA).
method was used (Livak & Schmittgen, 2001). The housekeeping genes β-actin or 18S ribosomal RNA were used as internal loading controls. For each pair of primers, a dissociation plot resulted in a single peak, indicating that a single PCR product was generated. Specific target amplification was confirmed by automatic sequencing (Macrogen Inc., Seoul, South Korea). All primers were from Sigma-Genosys (Houston, TX, USA).
Table 1.
Primer sets used for RT-qPCR
| Gene | Primers (5′ to 3′) | Amplicon (bp) |
|---|---|---|
| NIS (r) | Forward GCTGTGGCATTGTCATGTTC | 270 |
| Reverse TGAGGTCTTCCACAGTCACA | ||
| NIS (m) | Forward AGCTGCCAACACTTCCAGAGG | 145 |
| Reverse GCTGATGAGAGCACCACAAAG | ||
| ALP (r) | Forward CTGCTGAGCAGGAATCCACA | 364 |
| Reverse TAGTTGACATCACCACTCTC | ||
| ALP (m) | Forward TGGCTGTCAAAGCATCAGGGA | 146 |
| Reverse TCTACTGAGGGGTTTCGGTTGG | ||
| β-Actin (r) | Forward GGCACCACACTTTCTACAATG | 138 |
| Reverse TGGCTGGGGTGTTGAAGGT | ||
| β-Actin (m) | Forward CAGCCTTCCTTCTTGGGTATGG | 145 |
| Reverse CAGCAATGCCTGGGTACATGG | ||
| 18S | Forward GGCCTGCGGCTTAATTTGAC | 243 |
| Reverse GTGGCTGAACGCCACTTGTC | ||
| Neo | Forward ACAATCGGCTGCTCTGATG | 260 |
| Reverse GATACTTTCTCGGCAGGAGC | ||
| GFP | Forward ACCACATGAAGCAGCACGAC | 93 |
| Reverse TGTAGTTGCCGTCGTCCTTG |
The species of origin for primer sequences are indicated in parentheses following each primer pair (m, mouse; and r, rat).
Cloning and cDNA constructs
The complete rat NIS mRNA 3′ untranslated region (3′-UTR) (nucleotides +1858 to +2761, considering +1 to be the adenine of the ATG translation start codon) was amplified by PCR from genomic DNA using primers containing XhoI and EcoRI restriction sites (underlined), namely 5′- CCTCGAGATGAGGGTGGGGTCCAAGAAG-3′ (forward) and 5′- ACAGGCGAATTCACCTGAACACCTTCAGC-3′ (reverse). The sequence was cloned into the cytomegalovirus promoter-driven green fluorescent protein (GFP) expression vector pEGFP-C2 (Clontech) at the 3′ end of the GFP cDNA before the polyadenylation signal. To avoid translation of the cloned 3′-UTR sequence, a TGA stop codon (indicated in italic) was included in the forward primer in frame with the GFP open reading frame. The cloned DNA sequence was verified by sequencing analysis (Macrogen).
Genomic DNA fragment –2854 to +13 bp of the rat NIS promoter (pNIS-2.8) was as previously described (García & Santisteban, 2002). The pCMV-β-galactosidase normalization reporter was obtained from Promega (Madison, WI, USA).
Analysis of NIS protein half-life (t1/2)
IEC-6 cells that were either untreated or incubated with 100 μm I− for 24 h were treated with 3 μm cycloheximide (CHX) to block protein synthesis. Whole protein extracts were prepared at the time of CHX addition (t= 0) and 3, 6, 12, 18, 24 and 48 h later. The NIS protein levels were evaluated by Western blot. Ponceau S staining was used to control for equal protein loading. Densitometric measurements at each time point were plotted relative to the amount present at the time of CHX addition (considered as 100%) as the logarithm of the percentage of remaining NIS vs. time. The best-fitting equation to linear decay was determined in each case, and NIS half-life was calculated from the intersection at the point corresponding to 50% residual protein.
Analysis of NIS mRNA half-life
After I− treatment for 3 h, actinomycin D (ActD) (5 μg ml−1) was added to cultures to inhibit RNA synthesis completely (Guo et al. 2003; Zou et al. 2005). Total cellular RNA was isolated at the time of ActD addition (t= 0) and 2, 4, 8, 12 and 18 h later. The mRNA levels of NIS and alkaline phosphatase (ALP) were measured by RT-qPCR and normalized to expression of 18S rRNA. The mRNA levels at different times were expressed relative to the amount present immediately before ActD addition, arbitrarily considered as 100%. To calculate the mRNA decay rate in each of the conditions, the logarithm of the remaining mRNA expression was plotted vs. time. Linear regression was used to obtain the best-fitting equation to calculate the mRNA half-life.
Statistical analysis
Results are reported as the means ± SD or SEM, as indicated. Statistical analysis was performed using GraphPad Prism (GraphPad Software, San Diego, CA, USA) from at least three independent experiments, unless stated. Multiple group analysis was conducted by one-way ANOVA. As a post hoc test, the Newman–Keuls multiple comparison test was used. Comparisons between two groups were made using two-tailed Student's unpaired t test. Non-normally distributed data derived from RT-qPCR assays performed in rodent intestinal tissues are given as medians ± range. Results were analysed by the non-parametric Kruskal–Wallis and Dunn's multiple comparison post hoc tests. Differences were considered statistically significant at P < 0.05.
Results
Iodide decreases NIS-mediated I− transport in intestinal cells
Iodide is one of the main regulators of its own NIS-mediated transport in the thyroid (Wolff & Chaikoff, 1948; Braverman & Ingbar, 1963; Grollman et al. 1986), and also in at least one NIS-expressing extrathyroidal tissue, the small intestine, as evidenced by our recent report that an I−-rich diet lowers NIS expression in small intestine enterocytes in vivo (Nicola et al. 2009a). We studied the regulatory effect of I− on steady-state NIS-mediated I− uptake in the small intestine IEC-6 cell line in vitro. Cells were incubated with 100 μm I− for different times, ranging from 30 min (control) to 48 h. Although we observed significantly lower I− transport 3 h after I− treatment, inhibition of transport became more pronounced with longer incubation times (Fig. 1A). We then investigated the effect of increasing concentrations of I−, from 1 to 1000 μm (after 24 h), on NIS activity in IEC-6 cells, and observed a dose-dependent inhibitory effect, starting at 10 μm and becoming more pronounced as the I− concentration increased (Fig. 1B). Similar results were obtained in the small intestine IEC-18 cell line (Fig. 1C).
Figure 1. Iodide decreases Na+/I− symporter (NIS)-mediated I− uptake.
A, steady-state I− uptake in IEC-6 cells incubated with 100 μm I− for 3–48 h. Uptake was expressed as picomoles of I− per microgram of DNA. Each value represents the mean ± SD of five independent experiments done in triplicate. *P < 0.05, **P < 0.005 vs. control (time 0 h; ANOVA and Newman–Keuls test). B, steady-state I− uptake in IEC-6 cells treated with 1–1000 μm I− for 24 h. The results are expressed as a percentage of non-I−-treated uptake levels for each I− concentration. Each value represents the mean ± SD of three independent experiments done in triplicate. *P < 0.05, **P < 0.01 vs. control (time 0 h; ANOVA and Newman–Keuls test). C, steady-state I− uptake levels in IEC-18 cells incubated with 100 μm I− for 24 h. Each value represents the mean ± SD of two independent experiments performed in triplicate. *P < 0.005 vs. control (ANOVA and Newman–Keuls test). D, I− efflux in IEC-6 cells treated with 100 μm I− for 24 h. Results are plotted as a percentage of the remaining intracellular I−. Decay constants (k) for control and I−-treated cells are indicated. E, initial rates (2 min time points) of I− uptake at different I− concentrations (ranging from 0 to 160 μm) in the presence of a constant Na+ concentration (140 mm). Data were analysed using the following equation: v= (Vmax*[I−])/(Km+[I−]) and fitted by non-linear least squares using Gnuplot software. Kinetic parameters were determined in triplicate and expressed as means ± SD. *P < 0.05 vs. control Vmax (Student's unpaired t test). F, IEC-6 cells were incubated in buffered Hanks’ balanced salt solution containing 100 μm I− in the presence or absence of 1 mm ClO4− for 6 h, where osmolarity was maintained by either Na+ or choline+. After treatment, cells were extensively washed to remove remaining free I−, and steady-state I− transport was assessed. Each value represents the mean ± SD of three independent experiments done in triplicate. *P < 0.01 vs. 100 μm I− (140 mm choline+; ANOVA and Newman–Keuls test).
Steady-state I− uptake is the net result of NIS-mediated I− influx and I− efflux mediated by channels or transporters that have yet to be identified (Kaminsky et al. 1993; Dohán et al. 2003). To study the effect of I− on I− efflux, IEC-6 cells were incubated with 100 μm I− for 45 min (control) or 24 h, and any remaining extracellular I− was washed from the medium, thus leading intracellular I− to efflux as a result of the outwardly directed I− gradient. No significant differences in the decay constants (k) of I− efflux were observed between the control and the I−-treated cells (κc, 0.121 ± 0.009 min−1 vs.κI−, 0.132 ± 0.011 min−1; Fig. 1D), indicating that the ability of I− to inhibit its own uptake results from decreased NIS-mediated I− influx only, as previously suggested (Sherwin & Tong, 1974; Grollman et al. 1986), not from higher I− efflux. Therefore, we carried out a kinetic analysis of NIS-mediated I− transport by measuring initial rates (2 min) of I− uptake at different concentrations of I− (0–160 μm) in IEC-6 cells previously incubated (or not) with 100 μm I− for 24 h (Fig. 1E). Cells were extensively washed after I− treatment to ensure that no I− remained inside, as described in the Methods. Saturation was reached at approximately 80 μm I− in both conditions; however, the apparent Vmax value was significantly lower in the I−-treated than in the control cells [Vmax(c): 277.1 ± 7.6 pmol (2 min)−1 (μg DNA)−1; and Vmax(i−): 177.7 ± 6.5 pmol (2 min)−1 (μg DNA)−1]. In contrast, the calculated apparent affinity values for I− remained unchanged (Km(c): 37.6 ± 3.2 μmi; and Km(i−): 32.9 ± 3.9 μm). No difference was observed in the apparent affinity for Na+ either (Km(c): 65.2 ± 4.1 mm; and Km(i−): 63.5 ± 6.1 mm).
Iodide inhibition of I− transport requires that I− be accumulated into the cell
The NIS-mediated I− transport is inhibited by perchlorate (ClO4−), a well-known competitive inhibitor and substrate of NIS (Dai et al. 1996; Dohán et al. 2007; Paroder-Belenitsky et al. 2011). In order to prevent NIS-mediated I− uptake, IEC-6 cells were incubated in the presence of both ClO4− and I− for 6 h, and cells were then extensively washed to remove intracellular I− before performing steady-state I− uptake experiments. In these conditions, we observed no inhibition of NIS activity, indicating that I− needs to reach the cytoplasm for the inhibitory effect of I− on its own NIS-mediated transport to occur (Fig. 1F).
The Na+ gradient is the driving force responsible for NIS-mediated I− transport (Dai et al. 1996; Eskandari et al. 1997). IEC-6 cells were cultured in Na+- or choline+-containing isotonic HBSS in the presence of I− for 6 h. After extensive washing, steady-state I− transport was examined. A significant I−-induced reduction of NIS activity was observed in the presence but not in the absence of Na+ (Fig. 1F).
Iodide reduces the expression of NIS at the plasma membrane
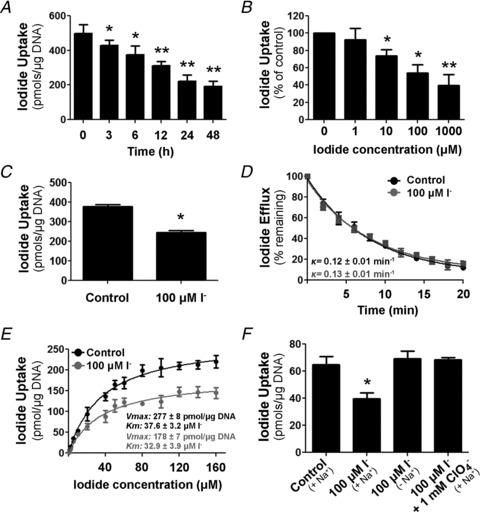
In order to analyse the mechanism of inhibition of NIS activity by I− further, IEC-6 cells were treated with 100 μm I− for different periods of time (3–48 h), and the total and plasma membrane NIS protein expression were assessed. Surface biotinylation assays revealed a significant time-dependent reduction of NIS expression at the plasma membrane 6 h after I− treatment (Fig. 2A, top panel); however, a decrease in total NIS protein was observed only 24 h after I− treatment (Fig. 2A, middle panel).
Figure 2. Iodide treatment decreases NIS plasma membrane expression.
A, IEC-6 cells were treated with 100 μm I− for different periods of time (0–48 h). Top panel, Western blot analysis of cell surface biotinylated proteins, showing NIS expression at the plasma membrane. Middle panel, immunoblot analysis of whole cell protein extract assayed for NIS expression. Bottom panel, Western blot assay of NIS levels in the intracellular fraction. Blots are representative of five independent experiments. Densitometric analysis was performed to determine relative NIS expression normalized to the loading control E-cadherin or α-tubulin, and indicated as fold change (FC) over NIS protein level in untreated cells, considered as 1.0. B, top panel, representative immunofluorescence staining of IEC-6 cells treated with 100 μm I− for the indicated periods of time. Merged images of NIS expression (green) and Na+–K+-ATPase (red), a well-characterized plasma membrane marker, are shown. Scale bars = 50μm. Bottom panel, Manders’ coefficient of co-localization of NIS and the Na+–K+-ATPase was determined for 10 randomly selected images per condition. *P < 0.05 vs. untreated cells (time 0 h; ANOVA and Newman–Keuls test). C, steady-state I− uptake in IEC-6 cells incubated with 1 mm I− for 6 h in the presence or absence of the endocytosis inhibitors chlorpromazine (CPZ), nystatin (Nys) or amiloride (AML). Uptake levels are expressed as picomoles of I− per microgram of DNA. Each value represents the mean ± SD of three independent experiments done in triplicate. *P < 0.05 vs. same condition in the absence of inhibitor; #P < 0.05 vs. control (ANOVA and Newman–Keuls test).
Interestingly, when assessed by immunofluorescence, NIS showed a time-dependent decrease in co-localization with the Na+–K+-ATPase and increased intracellular staining in response to excess I− (Fig. 2B). Given that only NIS at the plasma membrane accounts for I− uptake, these results provide a potential explanation for the rapid inhibition of NIS-mediated I− uptake by I−, i.e. that I− is likely to promote trafficking of plasma membrane NIS to intracellular compartments. We determined intracellular NIS protein levels by assaying the supernatant remaining after streptavidin-purified biotin-labelled surface proteins. Immunoblot analysis showed that NIS intracellular expression was augmented from 6 to 12 h after I− treatment. The amount of NIS decreased after 24 h, consistent with the observed reduction in total lysates (Fig. 2A, bottom panel). Next, we examined I−-induced NIS internalization in the presence or absence of different inhibitors of endocytic pathways, i.e. chlorpromazine (CPZ), nystatin (Nys) or amiloride (AML). We measured I− transport activity in cells treated with I− for 6 h (Fig. 2C). Treatment with AML increased I− transport in control cells, suggesting a constitutive internalization of NIS located at the plasma membrane through an AML-sensitive, macropinocytic mechanism. In addition, the inhibitory effect of I− on I− uptake was abolished by AML.
Iodide decreases intestinal NIS protein half-life
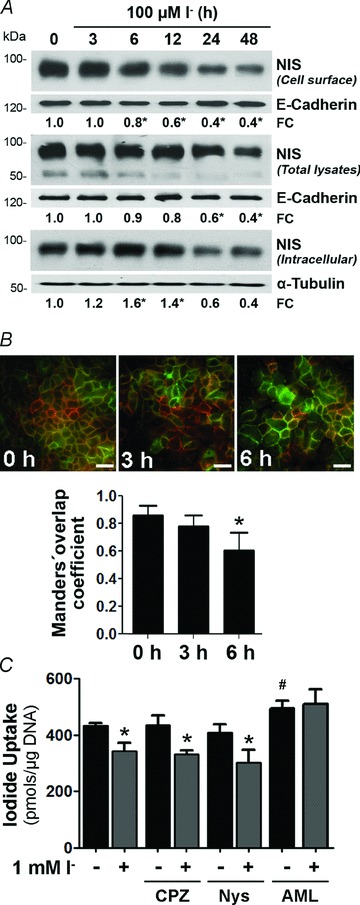
We assayed the effect of I− on NIS expression after cycloheximide (CHX) treatment in IEC-6 cells and found a reduction in NIS protein half-life in I−-treated cells compared with control cells (t1/2(i)−: 13.89 ± 4.66 h; and control t1/2(c): 21.65 ± 9.45 h; Fig. 3A), suggesting that an increase in NIS degradation is partly responsible for the lower I−-induced NIS protein expression in intestinal cells.
Figure 3. Iodide treatment increases NIS protein turnover through the lysosomal pathway.
A, IEC-6 cells were cultured in the presence of 100 μm I− for 24 h; thereafter, 3 μm cycloheximide (CHX) was added to the culture media, and total cell lysates were obtained at the indicated times. Top panel, representative Western blot analysis showing NIS expression after blocking protein synthesis. Bottom panel, values from the densitometric analysis were used to calculate the NIS protein half-life (t1/2). Results are expressed as the logarithm of the percentage of remaining NIS expression vs. time; NIS expression before CHX addition (t= 0) was taken as 100%. Ponceau S staining was used to correct loading differences (not shown). Each value represents the mean ± SEM of three independent experiments. *P < 0.05 vs. control t1/2 (Student's unpaired t test). B, representative Western blot of cell extracts. IEC-6 cells were treated with 5 μm MG132 (proteasome inhibitor) or 50 μm chloroquine (CQ; lysosomal inhibitor) in the presence of 100 μm I− for 24 h. Inhibitors were added to cell cultures 30 min before I−. C, steady-state I− uptake in IEC-6 cells treated with 5 μm MG132 for the indicated periods of time. Iodide uptake is expressed as picomoles of I− per microgram of DNA. Each value represents the mean ± SD of two independent experiments done in triplicate. *P < 0.05 vs. untreated cells (time 0 h; ANOVA and Newman–Keuls test).
In order to assess the proteolytic pathways involved in I−-induced NIS protein degradation, we incubated IEC-6 cells with lysosomal or proteasome inhibitors. The lysosomal inhibitor chloroquine did not have an effect on NIS levels, independently of the presence of I−. In contrast, the proteasome inhibitor MG132 markedly increased NIS protein expression levels, prevented the I−-induced reduction of NIS expression (Fig. 3B) and increased I− uptake (Fig. 3C).
Iodide reduces NIS mRNA levels in small intestine cells
The NIS mRNA levels were determined in IEC-6 cells after 100 μm I− treatment for different times (3–24 h). Quantitative PCR analysis demonstrated an I−-induced time-dependent reduction of NIS mRNA levels (Fig. 4A, left panel), without a significant change in the mRNA levels of the intestinal apical differentiation marker ALP (Fig. 4A, right panel).
Figure 4. Iodide decreases NIS mRNA levels in enterocytes.
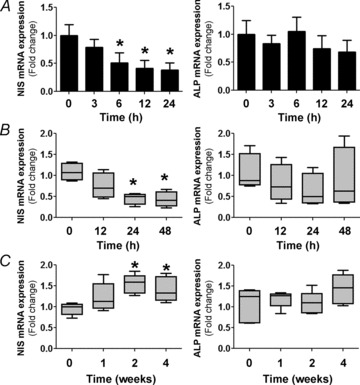
A, IEC-6 cell cultures were incubated with 100 μm I− for different periods of time. Quantitative PCR analysis was performed to quantify NIS and alkaline phosphatase (ALP) mRNA levels relative to those of β-actin. The expression level of untreated cells was set to 1. Values are indicated as fold change relative to the mRNA levels of untreated cells. *P < 0.01 vs. control (t= 0; ANOVA and Newman–Keuls test). B, male Sprague–Dawley rats (n= 6 per group) were treated with 0.05% I− in drinking water for the indicated periods of time. After treatment, villus tip small intestinal absorptive cells were isolated and qPCR analysis was performed to quantify mRNA levels relative to those of the loading control, β-actin. Data are indicated as the fold change relative to the mRNA levels of control animals (set as 1.0) and presented as box plots. *P < 0.05 vs. control (t= 0) (Kruskal–Wallis and Dunn's tests). C, male C57BL/6 mice (n= 5 per group) were subjected to an I−-deficient diet for 1–4 weeks. Quantitative mRNA analysis was performed as in B. *P < 0.05 vs. control (t= 0; Kruskal–Wallis and Dunn's tests).
We ascertained NIS mRNA expression in vivo in response to an I−-rich diet. Rats received drinking water supplemented with 0.05% I− for up to 48 h, whereas control rats received water without I− supplement. Small intestine epithelial cells from the villus tip were isolated as described previously (Nicola et al. 2009a) and further processed for total RNA extraction. Quantification of NIS mRNA levels showed a substantial reduction in enterocytes subjected to the I−-supplemented diet after 24 h (Fig. 4B, left panel), whereas I− had no effect on ALP mRNA expression (Fig. 4B, right panel), thus confirming the specificity of I− action as observed in IEC-6 cells. In a complementary experiment, we observed a significant increase in intestinal NIS mRNA levels in enterocytes of animals fed with an I−-deficient diet for 2 or 4 weeks (Fig. 4C, left panel), but no effect on ALP mRNA expression (Fig. 4C, right panel).
Intestinal NIS transcription is not modified by I−
We investigated a possible negative transcriptional effect exerted by I− on its own transporter. IEC-6 cells were transiently transfected with a luciferase reporter construct containing a 2854 bp DNA fragment of the rat NIS promoter (pNIS 2.8; García & Santisteban, 2002). Transiently transfected cells were treated with I− for 24 h. Although the NIS regulatory region showed a strong transcriptional activity in intestinal cells, I− did not modulate NIS expression at the transcriptional level (Fig. 5A).
Figure 5. Iodide regulates NIS mRNA levels at a post-transcriptional level.
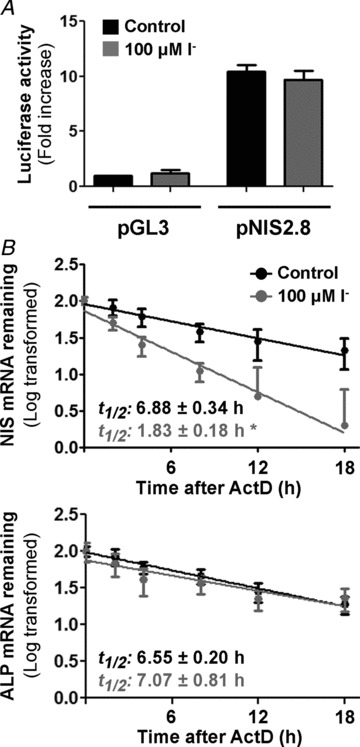
A, IEC-6 cells were transiently transfected with the empty vector pGL3 or the promoter construct pNIS-2.8 linked to the luciferase. Results are expressed as the fold change in luciferase activity, considering pGL3 levels as 1.0. β-Galactosidase activity was used to normalize transfection efficiency. Values are the means ± SD from triplicate samples of three independent experiments. *P < 0.001 vs. untreated pGL3-transfected cells (ANOVA and Newman–Keuls test). B, I−-treated IEC-6 cells were incubated with 5 μg ml−1 actinomycin D (ActD) for the indicated time periods. Total RNA was extracted and subjected to reverse transcription. Relative mRNA expression of NIS (top panel) and ALP (bottom panel) was analysed by RT-qPCR. 18S rRNA expression was used as a normalization control. Results are expressed as the logarithm of the percentage of remaining NIS mRNA expression vs. time; NIS expression before addition of ActD (t= 0) was defined as 1.0. Plotted values are the means ± SEM from three independent experiments. *P < 0.05 vs. control t1/2 (Student's unpaired t test).
Iodide strongly increases the NIS mRNA decay rate
Steady-state mRNA levels represent a balance between the rate of gene transcription and mRNA degradation. The reduction of NIS mRNA levels induced by I− was not due to a decrease in transcription. Thus, we tested NIS mRNA stability by assessing its half-life in IEC-6 cells. Cells were treated (or not) with 100 μm I− for 3 h and then incubated with the transcription inhibitor ActD for the indicated times. Total RNA was isolated, and NIS and ALP mRNA decay was followed by qPCR analysis. Consistently with our hypothesis, while I− significantly shortened NIS mRNA half-life by almost 75% (t1/2(c): 6.88 ± 0.34 h; and t1/2(i−): 1.83 ± 0.18 h; Fig. 5B, top panel), I− did not have an effect on the ALP mRNA (t1/2(c): 6.55 ± 0.20 h; and t1/2(i−): 7.07 ± 0.81 h; Fig. 5B, bottom panel).
Iodide-regulated NIS expression involves the NIS 3′ untranslated region
Untranslated regions of mRNAs play crucial roles in the post-transcriptional regulation of gene expression (Rana, 2007). In particular, 3′-UTRs harbour determinants that control mRNA stability and translation efficiency (Sonenberg & Hinnebusch, 2009; Jackson et al. 2010). We studied the effect of I− on a heterologous GFP reporter bearing the complete (nucleotides +1858 to +2761) NIS mRNA 3′-UTR sequence (pEGFP 3′-UTR) or on pEGFP as a control. IEC-6 cells were transiently transfected with the aforementioned reporters and treated with 100 μm I− for 24 h. Iodide had no effect on GFP expression in pEGFP-transfected cells but it markedly reduced GFP mRNA expression in pEGFP 3′-UTR-transfected cells (Fig. 6A). These findings were confirmed by Western blot analysis, showing that GFP protein expression also decreased in the pEGFP 3′-UTR-transfected cells compared with control cells in response to I− (Fig. 6B). Uniform transfection efficiency was ascertained by cotransfection with a β-galactosidase expression plasmid. These experiments demonstrate the functional importance of the NIS mRNA 3′-UTR sequence as a novel regulator of NIS mRNA stability.
Figure 6. Iodide-induced NIS repression targets NIS mRNA 3′ untranslated region (3′-UTR).
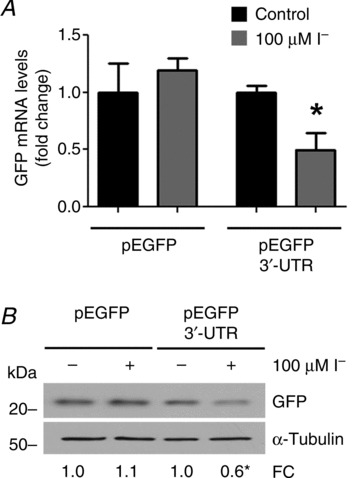
IEC-6 cells were transiently transfected with the heterologous GFP-based reporter pEGFP 3′-UTR, where NIS mRNA 3′-UTR sequence regulates GFP expression, or pEGFP empty vector as control. After transfection, cells were treated with 100 μm I− for 24 h. A, quantification of GFP mRNA levels relative to those of the pEGFP-encoded gene, neomycin resistance gene. The expression level of untreated cells was arbitrarily set to 1. Values representing the mean ± SD of three independent experiments are indicated as fold change relative to the mRNA levels of untreated cells. *P < 0.01 vs. untreated cells (Student's unpaired t test). B, representative Western blot analysis of whole cell extracts assayed for GFP expression. The housekeeping gene α-tubulin was used as loading control. β-Galactosidase activity was measured in order to evaluate equal transfection efficiency. Densitometric analysis was performed to determine the relative protein expression of GFP normalized to α-tubulin levels, and corrected by transfection efficiency. The relative protein expression of GFP in untreated IEC-6 cells was set arbitrarily to 1.0. The results represent the means of three independent experiments. *P < 0.05 vs. untreated cells (Student's unpaired t test).
Discussion
The absorption of I− in the small intestine is the first step in I− metabolism (Josefsson et al. 2002). We have reported that NIS, the mediator of active I− transport par excellence not only in the thyroid but also in other tissues, is functionally expressed on the apical surface of the small intestinal epithelium and, on the basis of this finding, we have suggested that NIS may play a key role in I− absorption in the small intestine (Nicola et al. 2009a). In agreement with our observation, proteomic analysis of mouse small intestine jejunum has revealed that NIS is a component of the jejunal brush border (Donowitz et al. 2007). The Na+–multivitamin transporter, a protein closely related to NIS that also transports I−, albeit with a lower affinity, has been proposed to provide a complementary pathway for I− absorption in the small intestine (de Carvalho & Quick, 2011).
The inhibition of NIS activity in the thyroid by high concentrations of I−, known as the ‘escape’ from the Wolff–Chaikoff effect, seems to be an adaptive response to reduce the intracellular I− content in thyrocytes and, in turn, to resume biosynthesis of thyroid hormones (interrupted by the Wolff–Chaikoff effect itself; Wolff & Chaikoff, 1948; Braverman & Ingbar, 1963; Grollman et al. 1986). We have previously reported that an I−-rich diet markedly decreases NIS protein expression and function in small intestine enterocytes in vivo (Nicola et al. 2009a). Here, we further examined the effect of I− on its own NIS-mediated uptake in the intestinal epithelial cell lines IEC-6 and IEC-18. We observed that I− treatment in these cells decreased NIS function in a dose-dependent fashion, like it does in thyroid cells (Grollman et al. 1986; Dohán et al. 2006). Our results indicate that the inhibition of NIS activity by I− results from lower I− influx rather than a modulation of I− efflux. Analysis of the kinetic parameters of I− transport demonstrated that I− at high concentrations did not affect the apparent affinity of NIS for either I− or Na+, but it did significantly lower the NIS Vmax (Fig. 1E), suggesting that the addition of high concentrations of I− results in a decreased number of functional NIS molecules at the plasma membrane.
In the thyroid, the presence of lower NIS-mediated I− uptake and the concomitant decrease in the intracellular I− concentration in the course of the ‘escape’ from the Wolff–Chaikoff effect suggest that the critical determinant for the escape is the intracellular rather than the blood concentration of I− (Raben, 1949; Serrano-Nascimento et al. 2010). We demonstrated that this is also the case in intestinal cells by showing that the inhibitory effect of I− on NIS activity was prevented by both the addition of the NIS inhibitor perchlorate and the absence of extracellular Na+.
Iodide at high concentrations has been reported to cause a decrease in NIS expression at the plasma membrane in thyroid cells (Dohán et al. 2006). Considering that I− uptake activity can be mediated only by NIS molecules located at the plasma membrane (Kaminsky et al. 1994; Riedel et al. 2001), a correlation can be discerned between the reduction in I− accumulation induced by I− and the corresponding lowering of NIS expression at the cell surface. Interestingly, we observed prompt recruitment of NIS from the plasma membrane to intracellular compartments upon I− treatment, supporting the existence of a rapid post-translational mechanism to reduce the number of functional NIS molecules at the plasma membrane. A similar phenomenon has been reported in thyroid cells in response to thyroid-stimulating hormone withdrawal (Riedel et al. 2001). Our data in intestinal cells are consistent with a pathway involving amiloride-sensitive endocytosis in the internalization of NIS induced by excess I−.
Reduced NIS protein expression in response to I− treatment has been demonstrated in different thyroid models (Eng et al. 1999, 2001; Li et al. 2007; Leoni et al. 2011; Serrano-Nascimento et al. 2012). Likewise, we observed a significant reduction in NIS protein in I−-treated IEC-6 cell lysates. Additionally, we demonstrated that I− reduced the NIS half-life, a mechanism that could be partly responsible for the significant decrease in NIS protein expression observed in IEC-6 cells in response to I− treatment (Fig. 3A). We estimated the half-life of NIS in untreated IEC-6 cells to be slightly shorter than 24 h, i.e. similar to that in the thyroid cell line FRTL-5 cells (Eng et al. 2001).
Little is known about the pathways that regulate NIS protein degradation in physiological conditions. Lysosome-mediated NIS protein degradation was reported in all-trans retinoic acid-stimulated MCF-7 human breast cancer cells (Beyer et al. 2011). Our results show that NIS protein turnover is regulated via the ubiquitin–proteasome system in enterocytes (Fig. 3B). Remarkably, blocking of proteasome-mediated degradation not only prevented the I−-induced reduction of NIS expression but also led to higher NIS protein levels and I− transport.
Several reports have demonstrated that excess I− promotes a reduction of NIS mRNA levels in thyroid cells in vivo and in vitro (Uyttersprot et al. 1997; Eng et al. 1999; Spitzweg et al. 1999; Leoni et al. 2008; Serrano-Nascimento et al. 2010, 2012; Leoni et al. 2011). Likewise, we observed a time-dependent reduction of NIS mRNA expression in response to I− treatment in IEC-6 cells. We have demonstrated that rats fed with a high-I− diet displayed lower and those fed a low-I− diet higher NIS mRNA expression in small intestine enterocytes in vivo (Fig. 4). These results indicate that a reduction of NIS mRNA is compatible with a decreased translational process, which in turn leads to reduced protein biosynthesis.
Transcriptional repression has been postulated to be responsible for the decrease in NIS mRNA levels induced by I− in thyroid cells (Eng et al. 1999; Leoni et al. 2008). However, and in agreement with recent observations in thyroid cells (Leoni et al. 2011), our results in IEC-6 cells demonstrate that I− does not regulate NIS transcriptional activity, although potential transcriptional regulation exerted outside the tested region cannot be entirely ruled out. As reported previously, thyroid-stimulating hormone-induced NIS expression is markedly reduced but not completely abolished in the presence of I− in thyroid cells, suggesting continued NIS biosynthesis even in the presence of high levels of I− (Eng et al. 2001).
Our data are consistent with the notion of a novel post-transcriptional regulation of NIS expression by its own substrate in enterocytes, because the half-life of the NIS mRNA in IEC-6 cells decreased from 6.9 to 1.8 h in the presence of I− (Fig. 5). Recent findings have suggested that a potential decrease in thyroid NIS mRNA stability or translation efficiency occurs as a result of a reduction of its poly-A tail length in response to I− administration (Serrano-Nascimento et al. 2010, 2012).
Iodide is like other trace elements that regulate genes encoding for proteins involved in their own transport or metabolism at the post-transcriptional level. For example, iron, selenium, zinc, calcium and phosphate regulate the mRNA abundance of transferrin receptor, glutathione peroxidase, zinc transporter ZnT5 and parathyroid hormone, respectively. The mechanisms that underlie this regulation involve protein interactions with mRNA UTRs, particularly 3′-UTRs (Owen & Kuhn, 1987; Bermano et al. 1996; Moallem et al. 1998; Erlitzki et al. 2002; Jackson et al. 2007; Nechama et al. 2009). Here, we used a GFP-NIS mRNA 3′-UTR (pEGFP 3′-UTR) chimeric gene to determine the possible role of I− in destabilizing the reporter gene. The insertion of NIS 3′-UTR resulted in a marked decrease in GFP mRNA and protein levels in I−-treated cells compared with control vector-transfected cells (pEGFP; Fig. 6), suggesting that the 3′-UTR sequence affected GFP mRNA stability rather than its transcription, given that both GFP-based reporters are controlled by the same promoter. Although the mechanism by which I− induces NIS mRNA decay remains unknown, our findings support the presence of functional cis-acting elements on the NIS mRNA 3′-UTR sequence related to the I− regulatory effect. In agreement with the growing importance of UTR sequences in the regulation of gene expression, we recently identified a homozygous –54C>T mutation in the NIS 5′-UTR region as being responsible for dishormonogenic congenital hypothyroidism due to reduced NIS mRNA translation efficiency (Nicola et al. 2011).
In conclusion, our data indicate that, by participating in a mechanism for tight control of NIS gene expression that is yet to be fully elucidated, I− plays an essential role in the physiology of enterocytes by controlling its own absorption and thus regulating the supply of I− to the body. In light of these findings, it is clear that NIS is a key molecule in the homeostatic management of inorganic I− in the organism.
Acknowledgments
We thank Dr Pilar Santisteban (Instituto de Investigaciones Biomédicas ‘Alberto Sols’, Madrid, Spain) and Dr Roberto Di Lauro (Università degli Studi di Napoli ‘Federico II’, Naples, Italy) for providing NIS promoter construct. We also thank Dr Cecilia Alvarez (CIBICI-CONICET) for providing pEGFP-C2 expression vector. We especially thank all members of our laboratories for providing technical assistance and many helpful discussions related to this work. This research was aided by awards from the Latin American Thyroid Society (to J.P.N.), and grants from Fondo Nacional de Ciencia y Tecnología (FONCyT), Secretaría de Ciencia y Tecnología de la Universidad Nacional de Córdoba (SeCyT), Agencia Córdoba Ciencia (to A.M.M.-R.) and NIH grant DK-41544 (to N.C.).
Glossary
- ActD
actinomycin D
- ALP
alkaline phosphatase
- AML
amiloride
- CHX
cycloheximide
- ClO4−
perchlorate
- CPZ
chlorpromazine
- GFP
green fluorescent protein
- HBSS
Hank's balanced salt solution
- IEC
intestinal epithelial cell line
- NIS
Na+/I− symporter
- Nys
nystatin
- 18S,
18S ribosomal RNA subunit
- t1/2
half-life
- 3′-UTR
3′ untranslated region
Author contributions
The experiments were performed at the Centro de Investigaciones en Bioquímica Clínica e Inmunología – Consejo Nacional de Investigaciones Científicas y Técnicas, Universidad Nacional de Córdoba and Department of Cellular and Molecular Physiology, Yale University School of Medicine by J.P.N. and A.R.-N. The study was designed by J.P.N., N.C. and A.M.M.-R. All authors wrote the paper and approved the final version of the manuscript.
Translational perspective
Iodide is an essential constituent of the thyroid hormones triiodothyronine (T3) and thyroxine (T4). Expression of the Na+/I− symporter (NIS) at the apical surface of the epithelium of the small intestine is a key component for I− absorption, the first step in I− metabolism. Here, we investigated the mechanism involved in the regulation of NIS expression by I− itself in enterocytes. We have discovered that I− regulates intestinal NIS expression, and thus its own intestinal absorption, by a complex array of post-transcriptional mechanisms. Excess I− downregulates NIS expression at the plasma membrane, increases NIS protein degradation and decreases NIS mRNA levels by reducing NIS mRNA stability and involving the 3′ untranslated region of NIS. This study underscores the physiological and regulatory significance of the apical localization of intestinal NIS, which contrasts with the basolateral localization observed in virtually all other tissues that express NIS, including the thyroid gland. It will be of interest to investigate whether the regulation of NIS-mediated I− absorption by I− itself in the small intestine is affected in patients with a variety of malabsorption syndromes, and to what extent NIS mutations that cause congenital I− transport defects in the thyroid impair the absorption of dietary I−.
References
- Altorjay A, Dohán O, Szilágyi A, Paroder M, Wapnir IL, Carrasco N. Expression of the Na+/I− symporter (NIS) is markedly decreased or absent in gastric cancer and intestinal metaplastic mucosa of Barrett esophagus. BMC Cancer. 2007;7:5. doi: 10.1186/1471-2407-7-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bermano G, Arthur JR, Hesketh JE. Role of the 3′ untranslated region in the regulation of cytosolic glutathione peroxidase and phospholipid-hydroperoxide glutathione peroxidase gene expression by selenium supply. Biochem J. 1996;320:891–895. doi: 10.1042/bj3200891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beyer S, Lakshmanan A, Liu YY, Zhang X, Wapnir I, Smolenski A, Jhiang S. KT5823 differentially modulates sodium iodide symporter expression, activity, and glycosylation between thyroid and breast cancer cells. Endocrinology. 2011;152:782–792. doi: 10.1210/en.2010-0782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bizhanova A, Kopp P. Minireview: The sodium-iodide symporter NIS and pendrin in iodide homeostasis of the thyroid. Endocrinology. 2009;150:1084–1090. doi: 10.1210/en.2008-1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braverman LE, Ingbar SH. Changes in thyroidal function during adaptation to large doses of iodide. J Clin Invest. 1963;42:1216–1231. doi: 10.1172/JCI104807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrasco N. Iodide transport in the thyroid gland. Biochim Biophys Acta. 1993;1154:65–82. doi: 10.1016/0304-4157(93)90017-i. [DOI] [PubMed] [Google Scholar]
- Dai G, Levy O, Carrasco N. Cloning and characterization of the thyroid iodide transporter. Nature. 1996;379:458–460. doi: 10.1038/379458a0. [DOI] [PubMed] [Google Scholar]
- de Carvalho FD, Quick M. Surprising substrate versatility in SLC5A6: Na+-coupled I− transport by the human Na+/multivitamin transporter (hSMVT) J Biol Chem. 2011;286:131–137. doi: 10.1074/jbc.M110.167197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dohán O, De la Vieja A, Carrasco N. Hydrocortisone and purinergic signaling stimulate sodium/iodide symporter (NIS)-mediated iodide transport in breast cancer cells. Mol Endocrinol. 2006;20:1121–1137. doi: 10.1210/me.2005-0376. [DOI] [PubMed] [Google Scholar]
- Dohán O, De la Vieja A, Paroder V, Riedel C, Artani M, Reed M, Ginter CS, Carrasco N. The sodium/iodide symporter (NIS): characterization, regulation, and medical significance. Endocr Rev. 2003;24:48–77. doi: 10.1210/er.2001-0029. [DOI] [PubMed] [Google Scholar]
- Dohán O, Portulano C, Basquin C, Reyna-Neyra A, Amzel LM, Carrasco N. The Na+/I− symporter (NIS) mediates electroneutral active transport of the environmental pollutant perchlorate. Proc Natl Acad Sci U S A. 2007;104:20250–20255. doi: 10.1073/pnas.0707207104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donowitz M, Singh S, Salahuddin FF, Hogema BM, Chen Y, Gucek M, Cole RN, Ham A, Zachos NC, Kovbasnjuk O, Lapierre LA, Broere N, Goldenring J, deJonge H, Li X. Proteome of murine jejunal brush border membrane vesicles. J Proteome Res. 2007;6:4068–4079. doi: 10.1021/pr0701761. [DOI] [PubMed] [Google Scholar]
- Eng PH, Cardona GR, Fang SL, Previti M, Alex S, Carrasco N, Chin WW, Braverman LE. Escape from the acute Wolff-Chaikoff effect is associated with a decrease in thyroid sodium/iodide symporter messenger ribonucleic acid and protein. Endocrinology. 1999;140:3404–3410. doi: 10.1210/endo.140.8.6893. [DOI] [PubMed] [Google Scholar]
- Eng PH, Cardona GR, Previti MC, Chin WW, Braverman LE. Regulation of the sodium iodide symporter by iodide in FRTL-5 cells. Eur J Endocrinol. 2001;144:139–144. doi: 10.1530/eje.0.1440139. [DOI] [PubMed] [Google Scholar]
- Erlitzki R, Long JC, Theil EC. Multiple, conserved iron-responsive elements in the 3′-untranslated region of transferrin receptor mRNA enhance binding of iron regulatory protein 2. J Biol Chem. 2002;277:42579–42587. doi: 10.1074/jbc.M207918200. [DOI] [PubMed] [Google Scholar]
- Eskandari S, Loo DD, Dai G, Levy O, Wright EM, Carrasco N. Thyroid Na+/I− symporter. Mechanism, stoichiometry, and specificity. J Biol Chem. 1997;272:27230–27238. doi: 10.1074/jbc.272.43.27230. [DOI] [PubMed] [Google Scholar]
- Fozzatti L, Vélez ML, Lucero AM, Nicola JP, Mascanfroni ID, Macció DR, Pellizas CG, Roth GA, Masini-Repiso AM. Endogenous thyrocyte-produced nitric oxide inhibits iodide uptake and thyroid-specific gene expression in FRTL-5 thyroid cells. J Endocrinol. 2007;192:627–637. doi: 10.1677/joe.1.06967. [DOI] [PubMed] [Google Scholar]
- García B, Santisteban P. PI3K is involved in the IGF-I inhibition of TSH-induced sodium/iodide symporter gene expression. Mol Endocrinol. 2002;16:342–352. doi: 10.1210/mend.16.2.0774. [DOI] [PubMed] [Google Scholar]
- Grollman EF, Smolar A, Ommaya A, Tombaccini D, Santisteban P. Iodine suppression of iodide uptake in FRTL-5 thyroid cells. Endocrinology. 1986;118:2477–2482. doi: 10.1210/endo-118-6-2477. [DOI] [PubMed] [Google Scholar]
- Guo H, Ray RM, Johnson LR. RhoA stimulates IEC-6 cell proliferation by increasing polyamine-dependent Cdk2 activity. Am J Physiol Gastrointest Liver Physiol. 2003;285:G704–G713. doi: 10.1152/ajpgi.00044.2003. [DOI] [PubMed] [Google Scholar]
- Jackson KA, Helston RM, McKay JA, O’Neill ED, Mathers JC, Ford D. Splice variants of the human zinc transporter ZnT5 (SLC30A5) are differentially localized and regulated by zinc through transcription and mRNA stability. J Biol Chem. 2007;282:10423–10431. doi: 10.1074/jbc.M610535200. [DOI] [PubMed] [Google Scholar]
- Jackson RJ, Hellen CU, Pestova TV. The mechanism of eukaryotic translation initiation and principles of its regulation. Nat Rev Mol Cell Biol. 2010;11:113–127. doi: 10.1038/nrm2838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josefsson M, Grunditz T, Ohlsson T, Ekblad E. Sodium/iodide-symporter: distribution in different mammals and role in entero-thyroid circulation of iodide. Acta Physiol Scand. 2002;175:129–137. doi: 10.1046/j.1365-201X.2002.00968.x. [DOI] [PubMed] [Google Scholar]
- Kaminsky SM, Levy O, Salvador C, Dai G, Carrasco N. The Na+/I− symporter of the thyroid gland. Soc Gen Physiol Ser. 1993;48:251–262. [PubMed] [Google Scholar]
- Kaminsky SM, Levy O, Salvador C, Dai G, Carrasco N. Na+/I− symport activity is present in membrane vesicles from thyrotropin-deprived non-I−-transporting cultured thyroid cells. Proc Natl Acad Sci U S A. 1994;91:3789–3793. doi: 10.1073/pnas.91.9.3789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leoni SG, Galante PA, Ricarte-Filho JC, Kimura ET. Differential gene expression analysis of iodide-treated rat thyroid follicular cell line PCCl3. Genomics. 2008;91:356–366. doi: 10.1016/j.ygeno.2007.12.009. [DOI] [PubMed] [Google Scholar]
- Leoni SG, Kimura ET, Santisteban P, De la Vieja A. Regulation of thyroid oxidative state by thioredoxin reductase has a crucial role in thyroid responses to iodide excess. Mol Endocrinol. 2011;25:1924–1935. doi: 10.1210/me.2011-0038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy O, Dai G, Riedel C, Ginter CS, Paul EM, Lebowitz AN, Carrasco N. Characterization of the thyroid Na+/I− symporter with an anti-COOH terminus antibody. Proc Natl Acad Sci U S A. 1997;94:5568–5573. doi: 10.1073/pnas.94.11.5568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Richard K, McKinnon B, Mortimer RH. Effect of iodide on human choriogonadotropin, sodium-iodide symporter expression, and iodide uptake in BeWo choriocarcinoma cells. J Clin Endocrinol Metab. 2007;92:4046–4051. doi: 10.1210/jc.2006-2358. [DOI] [PubMed] [Google Scholar]
-
Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the
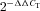 method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar] - Moallem E, Kilav R, Silver J, Naveh-Many T. RNA-Protein binding and post-transcriptional regulation of parathyroid hormone gene expression by calcium and phosphate. J Biol Chem. 1998;273:5253–5259. doi: 10.1074/jbc.273.9.5253. [DOI] [PubMed] [Google Scholar]
- Nechama M, Uchida T, Mor Yosef-Levi I, Silver J, Naveh-Many T. The peptidyl-prolyl isomerase Pin1 determines parathyroid hormone mRNA levels and stability in rat models of secondary hyperparathyroidism. J Clin Invest. 2009;119:3102–3114. doi: 10.1172/JCI39522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicola JP, Basquin C, Portulano C, Reyna-Neyra A, Paroder M, Carrasco N. The Na+/I− symporter mediates active iodide uptake in the intestine. Am J Physiol Cell Physiol. 2009a;296:C654–C662. doi: 10.1152/ajpcell.00509.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicola JP, Nazar M, Mascanfroni ID, Pellizas CG, Masini-Repiso AM. NF-κB p65 subunit mediates lipopolysaccharide-induced Na+/I− symporter gene expression by involving functional interaction with the paired domain transcription factor Pax8. Mol Endocrinol. 2010;24:1846–1862. doi: 10.1210/me.2010-0102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicola JP, Nazar M, Serrano-Nascimento C, Goulart-Silva F, Sobrero G, Testa G, Nunes MT, Muñoz L, Miras M, Masini-Repiso AM. Iodide transport defect: functional characterization of a novel mutation in the Na+/I− symporter 5′-untranslated region in a patient with congenital hypothyroidism. J Clin Endocrinol Metab. 2011;96:E1100–1107. doi: 10.1210/jc.2011-0349. [DOI] [PubMed] [Google Scholar]
- Nicola JP, Vélez ML, Lucero AM, Fozzatti L, Pellizas CG, Masini-Repiso AM. Functional Toll-like receptor 4 conferring lipopolysaccharide responsiveness is expressed in thyroid cells. Endocrinology. 2009b;150:500–508. doi: 10.1210/en.2008-0345. [DOI] [PubMed] [Google Scholar]
- Owen D, Kuhn LC. Noncoding 3′ sequences of the transferrin receptor gene are required for mRNA regulation by iron. EMBO J. 1987;6:1287–1293. doi: 10.1002/j.1460-2075.1987.tb02366.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paroder-Belenitsky M, Maestas MJ, Dohán O, Nicola JP, Reyna-Neyra A, Follenzi A, Dadachova E, Eskandari S, Amzel LM, Carrasco N. Mechanism of anion selectivity and stoichiometry of the Na+/I− symporter (NIS) Proc Natl Acad Sci U S A. 2011;108:17933–17938. doi: 10.1073/pnas.1108278108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raben MS. The paradoxical effects of thiocyanate and of thyrotropin on the organic binding of iodine by the thyroid in the presence of large amounts of iodide. Endocrinology. 1949;45:296–304. doi: 10.1210/endo-45-3-296. [DOI] [PubMed] [Google Scholar]
- Rana TM. Illuminating the silence: understanding the structure and function of small RNAs. Nat Rev Mol Cell Biol. 2007;8:23–36. doi: 10.1038/nrm2085. [DOI] [PubMed] [Google Scholar]
- Riedel C, Levy O, Carrasco N. Post-transcriptional regulation of the sodium/iodide symporter by thyrotropin. J Biol Chem. 2001;276:21458–21463. doi: 10.1074/jbc.M100561200. [DOI] [PubMed] [Google Scholar]
- Serrano-Nascimento C, Calil-Silveira J, Goulart-Silva F, Nunes MT. New insights about the posttranscriptional mechanisms triggered by iodide excess on sodium/iodide symporter (NIS) expression in PCCl3 cells. Mol Cell Endocrinol. 2012;349:154–161. doi: 10.1016/j.mce.2011.09.036. [DOI] [PubMed] [Google Scholar]
- Serrano-Nascimento C, Calil-Silveira J, Nunes MT. Posttranscriptional regulation of sodium-iodide symporter mRNA expression in the rat thyroid gland by acute iodide administration. Am J Physiol Cell Physiol. 2010;298:C893–C899. doi: 10.1152/ajpcell.00224.2009. [DOI] [PubMed] [Google Scholar]
- Sherwin JR, Tong W. The actions of iodide and TSH on thyroid cells showing a dual control system for the iodide pump. Endocrinology. 1974;94:1465–1474. doi: 10.1210/endo-94-5-1465. [DOI] [PubMed] [Google Scholar]
- Sonenberg N, Hinnebusch AG. Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell. 2009;136:731–745. doi: 10.1016/j.cell.2009.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitzweg C, Joba W, Morris JC, Heufelder AE. Regulation of sodium iodide symporter gene expression in FRTL-5 rat thyroid cells. Thyroid. 1999;9:821–830. doi: 10.1089/thy.1999.9.821. [DOI] [PubMed] [Google Scholar]
- Tazebay UH, Wapnir IL, Levy O, Dohan O, Zuckier LS, Zhao QH, Deng HF, Amenta PS, Fineberg S, Pestell RG, Carrasco N. The mammary gland iodide transporter is expressed during lactation and in breast cancer. Nat Med. 2000;6:871–878. doi: 10.1038/78630. [DOI] [PubMed] [Google Scholar]
- Uyttersprot N, Pelgrims N, Carrasco N, Gervy C, Maenhaut C, Dumont JE, Miot F. Moderate doses of iodide in vivo inhibit cell proliferation and the expression of thyroperoxidase and Na+/I− symporter mRNAs in dog thyroid. Mol Cell Endocrinol. 1997;131:195–203. doi: 10.1016/s0303-7207(97)00108-1. [DOI] [PubMed] [Google Scholar]
- Wapnir IL, van de Rijn M, Nowels K, Amenta PS, Walton K, Montgomery K, Greco RS, Dohán O, Carrasco N. Immunohistochemical profile of the sodium/iodide symporter in thyroid, breast, and other carcinomas using high density tissue microarrays and conventional sections. J Clin Endocrinol Metab. 2003;88:1880–1888. doi: 10.1210/jc.2002-021544. [DOI] [PubMed] [Google Scholar]
- Weiss SJ, Philp NJ, Grollman EF. Effect of thyrotropin on iodide efflux in FRTL-5 cells mediated by Ca2+ Endocrinology. 1984;114:1108–1113. doi: 10.1210/endo-114-4-1108. [DOI] [PubMed] [Google Scholar]
- Wolff J, Chaikoff IL. Plasma inorganic iodide as a homeostatic regulator of thyroid function. J Biol Chem. 1948;174:555–564. [PubMed] [Google Scholar]
- Wolff J, Chaikoff IL, Goldberg RC, Meier JR. The temporary nature of the inhibitory action of excess iodine on organic iodine synthesis in the normal thyroid. Endocrinology. 1949;45:504–513. doi: 10.1210/endo-45-5-504. [DOI] [PubMed] [Google Scholar]
- Zimmermann MB. Iodine deficiency. Endocr Rev. 2009;30:376–408. doi: 10.1210/er.2009-0011. [DOI] [PubMed] [Google Scholar]
- Zou T, Rao JN, Liu L, Marasa BS, Keledjian KM, Zhang AH, Xiao L, Bass BL, Wang JY. Polyamine depletion induces nucleophosmin modulating stability and transcriptional activity of p53 in intestinal epithelial cells. Am J Physiol Cell Physiol. 2005;289:C686–C696. doi: 10.1152/ajpcell.00085.2005. [DOI] [PubMed] [Google Scholar]