

Abstract
The increased vascular tone that defines essential hypertension is associated with depolarization of vascular smooth muscle cells (VSMCs) and involves a change in the expression profile of ion channels promoting arterial contraction. As a major regulator of VSMC resting membrane potential (VM), K+ channel activity is an important determinant of vascular tone and vessel diameter. However, hypertension-associated changes in the expression and/or modulation of K+ channels are poorly defined, due to their large molecular diversity and their bed-specific pattern of expression. Moreover, the impact of these changes on the integrated vessel function and their contribution to the development of altered vascular tone under physiological conditions need to be confirmed. Hypertensive (BPH) and normotensive (BPN) mice strains obtained by phenotypic selection were used to explore whether changes in the functional expression of VSMC inward rectifier K+ channels contribute to the more depolarized resting VM and the increased vascular reactivity of hypertensive arteries. We determined the expression levels of inward rectifier K+ channel mRNA in several vascular beds from BPN and BPH animals, and their functional contribution to VSMC excitability and vascular tone in mesenteric arteries. We found a decrease in the expression of Kir2.1, Kir4.1, Kir6.x and SUR2 mRNA in BPH VSMCs, and a decreased functional contribution of both KIR and KATP channels in isolated BPH VSMCs. However, only the effect of KATP channel modulators was impaired when exploring vascular tone, suggesting that decreased functional expression of KATP channels may be an important element in the remodelling of VSMCs in essential hypertension.
Key points
Essential hypertension involves an electrical remodelling of vascular smooth muscle cells (VSMCs), particularly relevant for K+ channels, as key determinants of resting membrane potential (VM) and excitability.
We explored mRNA expression levels of inward rectifier K+ channel genes in five different vascular beds from normotensive (BPN) and hypertensive (BPH) mice. In mesenteric VSMCs, their functional contribution to cell excitability and vascular reactivity was investigated.
BPH mesenteric VSMCs show a decreased functional expression of both classical inward rectifiers (KIR, Kir2 and Kir4 channels) and ATP-sensitive K+ channels (KATP, Kir6 channels). However, only the changes in the functional expression of KATP channels seem to contribute to the increased vascular reactivity of BPH arteries.
BPN and BPH mice are a useful model to provide integrated information of the impact in the pathophysiology of essential hypertension of the changes in ion channel functional expression.
Introduction
Vascular tone of small arteries and arterioles is the major determinant for peripheral resistance, affecting homeostatic control of blood pressure. Under dynamic conditions, arterial tone is regulated by multiple vasoactive stimuli that determine the degree of contraction of vascular smooth muscle cells (VSMCs) within the vessel wall, by controlling myofilament Ca2+ sensitivity (Somlyo & Somlyo, 2003), and/or intracellular [Ca2+]. Inasmuch as cytosolic [Ca2+] is tightly coupled to Ca2+ influx through voltage-dependent Ca2+ channels (VDCCs), there is a well-established correlation between vascular tone and resting membrane potential (VM) of VSMCs (Nelson & Quayle, 1995). The activity of K+ channels is a major regulator of VM and therefore an important determinant of vascular tone and blood vessel diameter.
VSMCs are able to integrate a plethora of intrinsic and extrinsic factors to determine the activity of the contractile machinery and consequently the arterial tone and diameter. One hallmark finding in all forms of hypertension is an anomalous vascular tone due to a rise in [Ca2+]i in VSMCs that contributes to the elevated peripheral vascular resistance. Systemic hypertension leads to an ‘electrical remodelling’ of VSMCs of the arterial vessels, meaning that there are coordinated changes in the expression and/or function of ion channels and transport mechanisms that ultimately generate a ‘disease-specific’ profile of vascular ion channels promoting arterial contraction and Ca2+ influx. In this electrical remodelling, changes contributing to sustain the higher levels of Ca2+ influx required for persistent arterial contraction coexist with adaptive responses aimed to compensate for the pro-hypertensive changes. To date, two fundamental events have been implicated in this process: VSMC membrane depolarization increasing the open probability of VDCCs (Harder et al. 1985; Cox & Rusch, 2002), and upregulation of the expression of the principal subunit (α1C subunit) of VDCCs, determining the increase in the number of functional VDCCs in VSMCs (Pesic et al. 2004).
Depolarization of the plasma membrane is a common feature of VSMCs in hypertensive animals. Since the pioneering studies reporting an abnormally low permeability of the plasma membrane to K+ ions in VSMCs from hypertensive animals (Harder et al. 1983), a loss of resting K+ efflux resulting in membrane depolarization is a common finding in VSMCs from many vascular beds under high blood pressure (Sonkusare et al. 2006). There is convincing evidence of the functional expression in VSMCs from different vascular beds of four classes of K+ channels: voltage-activated (Kv), large-conductance Ca2+-activated (BKCa), inward rectifiers (Kir, including the ATP-sensitive, KATP) and two-pore domain K+ (K2P) channels (Nelson & Quayle, 1995; Jackson, 2000; Cox & Rusch, 2002).
Molecular biology studies reveal a large diversity of K+ channel subtypes expressed in normal conditions in VSMCs, but there is little information available at the molecular level regarding regulation of the expression and function of K+ channels in essential hypertension. Moreover, the available data regarding the functional contribution of these channels to the hypertensive phenotype show clear discrepancies. Different changes in the functional expression of both BKCa and several Kv channels have been described in hypertensive models. In fact, there are contradictory reports even in the direction of the changes. In the case of BKCa channels, larger currents with increased Ca2+ sensitivity have been found in arteries from hypertensive rats (reviewed by Cox, 2002; Sobey, 2001), while decreased currents with lower Ca2+ sensitivity have been reported in other studies (Amberg et al. 2003; Moreno-Dominguez et al. 2009). Regarding Kv channels, changes in the expression of Kv1.x channels (up- and downregulations, Cox, 2002), decreased functional expression of Kv2 channels (Moreno-Dominguez et al. 2009) and downregulation of Kv7.4 channels (Jepps et al. 2011) are among the changes associated with essential hypertension. In addition to species differences, vascular-bed-specific differences and/or differences in the experimental models, several explanations can account for these discrepancies. First, our knowledge of the signalling pathways that regulate K+ channel expression and function in VSMCs is incomplete. Second, altered vascular K+ channel function during hypertension could be either a cause related to the pathogenesis of the disease or a compensatory mechanism directed to limit the progression of the disease. Finally, although specific gene-targeting studies demonstrate a clear association of these genes to elevated blood pressure in transgenic animals, it is not clear whether the reported changes are relevant for the naturally occurring forms of hypertension.
Using hypertensive and normotensive mice strains (BPH and BPN mice, for blood pressure high and normal, respectively) obtained by phenotypic selection after crossbreeding of eight different strains (Schlager & Sides, 1997) we have previously determined the functional expression of Kv channels and their contribution to VSMC excitability. Our results showed that in VSMCs from mesenteric arteries of BPH mice there is a remodelling of Kv2 currents leading to a decreased Kv current amplitude that contributes to the hypertensive phenotype in resistance arteries (Moreno-Dominguez et al. 2009). However, while VSMCs from BPH mice were significantly more depolarized than BPN VSMCs, we found no clear differences in the contribution of Kv channels (or BKCa channels) to setting resting VM between BPN and BPH cells. This observation implies that in addition to the decrease in both BKCa and Kv currents (Moreno-Dominguez et al. 2009), there must be changes in other non-Kv conductances in the BPH mesenteric VSMCs responsible for the more depolarized resting VM. Inward rectifier K+ channels (KIR and KATP channels) are more active at negative membrane potentials and therefore represent good candidates for regulating the VSMC membrane potential in arteries in the absence of extrinsic depolarizing influences such as pressure or vasoconstrictors. The expression and the functional role of KIR and KATP channels in VSMCs have been extensively characterized in many VSMC preparations (reviewed by Quayle et al. 1997; Sobey, 2001; Ko et al. 2008; Hibino et al. 2010).
KIR channels have been identified in small, resistance arteries (Smith et al. 2008), and their activity is a function of both membrane potential and extracellular K+ concentration (Nelson & Quayle, 1995). Kir2.1 gene expression in VSMCs is required for KIR currents and K+-induced dilations in cerebral arteries (Zaritsky et al. 2000). However, neither the changes in the expression nor their contribution to pathologies such as systemic hypertension has been explored. Although there is indirect evidence suggesting that this function of KIR channels is impaired in cerebral arteries of hypertensive animals (McCarron & Halpern, 1990) there are contradictory reports (reviewed by Sobey, 2001).
KATP channels are expressed in VSMCs from both small and large vessels, and their activity has been proposed to represent the functional link between cellular metabolism and membrane excitability. In addition, the channels are opened by vasodilators and closed by many vasoconstrictors, a modulation that is relevant for their physiological role. KATP provides a background K+ conductance important for the regulation of VM, and hence arterial tone and blood flow (Quayle et al. 1997; Hibino et al. 2010). However, KATP channel contribution to resting tone varies among vascular beds. In some cases (e.g. coronary, mesenteric and renal arteries), there is strong support for the channel contribution to the regulation of the VM of VSMCs, even in normoxia and in the absence of vasodilators. In contrast, KATP channel blockers such as glibenclamide generally do not affect vascular resistance, tone or membrane potential in cerebral arteries, although KATP channels are functional in these vessels (Quayle et al. 1997; Sobey, 2001). If KATP channels are inactive at basal conditions, increased vascular tone during chronic hypertension is unlikely to be related to impaired channel function (Kitazono et al. 1993). However, other studies suggest that KATP channels in hypertensive arteries are dysfunctional but can recover upon long-term treatment of high blood pressure (Ohya et al. 1996). Characterization of vascular function in transgenic mice with selective knockout of the two molecular constituents of KATP channels in VSMCs, namely SUR2B and Kir6.1, did not provide consistent results. Both transgenic models displayed sudden death from coronary artery vasospasm, highlighting the importance of KATP channels in the tonic regulation of vasomotion in coronary arteries, but while SUR2−/− animals were hypertensive (Chutkow et al. 2002), Kir6.1−/− mice had normal blood pressure (Miki et al. 2002).
Here we explore the expression levels of inward rectifier K+ channels in several vascular beds from normotensive (BPN) and hypertensive (BPH) mice. Using resistance (mesenteric) arteries, we also characterize the functional contribution of these channels to control VSMC excitability and vessel tone, measured in intact arteries with pressure myography. VSMCs from BPH mesenteric arteries exhibit a significant decrease in the expression of Kir2.1, Kir4.1, Kir6.x and SUR2 mRNA, and a decrease in the current amplitudes mediated by both KIR and KATP channels. Pharmacological characterization in current-clamp experiments shows a decreased contribution of KIR and KATP channels to VM in BPH cells, but only the response to KATP channel blockers and activators is impaired when arterial tone was tested. The data presented here indicate that the changes in KATP channels could be an important determinant of the hypertensive phenotype in resistance arteries, representing the main change responsible for mesenteric VSMC depolarization in BPH mice.
Methods
Ethical approval
BPN and BPH mice (The Jackson Laboratory, Bar Harbor, ME, USA) were maintained by inbreeding crossing in the animal facility of the School of Medicine of Valladolid, under temperature-controlled conditions (21°C) and with free access to water and food. All animal protocols were approved by the Institutional Care and Use Committee of the University of Valladolid, and are in accordance with the European Community guiding principles regarding the care and use of animals.
Blood pressure measurement
Blood pressure was measured in awake mice with a tail cuff pressure meter (LSI Letica Scientific Instruments, Barcelona, Spain). Repeated measurements were carried out in each animal in different sessions over several days to reduce stress and to obtain reliable values. We monitored systolic (SP) and diastolic (DP) pressure and calculated medium pressure (MP) as MP = DP + 0.33(SP – DP). SP values were (mean ± SEM) 134.9 ± 1.5 mmHg in BPN versus 155.8 ± 3.5 mmHg in BPH and DP values were 90.7 ± 2.8 mmHg in BPN versus 109.0 ± 3.6 mmHg in BPH. MP values averaged 105.0 ± 2.1 mmHg in BPN and 124.9 ± 3.7 mmHg in BPH (P < 0.001 in all cases, n= 15 animals in each group).
Isolation of VSMCs
Mice were killed by decapitation after isofluorane anaesthesia (5% at 2.5 l min−1 O2). VSMCs from mesenteric arteries were obtained as previously described (Moreno-Dominguez et al. 2009). Briefly, after careful dissection and cleaning of connective and endothelial tissues, the arteries were either frozen at −80°C to further extract RNA and proteins or used directly to obtain fresh dispersed VSMCs. Small pieces of mesenteric arteries were placed in smooth muscle dissociation solution (SMDS) Ca2+-free solution containing 0.4–0.6 mg ml−1 papain (Worthington, Lakewood, NJ, USA), 1 mg ml−1 BSA (Sigma-Aldrich, Spain) and 1 mg ml−1 1,4-dithioerythritol (DTE; Sigma) and incubated for 9–15 min at 37°C in a water bath. Next, a second 12–16 min incubation was performed with SMDS 10 μm Ca2+ with 0.4–0.8 mg ml−1 collagenase F (Sigma) and 1 mg ml−1 BSA solution. After two washings in SMDS containing 10 μm Ca2+, single cells were obtained by gentle trituration with a wide-bore glass pipette, stored at 4°C and used within the same day. The ionic composition of SMDS was (mm): 145 NaCl, 4.2 KCl, 0.6 KH2PO4, 1.2 MgCl2, 10 Hepes, glucose 11 (pH 7.4, adjusted with NaOH).
RNA isolation, RT and real-time PCR
Total RNA from arteries was isolated with a MELT Total RNA Isolation System Kit (Ambion, Life Technologies Corporation) following the manufacturer's instructions. Five or six mesenteric arteries (second and third order), the femoral and renal arteries and the thoracic aorta of five mice, and the basilar artery from 40–50 mice were employed for each assay. The quality of the RNA was assayed by optical density measurements at 260 and 280 nm and by electrophoresis on agarose gels. After DNAse I (Ambion) treatment, 500–750 ng of RNA was reverse transcribed with MuLvRT (2.5 u μl−1) in the presence of 1 u μl−1 of RNAse inhibitor, 2.5 μm random hexamers, 1× PCR buffer, 5 mm MgCl2 and 4 mm mixed dNTPs at 42°C for 60 min, to get cDNA (RT+). All reagents were from Applied Biosystems. From the same samples, 200–350 ng of total RNA was used as genomic control in reverse transcriptase reaction in the absence of MuLv and RNase inhibitor at 42°C for 60 min (RT−). A small fraction of these cDNAs were used for real-time amplifications of selected control genes (Gapdh and Gus) in a Rotor-Gene RG3000 (Corbett Life Science, Qiagen) thermocycler. Primer sets and TaqMan probes for these genes were:
mGapdh:
5′-TGTGTCCGTCGTGGATCTG-3′
5′- GATGCCTGCTTCACCACCTT-3′
5′-FAM-TGGAGAAACCTGCCAAGTATGATGACATCA-BHQ2-3′
mGus:
5′-CAATGGTACCGGCAGCC-3′
5′-AAGCTAGAAGGGACAGGCATGT-3′
5′-FAM-TACGGGAGTCGGGCCCAGTCTTG-BHQ2-3′
Amplifications were performed in a final volume of 20 μl, using 10 μl of Absolute QPCR mix (ABgene, Thermo Fisher Scientific Inc), 1 μl of each primer, 1 μl of the probe and 1 μl of cDNA. Cycling conditions were 15 min at 95°C, 40 cycles of 95°C for 15 s and 60°C for 1 min. These amplifications were used to compare the different samples, and also to check the efficiency of the DNase treatment by comparing expression levels of the gene between RT+ and RT− samples.
Real-time PCR was carried out with TaqMan Low Density Arrays (Applied Biosystems, Life Technologies Corporation) at the Genomic Service of the CNIC (Madrid, Spain), with an ABI Prism 7900HT Sequence detection system (Applied Biosystems). Data were acquires with SDS 2.1 software and analysed using the threshold cycle (Ct) relative quantification method (ΔΔCt, Livak & Schmittgen, 2001). Gene expression levels were normalized by the internal control rRNA 18S or Gapdh genes (see supplemental file). The relative abundance of the genes was calculated from 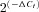 , where ΔCt=Ct,Channel–Ct18S. Differences between hypertensive and normotensive VSMCs were calculated from
, where ΔCt=Ct,Channel–Ct18S. Differences between hypertensive and normotensive VSMCs were calculated from 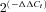 , where ΔΔCt=ΔCt(BPH)–ΔCt(BPN). For comparisons, ΔCt(BPN) was designated as the calibrator, and the data in BPH samples are presented as the fold change in gene expression. When changes are represented as log
, where ΔΔCt=ΔCt(BPH)–ΔCt(BPN). For comparisons, ΔCt(BPN) was designated as the calibrator, and the data in BPH samples are presented as the fold change in gene expression. When changes are represented as log 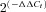 , as in Fig. 2B, a higher expression in BPH mice has a positive value, whilst a lower expression is negative, and absolute values of 0.3, 0.7 and 1 represent 2-, 5- and 10-fold changes respectively.
, as in Fig. 2B, a higher expression in BPH mice has a positive value, whilst a lower expression is negative, and absolute values of 0.3, 0.7 and 1 represent 2-, 5- and 10-fold changes respectively.
Figure 2. Relative abundances of Kir channel mRNA and their changes in hypertensive mice.
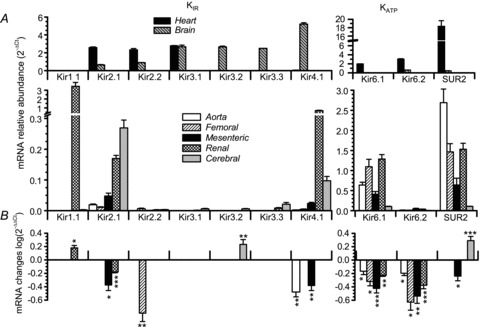
A, expression levels of all the channel genes explored were determined in two control tissues (heart and brain, upper panels) and five vascular beds (aorta, femoral, mesenteric, renal and cerebral). Expression levels are normalized with respect to RP18S. Relative abundance was expressed as 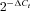 , where ΔCt=Ctchannel−Ct18S. B, the changes in Kir channel expression in BPH arteries (using BPN expression levels as calibrator) in all the vascular beds explored. The changes were calculated as
, where ΔCt=Ctchannel−Ct18S. B, the changes in Kir channel expression in BPH arteries (using BPN expression levels as calibrator) in all the vascular beds explored. The changes were calculated as 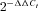 , where ΔΔCt=ΔCt(BPH)–ΔCt(BPN). Data are represented on log scale, so the absence of changes will give values close to 0, negative values mean decreased expression and positive values increased expression. Statistical analysis in this plot was by Student's t test for two independent samples (ΔCt(BPH) and ΔCt(BPN)) obtained with RP18S as endogenous control. For the shake of clarity, only statistically significant changes are plotted. Each bar shows the mean of 6–12 determinations obtained in 3–6 duplicate assays. *P < 0.05, **P < 0.01, ***P < 0.001.
, where ΔΔCt=ΔCt(BPH)–ΔCt(BPN). Data are represented on log scale, so the absence of changes will give values close to 0, negative values mean decreased expression and positive values increased expression. Statistical analysis in this plot was by Student's t test for two independent samples (ΔCt(BPH) and ΔCt(BPN)) obtained with RP18S as endogenous control. For the shake of clarity, only statistically significant changes are plotted. Each bar shows the mean of 6–12 determinations obtained in 3–6 duplicate assays. *P < 0.05, **P < 0.01, ***P < 0.001.
Data were obtained from duplicate determinations from at least three different assays. Where expression of a gene was not detected in one of the conditions, a Ct value of 40 was assigned to set a minimal expression value for comparisons.
A more detailed explanation of the statistical analysis used is provided as supplemental material.
The TaqMan Gene Expression assays (Applied Biosystems) employed were as follows: Kir1.1 (Kcnj1-Mm00444727_s1), Kir2.1 (Kcnj2-Mm00434616_m1), Kir2.2 (Kcnj12-Mm01237201_m1), Kir3.1 (Kcnj3-Mm00434618_m1), Kir3.2 (Kcnj6-Mm00440070_m1), Kir3.3 (Kcnj9-Mm00434622_m1), Kir4.1 (Kcnj10-Mm00445028_m1), Kir6.1 (Kcnj8-Mm00434620_m1), Kir6.2 (Kcnj11-Mm00440050_s1), Sur2 (Abcc9-Mm00441638_m1), Cnn1 (Cnn1-Mm00487032_m1), Nos3 (Nos3-Mm00435204_m1) and 18S rRNA (18S-Hs99999901_s1).
The Sur2 assay was designed at the 30–31 exon boundary, in a region conserved in all Sur2 isoforms.
Immunoblots
For total protein isolation, 20–25 mesenteric arteries obtained from five to six animals were placed in Trizol Reagent (Invitrogen, Life Technologies Corporation) and homogenized in a Precellys 24 homogenizator (Bertin Technologies, Montigny-le-Bretonneux, France) using ceramic beads (CK14) following the manufacturer's instructions. Protein samples were dissolved in 1% SDS. Samples of 10–20 μg were incubated with XT Reducing Agent and XT Sample Buffer (Bio-Rad Laboratories, Hercules, CA, USA) for 5 min at 95°C. Proteins were separated by SDS-PAGE (Criterion Precast Gel 10% Bis-Tris for Kir2.1, Kir4.1 and Kir6.1 or Criterion TGX 4–15% for SUR2 detection, Bio-Rad) and transferred onto polyvinylidene difluoride membrane. Membranes were blocked in either 5% non-fat dry milk (Kir6.1 and β-actin) or 0.5% Tropix® I-Block™ (Applied Biosystems, Kir2.1, Kir4.1 and SUR2) in TTBS (Tris-buffered saline with 0.1% Tween 20) for 1 h. Membranes were then incubated with the primary antibodies in blocking solution for 1 h. Antibodies used were mouse monoclonal anti-β-actin (1:1000, AbCam, Boston, MA, USA 8226) or rabbit polyclonal anti-Kir2.1 (1:333, Alomone Labs, Jerusalem, Israel, APC-026), anti-Kir4.1 (1:400, Alomone Labs, Jerusalem, Israel, APC-035), anti-Kir6.1 (1:1000, Alomone Labs, Jerusalem, Israel, APC-105) and anti-SUR2 (1:250, Santa Cruz Biotechnology, sc-25684). As in the case of the Sur2 TaqMan assay, anti-SUR antibody reacts with both SUR2A and SUR2B isoforms. Membranes were next incubated with horseradish peroxidase-conjugated anti-rabbit IgG (Santa Cruz Biotechnology, Santa Cruz, CA, USA) or anti-mouse IgG (Bio-Rad Laboratories) at a final concentration 1:10,000 for 1 h. Protein signals were detected in the VersaDoc 4000 Image System (Bio-Rad) with SuperSignal West Femto Maximum Sensitivity Substrate.
Quantification was carried out with the densitometric analysis of the bands for each antibody and normalized to their corresponding β-actin signals, using the Quantity One software (Bio-Rad).
Electrophysiological methods
Ionic currents were recorded at room temperature (20–25°C) using the whole cell or the perforated patch configurations of the patch-clamp technique. Freshly isolated VSMCs were placed directly in the recording chamber and allowed to settle for a few minutes before starting perfusion with the external solution. Patch pipettes were made from borosilicate glass (2.0 mm o.d., WPI) and double pulled (Narishige PP-83) to resistances ranging from 2 to 5 MΩ when filled with the internal solution. This solution was designed to amplify the magnitude of KATP currents in VSMCs, as previously described (Beech & Bolton, 1989) and contained (mm): 100 K-glutamate, 25 KCl, 2 MgCl2, 10 Hepes, 10 EGTA, 0.1 Na2GTP, 0.1 MgATP, 1 Na2GDP (pH 7.2, with KOH). The composition of the bath solution (Standarde) was (mm): 141 NaCl, 4.7 KCl, 1.2 MgCl2, 1.8 CaCl2, 10 glucose, 10 Hepes (pH 7.4, with NaOH). Whole-cell currents were recorded using an Axopatch 200 patch-clamp amplifier, filtered at 2 kHz (−3 dB, 4-pole Bessel filter), and sampled at 10 kHz. Recordings were digitized with a Digidata 1322A interface, driven by CLAMPEX 10 software (Axon Instruments, Molecular Devices, Sunnyvale, CA, USA). Current–voltage relationships were obtained from a holding potential of −80 mV in 2 s ramps from −150 to +30 mV.
VM measurements were performed at room temperature using the perforated-patch technique and recordings were obtained with an Axopatch 700A patch-clamp amplifier. Pipette tips were briefly dipped into a solution containing (in mm): 40 KCl, 95 K-glutamate, 8 CaCl2 and 10 Hepes (pH 7.2), and backfilled with the same solution containing amphotericin B (300 μg ml−1). Electrical access to cell cytoplasm was assessed by monitoring the increase in cell capacitance. When a stable value was reached, the amplifier was switched to current-clamp mode and VM was continuously recorded. The high Ca2+ content of the pipette solution ensures the correct performance of the perforate-patch technique, as accidental rupture of the patch (changing to whole-cell configuration) leads to a sudden Ca2+ load and cell death. The composition of the bath solution was the same as that indicated for the voltage-clamp experiments. The perforated-patch technique was also use in the voltage-clamp modality to explore the currents elicited by the same ramp protocol used in whole-cell experiments without manipulation of the intracellular medium.
Electrophysiological data analyses were performed with the Clampfit subroutine of the pCLAMP software (Axon Instruments) and with Origin 7.5 software (OriginLab Corp., Northampton, MA, USA). The linear fit of the ramps was used to determine the slope conductance of the cells and hence the total membrane resistance. Pooled data are expressed as mean ± standard error of the mean (SEM). Statistical comparisons between groups of data were carried out with the two-tailed Student's t test for unpaired data, and values of P < 0.05 were considered statistically different.
Myography recordings
Segments of 3rd order mesenteric arteries (unpressurized lumen diameters between 130 and 190 μm) were dissected out of surrounding tissues and mounted in the recording chamber of a pressure myograph system (Danish Myo Technology, Aarhus, Denmark). Arteries were cannulated at both ends with glass cannulae and secured with nylon filaments. The glass cannulae were filled with PSS solution which was maintained in the cannulae throughout all experiments. The chamber was placed on the stage of an inverted DMT microscope with a built-in CCD camera that allowed continuous measurement of lumen diameter using MyoView software. Arteries were superfused with physiological saline solution (PSS) at 37°C equilibrated with 5% CO2–95% air. The composition of this solution was (mm): 120 NaCl, 5 KCl, 2.5 CaCl2, 25 NaHCO3, 1.18 Na2HPO4, 1.17 MgSO4 and 10 glucose (pH 7.4). The endothelium was removed, prior to pressurization of the artery, by passing air bubbles through the lumen for a few seconds. Viability of mesenteric arteries was evaluated by their ability to constrict in response to 0.5–1 μm phenylephrine, and the endothelium denudation was confirmed by the absence of dilation in response to 10 μm acetylcholine. The mounted artery was pressurized to 70 mmHg and allowed to equilibrate for at least half an hour before starting the experiment. During this period, around 30–50% of the mounted arteries developed significant myogenic tone. When a dilatory response was tested, initial constriction was potentiated by adding 0.5–1 μm phenylephrine to facilitate analysis of the response. All chemical agents were added to the superfusate. At the end of each experiment, vessels were superfused either with a solution containing 10 μm nifedipine or with a 3 mm EGTA, Ca2+-free PSS solution to determine maximal vessel diameter upon relaxation.
For characterization of the myogenic response, pressure–diameter curves elicited by pressure steps from 20 to 140 mmHg were obtained in control conditions (active tone) and in the presence of 10 μm nifedipine (passive tone), and myogenic tone was defined as the difference between the two curves. For study of the contribution of KIR and KATP channels to vessel excitability, we determined the changes in diameter elicited by selective blockers and/or openers of the channels at least at two different pressure values. Data analysis was performed with Origin 7.5 software.
Results
Characterization of VSMC excitability in BPN and BPH mesenteric arteries
We first investigated the existence of differences between BPN and BPH VSMCs using freshly isolated cells to determine their electrical properties. Total ionic currents were characterized in isolated cells with the perforated patch configuration of the patch-clamp technique. Currents elicited with voltage ramps between −150 and +30 mV, in the presence of Standarde bath solution, showed clear differences between BPN and BPH VSMCs (Fig. 1A). BPH cells showed a decrease of the current elicited at hyperpolarizing potentials together with a decrease in the slope conductance of the current–voltage relationship at potentials around −80 mV (the estimated K+ reversal potential). There was also a more depolarized VM (membrane potential at I= 0), and around these values the slope conductance increased. When data from each individual cell were averaged, the differences in the slope conductances were statistically significant (Fig. 1B). Current-clamp measurements confirmed that VSMCs from BPH mesenteric arteries were depolarized when compared with BPN cells, as previously described (Moreno-Dominguez et al. 2009). Together, these data suggest that the more depolarized VM observed in VSMCs from BPH mice could be explained by a decrease in K+ conductance (reflected by the smaller slope conductance values around EK). Moreover, because the chord conductance at Ek is not statistically different between BPN and BPH cells (0.54 ± 0.046 nS in BPN vs. 0.60 ± 0.08 nS in BPH, P= 0.09), it is reasonable to assume that changes in other conductances may contribute minimally to the more depolarized VM observed in VSMCs from BPH mice.
Figure 1. Differences in excitability between BPN and BPH mesenteric arteries.
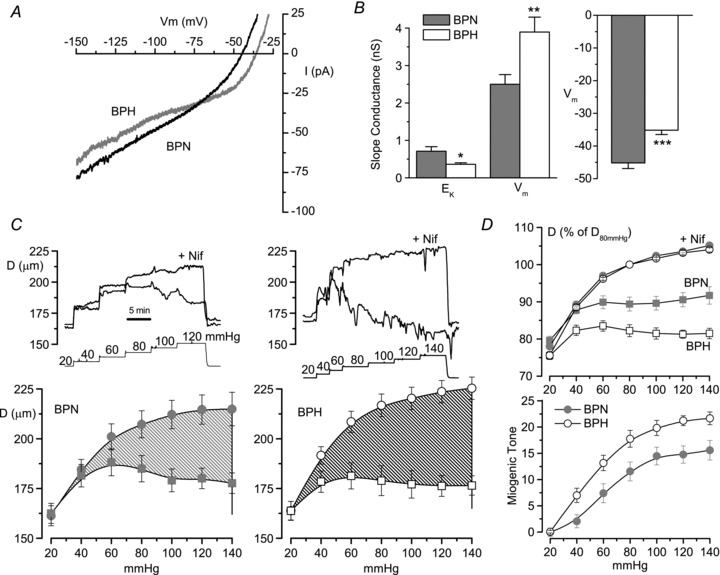
A, currents obtained in depolarizing ramps from −150 to +30 mV in acutely dissociated VSMCs from BPN and BPH mesenteric arteries. Currents were recorded with the perforated patch configuration of the patch-clamp technique while cells were bathed in standard saline solution. Each trace is the average current from 9–11 cells. B, slope conductance was calculated from the linear fit of the current traces obtained from each individual cell around (±5 mV) EK (−80 mV in our experimental conditions) and VM (V at I= 0). Each bar shows the mean ± SEM of 13–16 determinations in each group. *P < 0.05, **P < 0.01, ***P < 0.001, Student's t test for unpaired data. Resting VM was obtained with direct measurements from current-clamp experiments. Data are from 37 BPN and 34 BPH cells. C, pressure–diameter curves obtained from mesenteric BPN and BPH arteries mounted on a pressure myograph, and subjected to step pressure changes (in mmHg) with the protocol shown in the insets. For each artery, pressure protocol was applied in control conditions (active diameter, Dcontrol, squares) and in the presence of nifedipine (passive diameter, Dnif, circles). The average diameters obtained at the different pressures studied from 11 arteries in each group are shown in the lower panels. The shaded areas represent the difference between passive and active diameter, i.e. the myogenic tone. D, the upper panel shows passive diameter (nifedipine, circles) and active diameter (squares) after normalizing each artery to the passive diameter obtained at 80 mmHg. The average myogenic tone obtained from BPN and BPH arteries and measured as 100 × (Dnif–Dcontrol)/Dnif is represented in the lower panel. n= 11 and 12 for the BPN and BPH groups, respectively.
Pressure-induced changes in vessel diameter in both BPH and BPN mesenteric arteries were also explored with the protocol depicted in the insets to Fig. 1C. In each preparation, the pressure–diameter relationship was determined under control conditions (active diameter curves) and in the presence of 10 μm nifedipine (passive diameter curves). The difference between passive and active diameter at each pressure (the myogenic tone) in each preparation is plotted in Fig. 1D (lower graph). Myogenic tone was significantly larger at all pressures in BPH mesenteric arteries, suggesting an increased vascular reactivity in the hypertensive animals. This increased myogenic response in BPH arteries could be a consequence of the more depolarized VM of the VSMCs determined by the decreased K+ conductance in these cells. We examined this hypothesis by exploring the differences in expression of K+ channels that may contribute to setting VM, such as KIR and KATP channels.
mRNA expression profile of inward rectifier channel subunits
We analysed the expression profile of the channel subunits encoding inward rectifier K+ channels in mesenteric VSMCs as well as in several other vascular beds from BPN and BPH mice. The genes explored included members of the Kir1 to Kir6 subfamilies as well as the SUR2 subunit. Ten channel genes were studied, together with control genes such as calponin as a control for VSMCs, endothelial nitric oxide synthase (eNOS) as a control for endothelial cell contamination and ribosomal protein 18S (RP18S) as an endogenous control for the quantitative PCR. The preparations studied included VSMCs from aorta, femoral, renal, mesenteric and cerebral arteries of BPN and BPH animals, and two non-vascular preparations, cardiac muscle cells from the left ventricle and nervous tissue from the prefrontal cortex. In all cases, we explored both the relative abundance of the channel genes expressed in the normotensive preparations and the changes observed in BPH animals. The data are summarized in Fig. 2. Figure 2A shows the relative abundance of all the genes studied in both control tissues (heart and brain, upper panels) and vascular tissues (lower panels), and Fig. 2B depicts their changes upon hypertension only in VSMCs. No changes were detected when comparing heart and brain preparations from BPN and BPH mice (data not shown). Expression levels of the molecular constituents of KATP channels (Kir6.1, Kir6.2 channels and SUR2 receptor) are represented in separate plots (left panels) due to their higher expression levels (note the different scales).
Kir2.1 and Kir4.1 are expressed in all vascular beds studied, being more abundant in mesenteric, renal and cerebral vessels (Fig. 2A). Kir1.1 is also especially abundant in renal arteries whilst Kir2.2 and Kir3.x are minimally represented. As previously described, KIR channel genes are abundantly expressed in brain (all but Kir1.1) and in cardiac tissue (Kir2.x and Kir3.1). Regarding KATP channel genes, we detected high levels of mRNA expression for the Kir6.1 channel and SUR2 receptor, the main constituents of the vascular KATP channels (Seino & Miki, 2004) in all vessels studied. The more abundant expression of Kir6.2 mRNA in cardiac and neural tissues is also consistent with previous reports.
Expression changes in BPH are depicted in Fig. 2B. In this plot, positive values represent increased levels of expression compared with BPN whilst negative values represent decreased expression (see Methods). The most relevant changes were the lower expression of Kir2.1, Kir4.1 and SUR2 in BPH mesenteric arteries, as well as the general decrease in the expression of Kir6.1 in all BPH vascular beds tested. Other significant changes were the evident increase of Kir1.1 in renal arteries and Kir3.2 and SUR2 in cerebral arteries. Other changes, although statistically significant, may be considered less relevant if we take into account the low levels of expression.
Together, these data show that the hypertensive phenotype is associated with a decrease in the expression pattern of the mRNA encoding the most abundant vascular Kir channels (Kir2.1, Kir4.1 and Kir6.1 and SUR2 channels). Of note, all changes are found in the mesenteric bed, pointing to these arteries as a good model to study their functional impact.
Expression of KIR and KATP channel-encoding proteins in BPN and BPH VSMCs
We next determined whether the mRNA changes correlate with protein expression levels of VSMC extracts obtained from BPN and BPH mesenteric arteries. Figure 3 shows representative immunoblots for Kir2.1, Kir4.1, Kir6.1 and SUR2 proteins together with histograms showing their quantification from 3–5 different experiments. In agreement with the mRNA expression levels, we found a decreased expression of all the proteins in BPH VSMCs, although this decrease was not statistically significant for Kir2.1 protein. Changes in expression were particularly large in the case of Kir6.1 channels, in which protein levels were hard to detect in BPH samples.
Figure 3. Expression of Kir2.1, Kir4.1, Kir6.1 and SUR2 proteins in BPN and BPH mesenteric VSMCs.
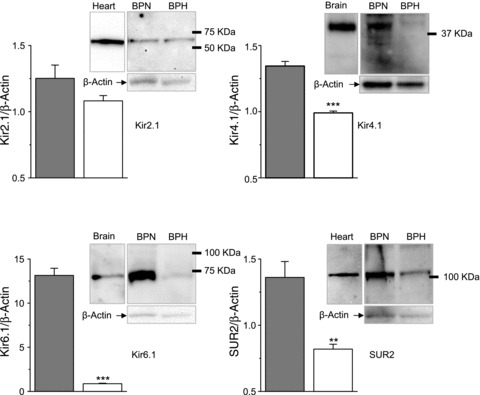
Representative immunoblots of mesenteric VSMC lysates from BPN and BPH arteries showing a band of the expected size for Kir2.1, Kir4.1, Kir6.1 and SUR2 as indicated. Loading control (β-actin) of the same membranes is also shown, as well as positive controls for the antibodies (brain lysates for Kir4.1 and Kir6.1 and heart homogenates for Kir2.1 and SUR2). Quantification of these data (bars plots) was obtained via densitometric analysis of 3–5 immunoblots for each antibody, normalized to their corresponding β-actin signals. **P < 0.01, ***P < 0.001.
Functional characterization of KIR and KATP channels in isolated VSMCs
The functional expression of inward rectifying K+ channels was evaluated with electrophysiological studies in freshly dispersed mesenteric VSMCs from BPN and BPH mice. Membrane currents were studied using a ramp-pulse protocol (Fig. 4). Total current density at −150 mV was significantly larger in BPN cells than in BPH cells (−9.6 ± 1.6 vs.−6.4 ± 0.69 pA pF−1, P < 0.05, n= 20 cells in each group). This decrease in current density reflects a small decrease in the total current amplitude in BPH cells (see averaged data in Fig. 1A) but more importantly an increase in the cell size of the VSMCs from BPH mesenteric arteries, as previously reported (Moreno-Dominguez et al. 2009). In the set of experiments reported in this paper, the average cell capacitance was 14.23 ± 0.77 pF in BPN cells versus 19.18 ± 1.12 pF in BPH cells, P < 0.001).
Figure 4. Pharmacological dissection of KIR and KATP currents in mesenteric VSMCs.
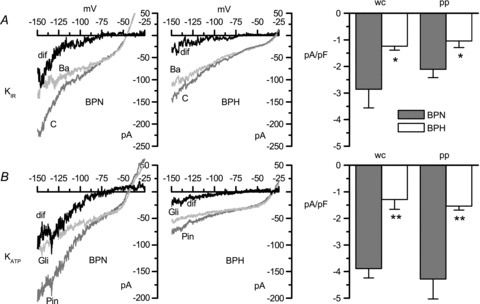
A, KIR currents were defined as the fraction of current sensitive to 100 μm BaCl2 (dif), and were obtained by subtracting current traces elicited by voltage ramps in the presence of BaCl2 (Ba) from traces in control conditions (C). Sample traces obtained with VSMCs from BPN and BPH mice using the perforated patch configuration are shown in the left panels. The KIR average current densities obtained at −150 mV, both with the perforated patch (pp) or the whole-cell configuration (wc) of the patch clamp technique, are depicted in the bar plot. B, KATP currents were defined as the difference (dif) between currents recorded in the presence of 100 μm pinacidil (Pin) and currents recorded in the presence of 20 μm glibenclamide (Gli). Current–voltage plots show representative traces obtained with the perforated patch configuration, and bar graphs depict the KATP average current densities obtained at −150 mV. Each bar, in both A and B, is the mean ± SEM of 7–10 cells. *P < 0.05, **P < 0.01.
A pharmacological approach was used to dissect the contribution of both KIR and KATP channels to the total current. KIR currents (Fig. 4A) were defined as the 100 μm BaCl2-sensitive fraction of the current, whereas KATP currents (Fig. 4B) were estimated as the difference between the current in the presence of 100 μm pinacidil and the current in the presence of 20 μm glibenclamide. Figure 4 shows representative examples of the effect of these drugs on the currents recorded from BPN and BPH cells in perforated-patch experiments, and the estimated KIR and KATP currents. In both cases, the small size of the currents and the inward rectification preclude a reliable estimation of the reversal potential of the subtracted currents, although in all cases this was around the estimated value for EK. The histograms on the right in Figure 4 represent summary data of current densities at −150 mV obtained both with perforated-patch and with whole-cell experiments. As shown, KATP currents in BPN cells represented a larger fraction of the total outward current than KIR currents (values in perforated patch were −4.28 ± 0.77 and −2.11 ± 0.32 pA pF−1, respectively). Also, in comparisons with BPH cells, the decrease in the fraction of the current due to KATP channels was larger than the decrease of KIR currents. Again, in perforated patch and at −150 mV, KATP current density in BPH cells represented 36% of the current in BPN cells, while KIR current density was reduced to 50% of the BPN current.
To complete the electrophysiological characterization, we explored the contribution of KIR and KATP to mesenteric VSMC excitability by analysing the changes in resting VM observed upon application of selective channel blockers and openers (Fig. 5). Figure 5A illustrates the effect of BaCl2 at two concentrations that are below the reported Kd of KATP channels for BaCl2 (Ko et al. 2008) and may be considered selective for KIR (30 and 100 μm). Figure 5B depicts the effects of pinacidil (100 μm) and glibenclamide (20 μm). The differences in the response of BPN and BPH cells were very evident, as shown in the representative traces (left) and in the averaged data (right). The depolarizing response to BaCl2 was decreased to 45–60% of that observed in BPN cells, but the changes in the response to KATP channel modulators in BPH cells were more prominent; glibenclamide-induced depolarization was reduced to 33% of that seen in BPN cells, while the hyperpolarizing response to pinacidil was absent in 7 of 22 cells, and represented on average 17% of the amplitude of the response in BPN cells. If we assume that pinacidil and glibenclamide open and close, respectively, all available channels, we can estimate the percentage of KATP channels open at rest as shown in the inset to Fig. 5B. Whilst there are around 50% of KATP channels open at rest in BPN cells, this percentage increases to almost 70% in BPH cells.
Figure 5. Contribution of KIR and KATP channels to resting VM in BPN and BPH mesenteric VSMCs.
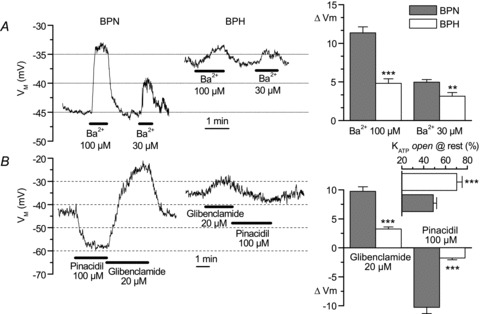
The left graphs show recordings illustrating the effects of two different BaCl2 concentrations (30 and 100 μm, A) and 100 μm pinacidil and 20 μm glibenclamide (B) in representative BPN and BPH cells. Both resting VM and the amplitude of the drug-induced changes are clearly different between BPN and BPH cells. These differences were consistent and significant, as shown in the summary data on the bar graphs. n= 9–18 cells in each group, Student's unpaired t test. The inset in B (horizontal bar plot) shows the fraction of KATP channels that are open at resting membrane potential, calculated as 100 ×ΔVM,glibenclamide/(ΔVM,glibenclamide+ΔVM,pinacidil). Each value is the mean ± SEM of 18 (BPN) and 16 (BPH) cells. **P < 0.01, ***P < 0.001
Functional characterization of KIR and KATP channels in pressurized mesenteric arteries from BPN and BPH mice
The functional data obtained in isolated VSMCs showed a good correlation with the expression data for Kir2.1, Kir4.1 and Kir6.x channels, suggesting that the decreased expression of KIR and KATP channels could be contributing to the hypertensive phenotype. To explore this hypothesis, we studied the effects of pharmacological modulation of these channels on the diameter of BPN and BPH mesenteric arteries pressurized in physiological conditions. The contribution of KIR channels to vascular tone was analysed by the changes in diameter observed in response to increasing BaCl2 concentrations (Fig. 6 shows the effects of 3, 30 and 300 μm) at two different pressures, 40 and 100 mmHg. The dose–response curve for the vasoconstriction induced by BaCl2 in BPN and BPH arteries is plotted in Fig. 6B. As expected from the well-established correlation between resting VM of VSMCs and arterial tone (Nelson & Quayle, 1995; Smith et al. 2008), the BaCl2-induced contraction was larger at lower vascular pressures. However, no significant differences were observed in the whole organ response between BPN and BPH arteries.
Figure 6. Contribution of KIR channels to vascular tone in mesenteric BPN and BPH arteries.
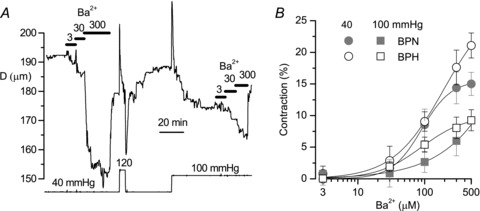
A, time course of the changes in diameter in a BPN artery subjected to the pressure protocol shown at the bottom. At each pressure value, increasing concentrations of BaCl2 were applied as indicated, to obtain a dose–response curve for the barium-induced contraction. Myogenic tone was clearly observed in this artery when pressure was raised from 40 to 120 or 100 mmHg. B, dose–response curves for barium are represented for the two experimental conditions (BPN, grey symbols; BPH, white symbols) and the two pressure values explored (40 mmHg, circles; 100 mmHg, squares). n= 7–8 arteries.
The contribution of KATP channels to vascular reactivity was explored by analysing the vasodilation in response to increasing concentrations of pinacidil, ranging from 0.1 to 100 μm. In these experiments, to facilitate the analysis of the vasodilatory effect, arteries were preconstricted with 0.5–1 μm phenylephrine and pressure was maintained through the entire experiment at 70 mmHg. In this group of experiments the differences in the response of BPH and BPN arteries were readily evident, as illustrated in Fig. 7. Both the EC50 for the pinacidil-induced relaxation and the maximal vasodilation observed were significantly reduced in BPH arteries, suggesting a decreased contribution of KATP channels to vascular tone in these arteries.
Figure 7. Contribution of KATP channels to vascular tone in mesenteric BPN and BPH arteries.
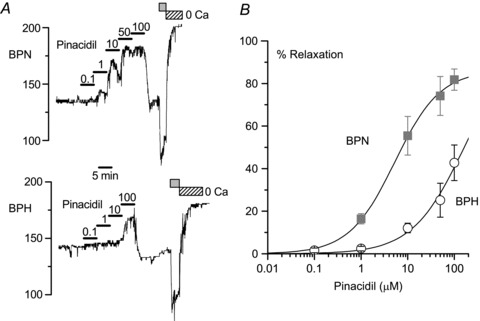
A, dose–response effect of pinacidil on the changes in diameter obtained in representative BPN and BPH mesenteric arteries. The arteries were pressurized to 70 mmHg in the presence of 0.5 μm phenylephrine to facilitate quantification of the pinacidil dilatory response. At the end of the experiments, the minimal and maximal arterial diameters were determined by sequential application of 10 μm phenylephrine (grey box) and a Ca2+-free solution (hatched box), respectively. B, summary of the effects of pinacidil at different concentrations in BPN (squares) and BPH (circles). Data are normalized to the maximal diameter values obtained in Ca2+-free solution and are expressed as a percentage of relaxation. Averaged data from five BPN and five BPH arteries were fitted to the sigmoidal function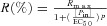 obtaining an apparent EC50 for pinacidil of 5.58 and 162.17 μm in BPN and BPH arteries, respectively.
obtaining an apparent EC50 for pinacidil of 5.58 and 162.17 μm in BPN and BPH arteries, respectively.
Discussion
In the present study, using a mouse model of essential hypertension, we aimed to determine changes in K+ conductances that could have an impact on the resting VM of VSMCs and hence participate in the changes in excitability observed in the arteries from hypertensive animals (Fig. 1). Pressure myography data obtained in BPN and BPH mesenteric arteries show that while passive changes in diameter in response to increased pressure are similar in the two groups, the active pressure-induced vasoconstriction and hence the myogenic tone are larger at all pressure values in BPH arteries (Fig. 1C and D), indicating an increased vascular reactivity. We hypothesize that this increased excitability could be related to the more depolarizing resting potential observed in BPH VSMCs (Fig. 1B) and explained by changes in the functional expression of K+ channels. We have chosen inward rectifier K+ channels as good candidates a priori for the role, as they are active at values near resting VM. Moreover, given the high resting input resistance of VSMCs (Fig. 1B; Nelson et al. 1990) it is plausible that small changes in the functional contribution of these K+ channels, such as a slight decrease in the number of available channels, could lead to a significant depolarization and then vasoconstriction.
Our data show that there is in fact a decrease in the expression of some molecular constituents of the classical KIR channels in mesenteric VSMCs (Kir2.1 and also Kir4.1) as well as in all the components of the KATP channels (albeit the decrease of SUR2 was moderate and failed to be statistically significant under all the conditions tested). These changes show a good correlation with the electrophysiological properties of VSMCs from BPN and BPH mice, as we found a decrease of both KIR and KATP currents and a diminished contribution of these channels to resting VM in BPH VSMCs.
The present work does not preclude the existence of changes in the functional expression of other ionic conductances that could also be participating in the changes in excitability observed in BPH cells, but it is clear that a decreased K+ conductance at values around and below resting VM is needed to explain the observed changes in excitability (Fig. 1). In fact, the average currents obtained in perforated patch experiments in BPN and BPH cells can be reasonably reproduced through simple calculations based on a parallel conductance model (Fig. 8). Assuming that at any voltage between −120 and −20 mV total membrane current is the arithmetic sum of currents through inward rectifying (KIR plus KATP), voltage-dependent K+ (KV) and Ca2+ (CaV) channels and a putative Na+-dependent inward current (mediated for example by TRP channels), actual data obtained in VSMCs from BPN mice can be reasonably reproduced. The calculation depicted for BPH currents includes the reduction of the KV current amplitude previously reported in BPH cells (Moreno-Dominguez et al. 2009) and a decrease of the current through inward rectifying K+ channels by 50%. As shown in Fig. 8, these changes generate a total current that closely mimics the changes observed in BPH cells, namely a reduction in the total current amplitude at −150 mV, a decrease of the slope conductance at potentials closed to EK and a shift of the resting VM towards more depolarized potentials. Actually, the fact that no other changes are needed is experimentally supported, as whole currents at EK are very similar in BPN and BPH mice (Fig. 1).
Figure 8. Minimal conductance model required to simulate the whole-cell I–V relationship in BPN and BPH mesenteric VSMCs.
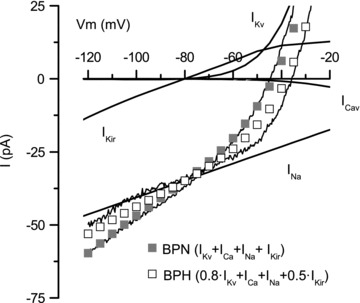
Whole-cell currents in the range between −120 and −20 mV are simply simulated by the addition of four parallel conductances mediated by inward rectifying channels (KIR plus KATP, IKir), voltage-dependent K+ (IKv) and Ca2+ (ICaV) channels and a voltage-independent inward cationic current (INa). The conductance mediating INa in the simulation has the required value to account for total current at the K+ reversal potential. Grey squares represent the simulated current for BPN currents, and open squares the simulated current for BPH currents. This latler simulation was obtained by decreasing Kir conductance to 50% and Kv conductance by 20%. The actual experimental data depicted in Fig. 1A are also represented in the figure.
Vascular bed distribution of inward rectifier K+ channels
We also provide a characterization of the expression pattern of the different molecular constituents of inward rectifier K+ channels in five different vascular beds both in normotensive and in hypertensive animals. Our results confirm the predominant expression of Kir2.1 transcripts in small vessels and the more homogeneous expression of the molecular components of KATP channels. Also, while hypertension induces significant changes in the expression of KATP channel transcripts in most vascular beds explored (Fig. 2), the changes in expression of Kir2.1 are very clearly restricted to renal and mesenteric arteries. We also detected a moderate expression of members of the G-protein gated inward rectifier K+ channels (GIRK, Kir3.x), especially in cerebral and renal vessels. Surprisingly, some members of the K+ transport channels (i.e. Kir1.x and Kir4.x channels) were detected in the mRNA expression studies. Kir4.1 mRNA was found in all vascular beds explored at levels similar to those of Kir2.1 transcripts, and its expression was significantly smaller in aorta and mesenteric arteries from BPH mice. These channels have been previously found in glial cells and in renal and gastric epithelium (Hibino et al. 2010), but this is the first report of their presence in VSMCs. Also, we have confirmed the expression of Kir4.1 protein in mesenteric VSMCs (Fig. 3). Lack of specific inhibitors has precluded further characterization of these channels, although their sensitivity to Ba2+ blockade suggests that these changes in expression could contribute to the overall changes in KIR currents reported in this work. Kir1.1 (first described as ROMK1) is expressed only in cerebral arteries and at very high levels in renal arteries and it is one of the few channels whose expression is upregulated in hypertension. Kir1.1 knockout causes a renal salt wasting phenotype associated with hyperaldosteronism and hypotension (Bartter syndrome; reviewed by Hibino et al. 2010). As in the case of Kir4.1, Kir1.1 channel expression has been described only in renal epithelium, and also in some cortical neurons, but it would be interesting to explore its possible function in VSMCs from those vascular beds.
Functional expression of KIR and KATP channels in mesenteric arteries
In mesenteric VSMCs, we found that current density due to KIR channels was smaller than current density through KATP channels in all the conditions studied (in perforated patch it represented 49% of KATP current density at −150 mV, see Fig. 4). In fact, while the contribution of KATP channels to VM and basal tone in mesenteric arteries has been reported in several studies (reviewed by Quayle et al. 1997), the functional role of KIR channels in this preparation is still debated. Several studies report expression of KIR transcripts (mainly Kir2.1) in mesenteric VSMCs (Bradley et al. 1999; Kim et al. 2005; Smith et al. 2008), and a role of these channels in VSMC excitability (either in isolated cells or in whole mesenteric arteries) was reported in some studies (Kim et al. 2005) but not in others (Crane et al. 2003; Brochet & Langton, 2006; Smith et al. 2008). These discrepancies could be attributed to differences in the experimental setting, the size of the arteries used or the species under study. Additionally, they could reflect a minor contribution of these channels to set resting VM in mesenteric arteries, as such discrepancies are absent in other preparations in which they are clearly relevant, such as cerebral or coronary arteries (Quayle et al. 1997).
Here, we detected expression of mRNA and protein for KIR channels in mesenteric VSMCs, and also found a consistent functional contribution of these channels both in isolated cells and in whole arteries in which endothelium has been removed. The role of the channels was explored pharmacologically by studying Ba2+-sensitive changes in current density, resting VM and contractile responses. BaCl2 is a rather unspecific blocker for many other potassium channels, although with higher Kd values, so that at concentrations up to 50–100 μm it can be considered as a specific inhibitor of the classical KIR channels in vascular smooth muscle (Quayle et al. 1997; Zaritsky et al. 2000; Brayden, 2002; Park et al. 2008). We found a significant effect at concentrations of 30 μm and even lower (see representative traces in Fig. 6A), supporting the presence of functional KIR channels in our preparation.
In contrast to KIR channels, the expression and functional role of KATP channels in mesenteric arteries is better established (Nelson et al. 1990; Nelson & Quayle, 1995; Hibino et al. 2010). The ability of glibenclamide to elicit a significant depolarization of mesenteric VSMCs is consistent with a major contribution of KATP channels to total K+ conductance (Jiang et al. 2007), and is also supported by the presence of glibenclamide and pinacidil-sensitive currents in these cells.
Changes in KIR and KATP channels in essential hypertension
We found a reduction in Kir2.1 and Kir4.1 mRNA transcripts upon hypertension, leading to a decrease of KIR channel current density in isolated mesenteric VSMCs. Interestingly, although the BaCl2 depolarizing response is clearly smaller in BPH cells, the contractile response of BPH mesenteric arteries to BaCl2 is not significantly different. As VSMCs from BPH arteries are depolarized, the lack of differences in their response to BaCl2 may simply reflect the fact that BPH arteries require smaller depolarizations to produce a Ca2+ entry equivalent to that obtained in BPN animals.
The available data regarding changes in KIR channels in other models of hypertension are limited to their expression in cerebral arteries, where several authors have reported a decrease of the Ba2+-sensitive vasodilator responses to moderate increases in extracellular K+ (McCarron & Halpern, 1990; Chrissobolis & Sobey, 2003), suggesting an impairment of cerebral KIR channels during chronic hypertension. However, there are no systematic studies of the changes of KIR channels in other vascular beds and their contribution to the increased vascular tone or the development of high blood pressure. It is interesting to note that the majority of the data on the function of KIR channels in the vasculature are derived from coronary and cerebral arteries. While these preparations are amenable for studying the role of ion channels in vascular tone control due to their relevant intrinsic control and their prominent myogenic tone, they may be less sensitive to the changes induced by essential hypertension, as their marked auto-regulation protects these vascular beds from systemic changes in blood pressure.
The data from BPH mesenteric arteries show a noticeable decrease in both the expression and the functional contribution of KATP channels to VSMC excitability. Our findings fit well with some previous reports indicating an impairment of the function of vascular KATP channels during hypertension (Sobey, 2001). However, decreased expression of Kir6.1/SUR2B protein in hypertensive aortas with preserved functional responses to KATP openers has also been reported (Blanco-Rivero et al. 2008). A reason for the discrepancy with our work may rely on the different vascular bed used, as the impact of changes in conductance vessels in the development of hypertension is expected to be minor. It is noteworthy that there is a change not only in the functional expression of KATP channels in BPH vessels, but also in the fraction of these channels that are active at rest (Fig. 5B). Our data in perforated patch using glibenclamide and pinacidil suggest that around half of the KATP channels expressed at the plasma membrane are active under basal conditions in normotensive VSMCs, but this fraction increases to more than 70% in BPH VSMCs (Fig. 5). This observation could explain previous reports indicating that KATP channel activators are less potent dilators in vivo in cerebral vessels of chronically hypertensive rats (Takaba et al. 1996), and is relevant for hypertension therapy, because it would imply that KATP channel dysfunction in hypertension may also interfere with vascular responsiveness to treatment with K+ channel openers.
Contribution of the BPN/BPH model for understanding the molecular basis of hypertension
The model used in this study (BPH mice) has a moderate hypertensive phenotype, obtained by phenotypic selection. This particular model closely resembles the most common forms of human hypertension, as a genome-wide differential analysis in BPH and BPN mice has shown the involvement of several systems in the pathology of hypertension, reinforcing its multifactorial and complex nature (Friese et al. 2005). For that reason, the characterization of KIR and KATP channels in this model allows us to draw some conclusions regarding the contribution of these channels to the development of essential hypertension, and also to frame the available data from knockout models. It could also provide a complementary approach for the study of the molecular determinants of essential hypertension, as so far there are no reports of genetic associations of Kir2.1, Kir4.1 or Kir6.1 genetic variants and hypertension in humans. While studies on Kir2.x−/− animals have not addressed directly the issue of their contribution to blood pressure, they indicate that Kir2.1 (but not Kir2.2) expression in VSMCs is required for KIR currents and K+-induced dilations in cerebral arteries (Zaritsky et al. 2000). With respect to knockout models of vascular KATP channels, there are two available Kir6.1−/− (Miki et al. 2002; Seino & Miki, 2004) and SUR2−/− (Chutkow et al. 2002). In both cases there is a complete loss of vascular KATP currents and a phenotype that highlights the critical role of this channel in regulating episodic vasomotor activity and vascular tone, especially in coronary arteries. However, only SUR2−/− mice show elevated blood pressure. In the light of our findings showing a decrease of both Kir6.1 and SUR2 mRNA in mesenteric VSMCs from BPH animals, we infer that SUR2 proteins may have other cellular partners that could compensate for the increased vascular tone observed in the absence of functional KATP channels in Kir6.1−/− animals.
In summary, we present here a characterization of inward rectifying K+ channels in normotensive and hypertensive mesenteric arteries that spans from the mRNA to the protein, their function in isolated cells and their contribution to the function of the pressurized arteries. Our data support the hypothesis that the changes in the functional expression of KIR and KATP channels are relevant to development of a hypertensive phenotype in a naturally occurring form of hypertension, as they could be key determinants of membrane depolarization in hypertensive VSMCs.
Acknowledgments
We thank Esperanza Alonso and Rodrigo de Pedro for excellent technical assistance. This work was supported by Ministerio de Sanidad, ISCIII grant R006/009 (Red Heracles), Ministerio de Ciencia e Innovación grant BFU2010-15898 (M.T.P.G.) and Junta de Castilla y León grant VA094A11-2 (J.R.L.L.). S.T. is a fellow of the Spanish MICINN.
Glossary
- BKCa
large-conductance Ca2+-activated K+ channels
- BPH
blood pressure high mice
- BPN
blood pressure normal mice
- Kir
inward rectifier K+ channels
- KATP channels
ATP-sensitive inward rectifier K+ channels (Kir6)
- KIR channels
classical inward rectifier K+ channels (Kir1–4)
- Kv
voltage-activated K+ channels
- VM
resting membrane potential
- VSMC
vascular smooth muscle cell
Author contributions
The experiments were performed at the Institute of Biology and Molecular Genetics (IBGM), School of Medicine, University of Valladolid. Study conception and design: P.C., M.T.P.G. and J.R.L.L. Manuscript editing: M.T.P.G. and J.R.L.L. All authors contributed to the collection, analysis and interpretation of the data, drafting the article or revising it critically, and approved the final version of the manuscript.
Author's present address
A. Moreno-Domínguez: Department of Physiology and Pharmacology, Faculty of Medicine, University of Calgary, Canada.
Supplementary material
Supporting Information
References
- Amberg GC, Bonev AD, Rossow CF, Nelson MT, Santana LF. Modulation of the molecular composition of large conductance, Ca2+ activated K+ channels in vascular smooth muscle during hypertension. J Clin Invest. 2003;112:717–724. doi: 10.1172/JCI18684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beech DJ, Bolton TB. A voltage-dependent outward current with fast kinetics in single smooth muscle cells isolated from rabbit portal vein. J Physiol. 1989;412:397–414. doi: 10.1113/jphysiol.1989.sp017623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco-Rivero J, Gamallo C, Aras-López R, Cobeño L, Cogolludo A, Pérez-Vizcaino F, Ferrer M, Balfagon G. Decreased expression of aortic KIR6.1 and SUR2B in hypertension does not correlate with changes in the functional role of KATP channels. Eur J Pharmacol. 2008;587:204–208. doi: 10.1016/j.ejphar.2008.03.039. [DOI] [PubMed] [Google Scholar]
- Bradley KK, Jaggar JH, Bonev AD, Heppner TJ, Flynn ER, Nelson MT, Horowitz B. Kir2.1 encodes the inward rectifier potassium channel in rat arterial smooth muscle cells. J Physiol. 1999;515:639–651. doi: 10.1111/j.1469-7793.1999.639ab.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brayden JE. Functional roles of KATP channels in vascular smooth muscle. Clin Exp Pharmacol Physiol. 2002;29:312–316. doi: 10.1046/j.1440-1681.2002.03650.x. [DOI] [PubMed] [Google Scholar]
- Brochet DX, Langton PD. Dual effect of initial [K] on vascular tone in rat mesenteric arteries. Pflugers Arch. 2006;453:33–41. doi: 10.1007/s00424-006-0106-1. [DOI] [PubMed] [Google Scholar]
- Chrissobolis S, Sobey CG. Inwardly rectifying potassium channels in the regulation of vascular tone. Curr Drug Targets. 2003;4:281–289. doi: 10.2174/1389450033491046. [DOI] [PubMed] [Google Scholar]
- Chutkow WA, Pu J, Wheeler MT, Wada T, Makielski JC, Burant CF, McNally EM. Episodic coronary artery vasospasm and hypertension develop in the absence of Sur2 KATP channels. J Clin Invest. 2002;110:203–208. doi: 10.1172/JCI15672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox RH, Rusch NJ. New expression profiles of voltage-gated ion channels in arteries exposed to high blood pressure. Microcirculation. 2002;9:243–257. doi: 10.1038/sj.mn.7800140. [DOI] [PubMed] [Google Scholar]
- Cox RH. Changes in the expression and function of arterial potassium channels during hypertension. Vasc Pharmacol. 2002;38:13–23. doi: 10.1016/s1537-1891(02)00122-2. [DOI] [PubMed] [Google Scholar]
- Crane GJ, Walker SD, Dora KA, Garland CJ. Evidence for a differential cellular distribution of inward rectifier K channels in the rat isolated mesenteric artery. J Vasc Res. 2003;40:159–168. doi: 10.1159/000070713. [DOI] [PubMed] [Google Scholar]
- Friese RS, Mahboubi P, Mahapatra NR, Mahata SK, Schork NJ, Schmid-Schonbein GW, O’Connor DT. Common genetic mechanisms of blood pressure elevation in two independent rodent models of human essential hypertension. Am J Hypertens. 2005;18:633–652. doi: 10.1016/j.amjhyper.2004.11.037. [DOI] [PubMed] [Google Scholar]
- Harder DR, Brann L, Halpern W. Altered membrane electrical properties of smooth muscle cells from small cerebral arteries of hypertensive rats. Blood Vessels. 1983;20:154–160. doi: 10.1159/000158469. [DOI] [PubMed] [Google Scholar]
- Harder DR, Smeda J, Lombard J. Enhanced myogenic depolarization in hypertensive cerebral arterial muscle. Circ Res. 1985;57:319–322. doi: 10.1161/01.res.57.2.319. [DOI] [PubMed] [Google Scholar]
- Hibino H, Inanobe A, Furutani K, Murakami S, Findlay I, Kurachi Y. Inwardly rectifying potassium channels: their structure, function, and physiological roles. Physiol Rev. 2010;90:291–366. doi: 10.1152/physrev.00021.2009. [DOI] [PubMed] [Google Scholar]
- Jackson WF. Ion channels and vascular tone. Hypertension. 2000;35:173–178. doi: 10.1161/01.hyp.35.1.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jepps TA, Chadha PS, Davis AJ, Harhun MI, Cockerill GW, Olesen SP, Hansen RS, Greenwood IA. Downregulation of Kv7.4 channel activity in primary and secondary hypertension. Circulation. 2011;124:602–611. doi: 10.1161/CIRCULATIONAHA.111.032136. [DOI] [PubMed] [Google Scholar]
- Jiang B, Wu L, Wang R. Sulphonylureas induced vasorelaxation of mouse arteries. Eur J Pharmacol. 2007;577:124–128. doi: 10.1016/j.ejphar.2007.09.007. [DOI] [PubMed] [Google Scholar]
- Kim MY, Liang GH, Kim JA, Park SH, Hah JS, Suh SH. Contribution of Na+–K+ pump and KIR currents to extracellular pH-dependent changes of contractility in rat superior mesenteric artery. Am J Physiol Heart Circ Physiol. 2005;289:H792–H800. doi: 10.1152/ajpheart.00050.2005. [DOI] [PubMed] [Google Scholar]
- Kitazono T, Heistad DD, Faraci FM. ATP-sensitive potassium channels in the basilar artery during chronic hypertension. Hypertension. 1993;22:677–681. doi: 10.1161/01.hyp.22.5.677. [DOI] [PubMed] [Google Scholar]
- Ko EA, Han J, Jung ID, Park WS. Physiological roles of K+ channels in vascular smooth muscle cells. J Smooth Muscle Res. 2008;44:65–81. doi: 10.1540/jsmr.44.65. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(–ΔΔCt) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- McCarron JG, Halpern W. Impaired potassium-induced dilation in hypertensive rat cerebral arteries does not reflect altered Na+,K+-ATPase dilation. Circ Res. 1990;67:1035–1039. doi: 10.1161/01.res.67.4.1035. [DOI] [PubMed] [Google Scholar]
- Miki T, Suzuki M, Shibasaki T, Uemura H, Sato T, Yamaguchi K, Koseki H, Iwanaga T, Nakaya H, Seino S. Mouse model of Prinzmetal angina by disruption of the inward rectifier Kir6.1. Nat Med. 2002;8:466–472. doi: 10.1038/nm0502-466. [DOI] [PubMed] [Google Scholar]
- Moreno-Dominguez A, Cidad P, Miguel-Velado E, Lopez-Lopez JR, Perez-Garcia MT. De novo expression of Kv6.3 contributes to changes in vascular smooth muscle cell excitability in a hypertensive mice strain. J Physiol. 2009;587:625–640. doi: 10.1113/jphysiol.2008.165217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson MT, Patlak JB, Worley JF, Standen NB. Calcium channels, potassium channels, and voltage dependence of arterial smooth muscle tone. Am J Physiol Cell Physiol. 1990;259:C3–18. doi: 10.1152/ajpcell.1990.259.1.C3. [DOI] [PubMed] [Google Scholar]
- Nelson MT, Quayle JM. Physiological roles and properties of potassium channels in arterial smooth muscle. Am J Physiol Cell Physiol. 1995;268:C799–C822. doi: 10.1152/ajpcell.1995.268.4.C799. [DOI] [PubMed] [Google Scholar]
- Ohya Y, Setoguchi M, Fujii K, Nagao T, Abe I, Fujishima M. Impaired action of levcromakalim on ATP-sensitive K+ channels in mesenteric artery cells from spontaneously hypertensive rats. Hypertension. 1996;27:1234–1239. doi: 10.1161/01.hyp.27.6.1234. [DOI] [PubMed] [Google Scholar]
- Park WS, Han J, Earm YE. Physiological role of inward rectifier K+ channels in vascular smooth muscle cells. Pflugers Arch. 2008;457:137–147. doi: 10.1007/s00424-008-0512-7. [DOI] [PubMed] [Google Scholar]
- Pesic A, Madden JA, Pesic M, Rusch NJ. High blood pressure upregulates arterial L-type Ca2+ channels: is membrane depolarization the signal. Circ Res. 2004;94:e97–104. doi: 10.1161/01.RES.0000131495.93500.3c. [DOI] [PubMed] [Google Scholar]
- Quayle JM, Nelson MT, Standen NB. ATP-sensitive and inwardly rectifying potassium channels in smooth muscle. Physiol Rev. 1997;77:1165–1232. doi: 10.1152/physrev.1997.77.4.1165. [DOI] [PubMed] [Google Scholar]
- Schlager G, Sides J. Characterization of hypertensive and hypotensive inbred strains of mice. Lab Anim Sci. 1997;47:288–292. [PubMed] [Google Scholar]
- Seino S, Miki T. Gene targeting approach to clarification of ion channel function: studies of Kir6.x null mice. J Physiol. 2004;554:295–300. doi: 10.1113/jphysiol.2003.047175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith PD, Brett SE, Luykenaar KD, Sandow SL, Marrelli SP, Vigmond EJ, Welsh DG. KIR channels function as electrical amplifiers in rat vascular smooth muscle. J Physiol. 2008;586:1147–1160. doi: 10.1113/jphysiol.2007.145474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobey CG. Potassium channel function in vascular disease. Arterioscler Thromb Vasc Biol. 2001;21:28–38. doi: 10.1161/01.atv.21.1.28. [DOI] [PubMed] [Google Scholar]
- Somlyo AP, Somlyo AV. Ca2+ sensitivity of smooth muscle and nonmuscle myosin II: modulated by G proteins, kinases, and myosin phosphatase. Physiol Rev. 2003;83:1325–1358. doi: 10.1152/physrev.00023.2003. [DOI] [PubMed] [Google Scholar]
- Sonkusare S, Palade PT, Marsh JD, Telemaque S, Pesic A, Rusch NJ. Vascular calcium channels and high blood pressure: pathophysiology and therapeutic implications. Vasc Pharmacol. 2006;44:131–142. doi: 10.1016/j.vph.2005.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takaba H, Nagao T, Ibayashi S, Kitazono T, Fujii K, Fujishima M. Altered cerebrovascular response to a potassium channel opener in hypertensive rats. Hypertension. 1996;28:143–146. doi: 10.1161/01.hyp.28.1.143. [DOI] [PubMed] [Google Scholar]
- Zaritsky JJ, Eckman DM, Wellman GC, Nelson MT, Schwarz TL. Targeted disruption of Kir2.1 and Kir2.2 genes reveals the essential role of the inwardly rectifying K+ current in K+-mediated vasodilation. Circ Res. 2000;87:160–166. doi: 10.1161/01.res.87.2.160. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.