

Abstract
β-Adrenergic signalling induces positive inotropic effects on the heart that associate with pro-arrhythmic spontaneous Ca2+ waves. A threshold level of sarcoplasmic reticulum (SR) Ca2+ ([Ca2+]SR) is necessary to trigger Ca2+ waves, and whether the increased incidence of Ca2+ waves during β-adrenergic stimulation is due to an alteration in this threshold remains controversial. Using the low-affinity Ca2+ indicator fluo-5N entrapped within the SR of rabbit ventricular myocytes, we addressed this controversy by directly monitoring [Ca2+]SR and Ca2+ waves during β-adrenergic stimulation. Electrical pacing in elevated extracellular Ca2+ ([Ca2+]o= 7 mm) was used to increase [Ca2+]SR to the threshold where Ca2+ waves were consistently observed. The β-adrenergic agonist isoproterenol (ISO; 1 μm) increased [Ca2+]SR well above the control threshold and consistently triggered Ca2+ waves. However, when [Ca2+]SR was subsequently lowered in the presence of ISO (by lowering [Ca2+]o to 1 mm and partially inhibiting sarcoplasmic/endoplasmic reticulum calcium ATPase with cyclopiazonic acid or thapsigargin), Ca2+ waves ceased to occur at a [Ca2+]SR that was higher than the control threshold. Furthermore, for a set [Ca2+]SR level the refractoriness of wave occurrence (Ca2+ wave latency) was prolonged during β-adrenergic stimulation, and was highly dependent on the extent that [Ca]SR exceeded the wave threshold. These data show that acute β-adrenergic stimulation increases the [Ca2+]SR threshold for Ca2+ waves, and therefore the primary cause of Ca2+ waves is the robust increase in [Ca2+]SR above this higher threshold level. Elevation of the [Ca2+]SR wave threshold and prolongation of wave latency represent potentially protective mechanisms against pro-arrhythmogenic Ca2+ release during β-adrenergic stimulation.
Key points
In the heart, Ca2+ waves are arrhythmogenic spontaneous sarcoplasmic reticulum (SR) Ca2+ release events that arise when the Ca2+ content in the SR reaches a critical threshold level.
β-Adrenergic signalling induces Ca2+ waves in cardiac myocytes, but it remains unclear if this is due to a decrease in the Ca2+ wave threshold or more simply due to an increase in SR Ca2+ content.
We used direct, dynamic measurement of SR Ca2+ levels to show that the Ca2+ wave threshold is unexpectedly increased during β-adrenergic stimulation.
Our data show that the primary cause of Ca2+ waves following acute β-adrenergic stimulation is the increase in SR Ca2+ content and not a decrease in the Ca2+ wave threshold.
We propose that the elevation of the Ca2+ wave threshold represents a protective mechanism against arrhythmogenic events during periods of β-adrenergic stimulation.
Introduction
Cardiac contraction is triggered via Ca2+-induced Ca2+ release (CICR), where influx of Ca2+ into the cardiomyocyte through L-type Ca2+ channels (LTCCs) opens ryanodine receptor (RyR) Ca2+ release channels in the junctional sarcoplasmic reticulum (SR) membrane and initiates coordinated release of Ca2+ from the SR Ca2+ store. SR Ca2+ release can also occur spontaneously during diastole. Diastolic Ca2+ release is detrimental to cardiac performance as it impairs relaxation, reduces the concentration of Ca2+ within the SR ([Ca2+]SR) and predisposes the heart to arrhythmia. One highly arrhythmogenic form of diastolic Ca2+ release is the Ca2+ wave, where Ca2+ that is spontaneously released from an SR Ca2+ release unit (CRU, formed by a cluster of RyRs) diffuses to neighbouring CRUs and triggers regenerative and propagating CICR independently of the cardiac action potential (AP) (Stern et al. 1988). Extrusion of Ca2+ that is released during a Ca2+ wave via the electrogenic Na+–Ca2+ exchange mechanism (Kass et al. 1978; Fedida et al. 1987) has a strong depolarizing effect on the diastolic membrane potential and underlies arrhythmogenic delayed afterdepolarizations (Capogrossi et al. 1987; Schlotthauer & Bers, 2000; Fujiwara et al. 2008). It is therefore of paramount importance to understand the mechanisms for Ca2+ wave generation in the heart.
A considerable amount of evidence has emerged that a distinct ‘overload’[Ca2+]SR is necessary to trigger Ca2+ waves, and this level has been termed the Ca2+ wave threshold (Díaz et al. 1997b; Venetucci et al. 2007) or store overload-induced Ca2+ release threshold (Jiang et al. 2004). The Ca2+ wave threshold is altered by agents that modulate RyR activity, with channel activators (e.g. caffeine) and inhibitors (e.g. tetracaine) decreasing (Venetucci et al. 2007; Kong et al. 2008) and increasing (Overend et al. 1997) the threshold, respectively. In animal models of heart failure where RyR channel activity is increased, the Ca2+ wave threshold is decreased (Belevych et al. 2012; Maxwell et al. 2012) and this may play a role in the arrhythmogenesis associated with these models. Furthermore, investigations of RyR mutations associated with catecholaminergic polymorphic ventricular tachycardia (CPVT) have shown that gain-of-function mutations (reviewed by Priori & Chen, 2011) result in a decrease in the Ca2+ wave threshold and increased incidence of arrhythmogenic Ca2+ release following increases in [Ca2+]SR[e.g. in response to cardiac glycosides (Sedej et al. 2010) or β-adrenergic stimulation (Kashimura et al. 2010)]. Interestingly, although the Ca2+ wave threshold is lower in cardiomyocytes from CPVT RyR R4496C mutant mice than in wild-type controls, β-adrenergic stimulation increases the Ca2+ wave threshold in R4496C myocytes (Kashimura et al. 2010). Thus, the likely factor triggering Ca2+ waves in response to catecholamines in this CPVT model is the sudden increase in [Ca2+]SR and not an acute lowering of the Ca2+ wave threshold. A similar increase in the Ca2+ wave threshold in response to β-adrenergic signalling was observed in wild-type mouse cardiomyocytes (Kashimura et al. 2010; Stokke et al. 2010a), suggesting that this effect is not unique to disease models. Contrasting with these data are investigations that show that β-adrenergic stimulation increases diastolic SR Ca2+ release in the form of Ca2+ sparks (Zhou et al. 2009; Ogrodnik & Niggli, 2010), SR Ca2+ leak (Curran et al. 2007; Bovo et al. 2012; Ullrich et al. 2012) and Ca2+ waves (Curran et al. 2010) at a similar or lower level of SR Ca2+ content (which intuitively should translate into a decrease in the Ca2+ wave threshold). Additionally, selective activation of signalling molecules acting downstream of β-adrenergic stimulation (Guo et al. 2006; Terentyev et al. 2008) or phosphomimetic mutation of target phosphorylation sites on the RyR (Shan et al. 2010; van Oort et al. 2010) have all been shown to augment SR Ca2+ release and predispose the heart to arrhythmia. Thus, it remains unclear and highly controversial whether Ca2+ waves during β-adrenergic stimulation are due to direct alterations in the Ca2+ wave threshold or more simply due to the increase in [Ca2+]SR that occurs concomitant with activation of the signalling pathway.
Measurement of the Ca2+ wave threshold in cardiomyocytes during β-adrenergic stimulation is experimentally challenging due to the enhanced activity of multiple Ca2+ handling proteins, which hinders targeted and controlled examination of the Ca2+ wave threshold. To gain new insight into the mechanisms underlying Ca2+ waves during β-adrenergic stimulation we utilized direct, dynamic fluorescent measurement of [Ca2+]SR and the Ca2+ wave threshold in acutely isolated rabbit ventricular myocytes. We found that β-adrenergic stimulation increases the Ca2+ wave threshold, which may serve to protect the heart from arrhythmogenic Ca2+ release as [Ca2+]SR increases during sympathetic nervous system activation.
Part of this work has been presented previously in abstract form (Domeier & Blatter, 2010; Domeier et al. 2011).
Methods
Solutions and chemicals
All reagents were purchased from Sigma-Aldrich (St. Louis, MO, USA) unless otherwise noted. Normal Tyrode solution contained (in mm): 135 NaCl, 4 KCl, 2 CaCl2, 1 MgCl2, 10 d-glucose and 10 Hepes (pH 7.4 with NaOH). Tyrode solutions with altered extracellular free calcium concentration ([Ca2+]o) were prepared using iso-osmotic substitution of CaCl2 and NaCl. Isoproterenol (ISO) was prepared daily as a stock solution of 100 mm (in H2O, maintained at 4°C), diluted in Tyrode solution immediately prior to experimental procedures and used within 30 min. Cyclopiazonic acid (CPA) and thapsigargin (TG) were dissolved in DMSO and diluted to working concentrations in Tyrode solution. All experiments were performed with appropriate vehicle controls. All experiments were conducted at room temperature (22–24°C).
Cardiac myocyte isolation
Left ventricular myocytes were isolated from male New Zealand White rabbits (30 animals, 2.5 kg, Myrtle's Rabbitry, Thompsons Station, TN, USA). Rabbits were anaesthetized with sodium pentobarbital (50 mg kg−1) and hearts were rapidly excised and retrogradely perfused with a nominally Ca2+-free Tyrode solution for 10 min, followed by a minimal essential Eagle's medium (MEM) solution with 20 μm Ca2+ and 45 μg ml−1 Liberase Blendzyme TH (Roche Applied Science, Indianapolis, IN, USA) for 20 min all at 37°C. The left ventricular free wall tissue was minced, filtered and washed in MEM solution containing 50 μm Ca2+ and 10 mg ml−1 BSA. Isolated myocytes were maintained in MEM solution with 50 μm Ca2+ at room temperature until Ca2+ indicator dye loading procedures. All protocols were approved by the Institutional Animal Care and Use Committee at Rush University, and comply with US and UK regulations on animal experimentation (Drummond, 2009).
Intra-SR Ca2+ measurements
Ventricular myocytes were incubated with 10 μm of membrane-permeant fluo-5N/AM (Molecular Probes-Life Technologies, Grand Island, NY, USA) for 2.5 h at 37°C to promote accumulation of dye within the SR (see Fig. 2A). Following a 30 min wash at 37°C, myocytes were equilibrated to room temperature and plated on laminin-coated coverslips for imaging experiments. Laser scanning fluorescence confocal microscopy was performed using the resonant scan head of a Nikon A1R system (Nikon Instruments Inc., Melville, NY, USA) in frame scan mode (60 frames s−1 at 256 × 512 pixels, 400 nm per pixel). Fluo-5N was excited using the 488 nm line of the argon ion laser and emission was recorded at 500–530 nm. Low laser excitation intensities and limited recording times were used to minimize photobleaching of the fluo-5N indicator. Although generally negligible, in some experiments with extended recording times (e.g. Fig. 3A) photobleaching did occur and these recordings were corrected using control bleach rates (single exponential fits) obtained from each cell under steady-state conditions. Changes in [Ca2+]SR are presented as (F–FMin)/(FMax–FMin) or calibrated as [Ca2+]SR=[400 μm× (F–FMin)/(FMax–F)] (Shannon et al. 2003; Zima et al. 2011). FMin is the quench-corrected (15%) fluorescence value following complete emptying of the SR with 10 mm caffeine, and FMax is taken as the diastolic fluorescence at 1 Hz pacing in the presence of 1 μm ISO.
Figure 2. Direct [Ca2+]SR measurements following isoproterenol application.
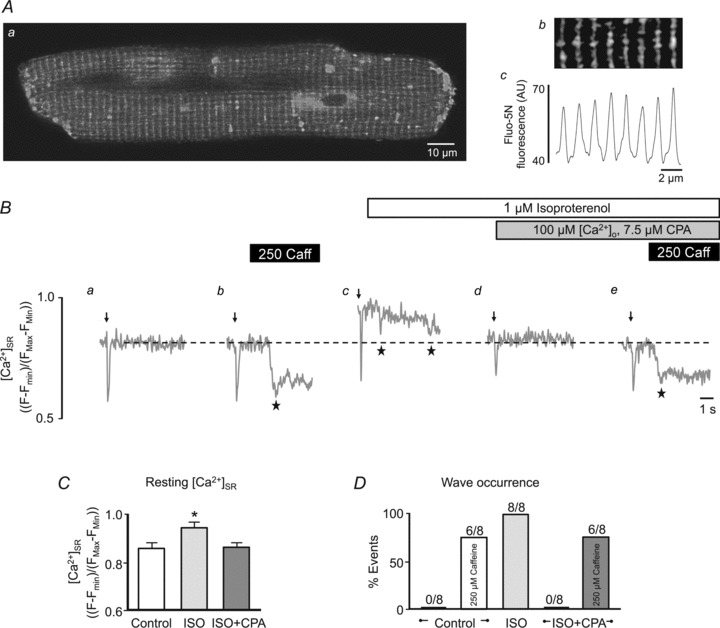
A, confocal fluorescence image (a) of a myocyte with the SR loaded with the low-affinity Ca2+ indicator fluo-5N. Enlarged region of image (b) and corresponding fluorescence profile (c) showing patterning of the junctional SR network. AU, arbitrary units of fluo-5N fluorescence. B, example [Ca2+]SR measurements with fluo-5N fluorescence showing an electrically induced Ca2+ depletion (arrow, 0.75 Hz) followed by rest under control conditions (a and b), in the presence of ISO (c) (a–c: [Ca2+]o= 2 mm), and in the presence of ISO with 7.5 μm CPA in 100 μm[Ca2+]o (d and e). In traces b and e 250 μm caffeine (250 Caff) was applied to pharmacologically sensitize RyRs and induce Ca2+ waves. Ca2+ waves are visible in fluo-5N fluorescence profiles as a non-triggered decrease in fluorescence (stars). Summary data of resting [Ca2+]SR (C) and Ca2+ wave occurrence (D) under the conditions shown in B. Ca2+ wave occurrence is presented as fraction (%) of cells showing waves, from a total of eight cells. *P < 0.05.
Figure 3. Ca2+ waves during sustained rest in the presence of isoproterenol.
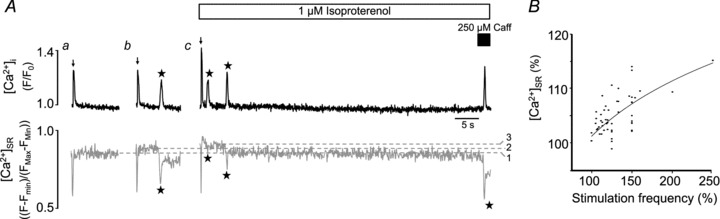
A, simultaneous [Ca2+]i (rhod-2, upper traces in black) and [Ca2+]SR (fluo-5N, lower traces in grey) measurements following rest from electrical stimulation under control conditions (7 mm[Ca2+]o, a= 1.1 Hz, b= 1.3 Hz) and in the presence of ISO (c: 2 mm[Ca2+]o, 1.3 Hz). Electrical stimulation frequency was incrementally increased to elevate [Ca2+]SR and determine the threshold level for Ca2+ waves. In the example shown, Ca2+ waves were only observed when [Ca2+]SR was higher than the control threshold level, unless RyRs were pharmacologically sensitized using 250 μm caffeine. Dashed lines: 1, no-wave threshold control; 2, wave threshold control; 3, wave threshold ISO. B, positive correlation between electrical stimulation frequency (range 0.5–1.3 Hz) and resting [Ca2+]SR measured using fluo-5N fluorescence. Frequency and [Ca2+]SR values are normalized to the respective values found at the lowest pacing frequency in each experimental trial.
Cytosolic Ca2+ ([Ca2+]i) measurements, indo-1
For ratiometric epifluorescence [Ca2+]i measurements, myocytes were loaded with 5 μm indo-1/AM (Molecular Probes-Life Technologies) for 10 min followed by a 30 min wash, all at room temperature. Single myocytes were exposed to 360 nm light to excite indo-1, and fluorescence emission was simultaneously monitored at 410 nm (F410) and 485 nm (F485). Fluorescence emission at each wavelength was background subtracted and [Ca2+]i is presented as the indo-1 ratio (R=F410/F485) or change in indo-1 ratio (ΔR=Rpeak–Rbaseline).
Cytosolic Ca2+ measurements, rhod-2
Myocytes preloaded with fluo-5N were subsequently loaded with the spectrally distinct Ca2+ indicator rhod-2/AM (Molecular Probes-Life Technologies, 5 μm, 10 min loading time, 10 min wash, all at room temperature) to monitor cytosolic Ca2+ waves simultaneously with [Ca2+]SR. Laser scanning fluorescence confocal microscopy was performed (Nikon A1R, 60 frames s−1) with rhod-2 excitation at 543 nm and fluorescence emission recorded at >600 nm. Rhod-2 fluorescence signals are presented as F/F0, where F0 is the diastolic fluorescence at the start of each experimental recording.
Experimental protocols to determine the Ca2+ wave threshold
Control conditions
Two series of experiments were conducted to determine the Ca2+ wave threshold. In one series, the Ca2+ wave threshold was determined based on direct measurement of [Ca2+]SR using fluo-5N, whereas the second series relied on cytosolic [Ca2+]i measurements (with indo-1) in response to caffeine challenge to estimate SR Ca2+ content. Because rabbit ventricular myocytes did not typically exhibit Ca2+ waves under control conditions ([Ca2+]o= 2 mm), a high extracellular Ca2+ solution was applied ([Ca2+]o= 7 mm). In both series of experiments rabbit ventricular myocytes underwent several consecutive experimental trials defined here as an episode consisting of electrical field stimulation of cells for a period of 30–60 s, followed by 8 s of rest. The stimulation–rest protocol was repeated at incrementally increasing pacing frequencies (0.05–0.2 Hz increments) to increase [Ca2+]SR (see Fig. 3B) until Ca2+ waves were observed during the rest period. In fluo-5N experiments the [Ca2+]SR where waves were observed was defined as the ‘wave threshold’ and the [Ca2+]SR at the preceding lower frequency where waves were not observed was defined as the ‘no-wave threshold’. Control measurements confirmed that the control wave and no-wave thresholds were stable and reproducible over time. Additionally, in experiments where waves were observed, SR Ca2+ refilling time and wave latency were calculated. SR Ca2+ refilling time was defined as the interval between the AP-induced depletion nadir (minimum [Ca2+]SR value) and the time point when [Ca2+]SR recovered to the [Ca2+]SR observed prior to the depletion (dark grey in Fig. 6A). Wave latency was calculated as the interval between the time point when [Ca2+]SR recovered from an AP-induced depletion and the onset of the wave (light grey in Fig. 6A). In indo-1 experiments the SR Ca2+ content associated with a particular stimulation frequency was determined in a separate pacing train using the amplitude of the Ca2+ transient in response to 10 mm caffeine. This value was determined immediately following cessation of pacing.
Figure 6. Wave latency is highly dependent on [Ca2+]SR.
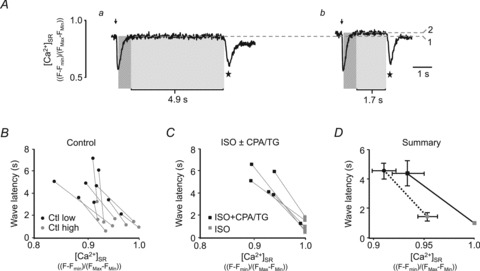
A, representative fluo-5N fluorescence traces from a cell with [Ca2+]SR at the wave threshold (a, threshold marked with dashed line 1) and above the wave threshold (b, steady-state [Ca2+]SR before wave marked with dashed line 2). In each trace, arrow denotes AP stimulation triggering [Ca2+]SR depletion, followed by SR Ca2+ refilling (dark grey) and a wave latency time (light grey) prior to the Ca2+ wave (marked by star). B, individual paired experiments obtained from nine cells, each yielding a wave latency measurement at two different [Ca2+]SR levels under control conditions (Ctl low, Ctl high). C, individual paired experiments obtained from six cells, each yielding a wave latency measurement in the presence of ISO (grey square) alone and in the combined presence of ISO plus CPA or TG (ISO+CPA/TG black square). D, summary data (averages from data shown in B and C) of the relationship between [Ca2+]SR and Ca2+ wave latency under control conditions at two different [Ca2+]SR levels (circles), in ISO (grey square) and in ISO+CPA/TG (black square). Statistical significance analysis of these data is presented in the text.
β-Adrenergic stimulation
In all experiments β-adrenergic stimulation (ISO, 5 min) increased [Ca2+]SR compared with control conditions and triggered Ca2+ waves. To determine the critical [Ca2+]SR threshold level where Ca2+ waves occurred (or ceased to occur when [Ca2+]SR was lowered) a protocol was applied that resulted in a gradual decrease of [Ca2+]SR levels after the initial increase, all in the maintained presence of ISO. For this, [Ca2+]o was lowered (as indicated) and sarcoplasmic/endoplasmic reticulum calcium ATPase (SERCA) was partially blocked by CPA or TG while β-adrenergic stimulation was maintained. In the fluo-5N experiments this protocol allowed for direct assessment of the [Ca2+]SR where waves occurred during β-adrenergic stimulation, with subsequent paired comparison with the respective control threshold levels. With the indo-1 experimental approach wave and no-wave thresholds under control conditions were determined as described above. However, in the presence of ISO+CPA only no-wave data points were used for analysis. This analysis design was implemented because the indirect assessment of SR Ca2+ content from the caffeine-induced cytosolic Ca2+ transient would have to occur after a Ca2+ wave. With CPA or TG present the amount of [Ca2+]SR remaining in the SR following a Ca2+ wave is significantly lower than that which preceded the wave due to the impaired reuptake by SERCA (Díaz et al. 1997a; Domeier et al. 2010; see Figs 2B, 3A and 4A), i.e. application of caffeine after a Ca2+ wave would grossly underestimate the wave threshold.
Figure 4. Isoproterenol increases the intra-SR Ca2+ wave threshold, determined by direct [Ca2+]SR measurements.
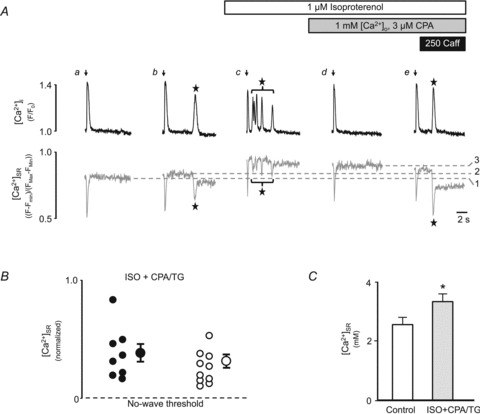
A, simultaneous [Ca2+]i (rhod-2, upper traces in black) and [Ca2+]SR (fluo-5N, lower traces in grey) measurements following rest from electrical stimulation (arrows) under control conditions (a: 7 mm[Ca2+]o, 0.7 Hz; b: 7 mm[Ca2+]o, 0.75 Hz), in the presence of ISO (c: 2 mm[Ca2+]o, 0.75 Hz) and in the presence of ISO with 3 μm CPA (d and e: 1 mm[Ca2+]o, 0.75 Hz). Dashed lines: 1, no-wave, threshold control; 2, wave threshold, control; 3, no-wave threshold, ISO. Subsequent application of 250 μm caffeine induced a Ca2+ wave (e). Waves are marked by stars. B, normalized [Ca2+]SR values from 18 individual experimental trials (from 12 cells) in ISO+CPA/TG separated by the criteria whether waves were observed (filled circle) or not (open circles), together with the mean ± SEM of [Ca2+]SR for the two groups. For each individual trial [Ca2+]SR was normalized to the maximal fluorescence observed in the presence of 1 μm ISO ([Ca2+]SR,normalized= 1) and the no-wave threshold under control conditions ([Ca2+]SR,normalized= 0). C, quantification of the [Ca2+]SR associated with the no-wave threshold under control conditions (open bar) and in the presence of ISO+CPA/TG (shaded bar). *P < 0.002 vs. control, n= 7 cells.
Statistics and data analysis
Only rod-shaped myocytes with clear striations were used for experimental protocols. Approximately 20% of myocytes exhibited visible damage during the repetitive stimulation/rest protocols (e.g. membrane blebbing or irreversible cellular contracture). These myocytes were not utilized for data analysis. Data are presented as individual observations from single myocytes or as mean ± standard error of the mean (SEM). Statistical comparisons were performed using Student's t test for paired or unpaired data, with significance set at P < 0.05. The number of individual cells or experimental trials is given as n as indicated.
Results
In this investigation we tested the hypothesis that β-adrenergic receptor stimulation alters the intra-SR Ca2+ threshold for spontaneous Ca2+ waves. For this, we monitored spontaneous Ca2+ waves that occurred during rest from steady-state electrical stimulation. A major difficulty associated with studies of SR Ca2+ release following activation of β-adrenergic stimulation is that intracellular Ca2+ handling is profoundly altered by this signalling pathway, most notably an increase in Ca2+ entry into the cell via the LTCC (Tsien et al. 1986) and an increase in SR Ca2+ content via effects on phospholamban and the SERCA pump (Kranias & Solaro, 1982; Lindemann et al. 1983). Because of these effects, it becomes experimentally difficult to control both [Ca2+]i and [Ca2+]SR, which is essential when monitoring SR Ca2+ release as RyR gating is influenced by both cytosolic and intra-luminal [Ca2+] (Fill & Copello, 2002). We therefore defined experimental conditions where Ca2+ cycling (in particular [Ca2+]i) was the same between control conditions and in the presence of β-adrenergic stimulation (1 μm ISO, 5 min), and reasoned that if during rest from electrical stimulation steady-state [Ca2+]i is equivalent between the two respective conditions then the [Ca2+]SR where Ca2+ waves occur will directly reflect the intra-SR Ca2+ wave threshold.
Acute application of ISO led to an increase in Ca2+ transient amplitude (Fig. 1A and C, arrows indicate electrically evoked Ca2+ transients), resting [Ca2+]i (Fig. 1A and D) and SR Ca2+ content as assessed from the amplitude of the [Ca2+]i increase in response to 10 mm caffeine (Fig. 1B and E). Under these conditions, spontaneous Ca2+ waves were frequently observed during the rest period following steady-state stimulation (Fig. 1A, star). Subsequent addition of a low extracellular Ca2+ solution ([Ca2+]o= 100 μm) containing ISO and the SERCA inhibitor CPA (7.5 μm) returned critical parameters of cellular Ca2+ handling to levels observed under control conditions, with Ca2+ transient amplitude (Fig. 1C), resting [Ca2+]i (Fig. 1D) and SR Ca2+ content (Fig. 1E) becoming similar to the original control conditions.
Figure 1. Effects of isoproterenol on intracellular Ca2+ handling.
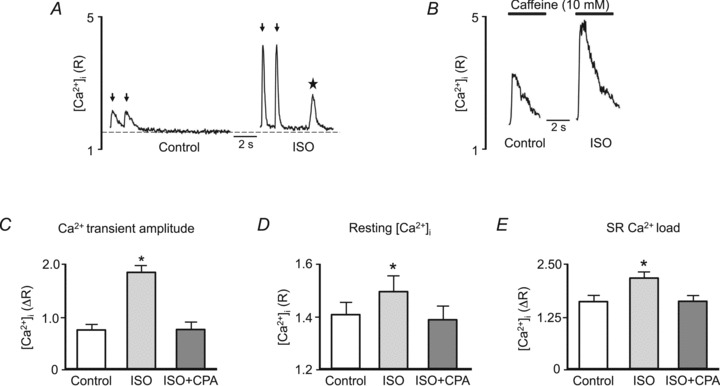
A, example cytosolic Ca2+ transients measured with indo-1 (0.75 Hz, electrical stimuli marked by arrows) followed by rest under control conditions ([Ca2+]o= 2 mm) and in the presence of 1 μm ISO. Control resting [Ca2+]i level is marked by dashed line. Note spontaneous Ca2+ wave that occurred during the rest period in the presence of ISO (star). B, example [Ca2+]i increases in response to 10 mm caffeine (black bars) used to assess SR Ca2+ content. Caffeine was applied immediately after identical 0.75 Hz pacing trains as shown in A, prior to the occurrence of spontaneous Ca2+ waves. Summary data of Ca2+ transient amplitude (0.75 Hz; C), resting [Ca2+]i (D) and SR Ca2+ load (E) under control conditions, in the presence of ISO, and in the presence of ISO with 7.5 μm cyclopiazonic acid (CPA) in 100 μm[Ca2+]o. *P < 0.05 versus control.
We next loaded the SR of cardiomyocytes with the low affinity Ca2+ indicator fluo-5N to directly and dynamically monitor the [Ca2+]SR when waves occur. Figure 2A shows the typical fluo-5N SR staining pattern. Based on observations of increased SR Ca2+ release in the form of Ca2+ sparks (Zhou et al. 2009; Ogrodnik & Niggli, 2010), Ca2+ leak (Curran et al. 2010; Bovo et al. 2012; Ullrich et al. 2012) and Ca2+ waves (Curran et al. 2007) in the presence of β-adrenergic stimulation, our initial hypothesis was that ISO would decrease the [Ca2+]SR threshold where waves occurred (i.e. Ca2+ waves would be observed in the presence of ISO at a [Ca2+]SR at or below the level where waves were not observed under control conditions). In rabbit ventricular myocytes Ca2+ waves were not typically observed under control conditions with 2 mm[Ca2+]o (Fig. 2Ba and D). However Ca2+ waves could be triggered using a low concentration (250 μm) of caffeine (Fig. 2Bb and D), a RyR agonist that sensitizes the release channel to Ca2+ and lowers the intra-SR Ca2+ threshold for Ca2+ waves (Venetucci et al. 2007; Kong et al. 2008). Application of ISO caused a large increase in [Ca2+]SR (Fig. 2Bc and C) and Ca2+ waves were observed (Fig. 2Bc and D). We then applied ISO+CPA ([Ca2+]o= 100 μm) to lower [Ca2+]SR. When [Ca2+]SR decreased to a similar level as under control conditions no Ca2+ waves were observed (Fig. 2Bd and D). Subsequent addition of 250 μm caffeine to myocytes treated with ISO+CPA triggered Ca2+ waves similarly to control conditions (Fig. 2Be and D). In summary, the data shown in Figs 1 and 2 indicate that under conditions of identical [Ca2+]i and [Ca2+]SR ISO stimulation failed to increase the propensity of Ca2+ waves and therefore does not appear to lower the Ca2+ wave threshold.
We next tested the alternative hypothesis that β-adrenergic stimulation increases the [Ca2+]SR threshold where waves occur. For this we designed experiments to observe Ca2+ waves under control conditions and to determine if, in the presence of ISO, Ca2+ waves were absent at the same [Ca2+]SR level or would require even higher [Ca2+]SR to occur. As spontaneous Ca2+ waves were not observed in normal [Ca2+]o of 2 mm (Fig. 2Ba and D), [Ca2+]o was raised to 7 mm and electrical stimulation frequency was incrementally increased to elevate [Ca2+]SR (Fig. 3B) until Ca2+ waves were observed during rest from stimulation (see Methods for details on the stimulation–rest protocol). The highest near steady-state [Ca2+]SR where Ca2+ waves were absent was defined as the control no-wave threshold (level 1 in Fig. 3Aa) while the lowest [Ca2+]SR that associated with Ca2+ waves was defined as the control wave threshold (level 2 in Fig. 3Ab). Across all experiments (n= 21 cells), the stimulation frequencies associated with the no-wave and wave thresholds were 0.83 ± 0.05 and 0.93 ± 0.05 Hz, respectively. We then applied ISO to the same cell which increased [Ca2+]SR due to its stimulatory effect on SERCA. As [Ca2+]SR subsequently declined during an extended period of rest we determined the lowest [Ca2+]SR that preceded a Ca2+ wave (ISO wave threshold; level 3 in Fig. 3Ac). As shown in Fig. 3A (trace c), Ca2+ waves were frequent with ISO but were never observed at a [Ca2+]SR below the control no-wave threshold (level 1), unless RyRs were sensitized with a low concentration of caffeine (250 μm).
We next supported these findings with a complementary experimental approach that allowed Ca2+ waves to be observed in a defined time interval (8 s) following rest from steady-state stimulation. For this the rest period where threshold levels were determined was preceded by a 30–60 s interval during which cells were electrically stimulated to establish identical steady-state conditions for the SR Ca2+ release mechanism. After control ([Ca2+]o= 7 mm) no-wave (level 1 in Fig. 4Aa) and wave (level 2 in Fig. 4Ab) thresholds were determined, ISO was applied for 5 min leading to an increase in [Ca2+]SR and frequent Ca2+ waves (Fig. 4Ac). [Ca2+]SR was then gradually reduced using application of ISO in the presence of low [Ca2+]o (1 mm) and the SERCA inhibitor CPA (3 μm). In the example shown waves were no longer observed following rest from stimulation (Fig. 4Ad), unless RyRs were pharmacologically sensitized with 250 μm caffeine (Fig. 4Ae). Level 3 in Fig. 4Ad represents the no-wave threshold in the presence of ISO. Additional experiments were performed using the alternative SERCA inhibitor TG (1 μm) with similar results, and these results were pooled to provide the summary data shown in Fig. 4B and C. From 12 individual myocytes measurements of the no-wave and wave thresholds, as well as [Ca2+]SR in the presence of ISO, followed by exposure to a SERCA blocker were obtained. In this set of experiments the no-wave threshold was 0.88 ± 0.01 [(F–FMin)/(FMax–FMin)] and the wave threshold was 0.90 ± 0.01 (P < 0.05, n= 12 cells, paired comparison). In the presence of ISO alone, all 12 cells exhibited Ca2+ waves, presumably due to the substantial increase of [Ca2+]SR above the control threshold with (F–FMin)/(FMax–FMin) values approaching 1. Myocytes were subsequently exposed to ISO+CPA or ISO+TG for examination of the [Ca2+]SR associated with waves (filled circles in Fig. 4B) or lack of waves (open circles). From each individual cell one or two measurements could be obtained for a total number of 18 experimental trials. In contrast to ISO alone, in the presence of ISO+CPA/TG waves were only observed in 8 of 18 experimental trials where [Ca2+]SR was above the control no-wave threshold. The observation that in a majority of experimental trials waves were absent in the presence of ISO at [Ca2+]SR levels where in control conditions waves were observed was indicative of an increase in the Ca2+ wave threshold. Quantitative analysis of the [Ca2+]SR associated with the no-wave threshold under the respective conditions showed that this level was significantly higher in the presence of ISO+CPA/TG (Fig. 4C). The no-wave threshold under control conditions was at [Ca2+]SR= 2.58 ± 0.24 mm (n= 7 cells) and increased to 3.39 ± 0.23 mm in the presence of ISO+CPA/TG (n= 7 cells; P= 0.002 vs. control). At [Ca2+]SR below the control no-wave threshold, Ca2+ waves were not observed with ISO+CPA/TG (data not shown), unless RyRs were sensitized pharmacologically with 250 μm caffeine. In the presence of 250 μm caffeine Ca2+ waves were observed in all experimental trials (eight Ca2+ waves in eight cells) at a [Ca2+]SR below the control no-wave threshold, indicative of a decrease in the Ca2+ wave threshold. Quantification of the no-wave threshold in the presence of 250 μm caffeine and ISO+CPA/TG showed that wave activity ceased when [Ca2+]SR dropped to 1.00 ± 0.11 mm (n= 5 cells), a level that was significantly (P < 0.001) below the control and ISO+CPA/TG no-wave thresholds.
The direct and dynamic [Ca2+]SR measurements clearly revealed an increase in the Ca2+ wave threshold during β-adrenergic stimulation. To further support these direct measurements we obtained additional measurements of the no-wave threshold in the presence of ISO+CPA using the amplitude of the [Ca2+]i increase in response to 10 mm caffeine to estimate SR Ca2+ content.
Figure 5A shows original traces to determine control no-wave (level 1) and wave (level 2) thresholds, followed by the no-wave threshold in the presence of ISO+CPA (level 3). As shown in example traces of Fig. 5A and summary data of Fig. 5B, the SR Ca2+ content associated with the disappearance of Ca2+ waves with ISO+CPA (ISO no-wave threshold) was significantly higher than the wave threshold observed under control conditions. Importantly, resting [Ca2+]i was similar under these experimental conditions (Fig. 5C; dotted line in Fig. 5A), which excluded the possibility that ISO-dependent changes in global cytosolic [Ca2+]i were responsible for the observed effects on the wave threshold. Thus, the observed differences in SR Ca2+ content (Figs 4A and 5B) where waves occurred reflect a true increase in the intra-SR Ca2+ wave threshold.
Figure 5. Isoproterenol increases the intra-SR Ca2+ wave threshold determined by cytosolic [Ca2+]i measurements.
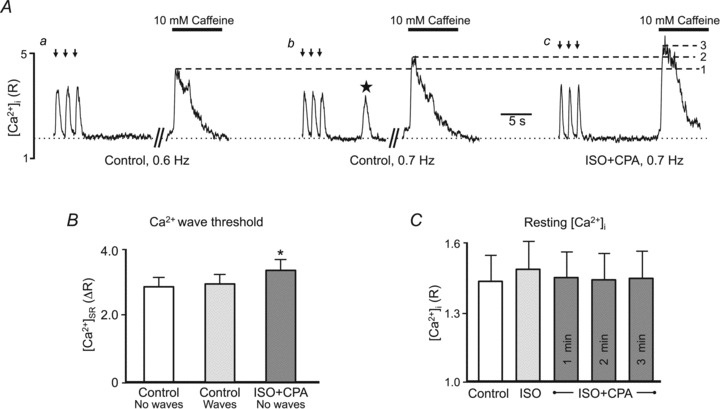
A, example indo-1 [Ca2+]i measurements following rest from electrical stimulation (arrows) under control conditions (7 mm[Ca2+]o, a= 0.6 Hz, b= 0.7 Hz) and in the presence of ISO with 3 μm CPA (c: 1 mm[Ca2+]o, 0.7 Hz). Caffeine (10 mm) was applied after rest from a separate pacing train to assess SR Ca2+ content at each pacing frequency under control conditions and to determine no-wave (dashed line 1) and wave (dashed line 2) threshold SR Ca2+ content. In the presence of ISO+CPA, when waves were not observed following rest from pacing, 10 mm caffeine was applied to assess the no-wave threshold in ISO (dashed line 3). Star denotes Ca2+ wave. Dotted line: resting [Ca2+]i. B, summary data of SR Ca2+ content under the conditions shown in A (n= 8 cells). C, average resting [Ca2+]i was similar under these experimental conditions (n= 7 cells). *P < 0.05.
Recent work has proposed that, in addition to the Ca2+ wave threshold, altered RyR ‘refractoriness’ may be a critical factor regulating SR Ca2+ release and the generation of Ca2+ waves (Ramay et al. 2011; Belevych et al. 2012). This concept is supported by direct measurements of [Ca2+]SR which show that after [Ca2+]SR recovers to a steady-state level following electrical stimulation a finite time interval (latency period) remains prior to Ca2+ wave initiation (Figs 2B, 3A and 4A; see also Domeier et al. 2010; Belevych et al. 2012; Maxwell et al. 2012). We therefore examined SR Ca2+ refilling, [Ca2+]SR and wave latency in experiments where waves were observed. In these experiments two measurements of [Ca2+]SR, SR Ca2+ refilling times and Ca2+ wave latency were obtained from an individual cell (nine cells total), one at the wave threshold (Fig. 6Aa) and one after [Ca2+]SR was increased above the control wave threshold (Fig. 6Ab). [Ca2+]SR was increased by increasing the pacing frequency prior to the rest period during which Ca2+ waves were observed. Under control conditions SR Ca2+ refilling time (marked by dark grey area in Fig. 6A) was independent of initial [Ca2+]SR (416 ± 25 ms at [Ca2+]SR= 0.95 ± 0.01 (F–FMin)/(FMax–FMin), versus 416 ± 28 ms at [Ca2+]SR= 0.91 ± 0.01; n= 9 cells). In the presence of ISO, SR Ca2+ refilling time was significantly shorter than under control conditions (227 ± 26 ms, n= 6 cells, P < 0.001 compared with control), and was slowed by subsequent SERCA inhibition with CPA or TG (367 ± 96 ms). The acceleration of SR refilling in the presence of ISO is explained by the stimulatory β-adrenergic effect on SERCA.
Analysis of wave latency revealed several interesting features. In control conditions (Fig. 6B; circles in Fig. 6D) as well as in the presence of ISO (Fig. 6C; squares in Fig. 6D), wave latency was highly dependent on [Ca2+]SR, with higher [Ca2+]SR leading to a significantly shorter wave latency. Furthermore, in the presence of ISO the dependence of wave latency on [Ca2+]SR shifted towards longer latencies, i.e. for a given SR Ca2+ content, the wave latency was prolonged in the presence of ISO compared with control conditions. For example, despite the fact during ISO treatment [Ca2+]SR was significantly higher than under control conditions [ISO (grey square) 1.0 ± 0 vs. control (grey circle) 0.95 ± 0.01; P < 0.001], latency times were similar (ISO 1.02 ± 0.22 s vs. control 1.44 ± 0.27 s; not significantly different at P= 0.38). Furthermore, comparison of control conditions (Fig. 6D, grey circle) with ISO+CPA/TG treatment (Fig. 6D, black square) showed that at a similar [Ca2+]SR (control 0.95 ± 0.01 vs. ISO+CPA 0.93 ± 0.02; not significantly different at P= 0.28) latency times were significantly shorter under control conditions (control 1.44 ± 0.27 s vs. ISO+CPA/TG 4.42 ± 0.85 s; P < 0.01).
In summary, our study shows that during β-adrenergic stimulation the propensity for Ca2+ waves is increased because of significantly increased [Ca2+]SR and SR Ca2+ overload. Counteracting the increase in [Ca2+]SR are changes in two [Ca2+]SR-dependent parameters, with an increase in Ca2+ wave threshold and a prolonged wave latency.
Discussion
During sympathetic nervous system activation the heart experiences chronotropic, inotropic and lusitropic effects that act in concert to facilitate an increase in cardiac output. At the level of the individual cardiomyocyte catecholamine binding to β-adrenoceptors causes an increase in cellular Ca2+ cycling that associates with Ca2+ waves, suggesting an acute lowering of the intra-SR Ca2+ wave threshold. However, our data using direct, dynamic measurements of this threshold clearly show that to the contrary, the threshold for Ca2+ waves is increased by acute β-adrenergic stimulation. The direct [Ca2+]SR measurements presented here support recent quantification of the Ca2+ wave threshold in mouse ventricular myocytes which show a similar effect of the Ca2+ wave threshold increasing following acute β-adrenergic stimulation (Kashimura et al. 2010; Stokke et al. 2010a).
The Ca2+ wave threshold is a useful experimental parameter that has greatly advanced our understanding of the mechanisms of cardiac arrhythmia, including those associated with diseases such as CPVT and heart failure. Experimental evidence and computational modelling implicate numerous intracellular factors in Ca2+ wave generation and propagation, including RyR open probability, release junction (sarcomere) spacing, SR Ca2+ uptake via SERCA, and cytosolic and intra-SR Ca2+ buffering processes (Izu et al. 2006; Ramay et al. 2010; Swietach et al. 2010). The appearance of Ca2+ waves as [Ca2+]SR increases can be explained by well-established RyR gating mechanisms, most notably that the RyR open probability increases at high [Ca2+]SR due to the channel's inherent sensitivity to [Ca2+]SR (Gyorke & Gyorke, 1998; Fill & Copello, 2002). Furthermore, because Ca2+ release terminates at a set level of [Ca2+]SR (Zima et al. 2008) Ca2+ sparks will be high-amplitude release events at high [Ca2+]SR, which will increase the ability of Ca2+ released during a spark to overcome cytosolic buffering and diffuse to adjacent release junctions and trigger CICR. These properties result in the appearance of Ca2+ waves at a defined [Ca2+]SR (the wave threshold), and make the time to wave initiation highly dependent on [Ca2+]SR (Fig. 6).
Direct measurements of [Ca2+]SR (Fig. 6) illustrate that Ca2+ waves do not occur immediately after [Ca2+]SR reaches a steady-state concentration (Domeier et al. 2010; Belevych et al. 2012; Maxwell et al. 2012) and therefore a time-dependent ‘refractory’ component of wave generation exists (Belevych et al. 2012). While this appears to conceptually challenge the Ca2+ wave threshold as being the critical parameter that determines whether a Ca2+ wave will be observed, our data suggest that changes in wave refractory (latency) time may simply be a manifestation of how high [Ca2+]SR is above the Ca2+ wave threshold. In an individual cell the Ca2+ wave threshold was highly reproducible among experimental trials, yet different wave latency times were recorded depending on [Ca2+]SR, with shorter times observed above the wave threshold than at the wave threshold (Fig. 6B). This relationship was similarly observed in canine ventricular myocytes (Belevych et al. 2012). Based on these data our working model (Fig. 7) is that the cell has a distinct intra-luminal [Ca2+]SR threshold for Ca2+ wave development (Díaz et al. 1997b; Jiang et al. 2004; Venetucci et al. 2007). At [Ca2+]SR below this threshold the probability for wave development following rest from steady-state stimulation is low and thus the cell has a wave latency period that approaches infinity (Fig. 7a). When [Ca2+]SR reaches the Ca2+ wave threshold the probability that a Ca2+ wave will occur increases, yet due to the stochastic nature of Ca2+ wave initiation there will be a finite latency period associated with the wave (Fig. 7b). As [Ca2+]SR continues to increase above this distinct wave threshold, the Ca2+ wave probability remains high but now the latency period will be shortened due to the stimulatory effect of luminal [Ca2+]SR on RyR open probability, increasing the likelihood of a spontaneous release event to trigger a Ca wave (Fig. 7c) (Gyorke & Gyorke, 1998). During β-adrenergic stimulation, despite the observation that the wave threshold is increased, the massive gain of Ca2+ in the SR results in a further shortening of the latency period (Fig. 7f). Overall, the latency period is inversely proportional (Fig. 6D) to the degree by which [Ca2+]SR exceeds the wave threshold. For a given [Ca2+]SR level, however, the latency period is longer in the presence of ISO (Fig. 7d) than under control conditions (Fig. 7c), and is similar when [Ca2+]SR exceeds the respective wave thresholds to the same degree (Fig. 7c vs. Fig. 7e). Thus, there may be an important distinction between the parameter of the Ca2+ wave threshold and that of the wave latency period. A cell may have multiple latency periods depending on how high [Ca2+]SR is above a distinct wave threshold (Fig. 6), and thus the wave threshold measurement is a more biophysically accurate way to assess the ability of the SR to sequester (or spontaneously release) Ca2+. Alternatively, however, the wave latency period may be the most physiologically relevant way to describe diastolic Ca2+ waves with respect to cellular SR Ca2+ content (i.e. inotropic state of heart) and rate of AP stimulation (i.e. chronotropic state of the heart). In vivo these complex relationships will determine if an arrhythmogenic Ca2+ wave will be observed during diastole in the beating heart.
Figure 7. Schematic diagram of proposed relationship between SR Ca2+ content, Ca2+ wave threshold and latency.
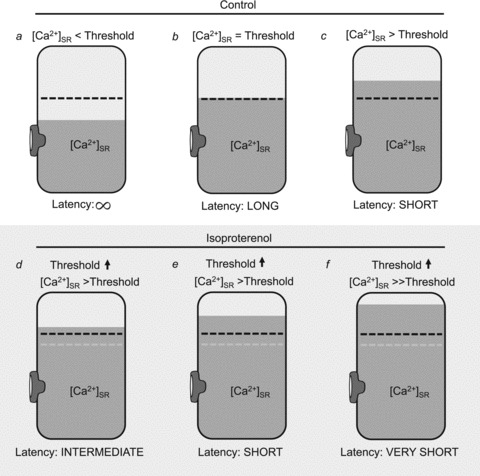
Schematic diagram of the SR showing the relationship between [Ca2+]SR (shaded area), the Ca2+ wave threshold (dashed black line) and Ca2+ wave latency when [Ca2+]SR is below (a), at (b) and above (c) the Ca2+ wave threshold. In the presence of ISO (d, e and f) the Ca2+ wave threshold (dashed black line) is increased when compared with control conditions (dashed grey line). When [Ca2+]SR is similar between control condition (c) and ISO condition (d), latency is shorter in control due to [Ca2+]SR being higher above the respective wave threshold. When [Ca2+]SR is elevated by the same relative amount above the wave threshold under control conditions (c) and with ISO (e) wave latencies are similar even though the total [Ca2+]SR is higher with ISO. Under most experimental conditions [Ca2+]SR increases dramatically in the presence of ISO, thus elevating [Ca2+]SR well above the respective Ca2+ wave threshold, resulting in a very short wave latency (f).
β-Adrenergic modulation of the RyR and Ca2+ waves
Alterations in RyR activity (e.g. via phosphorylation or redox modification) will lead to subsequent changes in the intra-SR Ca2+ wave threshold, as shown by RyR inhibitors (Overend et al. 1997) or activators (Venetucci et al. 2007; Kong et al. 2008) raising or lowering the threshold, respectively. The signalling events that regulate RyR activity downstream of β-adrenergic stimulation remain highly contentious, with conflicting reports throughout the literature (for example see commentaries by Yamaguchi & Meissner, 2007; Houser, 2010; Bers, 2012). Most investigations, however, report an increase in RyR activity which would result in a decrease in the Ca2+ wave threshold. The observation of an increased Ca2+ wave threshold is therefore counterintuitive. Discrepant results may emerge due to the multiple assays of RyR function which may not be mechanistically equivalent, particularly following activation of complex cellular signalling pathways (reviewed by George, 2008). As an example, systolic Ca2+ release in response to the AP, representing activation of RyRs via cytosolic Ca2+, may exhibit distinct regulation when compared to diastolic Ca2+ release triggered by SR Ca2+ overload and activation of RyRs by [Ca2+]SR. The properties of local diastolic Ca2+ release events (Ca2+ spark frequency, Ca2+ spark amplitude, non-spark-mediated SR Ca2+ leak) may also be differentially modulated (Zhou et al. 2009) and subsequently alter the ability of Ca2+ waves to initiate and propagate. Recently, novel genetic approaches have been utilized to ablate (Wehrens et al. 2006; Benkusky et al. 2007; Chelu et al. 2009) or chronically activate (Shan et al. 2010; van Oort et al. 2010) RyR phosphorylation sites in vivo, and gain additional insight into RyR regulation that may occur downstream of β-adrenergic signalling. However, this strategy may result in a cellular phenotype that is distinct from that observed when all cellular effectors are activated in parallel following receptor activation. Complex signal integration at the level of the RyR is expected given the multiple regulatory sites on the channel protein and the role of the macromolecular RyR complex as a subcellular signalling platform.
β-Adrenergic modulation of SERCA and Ca2+ waves
Although the Ca2+ wave threshold is primarily set by the regulation of RyR release channels, it is likely further tuned by altering the function of additional cellular proteins involved in wave generation. One of the main effects of β-adrenergic stimulation is to increase SERCA activity, which is accomplished via phosphorylation of the SERCA inhibitory protein phospholamban. Phospholamban phosphorylation, principally by protein kinase A although there is some evidence for a role of Ca2+/calmodulin-dependent protein kinase II, reduces its inhibitory interactions with SERCA and leads to a dramatic increase in SERCA activity and [Ca2+]SR. Indeed, SERCA-mediated Ca2+ uptake increasing [Ca2+]SR above the intra-SR Ca2+ wave threshold is one of the principal mechanisms by which β-adrenergic stimulation increases Ca2+ wave probability, and is supported by data showing that genetic ablation of phospholamban dramatically increases the propensity of Ca2+ waves (Huser et al. 1998). SERCA activity may also directly contribute to Ca2+ wave propagation and thus, in part, determine the intra-SR Ca2+ wave threshold. It has been shown experimentally that SERCA inhibition increases Ca2+ wave velocity (Lukyanenko et al. 1999), and computer simulations predict that increasing SERCA activity prevents Ca2+ wave propagation by decreasing [Ca2+]i and the ability of Ca2+ to diffuse between Ca2+ release junctions (Ramay et al. 2010). However, this cytosolic Ca2+ buffering effect may be countered by the ability of SERCA to locally increase [Ca2+]SR and create intra-SR ‘Ca2+ sensitization’ wavefronts which precede the cytosolic Ca2+ wave (Keller et al. 2007; Maxwell & Blatter, 2012). This provocative model is based on experimental evidence contradictory to that of Lukyanenko et al. (1999), which shows that SERCA inhibition decreases Ca2+ wave velocity (Keller et al. 2007). While this model of wave propagation can be reproduced by computer simulations (Ramay et al. 2010), it is critically dependent on the speed by which Ca2+ diffuses within the SR network, which is difficult to determine empirically and remains controversial (Swietach et al. 2008; Picht et al. 2011). Electrophysiological measurement of the Ca2+ wave threshold in rat myocytes showed that SERCA inhibition decreased the intra-SR Ca2+ wave threshold (O’Neill et al. 2004), and these data are supported by studies of transgenic mice with reduced SERCA abundance that also have a decreased Ca2+ wave threshold, although interpretation of data from this model may be complicated by concomitant compensatory changes in RyR function (Stokke et al. 2010a,b). However, in mouse ventricular myocytes where the Ca2+ wave threshold was increased following β-adrenergic stimulation, the Ca2+ wave threshold did not appear to correlate with SERCA activity (Kashimura et al. 2010), and it remains unclear if the increase in SERCA activity that accompanies β-adrenergic stimulation plays a role in increasing the intra-SR Ca2+ wave threshold.
β-Adrenergic modulation of cellular buffering and Ca2+ waves
As predicted by mathematical models, changes in the buffering properties of the cytosol and/or SR would also alter Ca2+ wave propagation and the intra-SR Ca2+ wave threshold. β-Adrenoceptor activation leads to a decrease in myofilament Ca2+ sensitivity which is believed in part to underlie the lusitropic effect of catecholamines on the heart. This change will likely lead to altered Ca2+ buffering properties and Ca2+ transport kinetics, although it remains unclear if these changes alter Ca2+ wave propagation. Recent evidence suggests that increased Ca2+ sensitivity promotes arrhythmogenesis, leading to the hypothesis that increased myofilament Ca2+ affinity may augment Ca2+ wave propagation by altering diastolic contracture and shortening the distance between release clusters (Izu et al. 2006; Chen-Izu et al. 2007). Changes in intra-SR Ca2+ buffering properties have also been shown to affect Ca2+ wave propagation (Kubalova et al. 2004). While there is experimental evidence that the intra-SR Ca2+ binding protein calsequestrin may be regulated by phosphorylation (Cala & Jones, 1991), and that phosphorylation increases Ca2+ binding capacity of calsequestrin (Beard et al. 2008), it is unclear if this phosphorylation occurs in response to acute activation of intracellular signalling pathways. Furthermore, the addition of exogenous intra-SR Ca2+ buffers alters Ca2+ wave frequency and amplitude without changing the intra-SR Ca2+ wave threshold (Kubalova et al. 2004), and thus it is unlikely that the acute effect of β-adrenergic stimulation on the intra-SR Ca2+ wave threshold is due to changes in intra-SR Ca2+ binding protein function.
Summary
β-Adrenergic stimulation enhances intracellular Ca2+ cycling and increases SR Ca2+ load to the extent that spontaneous and potentially detrimental Ca2+ waves occur. As shown in this study the β-adrenoceptor-mediated increase in SR Ca2+ load is also accompanied by an increase of the intra-SR Ca2+ wave threshold and a prolongation of the wave latency period. These two factors may be interpreted as a protective mechanism against diastolic arrhythmogenic Ca2+ release in the heart.
Acknowledgments
This work was supported by National Institutes of Health grants HL62231, HL80101 and HL101235 (to L.A.B.) and F32HL090211 (to T.L.D.) and the Leducq Foundation (to L.A.B.). The authors would also like to thank Dr Elisa Bovo, Stephen Shonts and Demetrio Santiago for assistance with myocyte isolation.
Glossary
- AP
action potential
- [Ca2+]i
cytosolic free calcium concentration
- [Ca2+]o
extracellular free calcium concentration
- [Ca2+]SR
intra-sarcoplasmic reticulum free calcium concentration
- CICR
calcium-induced calcium release
- CPA
cyclopiazonic acid
- CPVT
catecholaminergic polymorphic ventricular tachycardia
- CRU
calcium release unit
- DMSO
dimethyl sulfoxide
- ISO
isoproterenol
- LTCC
L-type calcium channel
- RyR
ryanodine receptor
- SERCA
sarcoplasmic/endoplasmic reticulum calcium ATPase
- SR
sarcoplasmic reticulum
- TG
thapsigargin
Author contributions
T.L.D., J.T.M. and L.A.B. contributed to the conception and design of the study, analysis and interpretation of data, and writing of the article, and have approved the final version of the manuscript. T.L.D. and J.T.M. performed the experimental work.
Author's present address
Timothy L. Domeier: Department of Medical Pharmacology and Physiology, University of Missouri School of Medicine, Columbia, MO 65212, USA.
References
- Beard NA, Wei L, Cheung SN, Kimura T, Varsanyi M, Dulhunty AF. Phosphorylation of skeletal muscle calsequestrin enhances its Ca2+ binding capacity and promotes its association with junctin. Cell Calcium. 2008;44:363–373. doi: 10.1016/j.ceca.2008.01.005. [DOI] [PubMed] [Google Scholar]
- Belevych AE, Terentyev D, Terentyeva R, Ho HT, Gyorke I, Bonilla IM, Carnes CA, Billman GE, Gyorke S. Shortened Ca2+ signalling refractoriness underlies cellular arrhythmogenesis in a postinfarction model of sudden cardiac death. Circ Res. 2012;110:569–577. doi: 10.1161/CIRCRESAHA.111.260455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benkusky NA, Weber CS, Scherman JA, Farrell EF, Hacker TA, John MC, Powers PA, Valdivia HH. Intactβ-adrenergic response and unmodified progression toward heart failure in mice with genetic ablation of a major protein kinase A phosphorylation site in the cardiac ryanodine receptor. Circ Res. 2007;101:819–829. doi: 10.1161/CIRCRESAHA.107.153007. [DOI] [PubMed] [Google Scholar]
- Bers DM. Ryanodine receptor S2808 phosphorylation in heart failure: smoking gun or red herring. Circ Res. 2012;110:796–799. doi: 10.1161/CIRCRESAHA.112.265579. [DOI] [PubMed] [Google Scholar]
- Bovo E, Lipsius SL, Zima AV. Reactive oxygen species contribute to the development of arrhythmogenic Ca2+ waves duringβ-adrenergic receptor stimulation in rabbit cardiomyocytes. J Physiol. 2012;590:3291–3304. doi: 10.1113/jphysiol.2012.230748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cala SE, Jones LR. Phosphorylation of cardiac and skeletal muscle calsequestrin isoforms by casein kinase II. Demonstration of a cluster of unique rapidly phosphorylated sites in cardiac calsequestrin. J Biol Chem. 1991;266:391–398. [PubMed] [Google Scholar]
- Capogrossi MC, Houser SR, Bahinski A, Lakatta EG. Synchronous occurrence of spontaneous localized calcium release from the sarcoplasmic reticulum generates action potentials in rat cardiac ventricular myocytes at normal resting membrane potential. Circ Res. 1987;61:498–503. doi: 10.1161/01.res.61.4.498. [DOI] [PubMed] [Google Scholar]
- Chelu MG, Sarma S, Sood S, Wang S, van Oort RJ, Skapura DG, Li N, Santonastasi M, Muller FU, Schmitz W, Schotten U, Anderson ME, Valderrabano M, Dobrev D, Wehrens XH. Calmodulin kinase II-mediated sarcoplasmic reticulum Ca2+ leak promotes atrial fibrillation in mice. J Clin Invest. 2009;119:1940–1951. doi: 10.1172/JCI37059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen-Izu Y, Ward CW, Stark W, Jr, Banyasz T, Sumandea MP, Balke CW, Izu LT, Wehrens XH. Phosphorylation of RyR2 and shortening of RyR2 cluster spacing in spontaneously hypertensive rat with heart failure. Am J Physiol Heart Circ Physiol. 2007;293:H2409–2417. doi: 10.1152/ajpheart.00562.2007. [DOI] [PubMed] [Google Scholar]
- Curran J, Brown KH, Santiago DJ, Pogwizd S, Bers DM, Shannon TR. Spontaneous Ca waves in ventricular myocytes from failing hearts depend on Ca2+-calmodulin-dependent protein kinase II. J Mol Cell Cardiol. 2010;49:25–32. doi: 10.1016/j.yjmcc.2010.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curran J, Hinton MJ, Rios E, Bers DM, Shannon TR. β-Adrenergic enhancement of sarcoplasmic reticulum calcium leak in cardiac myocytes is mediated by calcium/calmodulin-dependent protein kinase. Circ Res. 2007;100:391–398. doi: 10.1161/01.RES.0000258172.74570.e6. [DOI] [PubMed] [Google Scholar]
- Díaz ME, Trafford AW, O’Neill SC, Eisner DA. A measurable reduction of s.r. Ca content follows spontaneous Ca release in rat ventricular myocytes. Pflugers Arch. 1997a;434:852–854. doi: 10.1007/s004240050475. [DOI] [PubMed] [Google Scholar]
- Díaz ME, Trafford AW, O’Neill SC, Eisner DA. Measurement of sarcoplasmic reticulum Ca2+ content and sarcolemmal Ca2+ fluxes in isolated rat ventricular myocytes during spontaneous Ca2+ release. J Physiol. 1997b;501:3–16. doi: 10.1111/j.1469-7793.1997.003bo.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domeier TL, Blatter LA. Mechanisms of spontaneous calcium wave generation duringβ-adrenergic stimulation in rabbit ventricular myocytes. Biophys J. 2010;98:105a. [Google Scholar]
- Domeier TL, Blatter LA, Zima AV. Changes in intra-luminal calcium during spontaneous calcium waves following sensitization of ryanodine receptor channels. Channels. 2010;4:87–92. doi: 10.4161/chan.4.2.11019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domeier TL, Maxwell JT, Blatter LA. β-Adrenergic stimulation increases the intra-sarcoplasmic reticulum Ca threshold for spontaneous Ca waves. Biophys J. 2011;100:559a. [Google Scholar]
- Drummond GB. Reporting ethical matters in the Journal of Physiology: standards and advice. J Physiol. 2009;587:713–719. doi: 10.1113/jphysiol.2008.167387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedida D, Noble D, Rankin AC, Spindler AJ. The arrhythmogenic transient inward current iTI and related contraction in isolated guinea-pig ventricular myocytes. J Physiol. 1987;392:523–542. doi: 10.1113/jphysiol.1987.sp016795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fill M, Copello JA. Ryanodine receptor calcium release channels. Physiol Rev. 2002;82:893–922. doi: 10.1152/physrev.00013.2002. [DOI] [PubMed] [Google Scholar]
- Fujiwara K, Tanaka H, Mani H, Nakagami T, Takamatsu T. Burst emergence of intracellular Ca2+ waves evokes arrhythmogenic oscillatory depolarization via the Na+–Ca2+ exchanger: simultaneous confocal recording of membrane potential and intracellular Ca2+ in the heart. Circ Res. 2008;103:509–518. doi: 10.1161/CIRCRESAHA.108.176677. [DOI] [PubMed] [Google Scholar]
- George CH. Sarcoplasmic reticulum Ca2+ leak in heart failure: mere observation or functional relevance. Cardiovasc Res. 2008;77:302–314. doi: 10.1093/cvr/cvm006. [DOI] [PubMed] [Google Scholar]
- Guo T, Zhang T, Mestril R, Bers DM. Ca2+/calmodulin-dependent protein kinase II phosphorylation of ryanodine receptor does affect calcium sparks in mouse ventricular myocytes. Circ Res. 2006;99:398–406. doi: 10.1161/01.RES.0000236756.06252.13. [DOI] [PubMed] [Google Scholar]
- Gyorke I, Gyorke S. Regulation of the cardiac ryanodine receptor channel by luminal Ca2+ involves luminal Ca2+ sensing sites. Biophys J. 1998;75:2801–2810. doi: 10.1016/S0006-3495(98)77723-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houser SR. Does protein kinase A-mediated phosphorylation of the cardiac ryanodine receptor play any role in adrenergic regulation of calcium handling in health and disease. Circ Res. 2010;106:1672–1674. doi: 10.1161/CIRCRESAHA.110.221853. [DOI] [PubMed] [Google Scholar]
- Huser J, Bers DM, Blatter LA. Subcellular properties of [Ca2+]i transients in phospholamban-deficient mouse ventricular cells. Am J Physiol Heart Circ Physiol. 1998;274:H1800–1811. doi: 10.1152/ajpheart.1998.274.5.H1800. [DOI] [PubMed] [Google Scholar]
- Izu LT, Means SA, Shadid JN, Chen-Izu Y, Balke CW. Interplay of ryanodine receptor distribution and calcium dynamics. Biophys J. 2006;91:95–112. doi: 10.1529/biophysj.105.077214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang D, Xiao B, Yang D, Wang R, Choi P, Zhang L, Cheng H, Chen SR. RyR2 mutations linked to ventricular tachycardia and sudden death reduce the threshold for store-overload-induced Ca2+ release (SOICR) Proc Natl Acad Sci U S A. 2004;101:13062–13067. doi: 10.1073/pnas.0402388101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashimura T, Briston SJ, Trafford AW, Napolitano C, Priori SG, Eisner DA, Venetucci LA. In the RyR2(R4496C) mouse model of CPVT,β-adrenergic stimulation induces Ca waves by increasing SR Ca content and not by decreasing the threshold for Ca waves. Circ Res. 2010;107:1483–1489. doi: 10.1161/CIRCRESAHA.110.227744. [DOI] [PubMed] [Google Scholar]
- Kass RS, Lederer WJ, Tsien RW, Weingart R. Role of calcium ions in transient inward currents and aftercontractions induced by strophanthidin in cardiac Purkinje fibres. J Physiol. 1978;281:187–208. doi: 10.1113/jphysiol.1978.sp012416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller M, Kao JP, Egger M, Niggli E. Calcium waves driven by “sensitization” wave-fronts. Cardiovasc Res. 2007;74:39–45. doi: 10.1016/j.cardiores.2007.02.006. [DOI] [PubMed] [Google Scholar]
- Kong H, Jones PP, Koop A, Zhang L, Duff HJ, Chen SR. Caffeine induces Ca2+ release by reducing the threshold for luminal Ca2+ activation of the ryanodine receptor. Biochem J. 2008;414:441–452. doi: 10.1042/BJ20080489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kranias EG, Solaro RJ. Phosphorylation of troponin I and phospholamban during catecholamine stimulation of rabbit heart. Nature. 1982;298:182–184. doi: 10.1038/298182a0. [DOI] [PubMed] [Google Scholar]
- Kubalova Z, Gyorke I, Terentyeva R, Viatchenko-Karpinski S, Terentyev D, Williams SC, Gyorke S. Modulation of cytosolic and intra-sarcoplasmic reticulum calcium waves by calsequestrin in rat cardiac myocytes. J Physiol. 2004;561:515–524. doi: 10.1113/jphysiol.2004.073940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindemann JP, Jones LR, Hathaway DR, Henry BG, Watanabe AM. β-Adrenergic stimulation of phospholamban phosphorylation and Ca2+-ATPase activity in guinea pig ventricles. J Biol Chem. 1983;258:464–471. [PubMed] [Google Scholar]
- Lukyanenko V, Subramanian S, Gyorke I, Wiesner TF, Gyorke S. The role of luminal Ca2+ in the generation of Ca2+ waves in rat ventricular myocytes. J Physiol. 1999;518:173–186. doi: 10.1111/j.1469-7793.1999.0173r.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxwell JT, Blatter LA. Facilitation of cytosolic calcium wave propagation by local calcium uptake into the sarcoplasmic reticulum in cardiac myocytes. J Physiol. 2012;590:6037, 6045. doi: 10.1113/jphysiol.2012.239434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxwell JT, Domeier TL, Blatter LA. Dantrolene prevents arrhythmogenic Ca2+ release in heart failure. Am J Physiol Heart Circ Physiol. 2012;302:H953–963. doi: 10.1152/ajpheart.00936.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neill SC, Miller L, Hinch R, Eisner DA. Interplay between SERCA and sarcolemmal Ca2+ efflux pathways controls spontaneous release of Ca2+ from the sarcoplasmic reticulum in rat ventricular myocytes. J Physiol. 2004;559:121–128. doi: 10.1113/jphysiol.2003.058917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogrodnik J, Niggli E. Increased Ca2+ leak and spatiotemporal coherence of Ca2+ release in cardiomyocytes duringβ-adrenergic stimulation. J Physiol. 2010;588:225–242. doi: 10.1113/jphysiol.2009.181800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overend CL, Eisner DA, O’Neill SC. The effect of tetracaine on spontaneous Ca2+ release and sarcoplasmic reticulum calcium content in rat ventricular myocytes. J Physiol. 1997;502:471–479. doi: 10.1111/j.1469-7793.1997.471bj.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picht E, Zima AV, Shannon TR, Duncan AM, Blatter LA, Bers DM. Dynamic calcium movement inside cardiac sarcoplasmic reticulum during release. Circ Res. 2011;108:847–856. doi: 10.1161/CIRCRESAHA.111.240234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priori SG, Chen SR. Inherited dysfunction of sarcoplasmic reticulum Ca2+ handling and arrhythmogenesis. Circ Res. 2011;108:871–883. doi: 10.1161/CIRCRESAHA.110.226845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramay HR, Jafri MS, Lederer WJ, Sobie EA. Predicting local SR Ca2+ dynamics during Ca2+ wave propagation in ventricular myocytes. Biophys J. 2010;98:2515–2523. doi: 10.1016/j.bpj.2010.02.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramay HR, Liu OZ, Sobie EA. Recovery of cardiac calcium release is controlled by sarcoplasmic reticulum refilling and ryanodine receptor sensitivity. Cardiovasc Res. 2011;91:598–605. doi: 10.1093/cvr/cvr143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlotthauer K, Bers DM. Sarcoplasmic reticulum Ca2+ release causes myocyte depolarization. Underlying mechanism and threshold for triggered action potentials. Circ Res. 2000;87:774–780. doi: 10.1161/01.res.87.9.774. [DOI] [PubMed] [Google Scholar]
- Sedej S, Heinzel FR, Walther S, Dybkova N, Wakula P, Groborz J, Gronau P, Maier LS, Vos MA, Lai FA, Napolitano C, Priori SG, Kockskamper J, Pieske B. Na+-dependent SR Ca2+ overload induces arrhythmogenic events in mouse cardiomyocytes with a human CPVT mutation. Cardiovasc Res. 2010;87:50–59. doi: 10.1093/cvr/cvq007. [DOI] [PubMed] [Google Scholar]
- Shan J, Betzenhauser MJ, Kushnir A, Reiken S, Meli AC, Wronska A, Dura M, Chen BX, Marks AR. Role of chronic ryanodine receptor phosphorylation in heart failure andβ-adrenergic receptor blockade in mice. J Clin Invest. 2010;120:4375–4387. doi: 10.1172/JCI37649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon TR, Guo T, Bers DM. Ca2+ scraps: local depletions of free [Ca2+] in cardiac sarcoplasmic reticulum during contractions leave substantial Ca2+ reserve. Circ Res. 2003;93:40–45. doi: 10.1161/01.RES.0000079967.11815.19. [DOI] [PubMed] [Google Scholar]
- Stern MD, Capogrossi MC, Lakatta EG. Spontaneous calcium release from the sarcoplasmic reticulum in myocardial cells: mechanisms and consequences. Cell Calcium. 1988;9:247–256. doi: 10.1016/0143-4160(88)90005-x. [DOI] [PubMed] [Google Scholar]
- Stokke MK, Briston SJ, Jolle GF, Manzoor I, Louch WE, Oyehaug L, Christensen G, Eisner DA, Trafford AW, Sejersted OM, Sjaastad I. Ca2+ wave probability is determined by the balance between SERCA2-dependent Ca2+ reuptake and threshold SR Ca2+ content. Cardiovasc Res. 2010a;90:503–512. doi: 10.1093/cvr/cvr013. [DOI] [PubMed] [Google Scholar]
- Stokke MK, Hougen K, Sjaastad I, Louch WE, Briston SJ, Enger UH, Andersson KB, Christensen G, Eisner DA, Sejersted OM, Trafford AW. Reduced SERCA2 abundance decreases the propensity for Ca2+ wave development in ventricular myocytes. Cardiovasc Res. 2010b;86:63–71. doi: 10.1093/cvr/cvp401. [DOI] [PubMed] [Google Scholar]
- Swietach P, Spitzer KW, Vaughan-Jones RD. Ca2+-mobility in the sarcoplasmic reticulum of ventricular myocytes is low. Biophys J. 2008;95:1412–1427. doi: 10.1529/biophysj.108.130385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swietach P, Spitzer KW, Vaughan-Jones RD. Modeling calcium waves in cardiac myocytes: importance of calcium diffusion. Front Biosci. 2010;15:661–680. doi: 10.2741/3639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terentyev D, Gyorke I, Belevych AE, Terentyeva R, Sridhar A, Nishijima Y, de Blanco EC, Khanna S, Sen CK, Cardounel AJ, Carnes CA, Gyorke S. Redox modification of ryanodine receptors contributes to sarcoplasmic reticulum Ca2+ leak in chronic heart failure. Circ Res. 2008;103:1466–1472. doi: 10.1161/CIRCRESAHA.108.184457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsien RW, Bean BP, Hess P, Lansman JB, Nilius B, Nowycky MC. Mechanisms of calcium channel modulation byβ-adrenergic agents and dihydropyridine calcium agonists. J Mol Cell Cardiol. 1986;18:691–710. doi: 10.1016/s0022-2828(86)80941-5. [DOI] [PubMed] [Google Scholar]
- Ullrich ND, Valdivia HH, Niggli E. PKA phosphorylation of cardiac ryanodine receptor modulates SR luminal Ca2+ sensitivity. J Mol Cell Cardiol. 2012;53:33–42. doi: 10.1016/j.yjmcc.2012.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Oort RJ, McCauley MD, Dixit SS, Pereira L, Yang Y, Respress JL, Wang Q, De Almeida AC, Skapura DG, Anderson ME, Bers DM, Wehrens XH. Ryanodine receptor phosphorylation by calcium/calmodulin-dependent protein kinase II promotes life-threatening ventricular arrhythmias in mice with heart failure. Circulation. 2010;122:2669–2679. doi: 10.1161/CIRCULATIONAHA.110.982298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venetucci LA, Trafford AW, Eisner DA. Increasing ryanodine receptor open probability alone does not produce arrhythmogenic calcium waves: threshold sarcoplasmic reticulum calcium content is required. Circ Res. 2007;100:105–111. doi: 10.1161/01.RES.0000252828.17939.00. [DOI] [PubMed] [Google Scholar]
- Wehrens XH, Lehnart SE, Reiken S, Vest JA, Wronska A, Marks AR. Ryanodine receptor/calcium release channel PKA phosphorylation: a critical mediator of heart failure progression. Proc Natl Acad Sci U S A. 2006;103:511–518. doi: 10.1073/pnas.0510113103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi N, Meissner G. Does Ca2+/calmodulin-dependent protein kinase δc activate or inhibit the cardiac ryanodine receptor ion channel. Circ Res. 2007;100:293–295. doi: 10.1161/01.RES.0000259327.56377.55. [DOI] [PubMed] [Google Scholar]
- Zhou P, Zhao YT, Guo YB, Xu SM, Bai SH, Lakatta EG, Cheng H, Hao XM, Wang SQ. β-Adrenergic signalling accelerates and synchronizes cardiac ryanodine receptor response to a single L-type Ca2+ channel. Proc Natl Acad Sci U S A. 2009;106:18028–18033. doi: 10.1073/pnas.0906560106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zima AV, Bovo E, Bers DM, Blatter LA. Ca2+ spark-dependent and -independent sarcoplasmic reticulum Ca2+ leak in normal and failing rabbit ventricular myocytes. J Physiol. 2011;588:4743–4757. doi: 10.1113/jphysiol.2010.197913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zima AV, Picht E, Bers DM, Blatter LA. Termination of cardiac Ca2+ sparks: role of intra-SR [Ca2+], release flux, and intra-SR Ca2+ diffusion. Circ Res. 2008;103:e105–115. doi: 10.1161/CIRCRESAHA.107.183236. [DOI] [PMC free article] [PubMed] [Google Scholar]