

Abstract
The effects of vagal stimulation (VS) on cardiac energy substrate metabolism are unknown. We tested the hypothesis that acute VS alters the balance between free fatty acid (FFA) and carbohydrate oxidation and opposes the metabolic effects of β-adrenergic stimulation. A clinical-type selective stimulator of the vagal efferent fibres was connected to the intact right vagus in chronically instrumented dogs. VS was set to reduce heart rate by 30 beats min−1, and the confounding effects of bradycardia were then eliminated by pacing the heart at 165 beats min−1. [3H]Oleate and [14C]glucose were infused to measure FFA and glucose oxidation. The heart was subjected to β-adrenergic stress by infusing dobutamine at 5, 10 and 15 μg kg−1 min−1 before and during VS. VS did not significantly affect baseline cardiac performance, haemodynamics or myocardial metabolism. However, at peak dobutamine stress, VS attenuated the increase in left ventricular pressure–diameter area from 235.9 ± 72.8 to 167.3 ± 55.8%, and in cardiac oxygen consumption from 173.9 ± 23.3 to 127.89 ± 6.2% (both P < 0.05), and thus mechanical efficiency was not enhanced. The increase in glucose oxidation fell from 289.3 ± 55.5 to 131.1 ± 20.9%(P < 0.05), while FFA oxidation was not increased by β-adrenergic stress and fell below baseline during VS only at the lowest dose of dobutamine. The functional and in part the metabolic changes were reversed by 0.1 mg kg−1 atropine i.v. Our data show that acute right VS does not affect baseline cardiac metabolism, but attenuates myocardial oxygen consumption and glucose oxidation in response to adrenergic stress, thus functioning as a cardio-selective antagonist to β-adrenergic activation.
Key points
Whereas the effects of catecholamines on myocardial metabolism are well characterized, the potential role of the parasympathetic system is generally considered minor or absent.
We tested the hypothesis that acute stimulation of the right vagus nerve alters the balance between cardiac free fatty acid and carbohydrate oxidation and opposes the metabolic effects of beta-adrenergic stimulation.
Using a clinical-type selective stimulator of the vagal efferent fibers in dogs, we found that vagal stimulation did not significantly affect baseline cardiac performance, haemodynamics and myocardial metabolism.
During dobutamine stress, vagal stimulation attenuated the increase in left ventricular mechanical performance, cardiac oxygen consumption and myocardial glucose oxidation, while free fatty acid oxidation was affected only at low catecholamine dose.
Our results elucidate a previously unexplored parasympathetic function, indicating that selective vagal efferent stimulation antagonizes the effects of beta-adrenergic activation on myocardial metabolism.
Introduction
Cardiac metabolism is finely controlled by a complex network of intracellular feedback mechanisms and neuro-hormonal signals (Drake-Holland, 1983; Stanley et al. 2005). Among the extracellular signals, catecholamines released by sympathetic nerve endings exert a profound effect on myocardial energy turnover. Elegant quantitative studies performed in isolated heart preparations with controlled substrate delivery have shown that acute increases in contractile performance induced by β-adrenergic agonists stimulate myocardial glucose utilization to meet the higher metabolic demand (Goodwin et al. 1998; Doenst & Taegtmeyer, 1999). Because sympathetic system activation notoriously causes cardiac oxygen wasting (Ohgoshi et al. 1991), the selective reliance on glucose and not on the less efficient substrate free fatty acids (FFAs) (Korvald et al. 2000) during adrenergic stress might serve to limit oxygen consumption, while maintaining an adequate mechanical response. Conversely, carbohydrate oxidation is impaired in the chronically denervated heart, probably due to a fall in active pyruvate dehydrogenase (Van Der Vusse et al. 1998).
Whereas the effects of catecholamines on myocardial metabolism are well characterized, the potential role of the parasympathetic system is generally considered minor or absent, despite the fact that muscarinic receptors in the heart are more abundant than β-adrenoreceptors (Böhm et al. 1990). Nonetheless, classic in vivo studies found that electrical stimulation of the vagal efferents can reduce cardiac contractility (DeGeest et al. 1964) mainly by inhibiting noradrenaline release at presynaptic level (Daggett et al. 1967; Henning et al. 1990; Xenopoulos & Applegate, 1994) and can oppose the inotropic action of dobutamine (Hare et al. 1995). This negative inotropic action is mediated by nitric oxide (Hare et al. 1995), a molecule that is also implicated in the control of energy substrate utilization (Young & Leighton, 1998; Recchia et al. 2002; Lei et al. 2005), yet virtually no information is available on the effects of vagal output on the balance between FFA and carbohydrate oxidation, which is an important determinant of chemical conversion efficiency (Lopaschuk et al. 2010). A few studies have assessed myocardial oxygen consumption during vagal electrical stimulation in resting hearts and reported no significant changes (Daggett et al. 1967; Downing et al. 1977). However, the effects of vagal stimulation (VS) on oxygen use and energy substrate metabolism during concurrent β-adrenergic receptor stimulation remain unexplored.
Interest in cardiac parasympathetic innervation has recently been revitalized by experimental and clinical evidence of a beneficial effect of cervical right VS on failing hearts (Li et al. 2004; Zhang et al. 2009; De Ferrari et al. 2011). This approach is very appealing, as implantable vagal stimulators allow a selective therapeutic action on the target organ, thus avoiding systemic and untoward effects of pharmacological agents. The underlying mechanisms are likely to be numerous, but they remain largely unknown. It will be important first to characterize in more detail the consequences of superphysiological firing of vagal efferents on normal heart in the intact organism. In fact, one important limitation of previous studies was their invasive approach based on vagus severing and/or open chest preparations. In the present study, we used an electrical stimulator designed to generate unidirectional impulses along the efferent fibres of the intact nerve, which allowed us to test the effects of acute right VS on cardiac performance and energy substrate metabolism in dogs with chronically implanted probes and catheters. Our hypothesis was that vagal activation alters the balance between FFA and carbohydrate oxidation and opposes the metabolic effects of β-adrenergic stimulation, i.e. attenuates the oxygen demand, while preserving and possibly enhancing mechanical efficiency.
Methods
Surgical instrumentation
The surgical and experimental protocols were approved by the Institutional Animal Care and Use Committee of the New York Medical College and conform to the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health.
Fifteen adult male mongrel dogs (body weight 23–27 kg, purchased from Hodgins Kennels, Inc., Howell, MI, USA) were sedated with acepromazine maleate (1 mg kg−1 i.m.), anaesthetized with propofol (4 mg kg−1 i.v.) and ventilated with room air. An adequate level of gas anaesthesia was maintained by 1.5% isoflurane and monitored by checking somatic reflexes (corneal and toe pinch) and visceral reflexes [unexpected changes in heart rate (HR) and blood pressure], while arterial oxygen content was measured by pulse oximetry. The entire surgical procedure was carried out under aseptic conditions. A thoracotomy was performed in the left fifth intercostal space. One catheter was placed in the descending thoracic aorta, and a second catheter was placed in the coronary sinus with the tip leading away from the right atrium. A solid-state pressure gauge (P6.5; Konigsberg Instruments, Pasadena, CA, USA) was inserted into the left ventricle (LV) through the apex. A Doppler flow transducer (Craig Hartley, Houston, TX, USA) was placed around the left circumflex coronary artery, and a pair of 3 MHz piezoelectric crystals was fixed on opposing endocardial surfaces at the base of the LV. A human, screw-type, unipolar myocardial pacing lead was fixed in the LV wall. Wires and catheters were run subcutaneously to the intrascapular region, the chest was closed in layers and the pneumothorax was reduced. After surgery, dogs underwent analgesic therapy for 3 days (fentanyl, 75 μg kg−1 intradermal) and antibiotic therapy for 10 days (amoxicillin, 16 mg kg−1 day−1 i.m.). They responded very well to this therapeutic regimen, and therefore post-surgical fever and weight loss were only transient. Ten days was sufficient for full recovery, which was indicated by normal ambulation, appetite, and faecal and urine output. Once this phase was completed, the dogs were trained to lie quietly on the laboratory table. Rectal temperature was monitored daily for the entire duration of the protocol and, if found to be higher than 39°C, amoxicillin (16 mg kg−1) or cefazolin (40 mg kg−1) was given i.m. We have used these methods in previous studies (Recchia et al. 2002; Osorio et al. 2002).
Protocol
The experiments were conducted in 11 dogs lightly anaesthetized with propofol (rapid bolus of 4 mg kg−1 i.v., followed by 0.2 mg kg−1 min−1 i.v.) and in spontaneous respiration. Immediately after the propofol bolus, when the level of anaesthesia was deeper, a neck area along the right cervical line was infiltrated with 2% lidocaine. A 3 cm longitudinal cervical incision was then made and the right carotid sheath and right carotid nerve were rapidly exposed. The vagal nerve stimulator lead (CardioFit Stimulation Lead; BioControl Medical Ltd, Yehud, Israel) was then placed around the vagus nerve and secured by tightening the pre-existing tightening strings. The lead was then connected to the stimulator (CardioFit-X external stimulator, Model 5510; BioControl Medical Ltd), and the wound was closed around the lead using 2–0 silk suture. This procedure typically lasted 5 min. The experimental protocol is schematically depicted in Fig. 1. The isotopic tracers [9,10-3H]oleate and [U-14C]glucose were continuously infused for the duration of the experiment through a peripheral vein following a protocol previously established by us (Recchia et al. 2002). A 6-French, custom-made balloon catheter was introduced into a peripheral vein of a posterior leg and advanced to the inferior vena cava. After recording of systemic haemodynamics, coronary blood flow and LV diameter at spontaneous HR, occlusions of the inferior vena cava, separated by 5 min intervals, were performed by inflating the balloon to obtain end-systolic pressure–diameter curves. Once the baseline haemodynamics were re-established, the heart was paced at 165 beats min−1 via the LV leads, haemodynamics were recorded, and paired blood samples were taken from the aortic and the coronary sinus catheters. Cardiac pacing was used to eliminate the confounding effect of bradycardia on myocardial metabolism that could have affected the comparison between measurements obtained before and after VS. The heart was then subjected to β-adrenergic stress with dobutamine at 5, 10 and 15 μg kg−1 min−1 i.v. Haemodynamics were recorded and paired blood samples were taken from aorta and coronary sinus after each dose of dobutamine, once the effects had reached steady state (usually 5 min). Dobutamine infusion and pacing were stopped to re-establish baseline conditions (re-baseline) and record haemodynamics and pressure–diameter curves with vena cava occlusion. After completion of this step, the vagal stimulator was activated to reduce the spontaneous HR by 30 beats min−1. VS was delivered in asynchronous mode, using an asymmetric, biphasic, charge-balanced waveform, with a pulse width of 0.5 ms. In each animal, several current settings and number of pulses were first tested for a few seconds to find the best combination for the desired HR reduction. The average current used was 4.8 mA, with an average of 2.1 pulses per stimulation cycle. Haemodynamics and pressure–diameter curves were recorded again, cardiac pacing was then restarted and the heart was again subjected to β-adrenergic stress as described above. Finally, during infusion of the highest dose of dubutamine, 0.1 mg kg−1 atropine was administered as a bolus i.v. and the last haemodynamics and blood samples were taken at the peak of haemodynamic changes, which occurred after ∼1 min. Beyond that peak, haemodynamics became very unstable, and the experiment was considered concluded.
Figure 1.
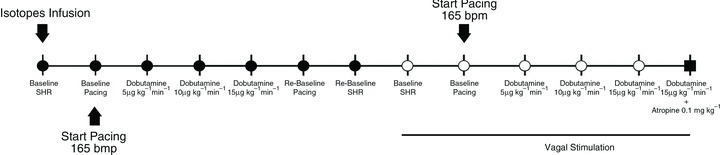
Experimental protocol
To rule out possible tachyphylaxis due to repeated dobutamine infusions, four additional dogs were subjected to the same protocol described above: the first series of infusions was followed by re-baseline and then by the second series of infusions, but in the absence of VS.
At the end of the protocol, the dogs were killed via an overdose of sodium pentobarbitone (100 mg kg−1 i.v.)
Haemodynamic and mechanoenergetic measurements
The aortic catheter was attached to a P23ID strain-gauge transducer for measurement of aortic pressure. LV pressure was measured using the solid-state pressure gauge. Blood flow in the left circumflex coronary artery was measured with a pulsed Doppler flowmeter (model 100; Triton Technology, San Diego, CA, USA). LV diameter was measured by connecting the implanted piezoelectric crystals to an ultrasonic dimension gauge. All signals were also digitally stored on an AT-type computer via an analog–digital interface (National Instruments, Austin, TX, USA) at a sampling rate of 250 Hz. Digitized data were analysed off-line by custom-made software. The parameters were determined during one respiratory cycle and comprised HR, mean aortic pressure, LV end-diastolic, peak systolic and end-systolic pressure, mean blood flow in the left circumflex coronary artery, the maximum and minimum of the first derivative of LV pressure, and end-diastolic and end-systolic LV internal diameters. The percentage shortening of the short axis diameter was calculated as the ratio of the difference between end-diastolic and end-systolic diameter (stroke dimension) to end-diastolic diameter (Saavedra et al. 2002; Post et al. 2003). The slope of the LV end-systolic pressure–diameter relationship (Ees) was calculated using the first 7–10 beats during inferior vena cava occlusion before any HR increase (Senzaki et al. 2000; Saavedra et al. 2002; Post et al. 2003). Effective arterial elastance (Ea), an index of LV afterload (Senzaki et al. 2000; Post et al. 2003), was calculated as the ratio of LV end-systolic pressure to stroke dimensions. An index of stroke work was determined by calculating the area of the LV pressure–diameter loop (pressure–diameter area, PDA) during a cardiac cycle (Senzaki et al. 2000; Saavedra et al. 2002; Post et al. 2003). The difference between end-diastolic and end-systolic LV diameter was used as a surrogate of stroke volume and multiplied by HR to obtain an index of cardiac output.
Blood gases were measured using a blood gas analyser (Instrumentation Laboratory, Bedford, MA, USA) and oxygen concentration was measured using a haemoglobin analyser (CO-Oximeter; Instrumentation Laboratory). LV myocardial oxygen consumption (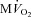 ) was calculated by multiplying the arterial–coronary sinus difference in oxygen content by total mean coronary blood flow, assumed to be double the mean blood flow in the left circumflex coronary artery (Recchia et al. 2002; Osorio et al. 2002).
) was calculated by multiplying the arterial–coronary sinus difference in oxygen content by total mean coronary blood flow, assumed to be double the mean blood flow in the left circumflex coronary artery (Recchia et al. 2002; Osorio et al. 2002).
Cardiac energy substrate metabolism
Total cardiac substrate concentrations were measured in arterial and coronary sinus blood samples, as previously described (Recchia et al. 2002). In brief, FFA concentration was determined spectrophotometrically in plasma. Glucose was measured in blood deproteinized with ice-cold 1 m perchloric acid (1:2 v/v) using spectrophotometric enzymatic assays. The concentrations of labelled metabolites were determined in arterial and coronary sinus blood samples. In particular, [3H]oleate activity was measured in plasma, whereas [14C]glucose activity was determined in blood deproteinized with ice-cold 1 m perchloric acid (1:2 v/v). 3H2O and 14CO2 activities were also measured in plasma and whole blood, respectively, and utilized to calculate the rate of FFA and glucose oxidation. Lactate concentrations were measured with the same cartridges utilized for blood gas analysis (Instrumentation Laboratory). The net chemical uptake of lactate by the heart was calculated based on arterial–coronary sinus difference and coronary blood flow and used as an index of oxidation, assuming that in the normo-oxygenated healthy heart, the fraction of lactate output remains constant.
Statistical analysis
All data are given as mean ± SEM. Data were compared using one-way ANOVA for repeated measurements or by two-way ANOVA followed by Dunnett or Tukey tests (SigmaStat 2.03; Systat Software Inc., Chicago, IL, USA). For all statistical analyses, significance was accepted at P < 0.05.
Results
Haemodynamics
Changes in haemodynamic parameters are shown in Fig. 2. Cardiac pacing was set at a rate predicted to be higher than the tachycardia induced by dobutamine infusion, in order to maintain HR constant throughout the protocol (Fig. 2A). In fact, only at the highest dobutamine dose, and before VS, was HR slightly but significantly higher than the pacing rate. VS reduced the spontaneous HR by approximately 30 beats min−1. Atropine caused an increase in HR that reached values significantly higher than the pacing rate. dP/dtmax, an index of contractility, increased significantly during dobutamine infusion, although VS significantly attenuated the contractile response to β-adrenergic stimulation (Fig. 2B). Atropine reversed the vagal effect. Similar responses before and after VS were observed for LV systolic pressure and mean coronary blood flow (Fig. 2C and D). Mean arterial pressure did not change significantly after VS and during dobutamine infusion (Table 1). Ea, an index of LV afterload, decreased significantly at the highest dose of dobutamine, but not during VS. In four additional dogs utilized to rule out the effect of dobutamine tachyphylaxis, we found the following changes in dP/dtmax, i.e. the parameter more characteristically affected by β-adrenoceptor stimulation. Considering for simplicity only baseline values and peak changes at 15 μg kg−1 min−1 dobutamine, dP/dtmax was, respectively: 2324.37 ± 129.48 and 4488.75 ± 575.62 mmHg s−1 (P < 0.05) during the first series of infusions and 2343.12 ± 324.53 and 4137.50 ± 486.01 mmHg s−1 during the second series, with no significant differences between the two phases of the protocol. Also other haemodynamic parameters were not significantly different during the second series of infusion compared with the first (data not shown).
Figure 2. Changes in heart rate (HR), dP/dtmax, LV systolic pressure (LVSP) and mean coronary blood flow (MCBF) in the left circumflex coronary artery during dobutamine infusion, before (filled circles) and during (open circles) vagal stimulation.
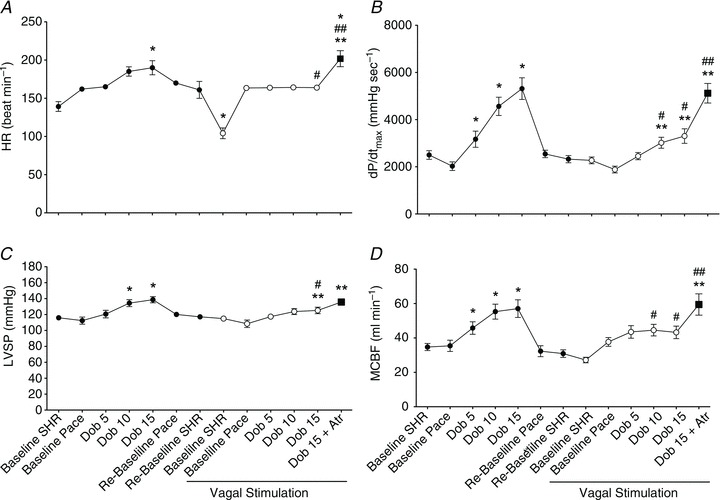
SHR, spontaneous heart rate; Pace, pacing at 165 beats min−1; Dob, dobutamine; Atr, atropine. Data are presented as mean ± SEM, n= 11. *P < 0.05 vs. pre-VS Baseline Pace; #P < 0.05 vs. corresponding time point pre-VS; **P < 0.05 vs. post-VS Baseline Pace; ##P < 0.05 Dob 15+atropine vs. Dob 15 pre-VS.
Table 1.
Changes in indexes of LV afterload and preload and in a calculated index of cardiac output with vagal stimulation (VS) on and off
| VS | Baseline | Dobutamine 5 μg kg min−1 | Dobutamine 10 μg kg min−1 | Dobutamine 15 μg kg min−1 | Re-baseline | Dobutamine 15+Atropine | |
|---|---|---|---|---|---|---|---|
| Mean arterial pressure (mmHg) | OFF | 95.5 ± 4.0 | 96.9 ± 4.1 | 104.6 ± 3.3 | 101.9 ± 2.6 | 101.3 ± 2.6 | |
| ON | 92.0 ± 4.8 | 94.4 ± 3.8 | 96.1 ± 3.6 | 92.2 ± 3.4 | 96.6 ± 2.9 | ||
| Ea (mmHg ml−1) | OFF | 34.20 ± 4.55 | 30.68 ± 2.00 | 28.55 ± 3.47 | 23.36 ± 2.08 | 38.13 ± 4.01 | |
| ON | 40.96 ± 4.69 | 34.38 ± 3.11 | 32.64 ± 3.05 | 32.45 ± 4.31** | 24.47 ± 3.08** | ||
| End-diastolic diameter (mm) | OFF | 42.62 ± 0.73 | 42.33 ± 0.73 | 42.89 ± 0.91 | 43.75 ± 1.01 | 42.57 ± 0.87 | |
| ON | 42.30 ± 0.88 | 41.84 ± 0.83 | 41.78 ± 0.86 | 41.63 ± 0.88 | 41.58 ± 1.01 | ||
| (EDD – ESD) × HR (mm min−1) | OFF | 451.45 ± 47.91 | 576.43 ± 61.22 | 766.20 ± 60.43 | 794.40 ± 52.35 | 491.08 ± 36.35 | |
| ON | 390.14 ± 43.71 | 426.32 ± 28.34 | 467.78 ± 36.65**# | 491.94 ± 43.91**# | 692.73 ± 93.65** |
Heart rate was fixed at 165 beats min−1. Ea, arterial elastance; EDD, end-diastolic diameter; ESD, LV end-systolic diameter. Values are mean ± SEM, n= 11. **P < 0.05 vs. VS baseline; #P < 0.05 vs. corresponding time point pre-VS.
Cardiac contractile performance, 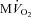 and mechanical efficiency
and mechanical efficiency
End-diastolic diameter did not change significantly under any of the experimental conditions (Table 1). Nonetheless, LV stroke work, as measured by PDA, increased significantly during dobutamine infusion; however, VS significantly attenuated the contractile response to β-adrenergic stimulation, while atropine reversed the vagal effect (Fig. 3A), although this response displayed a high degree of variability. Similarly, the calculated index of cardiac output increased significantly during dobutamine infusion, but only before VS, while atropine reversed the depressant effects of vagus (Table 1). Ees, an afterload-independent index of contractility measured at spontaneous HR, was 31.8 ± 4.7 mmHg mm−1 at baseline and 41.5 ± 4.9 mmHg mm−1 (n.s.) during the post-dobutamine re-baseline, indicating that, even if the other functional parameters had returned to resting values, contractility was still recovering. After VS, Ees was 32.4 ± 7.7 mmHg mm−1, not significantly different from the initial baseline.
Figure 3. Changes in LV pressure-diameter area (PDA), an index of stroke work, and in 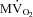 , before (filled circles) and during (open circles) vagal stimulation.
, before (filled circles) and during (open circles) vagal stimulation.
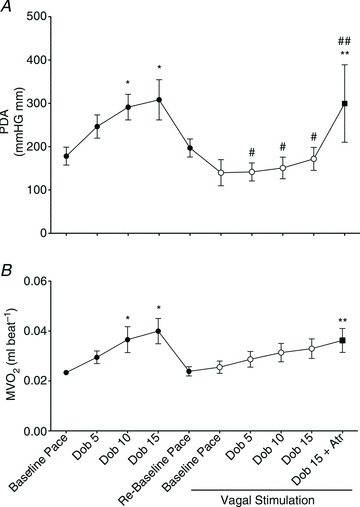
All measurements were taken with HR fixed at 165 beats min−1 (Pace). Data are presented as mean ± SEM, n= 7 for PDA, due to the low quality of the loops in some dogs. Note n= 8 for 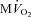 . *P < 0.05 vs. pre-VS Baseline Pace; #P < 0.05 vs. corresponding time point pre-VS; **P < 0.05 vs. post-VS Baseline Pace; ##P < 0.05 Dob 15+atropine vs. Dob 15 pre-VS.
. *P < 0.05 vs. pre-VS Baseline Pace; #P < 0.05 vs. corresponding time point pre-VS; **P < 0.05 vs. post-VS Baseline Pace; ##P < 0.05 Dob 15+atropine vs. Dob 15 pre-VS.
Changes in 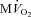 are shown in Fig. 3B. As expected,
are shown in Fig. 3B. As expected, 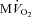 beat−1 increased significantly in response to adrenergic stress. VS did not affect
beat−1 increased significantly in response to adrenergic stress. VS did not affect 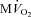 beat−1 at baseline, but significantly limited the enhanced oxygen consumption during dobutamine infusion, which was not significantly different from baseline, and atropine could not reverse this effect. When compared side by side in terms of percentage changes, PDA and
beat−1 at baseline, but significantly limited the enhanced oxygen consumption during dobutamine infusion, which was not significantly different from baseline, and atropine could not reverse this effect. When compared side by side in terms of percentage changes, PDA and 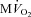 beat−1 were similarly attenuated by VS during dobutamine stress, indicating that the mechanical efficiency, i.e. the ratio between external work and oxygen use, remained unchanged (Fig. 4). Interestingly, however, atropine caused a marked rebound of PDA but not of
beat−1 were similarly attenuated by VS during dobutamine stress, indicating that the mechanical efficiency, i.e. the ratio between external work and oxygen use, remained unchanged (Fig. 4). Interestingly, however, atropine caused a marked rebound of PDA but not of 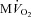 beat−1, i.e. the heart was able to perform an external work comparable with the pre-VS dobutamine response, while maintaining low oxygen consumption, indicating an acute increase in mechanical efficiency.
beat−1, i.e. the heart was able to perform an external work comparable with the pre-VS dobutamine response, while maintaining low oxygen consumption, indicating an acute increase in mechanical efficiency.
Figure 4. Percentage changes in LV pressure–diameter area (PDA), an index of stroke work, and in 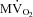 for a normalized comparison of these two parameters.
for a normalized comparison of these two parameters.
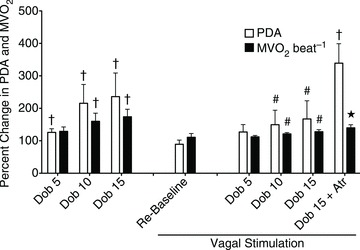
All measurements were taken with HR fixed at 165 beats min−1 (Pace). Data are presented as mean ± SEM, n= 7 for PDA and n= 8 for 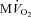 . *P < 0.05 vs. pre-VS Baseline Pace; #P < 0.05 vs. corresponding time point pre-VS; **P < 0.05 vs. post-VS Baseline Pace; †P < 0.05 PDA vs.
. *P < 0.05 vs. pre-VS Baseline Pace; #P < 0.05 vs. corresponding time point pre-VS; **P < 0.05 vs. post-VS Baseline Pace; †P < 0.05 PDA vs.
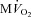 .
.
Cardiac energy substrate utilization
Due to technical problems, a full analysis of cardiac substrate utilization could be obtained only in 8 of the 11 dogs. Figure 5 shows the changes in FFA, glucose and lactate utilization. During dobutamine stress, glucose oxidation and lactate uptake increased significantly to meet the higher metabolic demand, while FFA oxidation did not display significant changes. The post-dobutamine re-baseline was characterized by a fall in FFA oxidation below baseline levels. VS did not affect significantly glucose oxidation and lactate uptake in the resting heart, while FFA oxidation returned to levels not significantly different from pre-VS baseline. During VS, β-adrenergic stress failed to increase significantly glucose oxidation and lactate uptake, which were indeed lower that control values. Interestingly, FFA oxidation fell significantly at the lowest dose of dobutamine (5 μg kg−1 min−1), and then returned to baseline values. Finally, atropine could not re-establish pre-VS values of FFA and glucose oxidation and lactate uptake.
Figure 5. Changes in cardiac glucose and free fatty acid (FFA) oxidation and in lactate uptake during dobutamine infusion, before (filled circles) and during (open circles) vagal stimulation.
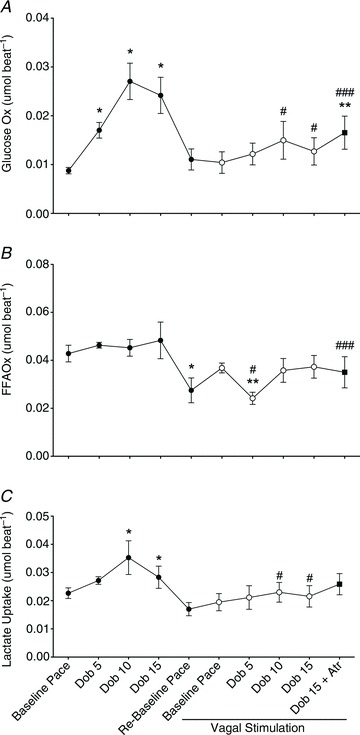
All measurements were taken with HR fixed at 165 beats min−1. Data are presented as mean ± SEM, n= 11. *P < 0.05 vs. pre-VS Baseline Pace; #P < 0.05 vs. corresponding time point pre-VS; **P < 0.05 vs. post-VS Baseline Pace; ###P < 0.05 vs. Dob 15+atropine vs. Dob 15 pre-VS.
Cardiac energy substrate utilization can be markedly affected by circulating levels of these metabolites. The only small but statistically significant changes were found before VS: an increase in FFA concentration at the highest dobutamine dose and a decrease in glucose concentration at the intermediate dobutamine dose (Table 2). Atropine did not affect the concentration of these substrates.
Table 2.
Changes in oxygen content and energy substrate concentration in arterial blood with vagal stimulation (VS) on and off
| VS | Baseline | Dobutamine 5 μg kg min−1 | Dobutamine 10 μg kg min−1 | Dobutamine 15 μg kg min−1 | Re-baseline | Dobutamine 15+Atropine | |
|---|---|---|---|---|---|---|---|
| Arterial O2 content (ml dl−1) | OFF | 17.02 ± 0.51 | 18.93 ± 0.50 | 21.56 ± 0.61 | 21.41 ± 0.69 | 19.37 ± 0.62 | |
| ON | 16.25 ± 1.06 | 18.39 ± 1.49 | 19.46 ± 1.16 | 19.97 ± 1.05 | 19.25 ± 1.15 | ||
| Arterial free fatty acids (mmol l−1) | OFF | 0.91 ± 0.09 | 1.03 ± 0.12 | 1.23 ± 0.14 | 1.47 ± 0.16 * | 1.18 ± 0.14 | |
| ON | 1.10 ± 0.11 | 1.04 ± 0.12 | 1.22 ± 0.14 | 1.29 ± 0.15 | 1.44 ± 0.20 | ||
| Arterial glucose (mmol l−1) | OFF | 4.80 ± 0.14 | 4.81 ± 0.16 | 4.71 ± 0.32 | 4.89 ± 0.25 | 4.62 ± 0.18 | |
| ON | 5.13 ± 0.25 | 5.41 ± 0.24 | 5.34 ± 0.26 # | 5.22 ± 0.26 | 5.34 ± 0.24 | ||
| Arterial lactate (mmol l−1) | OFF | 0.61 ± 0.10 | 0.67 ± 0.09 | 0.75 ± 0.13 | 0.82 ± 0.14 | 0.86 ± 0.13 | |
| ON | 0.83 ± 0.14 | 0.79 ± 0.13 | 0.88 ± 0.14 | 0.93 ± 0.14 | 0.98 ± 0.14 |
Values are mean ± SEM, n= 8. *P < 0.05 vs. pre-VS baseline; #P < 0.05 vs. corresponding time point pre-VS.
Discussion
Our study shows that acute electrical stimulation of the efferent fibres in the intact right vagus nerve does not affect baseline cardiac function and metabolism, but markedly attenuates mechanical and metabolic response to β-adrenergic stress. External work and oxygen consumption during dobutamine infusion were similarly mitigated, indicating that VS preserved but did not enhance mechanical efficiency. Moreover, vagal efferent firing did not affect the balance between carbohydrate and FFA utilization in the resting heart, whereas during β-adrenergic stress it lowered glucose and lactate consumption compared with control, consistent with the reduced metabolic demand. Interestingly, VS selectively reduced oxidation of the less efficient substrate FFA only at the lowest dose of dobutamine, which more closely approximates physiological levels of sympathetic activation. Atropine reversed many of the functional effects of vagal stimulation, especially stroke work, which showed a marked rebound not paralleled by a similar increase in oxygen consumption and in carbohydrate utilization. As blood was sampled approximately 1 min after atropine administration to avoid the ensuing haemodynamic destabilization, it is possible that we missed the complete reversal of at least some metabolic changes which might have been slower than the functional reversal.
In our experiments, the confounding effects of bradycardia and changes in preload, the latter quantified as LV end-diastolic diameter, were prevented by pacing the heart at a fixed rate. Thus we can safely affirm that the described metabolic changes were independent of HR and preload. The neurostimulator employed in the present study was set to deliver currents in the range 2.5–5 mA, which recruit mainly the efferent fibres (Anholt et al. 2011), also known as B-fibres. We cannot exclude that the afferent fibres (A-fibres) were in part recruited too, although, if relevant, the expected consequence would have been a reflex sympathetic withdrawal. The absence of significant changes in ventricular afterload, as quantified by Ea, indicated that the afferent fibre stimulation, if any, was probably very small. Moreover, the rapid reversal following atropine administration provided further evidence of the involvement of the vagal efferent fibres acting on cardiac muscarinic receptors.
Classic studies in humans and animal models previously found that VS depresses cardiac contractility (DeGeest et al. 1964; Daggett et al. 1967; Henning et al. 1990; Lewis et al. 2001; Xenopoulos & Applegate, 1994). Just a few have focused also on cardiac oxygen consumption (Daggett et al. 1967; Downing et al. 1977), albeit in the absence of β-adrenergic stress, and none on the modulation of energy substrate utilization. The normo-perfused, healthy heart utilizes FFA as the preferential energy substrate. Under pathological conditions, such as ischaemia and failure, the balance between FFA and carbohydrate oxidation changes dramatically and this alteration may play an important role in the pathophysiological processes (Stanley et al. 2005). For instance, we have previously shown that the oxidation of glucose in the pentose phosphate pathway might fuel superoxide generation in myocardial tissue harvested from canine and human failing hearts (Gupte et al. 2006, 2007). It is difficult, based on the present data, to establish whether the inhibition of glucose and lactate consumption found during VS in stressed hearts was the cause or the consequence of reduced oxygen utilization. In both cases, our results are consistent with the direct inhibitory effects of nitric oxide, i.e. the mediator of the vagal negative inotropic action (Hare et al. 1995), on myocardial respiration and glucose utilization (Loke et al. 1999; Lei et al. 2005). As the stimulus intensity applied to the right vagus was very strong (reducing HR by 30 beats min−1), another question is whether this metabolic control is physiologically relevant. We purposely tested extreme experimental conditions, such as superphysiological vagal firing and pharmacological β-adrenoreceptor activation, to amplify the measurable metabolic effects, but it is conceivable that the same mechanisms operate on a smaller scale as part of the physiological parasympathetic–sympathetic antagonism.
While our study provides further evidence that an exogenous activation of the vagal efferent branch minimally affects cardiac function under resting conditions, the interesting mechanisms occurring during β-adrenergic stress can be exploited for clinical use. The potential therapeutic implications of superphysiological vagal efferent output have been recently highlighted by studies in animal models of heart failure and pilot clinical trials (De Ferrari & Schwartz, 2011). Vagus nerve activity is notoriously impaired in heart failure (Chen et al. 1991; Bibevski & Dunlap, 2011). In rats subjected to coronary ligation, VS markedly improved long-term survival through the prevention of pumping failure and cardiac remodelling (Li et al. 2004). In a pilot study, we chronically implanted the CardioFit stimulator in dogs with microembolism-induced heart failure and found improved LV function and biomarkers (Sabbah et al. 2010). Consistently, other investigators have documented a marked attenuation of cardiac dysfunction in dogs with tachypacing-induced heart failure when chronic stimulation of the cervical vagus was concurrently applied (Zhang et al. 2009). Finally, a multi-centre, open-labelled phase II study in New York Heart Association class II–IV heart failure patients showed that right cervical VS was well tolerated and produced significant improvements in quality of life, walk test and LV ejection fraction, maintained at 1 year (De Ferrari et al. 2011). This first trial might open a new avenue for the treatment of heart failure and calls for more extensive evaluations of VS.
The mechanisms underlying the beneficial effects of VS are multiple and only partially understood. Based on previous investigations and on the present one, this type of therapeutic intervention can be envisaged as a cardioselective β-blockade, devoid of systemic side effects. In particular, our results suggest that VS can buffer metabolic peaks following episodes of pronounced sympathetic discharge, a mechanism that should be protective in cardiac ischaemia and/or failure.
Several limitations of the present study should be noted. First, the use of light propofol anaesthesia: VS causes discomfort when initially activated and we opted for an acute implantation of the stimulator on the day of the experiment, and hence we could exploit only in part the advantages of chronic animal preparations. A second limitation was the specific focus on right vagus, which may not account for other actions exerted by the left nerve. However, other authors have compared the stimulation of the right and left vagus and found no differences in their negative inotropic effects (DeGeest et al. 1964; Daggett et al. 1967). Third, we used atropine to block the cardiac muscarinic acetylcholine M2 receptors, although its partial efficacy in reversing metabolic changes might suggest the involvement of different acetylcholine receptors, such as the α7 nicotinic receptor, whose protective role in the heart has been recently shown in a rat model of ischaemia/reperfusion (Calvillo et al. 2011). Unfortunately, this potential mechanism is hardly testable in intact dog preparations, as the infusion of pharmacological inhibitors of nicotinic receptors would cause confounding effects due to parasympathetic ganglionic transmission (Bibevski et al. 2000).
In conclusion, we performed a systematic study on a specific vagal function that was previously unexplored, namely the control of cardiac metabolism in relation to various functional states. While our results do not support appreciable effects in the resting heart, they suggest a significant role under conditions of β-adrenergic stress. Besides the physiological implications, our findings may contribute to better understanding the therapeutic effects of VS, recently proposed for the treatment of heart failure.
Acknowledgments
The authors thank Dr Wenhong Xu for performing the analysis of metabolites. This study was supported by NIH grant P01 HL-74237 (F.A.R., W.C.S. and H.N.S.) and by an Established Investigator Award of the American Heart Association (F.A.R.).
Glossary
- FFA
free fatty acid
- LV
left ventricle/left ventricular
- PDA
pressure–diameter area
- VS
vagal stimulation
Author contributions
Conception and design of the experiments: C.V., H.N.S. and F.A.R. Collection, analysis and interpretation of data K.Q., I.I., G.M. and R.S. Drafting/revising the article: C.V., W.C.S., H.N.S. and F.A.R. All authors approved the final version. I.I. is a full time employee of BioControl Medical, Ltd. The entire study was performed at the New York Medical College, Valhalla, NY.
References
- Anholt TA, Ayal S, Goldberg JA. Recruitment and blocking properties of the CardioFit stimulation lead. J Neural Eng. 2011;8:034004. doi: 10.1088/1741-2560/8/3/034004. [DOI] [PubMed] [Google Scholar]
- Bibevski S, Zhou Y, McIntosh JM, Zigmond RE, Dunlap ME. Functional nicotinic acetylcholine receptors that mediate ganglionic transmission in cardiac parasympathetic neurons. J Neurosci. 2000;20:5076–5082. doi: 10.1523/JNEUROSCI.20-13-05076.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bibevski S, Dunlap ME. Evidence for impaired vagus nerve activity in heart failure. Heart Fail Rev. 2011;16:129–135. doi: 10.1007/s10741-010-9190-6. [DOI] [PubMed] [Google Scholar]
- Böhm M, Gierschik P, Jakobs KH, Pieske B, Schnabel P, Ungerer M, Erdmann E. Increase of Gi alpha in human hearts with dilated but not ischemic cardiomyopathy. Circulation. 1990;82:1249–1265. doi: 10.1161/01.cir.82.4.1249. [DOI] [PubMed] [Google Scholar]
- Calvillo L, Vanoli E, Andreoli E, Besana A, Omodeo E, Gnecchi M, Zerbi P, Vago G, Busca G, Schwartz PJ. Vagal stimulation, through its nicotinic action, limits infarct size and the inflammatory response to myocardial ischemia and reperfusion. J Cardiovasc Pharmacol. 2011;58:500–507. doi: 10.1097/FJC.0b013e31822b7204. [DOI] [PubMed] [Google Scholar]
- Chen JS, Wang W, Bartholet T, Zucker IH. Analysis of baroreflex control of heart rate in conscious dogs with pacing-induced heart failure. Circulation. 1991;83:260–267. doi: 10.1161/01.cir.83.1.260. [DOI] [PubMed] [Google Scholar]
- Daggett WM, Nugent GC, Carr PW, Powers PC, Harada Y. Influence of vagal stimulation on ventricular contractility, O2 consumption, and coronary flow. Am J Physiol. 1967;212:8–18. doi: 10.1152/ajplegacy.1967.212.1.8. [DOI] [PubMed] [Google Scholar]
- De Ferrari GM, Crijns HJ, Borggrefe M, Milasinovic G, Smid J, Zabel M, Gavazzi A, Sanzo A, Dennert R, Kuschyk J, Raspopovic S, Klein H, Swedberg K, Schwartz PJ CardioFit Multicenter Trial Investigators. Chronic vagus nerve stimulation: a new and promising therapeutic approach for chronic heart failure. Eur Heart J. 2011;32:847–855. doi: 10.1093/eurheartj/ehq391. [DOI] [PubMed] [Google Scholar]
- De Ferrari GM, Schwartz PJ. Vagus nerve stimulation: from pre-clinical to clinical application: challenges and future directions. Heart Fail Rev. 2011;16:195–203. doi: 10.1007/s10741-010-9216-0. [DOI] [PubMed] [Google Scholar]
- DeGeest H, Levy MN, Zieske H. Negative inotropic effect of the vagus nerves upon the canine ventricle. Science. 1964;144:1223–1225. doi: 10.1126/science.144.3623.1223. [DOI] [PubMed] [Google Scholar]
- Doenst T, Taegtmeyer H. Alpha-adrenergic stimulation mediates glucose uptake through phosphatidylinositol 3-kinase in rat heart. Circ Res. 1999;84:467–474. doi: 10.1161/01.res.84.4.467. [DOI] [PubMed] [Google Scholar]
- Drake-Holland AJ. The influence of innervation on cardiac metabolism. In: Drake-Holland AJ, Noble MIM, editors. Cardiac Metabolism. Chichester, UK: John Wiley and Sons; 1983. pp. 487–504. [Google Scholar]
- Downing SE, Lee JC, Taylor JF. Cardiac function and metabolism during cholinergic stimulation in the newborn lamb. Am J Physiol Heart Circ Physiol. 1977;233:H451–H471. doi: 10.1152/ajpheart.1977.233.4.H451. [DOI] [PubMed] [Google Scholar]
- Goodwin GW, Ahmad F, Doenst T, Taegtmeyer H. Energy provision from glycogen, glucose, and fatty acids on adrenergic stimulation of isolated working rat hearts. Am J Physiol Heart Circ Physiol. 1998;274:H1239–H1247. doi: 10.1152/ajpheart.1998.274.4.H1239. [DOI] [PubMed] [Google Scholar]
- Gupte SA, Levine RJ, Gupte RS, Young ME, Lionetti V, Labinskyy V, Floyd BC, Ojaimi C, Bellomo M, Wolin MS, Recchia FA. Glucose-6-phosphate dehydrogenase-derived NADPH fuels superoxide production in the failing heart. J Mol Cell Cardiol. 2006;41:340–349. doi: 10.1016/j.yjmcc.2006.05.003. [DOI] [PubMed] [Google Scholar]
- Gupte RS, Vijay V, Marks B, Levine RJ, Sabbah HN, Wolin MS, Recchia FA, Gupte SA. Upregulation of glucose-6-phosphate dehydrogenase and NAD(P)H oxidase activity increases oxidative stress in failing human heart. J Card Fail. 2007;13:497–506. doi: 10.1016/j.cardfail.2007.04.003. [DOI] [PubMed] [Google Scholar]
- Hare JM, Keaney JF, Jr, Balligand JL, Loscalzo J, Smith TW, Colucci WS. Role of nitric oxide in parasympathetic modulation of beta-adrenergic myocardial contractility in normal dogs. J Clin Invest. 1995;95:360–366. doi: 10.1172/JCI117664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henning RJ, Khalil IR, Levy MN. Vagal stimulation attenuates sympathetic enhancement of left ventricular function. Am J Physiol. 1990;258:H1470–H1475. doi: 10.1152/ajpheart.1990.258.5.H1470. [DOI] [PubMed] [Google Scholar]
- Korvald C, Elvenes OP, Myrmel T. Myocardial substrate metabolism influences left ventricular energetics in vivo. Am J Physiol Heart Circ Physiol. 2000;278:H1345–H1351. doi: 10.1152/ajpheart.2000.278.4.H1345. [DOI] [PubMed] [Google Scholar]
- Lei B, Matsuo K, Labinskyy V, Sharma N, Chandler MP, Ahn A, Hintze TH, Stanley WC, Recchia FA. Exogenous nitric oxide reduces glucose transporters translocation and lactate production in ischemic myocardium in vivo. Proc Natl Acad Sci U S A. 2005;102:6966–6971. doi: 10.1073/pnas.0500768102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis ME, Al-Khalidi AH, Bonser RS, Clutton-Brock T, Morton D, Paterson D, Townend JN, Coote JH. Vagus nerve stimulation decreases left ventricular contractility in vivo in the human and pig heart. J Physiol. 2001;534:547–552. doi: 10.1111/j.1469-7793.2001.00547.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Zheng C, Sato T, Kawada T, Sugimachi M, Sunagawa K. Vagal nerve stimulation markedly improves long-term survival after chronic heart failure in rats. Circulation. 2004;109:120–124. doi: 10.1161/01.CIR.0000105721.71640.DA. [DOI] [PubMed] [Google Scholar]
- Loke KE, McConnell PI, Tuzman JM, Shesely EG, Smith CJ, Stackpole CJ, Thompson CI, Kaley G, Wolin MS, Hintze TH. Endogenous endothelial nitric oxide synthase-derived nitric oxide is a physiological regulator of myocardial oxygen consumption. Circ Res. 1999;84:840–845. doi: 10.1161/01.res.84.7.840. [DOI] [PubMed] [Google Scholar]
- Lopaschuk GD, Ussher JR, Folmes CD, Jaswal JS, Stanley WC. Myocardial fatty acid metabolism in health and disease. Physiol Rev. 2010;90:207–258. doi: 10.1152/physrev.00015.2009. [DOI] [PubMed] [Google Scholar]
- Ohgoshi Y, Goto Y, Futaki S, Yaku H, Suga H. Sensitivities of cardiac O2 consumption and contractility to catecholamines in dogs. Am J Physiol Heart Circ Physiol. 1991;261:H196–H205. doi: 10.1152/ajpheart.1991.261.1.H196. [DOI] [PubMed] [Google Scholar]
- Osorio JC, Stanley WC, Linke A, Castellari M, Diep QN, Panchal AR, Hintze TH, Lopaschuk GD, Recchia FA. Impaired myocardial fatty acid oxidation and reduced protein expression of retinoid X receptor-alpha in pacing-induced heart failure. Circulation. 2002;106:606–612. doi: 10.1161/01.cir.0000023531.22727.c1. [DOI] [PubMed] [Google Scholar]
- Post H, d’Agostino C, Lionetti V, Castellari M, Kang EY, Altarejos M, Xu X, Hintze TH, Recchia FA. Reduced left ventricular compliance and mechanical efficiency after prolonged inhibition of NO synthesis in conscious dogs. J Physiol. 2003;552:233–239. doi: 10.1113/jphysiol.2003.048769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Recchia FA, Osorio JC, Chandler MP, Xu X, Panchal AR, Lopaschuk GD, Hintze TH, Stanley WC. Reduced synthesis of NO causes marked alterations in myocardial substrate metabolism in conscious dogs. Am J Physiol Endocrinol Metab. 2002;282:E197–E206. doi: 10.1152/ajpendo.2002.282.1.E197. [DOI] [PubMed] [Google Scholar]
- Saavedra WF, Paolocci N, St John ME, Skaf MW, Stewart GC, Xie JS, Harrison RW, Zeichner J, Mudrick D, Marban E, Kass DA, Hare JM. Imbalance between xanthine oxidase and nitric oxide synthase signalling pathways underlies mechanoenergetic uncoupling in the failing heart. Circ Res. 2002;90:297–304. doi: 10.1161/hh0302.104531. [DOI] [PubMed] [Google Scholar]
- Sabbah HN, Wang M, Jiang A, Ruble SB, Hamann JJ. Right vagus nerve stimulation improves left ventricular function in dogs with heart failure. J Am Coll Cardiol. 2010;55(Suppl):A16.E151. [Google Scholar]
- Senzaki H, Isoda T, Paolocci N, Ekelund U, Hare JM, Kass DA. Improved mechanoenergetics and cardiac rest and reserve function of in vivo failing heart by calcium sensitizer EMD-57033. Circulation. 2000;101:1040–1048. doi: 10.1161/01.cir.101.9.1040. [DOI] [PubMed] [Google Scholar]
- Stanley WC, Recchia FA, Lopaschuk GD. Myocardial substrate metabolism in the normal and failing heart. Physiol Rev. 2005;85:1093–1129. doi: 10.1152/physrev.00006.2004. [DOI] [PubMed] [Google Scholar]
- Van Der Vusse GJ, Dubelaar ML, Coumans WA, Seymour AM, Clarke SB, Bonen A, Drake-Holland AJ, Noble MI. Metabolic alterations in the chronically denervated dog heart. Cardiovasc Res. 1998;37:160–170. doi: 10.1016/s0008-6363(97)00220-4. [DOI] [PubMed] [Google Scholar]
- Xenopoulos NP, Applegate RJ. The effect of vagal stimulation on left ventricular systolic and diastolic performance. Am J Physiol Heart Circ Physiol. 1994;266:H2167–H2173. doi: 10.1152/ajpheart.1994.266.6.H2167. [DOI] [PubMed] [Google Scholar]
- Young ME, Leighton B. Fuel oxidation in skeletal muscle is increased by nitric oxide/cGMP–evidence for involvement of cGMP-dependent protein kinase. FEBS Lett. 1998;424:79–83. doi: 10.1016/s0014-5793(98)00143-4. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Popovic ZB, Bibevski S, Fakhry I, Sica DA, Van Wagoner DR, Mazgalev TN. Chronic vagus nerve stimulation improves autonomic control and attenuates systemic inflammation and heart failure progression in a canine high-rate pacing model. Circ Heart Fail. 2009;2:692–699. doi: 10.1161/CIRCHEARTFAILURE.109.873968. [DOI] [PubMed] [Google Scholar]