

Abstract
Troponin I (TnI) and myosin heavy chain (MHC) are two contractile regulatory proteins that undergo major shifts in isoform expression as cardiac myocytes mature from embryonic to adult stages. To date, many studies have investigated individual effects of embryonic vs. cardiac isoforms of either TnI or MHC on cardiac muscle function and contractile dynamics. Thus, we sought to determine whether concomitant expression of the embryonic isoforms of both TnI and MHC had functional effects that were not previously observed. Adult transgenic (TG) mice that express the embryonic isoform of TnI, slow skeletal TnI (ssTnI), were treated with propylthiouracil (PTU) to revert MHC expression from adult (α-MHC) to embryonic (β-MHC) isoforms. Cardiac muscle fibres from these mice contained ∼80%β-MHC and ∼34% ssTnI of total MHC or TnI, respectively, allowing us to test the functional effects of ssTnI in the presence of β-MHC. Detergent-skinned cardiac muscle fibre bundles were used to study how the interplay between MHC and TnI modulate muscle length-mediated effect on crossbridge (XB) recruitment dynamics, Ca2+-activated tension, and ATPase activity. One major finding was that the model-predicted XB recruitment rate (b) was enhanced significantly by ssTnI, and this speeding effect of ssTnI on XB recruitment rate was much greater (3.8-fold) when β-MHC was present. Another major finding was that the previously documented ssTnI-mediated increase in myofilament Ca2+ sensitivity (pCa50) was blunted when β-MHC was present. ssTnI expression increased pCa50 by 0.33 in α-MHC fibres, whereas ssTnI increased pCa50 by only 0.05 in β-MHC fibres. Our study provides new evidence for significant interplay between MHC and TnI isoforms that is essential for tuning cardiac contractile function. Thus, MHC–TnI interplay may provide a developmentally dependent mechanism to enhance XB recruitment dynamics at a time when Ca2+-handling mechanisms are underdeveloped, and to prevent excessive ssTnI-dependent inotropy (increased Ca2+ sensitivity) in the embryonic myocardium.
Key points
Slow skeletal troponin I (ssTnI) transgenic (TG) mice were treated with propylthiouracil (PTU) to induce a shift in myosin heavy chain (MHC) from the α- to β-MHC isoform, to understand how concomitant expression of these proteins affects cardiac muscle function.
Following PTU treatment, β-MHC expression increased to ∼80%, relative to α-MHC while TG ssTnI expression persisted at a level of ∼34% of total TnI.
ssTnI sped XB recruitment dynamics, and this increase was enhanced ∼3.8-fold in the presence of β-MHC when compared to ssTnI effects against α-MHC.
The ssTnI effect to increase myofilament Ca2+ sensitivity was blunted in the presence of β-MHC.
Our results provide new evidence for significant TnI–MHC interactions in their effects on cardiac function, which has major implications for coupling between concerted expression of contractile regulatory isoforms and the thick and thin filament-mediated tuning of cardiac contractile function.
Introduction
Myocardial contraction is initiated and shaped by highly regulated interactions between proteins in the thin-filament regulatory unit – consisting of actin, tropomyosin, and troponin (Tn) subunits – and cyclic interactions of myosin crossbridges (XBs) with actin. Because of such precisely coordinated interactions between Tn actions and XB cycling, myocardial contraction and relaxation dynamics can be significantly affected by alterations in Tn and myosin heavy chain (MHC) isoform expression. For example, we recently demonstrated that interactions between myosin heavy chain (MHC) and Tn isoforms play an important role in modulating myofilament contractile dynamics and cardiac contractile function (Chandra et al. 2006; Tschirgi et al. 2006; Chandra et al. 2007). This concept has major functional implications when considering that changes in cardiac Tn and MHC isoform expression exist across different species (Chandra et al. 2007), across muscle types within a species (Clemmens et al. 2005; Chandra et al. 2006), and also within a species during development. The latter cases have been studied, in part, using transgenic (TG) models to understand how expression of the slow skeletal isoform of troponin I (TnI) influences contractile function in adult mouse hearts (Fentzke et al. 1999; Arteaga et al. 2000; Wolska et al. 2001, 2002; Konhilas et al. 2003).
The slow skeletal isoform of TnI (ssTnI) is found in the mammalian heart during development from embryonic to neonatal stages (Sabry & Dhoot, 1989; Saggin et al. 1989; Bhavsar et al. 1991; Martin et al. 1991; Gao et al. 1995). The effects of ssTnI expression in the heart were studied by several investigators using a TG model in which the ssTnI isoform is overexpressed in the adult mouse heart. This TG mouse model was used to demonstrate that ssTnI expression in adult mouse hearts increases myofilament Ca2+ sensitivity (Fentzke et al. 1999), blunts muscle fibre length-induced increase in myofilament Ca2+ sensitivity (Arteaga et al. 2000; Konhilas et al. 2003), abolishes the PKA phosphorylation-induced decrease in Ca2+ sensitivity in cardiac muscle fibres (Konhilas et al. 2003), and reduces β-agonist induced enhancement of relaxation in intact hearts and isolated cardiac myocytes (Wolska et al. 2002; Peña & Wolska, 2004).
TnI, among other contractile proteins including MHC (Lompréet al. 1984; Siedner et al. 2003), undergoes major changes during the development of the mammalian heart. Investigators have studied whether developmental switching of other regulatory proteins, such as troponin T (Reiser et al. 1994; Gomes et al. 2004), and titin (Krüger et al. 2006), further modulates the functional effects of developmental switching of TnI. Previous studies suggest that, of the thin filament regulatory proteins, ssTnI imparts the dominant effect on myofilament responsiveness to Ca2+ (Metzger et al. 2003). Other studies suggest that MHC isoforms have a dominant effect on XB cycling dynamics (Fitzsimons et al. 1998b; Rundell et al. 2005b; Chandra et al. 2007). Collectively, these findings suggest that isoform-specific expression of thin- and thick-filament regulatory proteins act to regulate Ca2+- and XB-mediated contractile activation. Whether there is a meaningful interplay between the effects elicited by TnI and MHC on thin filaments, and how that relates to developmentally mediated tuning of heart function remain unaddressed.
To date, there is little understanding as to how developmental switching of MHC isoforms from slow β-MHC to fast α-MHC influences cardiac function in the context of developmental switching from ssTnI to cardiac TnI (cTnI). Such developmentally regulated changes in MHC isoform expression may bring about dynamic complementarity such that cardiac muscle contractile dynamics are tuned to match cardiac output requirements. For example, the slow cycling MHC isoform (β-MHC) predominates in embryonic and neonatal mouse hearts (Lompréet al. 1984), which have slow heart rates (60–120 bpm). The fast cycling MHC isoform (α-MHC) predominates in adult mouse hearts, which have fast heart rates (650–800 bpm). Since developmental switching of MHC isoform expression is accompanied by a major shift in TnI isoform expression (Saggin et al. 1989; Siedner et al. 2003), we sought to determine whether concomitant expression of β-MHC and ssTnI influenced the outcome of contractile function.
Given that the different isoforms of MHC differently influence Tn-dependent (Chandra et al. 2007) and disease-related (Lowey et al. 2008; Rice et al. 2010) effects on contractile dynamics and function, the main objective of this work was to study the effect of β-MHC-ssTnI interactions on contractile function and dynamics. The slower cycling β-MHC may amplify thin filament cooperativity through increased XB-dwell time in the strongly bound state. However, the cooperative/allosteric mechanisms in the thin filament are also strongly dependent on the nature of the troponin subunits (Chandra et al. 2006, 2007; Ford et al. 2010). Therefore, we tested the hypothesis that the ssTnI effect on myofilament contractile dynamics would be affected differently by a shift in MHC isoforms. ssTnI TG mice were treated with propylthiouracil (PTU) to shift the MHC isoform background from predominantly α-MHC to predominantly β-MHC in the mouse heart. Fortuitously for us, the level of ssTnI in cardiomyocytes remained high (34%), despite reemergence of cTnI expression (66%). This level of ssTnI expression is above that reported previously for ssTnI to confer its major effects on myofilament Ca2+ sensitivity (Metzger et al. 2003). Thus, muscle fibres from PTU-treated TG mouse hearts enabled us to study how ssTnI effects on contractile function are further affected by interplay with MHC.
One major finding in our study is that myofilament Ca2+ sensitizing effect of ssTnI is attenuated after the MHC isoform is shifted from α- to β-MHC. Another major finding is that the effect of ssTnI on the rate of muscle length-mediated XB recruitment is much more pronounced in the presence of β-MHC than it is in the presence of α-MHC. Collectively, these studies provide evidence of significant interplay between MHC and TnI isoform expression that is essential for matching and tuning of cardiac contractile function.
Methods
Ethical approval/animal treatment protocols
The treatment of animals used in these experiments followed the established guidelines of the Washington State University Institutional Animal Care and Use Committee. ssTnI TG mice (Fentzke et al. 1999; Kentish et al. 2001) were gifted by Dr John Solaro of The Department of Physiology and Biophysics, University of Illinois at Chicago. Breeding heterozygous mice resulted in a mixed litter of non-transgenic (NTG) and TG mice, which were genotyped using PCR. Both NTG (cTnI) and TG (ssTnI) young adults from this colony were either: (1) normal, non-treated mice that expressed native α-MHC isoform in the left ventricle, or (2) propylthiouracil-treated (PTU) mice (PTU administered in food (Harlan Teklad, Indianapolis, IN, USA) and water for 5–6 weeks), consequently resulting in the expression of β-MHC in the left ventricle.
Preparation of detergent-skinned cardiac muscle fibre bundles
Mice were anaesthetized by inhalation of isoflurane and hearts were quickly excised and placed into an ice cold relaxing solution containing 50 mm Bes, pH 7.0, 30.83 mm potassium propionate, 10 mm sodium azide, 20 mm EGTA, 6.29 mm MgCl2, 6.09 mm Na2ATP, 1.0 mm DTT, and 20 mm 2,3-butanedione monoxime (BDM). A fresh cocktail of protease inhibitors (4 μm benzamidine-HCl, 5 μm bestatin, 2 μm E-64, 10 μm leupeptin, 1 μm pepstatin and 200 μm PMSF) was added to all buffered solutions. Papillary muscle bundles were then carefully removed from the left ventricles of normal or PTU-treated NTG or ssTnI TG mouse hearts. Very small muscle fibre bundles (∼150–200 μm in width and 2.0 mm in length) were dissected from the papillary bundles and skinned overnight using 1% Triton X-100 in relaxing solution.
Polyacrylamide gel electrophoresis, Western blotting and examination of phospho-protein content
Protein samples from normal or PTU-treated NTG or ssTnI TG mouse left ventricles were prepared and separated using SDS-PAGE (Laemmli, 1970; Rundell et al. 2005b). Large, 6.5% polyacrylamide gels were used to assess MHC isoform content in normal and PTU-treated mice, and small 15% polyacrylamide gels were used to separate TnI expression. TnI was transferred from the 15% gel to a PVDF membrane, which was probed using an anti-TnI primary antibody (Fitzgerald M8010509), followed by anti-mouse secondary antibody (Amersham NIF825). Densitometric analysis was used to estimate the relative quantity of ssTnI expression in the normal and PTU-treated TG mouse hearts. To estimate phosphorylation status of myofibrillar proteins, 12.5% gels were stained with ProQ diamond staining solution (Invitrogen P33300, stain; P33310, destain). Gels were imaged using UV transillumination and Bio-Rad Chemi Doc XRS camera. ImageJ was used to compare band intensities to determine the phosphorylation status of the myofibrillar proteins in normal and PTU-treated TG and NTG mouse hearts.
Simultaneous measurement of isometric force and ATPase activity in detergent-skinned cardiac muscle fibres
Muscle fibres were mounted between a motor arm and force transducer using aluminum T-clips. Muscle fibre sarcomere length (SL) was determined using He–Ne laser diffraction measurements and adjusted to the desired SL of 2.2 μm following two to three repetitions of maximal contraction and relaxation. SL was monitored again by measuring laser diffraction after these repetitions. Next, the muscle fibre was activated by immersion in a 15 μl chamber (temperature controlled to 20°C) loaded with activation solutions that subsequently varied in pCa levels (pCa =−log10[Ca2+]). Motor-driven vibration of a membrane at the bottom of the bath kept the activation solution continuously stirred during muscle fibre contractions. Maximal activating solution (pCa 4.3) contained the following: 10 mm EGTA, 10.11 mm CaCl2, 6.61 mm MgCl2, 5.95 mm Na2ATP, 30.83 mm potassium propionate, 10 mm phosphoenolpyruvate, 0.5 mg ml−1 pyruvate kinase (∼350 U/mg, MP Biomedicals 151999), 0.05 mg ml−1 lactate dehydrogenase (∼700-1,200 U/mg, SIGMA L1254), 20 μm A2P5, and a cocktail of protease inhibitors. Relaxing solution (pCa 9.0) contained the same make-up as the pCa 4.3 solution except the following: 0.02416 mm CaCl2, 6.87 mm MgCl2, 5.83 mm Na2ATP, 51.14 mm potassium propionate.
Isometric tension production was measured during steady-state activation using a SensorOne AE-801 (Sausalito, CA, USA) force transducer and, simultaneously, ATPase activity was estimated as previously described (de Tombe & Stienen, 1995; Chandra et al. 2006). In brief, near-UV light was projected through a window in the activation chamber and the emergent beam was split for intensity detection at 340 nm and at 410 nm wavelengths. Light intensity of the emergent beam at 340 nm is sensitive to [NADH] in the bath, and served as an ATPase-dependent signal via an enzymatic coupling of ATP use and regeneration, and NADH oxidation reactions. Light intensity at 410 nm is insensitive to changes in [NADH], and served as a reference signal to the 340 nm intensity measurements. Intensity measurements were processed through an analog divider and a log amplifier to produce a signal proportional to the rate at which ATP was consumed by the muscle fibre during activation.
Muscle fibre mechano-dynamics
We studied the dynamic properties of the force–length relationship (FLR) during steady-state activation, by implementing a protocol which we have previously used successfully to elicit dynamic features of constantly activated muscle (Campbell et al. 2004; Chandra et al. 2006). Muscle length (ML) was perturbed using a constant-amplitude (±0.5% of ML) sinusoid with increasing frequency over time (chirp). The force response (ΔF(t); Fig. 1A) to changes in ML (ΔL(t); Fig. 1B) was measured and then fitted using a model described previously (Campbell et al. 2004; Chandra et al. 2007) to estimate contractile dynamics of the muscle. To emphasize low and high frequency force responses, two different chirps were administered during two sequential time periods. The first frequency sweep emphasized low frequencies (0.1 to 2 Hz) over a period of 40 s and the second frequency sweep emphasized higher frequencies (1 to 40 Hz) over a period of 5 s.
Figure 1. Representative chirp response data.
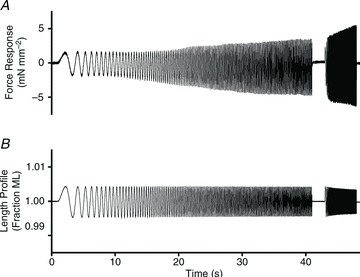
Representative force response (A) to chirp-length perturbation (B) from a maximally activated muscle fibre from control (untreated NTG) mouse cardiac muscle fibre
As we have done successfully in the past (Campbell et al. 2004; Chandra et al. 2007), we used a recruitment-distortion model of muscle contraction to analyse muscle fibre force responses to dynamic changes in ML. For the current study, we used the previously modified model which includes the additional distortion component capable of accounting for the consistently small, yet systematic error associated with length changes at higher frequencies (described in detail in Chandra et al. 2007). The model predicts of changes in force, ΔF(t), of a constantly activated muscle fibre in response to changes in length, ΔL(t), based on the following equation:
| (1) |
where η(t) is a variable that describes the dynamics of XB recruitment with changes in length, and x1(t) and x2(t) are strong- and weak-XB distortion variables, respectively. Each of these variables possesses units of length. E0, E∞, and D are scaling coefficients and possess the units of stiffness.
The dynamic XB recruitment variable, η(t), of the model-predicted ΔF(t) responds to ΔL(t) according to the differential equation:
| (2) |
where b is the rate constant associated with the speed of XB recruitment.
XB distortion variables, x1(t) and x2(t), respond dynamically to the first time derivative of muscle length, 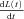 , according to the differential equations:
, according to the differential equations:
| (3) |
| (4) |
In brief, the three-component model was fitted to the entire record of ΔF(t) (Fig. 1A) using ΔL(t) (Fig. 1B) as an input and a non-linear regression technique was used to minimize the sum of square errors between the model prediction and the measured ΔF(t) record. A representative overall model prediction is shown in Fig. 2A and the individual contributions of each model component are shown in Fig. 2B–D.
Figure 2. Representative chirp response model prediction.
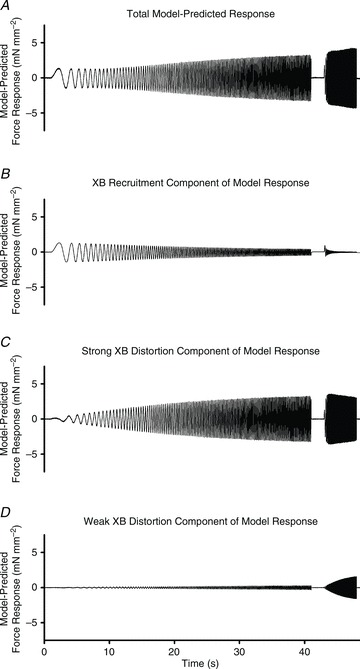
Representative model-predicted force response (A) to chirp-perturbation of the same responses from the WT fibre data shown in Fig. 1. Model components are shown to illustrate: the slow phase, XB-recruitment component of the force response (B); the fast phase, strong XB-distortion component of the response (C); and the fastest phase, weak XB-distortion component of the response (D). The shape and magnitude of the response shown in panel A represents the summation of each of the model components. The shape and magnitude of the components shown in panels B, C and D are dependent on the values of their respective parameters.
The E0η(t) model component is termed the recruitment component because at very low frequencies of perturbation, the velocity of length change is sufficiently slow to allow the fibre to recruit additional strong XBs in response to ΔL(t). This XB recruitment component, E0η(t), dominated ΔF(t) at the lowest frequencies (<1 Hz), but its contribution to the total response progressively decreased as frequency increased (Fig. 2B). Therefore, E0 is considered proportional to the number of strong XBs added to the cycling pool in response to length change, and b the rate by which XB recruitment takes place. As the frequency of perturbation increases, the slower XB recruitment mechanisms cannot match the speed of length change, and the contribution of XB recruitment diminishes (Fig. 2B).
The E∞x1(t) model component is termed the XB distortion component because at high frequencies of perturbation, the velocity of perturbation is sufficiently high that bound XBs are unable to cycle and experience strain. This XB distortion component, E∞x1(t), dominated ΔF(t) at frequencies >2 Hz and its contribution rose until reaching a near plateau at the 40 Hz end of the second chirp (Fig. 2C). This eventual rise and plateau is attributed to the assumption that at low frequencies of perturbation, most XBs detach and recycle before ΔL(t) can impose XB distortion, but at high frequencies of perturbation, bound XBs experience distortion and contribute to ΔF(t). Therefore, E∞ approximates the number and stiffness of strong-XBs, and c approximates the frequency at which XB distortion begins to contribute to ΔF(t). Because c determines the characteristic frequency of the rise in XB distortion, it is thought to reflect the intrinsic rate of XB detachment (Campbell et al. 2004; Chandra et al. 2007).
At the highest frequencies of perturbation, a small, but systematic rise in ΔF(t) was attributed to the distortion of weak XBs. The Dx2(t) XB distortion component made only small contributions to the force response and this contribution was prominent only at the highest frequencies (at frequencies approaching 40 Hz; Fig. 2D). In this study, however, the Dx2(t) weak XB distortion component contributed little to the prediction of the force response in the range of frequencies of interest (e.g. ∼0.1–2 Hz). As a result, the values of parameters E0, b, E∞ and c were largely unaffected by whether or not the Dx2(t)-distortion component was made part of the model, and we therefore report only the E0, b, E∞, c parameters, although parameters D and d were used as per eqns (1) and (4) for model fitting. Model parameters (E0, b, E∞, c) were typically estimated with less than 1% standard error, indicating that these estimated parameter values were uniquely estimated and reliable for comparing one muscle fibre with another. Model fits were routinely good (R2 > 0.9 in most cases), with normally distributed residuals, providing correlation between model-predicted and observed ΔF(t) that was consistently significant (P < 0.001 in most cases).
Rate of tension redevelopment
The rate constant of tension redevelopment (ktr), following rapid slackening and re-stretching of an activated muscle fibre, was as described previously (Brenner & Eisenberg, 1986; Chandra et al. 2007). During steady-state activation, the muscle fibre was slackened by 10% of ML within 1 ms and then held for 25 ms using a high-speed length control device (Aurora Scientific Inc., Aurora, ON, Canada). After this brief shortening period, the fibre was rapidly stretched past its original length by 10% for 1 ms to break any residually bound XBs. The fibre was then returned to its original ML within 1 ms, from which force began to redevelop. Tension redevelopment was fitted using a mono-exponential equation to give the rate constant ktr (s−1):
| (5) |
where F(t) is the force at time t, Fobs is the steady state redeveloped force, and F0 is the residual force from which tension begins to redevelop.
Data analysis
Mechano-dynamic parameters (E0, b, E∞, c and ktr) and contractile function parameters (Ca2+-activated maximal tension, maximal ATPase activity, pCa50, Hill coefficient (nH) and tension cost) of fibres from normal and PTU-treated NTG and TG mouse hearts were compared using two-way analysis of variance (ANOVA); one factor in this test was the strain of mouse (NTG = cTnI vs. TG = ssTnI) and the other was PTU treatment (normal =α-MHC vs. PTU-treated =β-MHC). Following two-way ANOVA, planned multiple pairwise comparisons were made using Fisher's LSD method. Comparisons were made to determine the effects of TnI isoform expression and PTU treatment on individual contractile function or contractile dynamic parameters. Hill's equation was fitted to data from normalized pCa–tension measurements using a least-square regression procedure to obtain pCa50 and nH. The XB recruitment–distortion model was fitted to force response data from the chirp-length perturbation as described above. Each contractile parameter was determined independently for each muscle fibre experiment and reported values are the mean of fitted parameters ± SEM, unless otherwise noted. The number of determinants (fibres) was at least 10, isolated from around three hearts, in each group. Asterisks in figure legends indicate significance from planned post hoc cell-wise comparisons.
Results
During development from prenatal to adult stages, mice undergo changes in expression of TnI and MHC isoforms; this change in protein expression coincides with a heart rate that increases as the mice mature. In this study, we hypothesized that these differences in isoform expression of TnI and MHC are linked to the contractile functional and dynamic changes that coincide with the change in cardiac output demands. To address our hypothesis, we studied the mechanical properties (i.e. pCa–tension responses, maximal tension/ATPase relationships, and contractile dynamics from frequency-dependent stiffness measurements) of cardiac muscle fibres from two groups of cardiac muscle fibres. The first group of fibres was from native NTG mice and ssTnI-expressing TG mice to determine how ssTnI isoform expression affects contractile function and dynamics against a background of α-MHC (to serve as a control). The second group of fibres was from PTU-treated NTG and TG mice to determine how the effect of ssTnI on muscle function and dynamics is affected when MHC isoform switches from α-MHC to β-MHC.
Effect of PTU treatment on MHC and ssTnI isoform expression and phosphorylation status of sarcomeric proteins in NTG and TG mouse hearts
In order to probe the effects of concomitant expression of ssTnI and β-MHC, we treated ssTnI TG mice with PTU to produce cardiac muscle where ssTnI is expressed against a background of β-MHC. PTU treatment in rodents disrupts thyroid function by inhibiting T3 and T4 thyroid hormone formation (de Tombe & ter Keurs, 1991; Rundell et al. 2005b), developmentally dependent transcription factors that up-regulate α-MHC expression and down-regulate β-MHC expression in adult hearts (Chizzonite & Zak, 1984; Haddad et al. 2010). Therefore, the thyrotoxic effect of PTU treatment down-regulates expression of the α-MHC promoter (predominantly expressed in adult mice), thereby causing re-expression of β-MHC in the myocardium (Pope et al. 1980; Rundell et al. 2005b; Chandra et al. 2007).
As shown in Fig. 3A, PTU treatment resulted in a switch from predominantly α-MHC to predominantly β-MHC in both NTG and TG mice; densitometric estimation of the relative expression of β-MHC was ∼75% total MHC in both PTU-treated NTG and TG hearts. Since the α-MHC promoter is used to control ssTnI expression in TG mouse hearts, we explored the possibility that PTU treatment would result in suppression of ssTnI expression in the TG mouse hearts. While re-expression of the cTnI isoform was seen in the PTU-treated ssTnI TG mouse hearts, fortuitously for us, ssTnI expression substantially persisted (Fig. 3B). Densitometric estimation of the comparative levels of cTnI:ssTnI (vs. total TnI) in the normal ssTnI TG mice was ∼15%:85%, whereas in PTU-treated TG mice, this ratio was ∼66%:34%. Thus, while cTnI isoform re-emergence was observed, significant amounts of ssTnI remained in PTU-treated ssTnI TG mice. Next, we examined whether PTU treatment or ssTnI TG expression had any effects on the phosphorylation status of other sarcomeric proteins (Fig. 3C). Pro-Q Diamond staining revealed that the phosphorylation status of other proteins (e.g. cardiac myosin binding protein C, cTnT, tropomyosin, myosin light chain 1 (MLC1), MLC2, or cardiac TnC) was not different in normal and PTU-treated mice. An exception was found in the re-emergence of phospho-cTnI in PTU-treated TG mice, but this was consistent with the ∼66% re-emergence of cTnI. For convenience, normal NTG or TG groups will be referred to as α-MHC(cTnI) or α-MHC(ssTnI), and PTU-treated NTG or TG groups will be referred to as β-MHC(cTnI) or β-MHC(ssTnI), respectively.
Figure 3. Assessment of sarcomeric protein content and phosphorylation status.
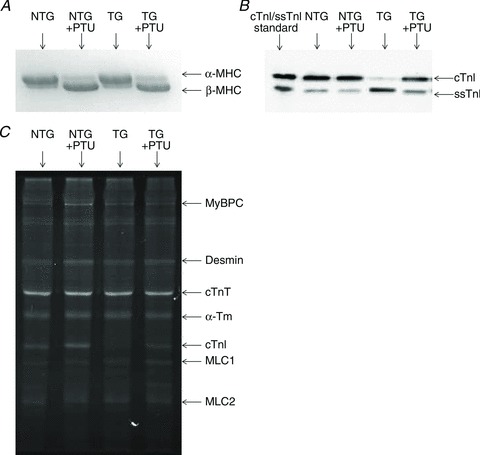
SDS-PAGE to determine the expression profiles of MHC isoform (A) and Western blot analysis to determine level of TnI isoform expression (B) of hearts from normal (α-MHC) or PTU-treated (β-MHC) cTnI NTG or ssTnI TG mice. Pro-Q Diamond staining (C) was done to determine the effects of PTU treatment or TG ssTnI expression on phosphorylation of sarcomeric proteins. Images are representative data from samples collected from at least 3 hearts in each group.
Effect of ssTnI on XB recruitment dynamics in α- vs. β-MHC fibres
We sought to characterize the effect of ssTnI expression on myofilament contractile dynamics and determine how such effects might be further influenced by a shift in MHC isoform following PTU treatment. A shift from α-MHC to β-MHC isoform expression has been shown to slow XB cycling dynamics (Fitzsimons et al. 1998b; Rundell et al. 2005b; Chandra et al. 2007) due to the slower enzymatic rate of the β-MHC isoform. However, the effect of TnI isoform switching on myofilament contractile dynamics is not well understood. To study these effects, the dynamic force responses to sinusoidal length perturbations of increasing frequency (i.e. chirp protocol, as shown in Fig. 1) were collected from constantly activated muscle fibres from each group of mouse hearts. Contractile dynamic behaviour was approximated using a previously established model (Campbell et al. 2004; Chandra et al. 2007) and fitted model parameters were subsequently used to determine the effects of ssTnI on myofilament contractile dynamics. Model fits were similarly good in α-MHC(cTnI), β-MHC(cTnI), α-MHC(ssTnI), and β-MHC(ssTnI) groups. R2 values were routinely >0.99, and residuals were normally distributed along the entire range of F(t), providing significant correlations (P < 0.001) in all groups of fitted data. This permitted interpretation of changes seen in model parameters as being due to changes in MHC or TnI, and not due to systematic errors in our model fitting. Fitted model predictions of force responses versus the frequency sweep of length perturbation are shown in Fig. 4A and B.
Figure 4. Relationship between frequency of length perturbation and magnitude of force response.
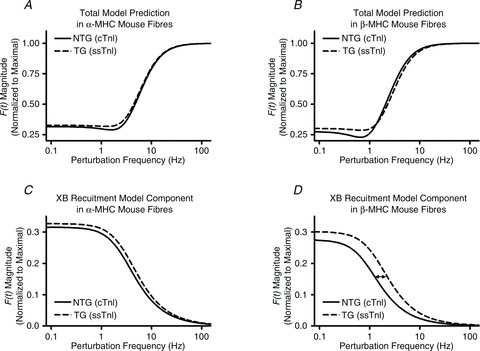
Model-predicted force responses to sinusoidal length perturbations of increasing frequency are shown from fibres of α-MHC (A) or β-MHC (B) NTG and TG mouse hearts. Low-frequency model components are shown for fibres from α-MHC (C) or β-MHC (D) mouse hearts to illustrate the ssTnI effect on the model component dominated by XB-recruitment dynamics. The magnitude of F(t) was normalized to the maximal value observed as frequency approached infinity.
Interestingly, we noted that ssTnI had an apparent effect on the shape of the force response and that this effect was most evident at lower frequencies. To illustrate this ssTnI effect, the low-frequency component of the model, which is attributed to the slower recruitment phase of the XB cycle, was plotted in Fig. 4C and D. Figure 4C and D illustrates an apparent enhancement in the ssTnI-speeding of XB recruitment dynamics following PTU-treatment (compare increased rightward-shift in the responses of ssTnI fibres in Fig. 4D to that of Fig. 4C). These effects were further quantified in the model parameter corresponding to the rate constant of XB recruitment, b (Fig. 5A).
Figure 5. Estimation of the rate of XB recruitment and tension redevelopment.
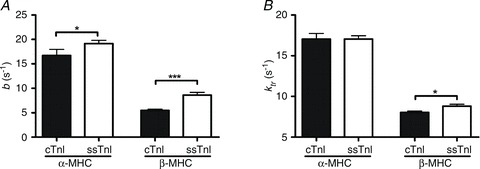
A, the model-predicted XB recruitment rate constant determined by fits to chirp data from α-MHC or β-MHC cTnI NTG or ssTnI TG mouse fibres. B, the rate constant of tension redevelopment determined by a rapid release and restretch protocol at maximal Ca2+ activation. Parameter values are represented as mean + SEM. Number of determinants was at least 10 for each group. *P < 0.05; ***P < 0.001.
The model-predicted rate of XB recruitment, b, was only slightly (∼15%) faster in α-MHC(ssTnI) fibres when compared to values from α-MHC(cTnI) fibres. On the other hand, b was much (∼57%) faster in β-MHC(ssTnI) fibres when compared to values from β-MHC(cTnI) fibres (Fig. 5A). Thus, the ssTnI effect on speeding XB recruitment dynamics was 3.8 times greater against a background of β-MHC, suggesting interplay between MHC and TnI on XB recruitment dynamics. Similarly, the rate constant of tension redevelopment, ktr, was significantly sped (by ∼10%) by ssTnI in β-MHC (P < 0.05), but not α-MHC fibres (Fig. 5B). These data suggest that the interplay between MHC and TnI has distinct regulatory effects, which go beyond that of the individual effects exerted by MHC or TnI. This is consistent with the notion that interplay between key regulatory proteins is essential for tuning cardiac contractile dynamics during development.
While ktr at maximal activation reflects the sum of XB attachment, f, and detachment, g, rate constants, ktr at very low levels of activation reflects g (Brenner & Eisenberg, 1986). Studies of the activation dependence of ktr show a curvilinear relationship, and extrapolating this relationship to a tension ∼0 predicts gapp, as proposed by Brenner & Eisenberg (Baker et al. 1998; Palmer & Kentish, 1998; Fitzsimons et al. 2001; Tesi et al. 2002; de Tombe & Stienen, 2007). Likewise, the relationship between ktr and the level of activation in our studies showed a curvilinear trend. To linearize the trend, we plotted the natural logarithm of ktr against tension (Fig. 6). The regression of this relationship was used to estimate ktr at low levels of activation, and thus allowed an approximation of gapp. The MHC–TnI interaction effect on gapp was not significant. However, post hoc analyses suggested that ssTnI expression had differential effects on gapp. As shown in Table 1 and Fig. 6A, gapp was not different between α-MHC(cTnI) and α-MHC(ssTnI) fibres. However, as shown in Table 1 and Fig. 6B, gapp was slightly, but statistically greater in β-MHC(ssTnI) fibres when compared to β-MHC(cTnI) fibres.
Figure 6. Rate constant of tension redevelopment, ktr, plotted as a function of level of activation (tension as normalized to maximal fibre tension, Tens/TensMax).
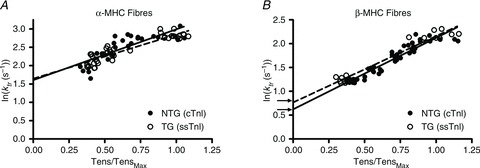
Because ktr exhibits an exponential trend with an increase in the level of activation, the logarithm of ktr vs. activation was plotted to linearize the trend. Regression analysis determined the intercept of the relationship of ktr vs. activation, which approximates the apparent rate of XB detachment, gapp (Baker et al. 1998; Palmer & Kentish, 1998; Fitzsimons et al. 2001; Tesi et al. 2002; de Tombe & Stienen, 2007). As show in panel A, the predicted intercept between α-MHC(cTnI) (continuous line) and α-MHC(ssTnI) (dashed line) fibres was not different. As show in panel B, the predicted intercept was slightly lower in β-MHC(cTnI) fibres (continuous line) when compared to β-MHC(ssTnI) fibres (dashed line) was slightly different. This difference in the intercept was significant, suggesting that gapp was slightly faster in β-MHC(ssTnI) vs. β-MHC(cTnI) fibres (Table 1).
Table 1.
Estimates of XB cycling kinetics
| Fibres from normal mice (α-MHC) | Fibres from PTU-treated mice (β-MHC) | |||
|---|---|---|---|---|
| α-MHC(cTnI) | α-MHC(ssTnI) | β-MHC(cTnI) | β-MHC(ssTnI) | |
| ktr | 17.0 ± 0.67 | 17.0 ± 0.41 | 8.04 ± 0.17a | 8.87 ± 0.24b,c |
| gapp | 5.02 ± 0.54 | 5.12 ± 0.35 | 1.73 ± 0.12a | 2.06 ± 0.08b,c |
| fapp | 11.9 ± 0.75 | 11.9 ± 0.59 | 6.32 ± 0.23a | 6.72 ± 0.26c |
| Tension cost | 7.58 ± 0.32 | 7.16 ± 0.19 | 2.99 ± 0.12a | 3.54 ± 0.10b,c |
| c | 44.7 ± 2.98 | 43.1 ± 2.40 | 19.8 ± 0.77a | 22.0 ± 1.18c |
The rate constant of tension redevelopment, ktr (s−1), was determined based on a rapid slack/restretch protocol, and is reported here at maximal Ca2+ activation. The intercept of the exponential trend of ktr vs. activation level (Fig. 6) was used to determine gapp (s−1). fapp (s−1) was determined by taking the difference of ktr and gapp, since at max activation ktr≈fapp+gapp (Brenner & Eisenberg, 1986). Tension cost (pmol mN−1 mm−1 s−1) is reported as the slope of the regression from the relationship between tension and ATPase activity. c (s−1) is the model-predicted rate constant of XB detachment, and correlates with tension cost. Values are means ± SEM. Number of determinants is at least 10 for each group. aP < 0.05 vs. α-MHC(cTnI); bP < 0.05 vs. β-MHC(cTnI); cP < 0.05 vs. α-MHC(ssTnI).
ktr at maximal activation is assumed to be proportional to the sum of gapp+fapp (Brenner & Eisenberg, 1986). Therefore, we predicted fapp by subtracting gapp estimates from ktr at maximal activation. As shown in Table 1, fapp was not affected by ssTnI in either α-MHC or β-MHC fibres, and as a result, the MHC–TnI interaction effect on fapp was not significant. However, β-MHC expression significantly slowed fapp in both NTG and TG fibres (P < 0.001). Together with our estimations from gapp, these results suggest ssTnI increased ktr in β-MHC, but not α-MHC fibres as a result of an increased gapp, but not fapp.
The model-estimated XB detachment rate, c, showed a similar trend to that of gapp, where ssTnI increased c by ∼10% in β-MHC, but not α-MHC fibres (Table 1). The MHC–TnI interaction effect on the model parameter c was not significant. β-MHC expression was the only factor that significantly influenced the XB distortion rate constant, slowing c by a similar amount in both NTG and TG mouse fibres (to 51% and 44% of values from α-MHC mice, respectively; P < 0.001). c was slightly (∼11%) faster in β-MHC(ssTnI) fibres when compared to β-MHC(cTnI) fibres, but this difference was not significant. Thus, XB distortion dynamics were not affected by ssTnI, but were significantly slowed by the presence of β-MHC.
E0 is the model-predicted low-frequency stiffness parameter that corresponds to the recruitment of additional XBs in response to change in muscle length, ΔL(t). E∞ is the high-frequency stiffness parameter that corresponds to the number of strongly bound XBs prior to ΔL(t). Thus, the ratio E0/E∞ can be used as an index of the fraction of new XBs that are recruited in response to changes in ΔL(t), with respect to the population prior to ΔL(t). This muscle length-mediated increase in recruitment of additional force-bearing XBs (E0/E∞) was not influenced by the interplay between TnI and MHC (Table 2). Furthermore, neither MHC isoform nor TnI isoform had any significant effects on E0/E∞.
Table 2.
Parameter estimates of muscle fibre contractile function
| Fibres from normal mice (α-MHC) | Fibres from PTU-treated mice (β-MHC) | |||
|---|---|---|---|---|
| α-MHC(cTnI) | α-MHC(ssTnI) | β-MHC(cTnI) | β-MHC(ssTnI) | |
| Max Tens | 43.8 ± 1.53 | 48.0 ± 0.76a | 52.8 ± 2.37a | 47.7 ± 1.56 |
| Max ATPase | 320 ± 18.6 | 359 ± 24.2 | 146 ± 5.19a | 169 ± 7.73b,c |
| E∞ | 595 ± 26.8 | 651 ± 19.1 | 749 ± 32.4a | 705 ± 27.4 |
| E0/E∞ | 0.32 ± 0.02 | 0.33 ± 0.01 | 0.27 ± 0.01a | 0.30 ± 0.01 |
| pCa50 | 5.78 ± 0.01 | 6.11 ± 0.01a | 5.85 ± 0.01a | 5.90 ± 0.01b,c |
| nH | 2.16 ± 0.09 | 1.80 ± 0.04a | 2.46 ± 0.06a | 2.62 ± 0.05c |
Maximal tension production (mN mm−2) and rate of ATPase activity (pmol mm−3 s−1) were measured from α-MHC and β-MHC NTG and ssTnI TG mice. E∞ (mN mm−3) is the stiffness approximated at infinite frequency of chirp perturbation, and correlates with maximal tension production. E0/E∞ (unitless) is an approximation of the magnitude of length-mediated XB recruitment as a fraction of the number of strongly bound XB prior to perturbation. Hill's equation was fitted to pCa–tension relationships (Fig. 7) to determine parameters for myofilament Ca2+ sensitivity, pCa50, and cooperativity, nH. Values are means ± SEM. Number of determinants is at least 10 for each group. aP < 0.05 vs. α-MHC(cTnI); bP < 0.05 vs. β-MHC(cTnI); cP < 0.05 vs. α-MHC(ssTnI).
ssTnI effect on Ca2+-activated maximal tension, ATPase activity, and tension cost in α- vs. β-MHC fibres
Table 2 summarizes the maximally activated tension and ATPase activity measured in fibres from ssTnI TG or NTG mice. The MHC–TnI interaction effect on maximal tension was significant. This effect is attributed to the fact that, when compared to α-MHC(cTnI) fibres, β-MHC(cTnI) fibres showed an unexpected increase in tension, but this rise in tension was not seen in β-MHC(ssTnI) fibres. While this increase in tension was different when compared to other studies, this discrepancy may be attributed, in part, to differences in preparation of cardiac samples, in the length of PTU treatment, and/or in the ionic strengths of activating solutions used in ours vs. others’ studies (Fitzsimons et al. 1998a; Herron et al. 2001; Rundell et al. 2005a; Stelzer et al. 2007).
The MHC–TnI interaction had no significant effect on Ca2+-activated maximal ATPase activity. ssTnI resulted in a slight, but insignificant increase in maximal ATPase activity, a trend that was roughly the same in both α-MHC and β-MHC mouse fibres. As expected, β-MHC expression had a strong effect to slow ATPase activity; maximal ATPase rates from β-MHC(ssTnI) and β-MHC(cTnI) fibres were ∼46% and ∼47% of those from α-MHC(ssTnI) and α-MHC(cTnI) fibres, respectively (P < 0.001).
Tension cost was determined from the slope of the relationship between tension production and ATPase activity observed at each level of pCa activation (values reported in Table 1). We observed small but significant MHC–TnI interaction effect on tension cost (P= 0.023). Post hoc tests revealed that tension cost was not different between α-MHC(ssTnI) and α-MHC(cTnI) fibres. However, a slight (∼18%) but significant increase in tension cost was observed in β-MHC(ssTnI) fibres when compared to β-MHC(cTnI). The effects on tension cost correlated with the general trend observed in the model-predicted rate constant of XB detachment, c, which showed very little change when ssTnI was expressed against α-MHC, but a slight increase when expressed against β-MHC. As was seen in c, β-MHC(cTnI) and β-MHC(ssTnI) fibres exhibited significantly lower tension cost (P < 0.001) than α-MHC(cTnI) or α-MHC(ssTnI) fibres by ∼40% and ∼49%, respectively. This correlation between tension cost and c is expected because, like c, tension cost can be used to approximate XB detachment kinetics (Brenner, 1988).
Effect of ssTnI on cardiac myofilament Ca2+ sensitivity in α- vs. β-MHC fibres
Myofilament Ca2+ sensitivity and cooperativity of tension development were determined by fitting Hill's equation to the pCa–tension relationship obtained from each muscle fibre (Fig. 7). pCa50, the pCa value required to reach half-maximal activation, was used as a determinant of myofilament Ca2+ sensitivity. In agreement with several other studies (Fentzke et al. 1999; Arteaga et al. 2000; Konhilas et al. 2003), we observed a leftward shift in the pCa–tension relationship in α-MHC(ssTnI) fibres, indicating an increase in myofilament Ca2+ sensitivity when compared to that of α-MHC(cTnI) fibres (note the leftward shifted curve from TG fibres, Fig. 7A). pCa50 was higher in fibres from α-MHC(ssTnI) fibres (pCa50= 6.11 in α-MHC(ssTnI) vs. pCa50= 5.78 in α-MHC(cTnI) fibres; ΔpCa50= 0.33; Fig 7A), indicating that the [Ca2+]Free needed to reach half-maximal activation in α-MHC(ssTnI) fibres was ∼54% less than that observed in α-MHC(cTnI) fibres. However, this ssTnI-mediated increase in pCa50 was blunted in fibres from β-MHC mice (pCa50= 5.85 in β-MHC(ssTnI) fibres vs. pCa50= 5.80 in β-MHC(cTnI) fibres; ΔpCa50= 0.05; Fig 7B), indicating that the [Ca2+]Free needed to reach half-maximal activation in β-MHC(ssTnI) fibres was ∼11% less than that observed in β-MHC(cTnI) fibres. Thus, the extent of this ssTnI-associated increase in Ca2+ sensitivity was different in α-MHC and β-MHC mouse fibres. As a result, the MHC–TnI interaction effect on pCa50 was highly significant (P < 0.001), suggesting that the ssTnI effect on myofilament Ca2+ sensitivity seen in α-MHC fibres was blunted by β-MHC expression.
Figure 7. pCa-tension relationships in fibres from α-MHC (A) or β-MHC (B) mice from cTnI NTG groups (•, continuous lines are Hill model fits) and ssTnI TG groups (○, dashed lines are Hill model fits).
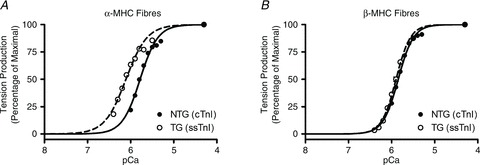
Data were normalized to maximal tension produced (pCa 4.3) by each respective fibre. Normalized curves were fitted to Hill's equation to determine the pCa50 and Hill's cooperativity coefficient, nH, for each group of fibres. As shown in Table 2, pCa50 was 5.78 ± 0.01 and 6.11 ± 0.01 for α-MHC(cTnI) and α-MHC(ssTnI) fibres, respectively, and 5.85 ± 0.01 and 5.90 ± 0.01 for β-MHC(cTnI) and β-MHC(ssTnI) fibres, respectively. nH was 2.16 ± 0.09 and 1.80 ± 0.04 for α-MHC(cTnI) and α-MHC(ssTnI) fibres, respectively, and 2.46 ± 0.06 and 2.62 ± 0.05 for β-MHC(cTnI) and β-MHC(ssTnI) fibres, respectively. Each point represents the average normalized tension produced from all fibres in each group ± SEM. Number of determinants was at least 10 for each group.
The apparent cooperativity of Ca2+-activated tension development, as estimated by Hill's coefficient, nH, was much lower in the α-MHC(ssTnI) fibres than in the α-MHC(cTnI) fibres (nH= 1.80 vs. nH= 2.16), but this trend was not seen in the comparison between β-MHC(ssTnI) and β-MHC(cTnI) fibres (Table 1). Thus, the TnI isoform affected nH differently in α-MHC and β-MHC mouse fibres, resulting in a significant MHC–TnI interaction effect on nH (P < 0.001). This further suggests that the interplay between MHC and TnI isoform had an effect whereby ssTnI increased thin filament cooperativity when α-MHC was present, but had no effect when β-MHC was present.
Discussion
Developmentally regulated shifts in contractile protein isoforms in mice are a well-documented phenomenon; β-MHC and ssTnI are expressed exclusively in the embryonic stage, whereas α-MHC and cTnI are expressed exclusively in the adult stage of cardiogenesis. Such changes in two key contractile regulatory proteins coincide with increases in heart rate as the mice mature. Therefore, we hypothesized that concomitant expression of β-MHC and ssTnI isoforms influence the tuned operation of the cardiac myofilament system. While the mice used here are not intended to explicitly represent an embryonic stage of development, it allowed for the investigation as to how α-MHC or β-MHC influenced ssTnI effects on contractile function. Our study is the first explicit attempt to characterize the effects of ssTnI expression on cardiac contractile dynamics, and to demonstrate that a shift in MHC isoform modulates the ssTnI effect on cardiac contractile function and dynamics. Because PTU treatment did not alter the phosphorylation status of other sarcomeric proteins, our results could be interpreted in terms of the β-MHC and ssTnI interplay effects on contractile function.
ssTnI effect to speed XB recruitment dynamics is enhanced by the presence of β-MHC
We have previously demonstrated that MHC isoform switching influences how alterations in troponin T – and how species-specific expression of the Tn complex as a whole – regulates cardiac contractile function and dynamics (Chandra et al. 2006; Tschirgi et al. 2006; Chandra et al. 2007). The present study offers further evidence that the TnI effects on cardiac contractile function and dynamics depend on MHC isoform expression.
A major finding in our study is that ssTnI increased the rate of XB recruitment, b, in a way that was more pronounced against a background of β-MHC (Fig. 5A). This suggests that ssTnI has an effect of speeding the recruitment of XBs more effectively when XBs are of the slower β-MHC isoform. This finding was supported by the observation that ktr was significantly higher in ssTnI fibres against β-MHC, but not against α-MHC (Fig. 5B).
Since XBs influence thin-filament activation (McKillop & Geeves, 1993; Vibert et al. 1997), it is possible that the longer duty ratio of β-MHC XBs allows for different allosteric modulation of ssTnI-effects on contractile activation, as suggested by b and ktr. In a simple two-state model of the XB cycle, ktr is proportional to the sum of the forward, f, and reverse, g, rate constants (Brenner & Eisenberg, 1986). Based on three separate experimental estimations of XB detachment rates (c, gapp, and tension cost), our results suggest that g was slightly higher in β-MHC(ssTnI) vs. β-MHC(cTnI) fibres. Thus, we can attribute the 10% increase in ktr to the comparable ∼11–19% increase in g. These findings further suggest that f is not affected by ssTnI in either α-MHC or β-MHC fibres, but that g is increased by ssTnI only in β-MHC fibres, as supported by gapp and fapp estimations shown in Table 1. While ktr approximates f and g of the XB cycle, b includes the regulatory processes that underlie the regulatory unit on/off kinetics that are activated in response to ΔL (Campbell et al. 2004). The much greater increase in b, therefore, implies an increase in cooperative mechanisms, whereby the slower β-MHC enhances the ssTnI-induced increase in regulatory unit on/off kinetics.
An important question then is: what functional advantage would β-MHC-ssTnI interplay confer on embryonic myocytes? The answer may lie in the observation that faster basal contractile dynamics are essential to an immature embryonic cardiomyocyte that has underdeveloped sarcoplasmic reticulum (SR), expresses lower levels of ryanodine receptors (RyRs), SR Ca2+-pumps (SERCA) and phospholamban (PLB), and lacks a well-developed β-adrenergic system. Thus, the faster contractile dynamics conferred by ssTnI and β-MHC may tune these cardiac cells to a beat frequency of 120 Hz. On the other hand, switching to the cTnI isoform is advantageous to adult cardiomyocytes because this isoform contains two key phosphorylation sites, Ser23 and Ser24 (Solaro et al. 1976; Mittmann et al. 1990). Phosphorylation of these residues in cTnI allows adult cardiomyocytes to adapt to varying haemodynamic demands by adjusting the speed of Ca2+ dissociation from troponin C and contractile dynamics, and thus having an overall inotropic effect on cardiac muscle fibres (for review, see Layland et al. 2005b). Our observations suggest that ssTnI may act to speed XB recruitment to match heart rate when XB cycling is limited by the slower β-MHC isoform. This may continue to be important until the faster contractile dynamics are conferred by the expression of α-MHC, and the maturing of cardiomyocytes with respect to SR density, RyR, SERCA, PLB and the β-adrenergic system.
ssTnI effect on cardiac myofilament Ca2+ sensitivity is blunted in the presence of β-MHC
Another major finding in our study is that the enhancing effect of ssTnI on myofilament Ca2+ sensitivity is blunted to a large extent in the presence of β-MHC. This finding could be attributed to the presence of β-MHC, or to the reemergence of some cTnI following PTU treatment. Our data suggest that it is most likely a reflection of both.
Findings by Metzger et al. (2003) suggest a dominant effect of ssTnI expression on myofilament Ca2+ sensitivity. In their study (Metzger et al. 2003), adult cardiomyocytes were transfected with the ssTnI gene, which resulted in ssTnI expression and incorporation into the sarcomere. At 2–3 days after transfection, ssTnI expression reached a level 14–29% of the total TnI, yet ΔpCa50 (ssTnI vs. cTnI) remained close to that reported at levels of >90% ssTnI expression (ΔpCa50∼0.26). Therefore, if ssTnI expression was the dominant factor in determining Ca2+ sensitivity, we would expect to see a similar effect on ΔpCa50 since our observed percentage ssTnI expression was even higher than that observed at 2–3 days after ssTnI gene transfection. Instead, we observed a ΔpCa50 of only 0.05 (β-MHC(ssTnI) vs. β-MHC(cTnI)), which substantiates our conclusion that interplay with β-MHC modulates the ssTnI effect on Ca2+ sensitivity and that ssTnI is not the sole determinant of myofilament Ca2+ sensitivity.
Developmentally regulated expression of cTnI in guinea pigs has been shown by Krüger et al. (2006) to have a significant effect on ablating the ssTnI effect on myofilament Ca2+ sensitivity. For example, at 55 days of embryonic development in the guinea pig, when cTnI expression is greater than ssTnI expression, Krüger et al. reported a ∼0.14 increase in pCa50 when compared to normal adults expressing predominantly cTnI. The ratio of cTnI:ssTnI in our PTU-treated mice is comparable to that of the 55-day-old guinea pig, and we also observed a ∼0.12 increase in pCa50 when comparing calcium sensitivity of β-MHC(ssTnI) mouse with that of α-MHC(cTnI) mouse fibres. Thus, we observed a similar increase in myofilament Ca2+ sensitivity in β-MHC(ssTnI) vs. α-MHC(cTnI) fibres when compared to the trend reported in peri-natal vs. adult guinea pig fibres (Krüger et al. 2006). Our unique observation, however, was that the effect of ssTnI on myofilament Ca2+ sensitivity against β-MHC was less pronounced. PTU treatment itself also resulted in a significant increase in pCa50 in fibres from NTG mouse hearts. Taken together, these findings suggest that the cTnI reemergence alone cannot explain the effects on myofilament Ca2+ sensitivity we see in β-MHC containing fibres.
Thus, our study demonstrates that MHC isoform switching plays a role in modulating the TnI effect on myofilament Ca2+ sensitivity, which would account for the attenuation of the ssTnI-induced increase in pCa50 in β-MHC fibres. This further implies that the ssTnI-induced increase in myofilament Ca2+ sensitivity may not be as pronounced in the embryonic myocardium, which expresses predominantly β-MHC. While ssTnI sensitizes the myofilaments to Ca2+– against either α-MHC or β-MHC – the ssTnI effect is much less pronounced against β-MHC. While ssTnI may decrease myofilament responsiveness to inotropic agents (Arteaga et al. 2000), β-MHC may further alter the effect of ssTnI on submaximal contractile activation in the developing heart to prevent excessive inotropy. The MHC–TnI interaction effect may have further implications for the ssTnI impact on pH sensitivity (Westfall et al. 1997) or on cardio-protective mechanisms (Arteaga et al. 2005; Layland et al. 2005a; Pinto et al. 2011; Pound et al. 2011) observed in α-MHC(ssTnI) mice.
PTU treatment-related reemergence of cTnI in TG mouse hearts (Fig. 3B; Huang et al. 2000; Riedel et al. 2005) may explain why TnI-dependent effects on myofilament Ca2+ responsiveness were attenuated in β-MHC fibres. However, the same logic cannot explain why β-MHC(ssTnI) fibres showed higher rates of tension cost and XB recruitment (b). In our study, ssTnI expression remained at ∼34% in PTU-treated TG mice, still allowing for plausible inference as to the effects of ssTnI against a background of β-MHC. For example, this ∼34% expression of ssTnI resulted in a ∼60% increase in the rate constant of XB recruitment (b), implying that the functional effects due to the presence of ssTnI in PTU-treated animals was still measurable and highly significant. In fact, the ssTnI-dependent increase in b was more pronounced against β-MHC, despite the relatively lower level of ssTnI expression in β-MHC fibres. These observations lead to the conclusion that at least some of the ssTnI expressed in the myocardium of PTU-treated mice resides in the overlap region of the cardiac thin filament. If evenly distributed, ssTnI would occupy one regulatory unit to every two occupied by cTnI, and the ssTnI effects could be proximally transmitted to near-neighbor regulatory units through allosteric interactions. Such effects have been reported in other TG lines, and in some cases of pathological cardiomyopathies, where expression as low as 10% or less of a protein variant can lead to significant changes in phenotypes and in contractile function (e.g. Tardiff et al. 1998; Stelzer et al. 2004; Kirk et al. 2009).
In conclusion, our study is the first attempt an understanding of how the concomitant switching of ssTnI to cTnI and β-MHC to α-MHC imparts a regulatory interplay in cardiac muscle function. The significance of our study is that it allows for a better understanding of how the regulatory proteins TnI and MHC, which both undergo major shifts in isoform expression during mammalian development, modulate heart function to meet developmental requirements. These findings hold further significance in understanding the diseased heart, where MHC shifts to nearly complete β-MHC (Nakao et al. 1997; Miyata et al. 2000) coincide with mutations in cTnI and other thin filament proteins (Thierfelder et al. 1994; Willott et al. 2010). Interestingly, we found that myofilament responsiveness to Ca2+ and XB recruitment dynamics were affected differently by ssTnI when α-MHC was replaced by β-MHC. These findings may have implications in understanding diseased hearts, where β-MHC expression may be a mechanism to compensate for functional effects imposed by disease-related alterations in other sarcomeric proteins. For example, it is possible that β-MHC expression may act to regulate changes in myofilament Ca2+ sensitivity or XB recruitment kinetics imposed by disease-related mutations in sarcomeric proteins, in a way similar to the effects observed in our study. This effect may be ascribed to an increase in XB dwell time, which could amplify allosteric mechanisms that allow β-MHC to modulate actions of TnI or other thin filament regulatory proteins. Thus, our study provides a molecular basis for the tuning and matching of the isoforms of TnI and MHC to optimize contractile function and dynamics in a context that can be applied to understanding developing myocardium as well as healthy and diseased states of the heart.
Acknowledgments
This work was sponsored, in part, by the National Heart, Lung, and Blood Institute grant R01-HL75643 (to M.C.), the American Heart Association fellowship 10PRE3480045 and an ARCS fellowship (to S.J.F.). The authors would like to thank Dr R. John Solaro for the gifting of the TG mice and for the comments on the manuscript, and Ranganath Mamidi and Dr Sampath Gollapudi for their thoughtful revisions.
Glossary
- cTnI
cardiac troponin I
- MHC
myosin heavy chain
- NTG
non-transgenic
- PTU
propylthiouracil
- PLB
phospholamban
- RyR
ryanodine receptor
- SERCA
sarco/endoplasmic reticulum calcium-ATPase pump
- SR
sarcoplasmic reticulum
- ssTnI
slow skeletal troponin I
- TG
transgenic
- Tn
troponin
- TnI
troponin I
- XB(s)
crossbridge(s)
Author contributions
S.J.F.: collection, analysis and interpretation of data, and drafting the article and revising the manuscript. M.C.: conception and design of the experiments, interpretation of the data, and revising the manuscript. Both authors approved the submitted version of the manuscript.
References
- Arteaga GM, Palmiter KA, Leiden JM, Solaro RJ. Attenuation of length dependence of calcium activation in myofilaments of transgenic mouse hearts expressing slow skeletal troponin I. J Physiol. 2000;526:541–549. doi: 10.1111/j.1469-7793.2000.t01-1-00541.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arteaga GM, Warren CM, Milutinovic S, Martin AF, Solaro RJ. Specific enhancement of sarcomeric response to Ca2+ protects murine myocardium against ischemia-reperfusion dysfunction. Am J Physiol Heart Circ Physiol. 2005;289:H2183–2192. doi: 10.1152/ajpheart.00520.2005. [DOI] [PubMed] [Google Scholar]
- Baker AJ, Figueredo VM, Keung EC, Camacho SA. Ca2+ regulates the kinetics of tension development in intact cardiac muscle. Am J Physiol Heart Circ Physiol. 1998;275:H744–750. doi: 10.1152/ajpheart.1998.275.3.H744. [DOI] [PubMed] [Google Scholar]
- Bhavsar PK, Dhoot GK, Cumming DV, Butler-Browne GS, Yacoub MH, Barton PJ. Developmental expression of troponin I isoforms in fetal human heart. FEBS Lett. 1991;292:5–8. doi: 10.1016/0014-5793(91)80820-s. [DOI] [PubMed] [Google Scholar]
- Brenner B. Effect of Ca2+ on cross-bridge turnover kinetics in skinned single rabbit psoas fibers: implications for regulation of muscle contraction. Proc Natl Acad Sci U S A. 1988;85:3265–3269. doi: 10.1073/pnas.85.9.3265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner B, Eisenberg E. Rate of force generation in muscle: correlation with actomyosin ATPase activity in solution. Proc Natl Acad Sci U S A. 1986;83:3542–3546. doi: 10.1073/pnas.83.10.3542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell KB, Chandra M, Kirkpatrick RD, Slinker BK, Hunter WC. Interpreting cardiac muscle force-length dynamics using a novel functional model. Am J Physiol Heart Circ Physiol. 2004;286:H1535–1545. doi: 10.1152/ajpheart.01029.2003. [DOI] [PubMed] [Google Scholar]
- Chandra M, Tschirgi ML, Ford SJ, Slinker BK, Campbell KB. Interaction between myosin heavy chain and troponin isoforms modulate cardiac myofiber contractile dynamics. Am J Physiol Regul Integr Comp Physiol. 2007;293:R1595–1607. doi: 10.1152/ajpregu.00157.2007. [DOI] [PubMed] [Google Scholar]
- Chandra M, Tschirgi ML, Rajapakse I, Campbell KB. Troponin T modulates sarcomere length-dependent recruitment of cross-bridges in cardiac muscle. Biophys J. 2006;90:2867–2876. doi: 10.1529/biophysj.105.076950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chizzonite RA, Zak R. Regulation of myosin isoenzyme composition in fetal and neonatal rat ventricle by endogenous thyroid hormones. J Biol Chem. 1984;259:12628–12632. [PubMed] [Google Scholar]
- Clemmens EW, Entezari M, Martyn DA, Regnier M. Different effects of cardiac versus skeletal muscle regulatory proteins on in vitro measures of actin filament speed and force. J Physiol. 2005;566:737–746. doi: 10.1113/jphysiol.2005.084194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Tombe PP, Stienen GJ. Protein kinase A does not alter economy of force maintenance in skinned rat cardiac trabeculae. Circ Res. 1995;76:734–741. doi: 10.1161/01.res.76.5.734. [DOI] [PubMed] [Google Scholar]
- de Tombe PP, Stienen GJ. Impact of temperature on cross-bridge cycling kinetics in rat myocardium. J Physiol. 2007;584:591–600. doi: 10.1113/jphysiol.2007.138693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Tombe PP, ter Keurs HE. Lack of effect of isoproterenol on unloaded velocity of sarcomere shortening in rat cardiac trabeculae. Circ Res. 1991;68:382–391. doi: 10.1161/01.res.68.2.382. [DOI] [PubMed] [Google Scholar]
- Fentzke RC, Buck SH, Patel JR, Lin H, Wolska BM, Stojanovic MO, Martin AF, Solaro RJ, Moss RL, Leiden JM. Impaired cardiomyocyte relaxation and diastolic function in transgenic mice expressing slow skeletal troponin I in the heart. J Physiol. 1999;517:143–157. doi: 10.1111/j.1469-7793.1999.0143z.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzsimons DP, Patel JR, Moss RL. Role of myosin heavy chain composition in kinetics of force development and relaxation in rat myocardium. J Physiol. 1998a;513:171–183. doi: 10.1111/j.1469-7793.1998.171by.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzsimons DP, Patel JR, Moss RL. Role of myosin heavy chain composition in kinetics of force development and relaxation in rat myocardium. J Physiol. 1998b;513:171–183. doi: 10.1111/j.1469-7793.1998.171by.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzsimons DP, Patel JR, Moss RL. Cross-bridge interaction kinetics in rat myocardium are accelerated by strong binding of myosin to the thin filament. J Physiol. 2001;530:263–272. doi: 10.1111/j.1469-7793.2001.0263l.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford SJ, Chandra M, Mamidi R, Dong W, Campbell KB. Model representation of the nonlinear step response in cardiac muscle. J Gen Physiol. 2010;136:159–177. doi: 10.1085/jgp.201010467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao L, Kennedy JM, Solaro RJ. Differential expression of TnI and TnT isoforms in rabbit heart during the perinatal period and during cardiovascular stress. J Mol Cell Cardiol. 1995;27:541–550. doi: 10.1016/s0022-2828(08)80049-1. [DOI] [PubMed] [Google Scholar]
- Gomes AV, Venkatraman G, Davis JP, Tikunova SB, Engel P, Solaro RJ, Potter JD. Cardiac troponin T isoforms affect the Ca2+ sensitivity of force development in the presence of slow skeletal troponin I: insights into the role of troponin T isoforms in the fetal heart. J Biol Chem. 2004;279:49579–49587. doi: 10.1074/jbc.M407340200. [DOI] [PubMed] [Google Scholar]
- Haddad F, Jiang W, Bodell PW, Qin AX, Baldwin KM. Cardiac myosin heavy chain gene regulation by thyroid hormone involves altered histone modifications. Am J Physiol Heart Circ Physiol. 2010;299:H1968–1980. doi: 10.1152/ajpheart.00644.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herron TJ, Korte FS, McDonald KS. Loaded shortening and power output in cardiac myocytes are dependent on myosin heavy chain isoform expression. Am J Physiol Heart Circ Physiol. 2001;281:H1217–1222. doi: 10.1152/ajpheart.2001.281.3.H1217. [DOI] [PubMed] [Google Scholar]
- Huang X, Lee KJ, Riedel B, Zhang C, Lemanski LF, Walker JW. Thyroid hormone regulates slow skeletal troponin I gene inactivation in cardiac troponin I null mouse hearts. J Mol Cell Cardiol. 2000;32:2221–2228. doi: 10.1006/jmcc.2000.1249. [DOI] [PubMed] [Google Scholar]
- Kentish JC, McCloskey DT, Layland J, Palmer S, Leiden JM, Martin AF, Solaro RJ. Phosphorylation of troponin I by protein kinase A accelerates relaxation and crossbridge cycle kinetics in mouse ventricular muscle. Circ Res. 2001;88:1059–1065. doi: 10.1161/hh1001.091640. [DOI] [PubMed] [Google Scholar]
- Kirk JA, MacGowan GA, Evans C, Smith SH, Warren CM, Mamidi R, Chandra M, Stewart AFR, Solaro RJ, Shroff SG. Left ventricular and myocardial function in mice expressing constitutively pseudophosphorylated cardiac troponin I. Circ Res. 2009;105:1232–1239. doi: 10.1161/CIRCRESAHA.109.205427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konhilas JP, Irving TC, Wolska BM, Jweied EE, Martin AF, Solaro RJ, de Tombe PP. Troponin I in the murine myocardium: influence on length-dependent activation and interfilament spacing. J Physiol. 2003;547:951–961. doi: 10.1113/jphysiol.2002.038117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krüger M, Kohl T, Linke WA. Developmental changes in passive stiffness and myofilament Ca2+ sensitivity due to titin and troponin-I isoform switching are not critically triggered by birth. Am J Physiol Heart Circ Physiol. 2006;291:H496–506. doi: 10.1152/ajpheart.00114.2006. [DOI] [PubMed] [Google Scholar]
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Layland J, Cave AC, Warren C, Grieve DJ, Sparks E, Kentish JC, Solaro RJ, Shah AM. Protection against endotoxemia-induced contractile dysfunction in mice with cardiac-specific expression of slow skeletal troponin I. FASEB J. 2005a;19:1137–1139. doi: 10.1096/fj.04-2519fje. [DOI] [PubMed] [Google Scholar]
- Layland J, Solaro RJ, Shah AM. Regulation of cardiac contractile function by troponin I phosphorylation. Cardiovasc Res. 2005b;66:12–21. doi: 10.1016/j.cardiores.2004.12.022. [DOI] [PubMed] [Google Scholar]
- Lompré AM, Nadal-Ginard B, Mahdavi V. Expression of the cardiac ventricular α- and β-myosin heavy chain genes is developmentally and hormonally regulated. J Biol Chem. 1984;259:6437–6446. [PubMed] [Google Scholar]
- Lowey S, Lesko LM, Rovner AS, Hodges AR, White SL, Low RB, Rincon M, Gulick J, Robbins J. Functional effects of the hypertrophic cardiomyopathy R403Q mutation are different in an α- or β-myosin heavy chain backbone. J Biol Chem. 2008;283:20579–20589. doi: 10.1074/jbc.M800554200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin AF, Ball K, Gao LZ, Kumar P, Solaro RJ. Identification and functional significance of troponin I isoforms in neonatal rat heart myofibrils. Circ Res. 1991;69:1244–1252. doi: 10.1161/01.res.69.5.1244. [DOI] [PubMed] [Google Scholar]
- McKillop DF, Geeves MA. Regulation of the interaction between actin and myosin subfragment 1: evidence for three states of the thin filament. Biophys J. 1993;65:693–701. doi: 10.1016/S0006-3495(93)81110-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzger JM, Michele DE, Rust EM, Borton AR, Westfall MV. Sarcomere thin filament regulatory isoforms. Evidence of a dominant effect of slow skeletal troponin I on cardiac contraction. J Biol Chem. 2003;278:13118–13123. doi: 10.1074/jbc.M212601200. [DOI] [PubMed] [Google Scholar]
- Mittmann K, Jaquet K, Heilmeyer LM. A common motif of two adjacent phosphoserines in bovine, rabbit and human cardiac troponin I. FEBS Lett. 1990;273:41–45. doi: 10.1016/0014-5793(90)81046-q. [DOI] [PubMed] [Google Scholar]
- Miyata S, Minobe W, Bristow MR, Leinwand LA. Myosin heavy chain isoform expression in the failing and nonfailing human heart. Circ Res. 2000;86:386–390. doi: 10.1161/01.res.86.4.386. [DOI] [PubMed] [Google Scholar]
- Nakao K, Minobe W, Roden R, Bristow MR, Leinwand LA. Myosin heavy chain gene expression in human heart failure. J Clin Invest. 1997;100:2362–2370. doi: 10.1172/JCI119776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer S, Kentish JC. Roles of Ca2+ and crossbridge kinetics in determining the maximum rates of Ca2+ activation and relaxation in rat and guinea pig skinned trabeculae. Circ Res. 1998;83:179–186. doi: 10.1161/01.res.83.2.179. [DOI] [PubMed] [Google Scholar]
- Peña JR, Wolska BM. Troponin I phosphorylation plays an important role in the relaxant effect of β-adrenergic stimulation in mouse hearts. Cardiovasc Res. 2004;61:756–763. doi: 10.1016/j.cardiores.2003.12.019. [DOI] [PubMed] [Google Scholar]
- Pinto JR, Yang SW, Hitz M-P, Parvatiyar MS, Jones MA, Liang J, Kokta V, Talajic M, Tremblay N, Jaeggi M, Andelfinger G, Potter JD. Fetal cardiac troponin isoforms rescue the increased Ca2+ sensitivity produced by a novel double deletion in cardiac troponin T linked to restrictive cardiomyopathy: A clinical, genetic, and functional approach. J Biol Chem. 2011;286:20901–20912. doi: 10.1074/jbc.M111.234336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pope B, Hoh JF, Weeds A. The ATPase activities of rat cardiac myosin isoenzymes. FEBS Lett. 1980;118:205–208. doi: 10.1016/0014-5793(80)80219-5. [DOI] [PubMed] [Google Scholar]
- Pound KM, Arteaga GM, Fasano M, Wilder T, Fischer SK, Warren CM, Wende AR, Farjah M, Abel ED, Solaro RJ, Lewandowski ED. Expression of slow skeletal TnI in adult mouse hearts confers metabolic protection to ischemia. J Mol Cell Cardiol. 2011;51:236–243. doi: 10.1016/j.yjmcc.2011.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiser PJ, Westfall MV, Schiaffino S, Solaro RJ. Tension production and thin-filament protein isoforms in developing rat myocardium. Am J Physiol Heart Circ Physiol. 1994;267:H1589–1596. doi: 10.1152/ajpheart.1994.267.4.H1589. [DOI] [PubMed] [Google Scholar]
- Rice R, Guinto P, Dowell-Martino C, He H, Hoyer K, Krenz M, Robbins J, Ingwall JS, Tardiff JC. Cardiac myosin heavy chain isoform exchange alters the phenotype of cTnT-related cardiomyopathies in mouse hearts. J Mol Cell Cardiol. 2010;48:979–988. doi: 10.1016/j.yjmcc.2009.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riedel B, Jia Y, Du J, Akerman S, Huang X. Thyroid hormone inhibits slow skeletal TnI expression in cardiac TnI-null myocardial cells. Tissue Cell. 2005;37:47–51. doi: 10.1016/j.tice.2004.10.002. [DOI] [PubMed] [Google Scholar]
- Rundell VL, Manaves V, Martin AF, de Tombe PP. Impact of β-myosin heavy chain isoform expression on cross-bridge cycling kinetics. Am J Physiol Heart Circ Physiol. 2005a;288:H896–903. doi: 10.1152/ajpheart.00407.2004. [DOI] [PubMed] [Google Scholar]
- Rundell VLM, Manaves V, Martin AF, de Tombe PP. Impact of β-myosin heavy chain isoform expression on cross-bridge cycling kinetics. Am J Physiol Heart Circ Physiol. 2005b;288:H896–903. doi: 10.1152/ajpheart.00407.2004. [DOI] [PubMed] [Google Scholar]
- Sabry MA, Dhoot GK. Identification and pattern of expression of a developmental isoform of troponin I in chicken and rat cardiac muscle. J Muscle Res Cell Motil. 1989;10:85–91. doi: 10.1007/BF01739858. [DOI] [PubMed] [Google Scholar]
- Saggin L, Gorza L, Ausoni S, Schiaffino S. Troponin I switching in the developing heart. J Biol Chem. 1989;264:16299–16302. [PubMed] [Google Scholar]
- Siedner S, Krüger M, Schroeter M, Metzler D, Roell W, Fleischmann BK, Hescheler J, Pfitzer G, Stehle R. Developmental changes in contractility and sarcomeric proteins from the early embryonic to the adult stage in the mouse heart. J Physiol. 2003;548:493–505. doi: 10.1113/jphysiol.2002.036509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solaro RJ, Moir AJ, Perry SV. Phosphorylation of troponin I and the inotropic effect of adrenaline in the perfused rabbit heart. Nature. 1976;262:615–617. doi: 10.1038/262615a0. [DOI] [PubMed] [Google Scholar]
- Stelzer JE, Brickson SL, Locher MR, Moss RL. Role of myosin heavy chain composition in the stretch activation response of rat myocardium. J Physiol. 2007;579:161–173. doi: 10.1113/jphysiol.2006.119719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stelzer JE, Patel JR, Olsson MC, Fitzsimons DP, Leinwand LA, Moss RL. Expression of cardiac troponin T with COOH-terminal truncation accelerates cross-bridge interaction kinetics in mouse myocardium. Am J Physiol Heart Circ Physiol. 2004;287:H1756–1761. doi: 10.1152/ajpheart.00172.2004. [DOI] [PubMed] [Google Scholar]
- Tardiff JC, Factor SM, Tompkins BD, Hewett TE, Palmer BM, Moore RL, Schwartz S, Robbins J, Leinwand LA. A truncated cardiac troponin T molecule in transgenic mice suggests multiple cellular mechanisms for familial hypertrophic cardiomyopathy. J Clin Invest. 1998;101:2800–2811. doi: 10.1172/JCI2389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tesi C, Piroddi N, Colomo F, Poggesi C. Relaxation kinetics following sudden Ca2+ reduction in single myofibrils from skeletal muscle. Biophys J. 2002;83:2142–2151. doi: 10.1016/S0006-3495(02)73974-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thierfelder L, Watkins H, MacRae C, Lamas R, McKenna W, Vosberg HP, Seidman JG, Seidman CE. α-Tropomyosin and cardiac troponin T mutations cause familial hypertrophic cardiomyopathy: a disease of the sarcomere. Cell. 1994;77:701–712. doi: 10.1016/0092-8674(94)90054-x. [DOI] [PubMed] [Google Scholar]
- Tschirgi ML, Rajapakse I, Chandra M. Functional consequence of mutation in rat cardiac troponin T is affected differently by myosin heavy chain isoforms. J Physiol. 2006;574:263–273. doi: 10.1113/jphysiol.2006.107417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vibert P, Craig R, Lehman W. Steric-model for activation of muscle thin filaments. J Mol Biol. 1997;266:8–14. doi: 10.1006/jmbi.1996.0800. [DOI] [PubMed] [Google Scholar]
- Westfall MV, Rust EM, Metzger JM. Slow skeletal troponin I gene transfer, expression, and myofilament incorporation enhances adult cardiac myocyte contractile function. Proc Natl Acad Sci U S A. 1997;94:5444–5449. doi: 10.1073/pnas.94.10.5444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willott RH, Gomes AV, Chang AN, Parvatiyar MS, Pinto JR, Potter JD. Mutations in troponin that cause HCM, DCM AND RCM: what can we learn about thin filament function. J Mol Cell Cardiol. 2010;48:882–892. doi: 10.1016/j.yjmcc.2009.10.031. [DOI] [PubMed] [Google Scholar]
- Wolska BM, Arteaga GM, Peña JR, Nowak G, Phillips RM, Sahai S, de Tombe PP, Martin AF, Kranias EG, Solaro RJ. Expression of slow skeletal troponin I in hearts of phospholamban knockout mice alters the relaxant effect of β-adrenergic stimulation. Circ Res. 2002;90:882–888. doi: 10.1161/01.res.0000016962.36404.04. [DOI] [PubMed] [Google Scholar]
- Wolska BM, Vijayan K, Arteaga GM, Konhilas JP, Phillips RM, Kim R, Naya T, Leiden JM, Martin AF, de Tombe PP, Solaro RJ. Expression of slow skeletal troponin I in adult transgenic mouse heart muscle reduces the force decline observed during acidic conditions. J Physiol. 2001;536:863–870. doi: 10.1111/j.1469-7793.2001.00863.x. [DOI] [PMC free article] [PubMed] [Google Scholar]