

Abstract
Alterations in BRAF have been discovered in the majority of pediatric low-grade gliomas. Because the field has moved quickly over the past few years, there is not yet widespread awareness about what B-Raf normally does, how the BRAF gene is modified in gliomas, why mutant proteins promote gliomagenesis, and what an abnormal BRAF result means for diagnosis, prognosis, and treatment. Depending on the data from ongoing clinical trials, however, BRAF mutation screening could quickly become mandatory for all pediatric gliomas, and perhaps even a subset of adult gliomas. Herein, these topics and different methods of testing for BRAF fusions and V600E point mutations are reviewed.
Keywords: BRAF, FISH, Fusion, Ganglioglioma, Pilocytic astrocytoma, Pleomorphic xanthoastrocytoma, V600E
BRAF in Neurooncology
Other than isocitrate dehydrogenases 1 and 2 (IDH1/2), no molecule has been of greater recent interest to the neurooncology world than BRAF. Because it is a key gene altered in most pediatric low-grade gliomas, BRAF reigns supreme in the pediatric neurooncology world. B-Raf has been identified as a highly “druggable” target protein (i.e. capable of being bound by drug-like compounds) in melanomas (1), analogous clinical trials in pediatric gliomas have already begun. Consequently, pediatric neurooncologists are requesting BRAF testing more frequently. This review, therefore, aims to update both neuropathologists and neurooncologists in this rapidly moving field. Herein, I will briefly review the signaling pathway in which B-Raf resides and study the 2 types of BRAF alterations in gliomas, specifically, in what sort of tumors they occur, how to test for them, and what each test result means for the patient.
BRAF and the MAPK Signaling Pathway
B-Raf is an intracellular serine/threonine kinase component of the mitogen-activated protein kinase (MAPK) pathway (Fig. 1A) (2). This pathway normally begins with activation of a transmembrane receptor tyrosine kinase, which binds, phosphorylates, and activates Ras, which in turn activates a Raf kinase, in turn activating MEK1/2, leading to activation of the ERK1/2 transcription complex. Like most signaling pathways, the MAPK cascade has a wide range of effects, some of which appear contradictory. For example, MAPK activation can result in proliferation, survival, and tumorigenesis, but can also trigger cell differentiation and senescence (3, 4). This duality of the MAPK pathway might help explain why most gliomas with B-Raf activation are low grade and tend to stay that way, unless other genetic alterations also occur.
Figure 1.

MAPK signaling pathway and B-Raf derangements in gliomas. (A) Under normal circumstances Raf proteins require activation by Ras before activating MEK, which ultimately leads to promotion of cell division, survival, and in certain circumstances, differentiation. B-Raf is a more potent activator of MEK than C-Raf, which in turn is more potent than A-Raf. GF = growth factor; RTK = receptor tyrosine kinase. (B) When the N-terminal Ras-binding portion of B-Raf is lost in a BRAF:KIAA1549 (B–K) fusion, the mutant protein can activate MEK without first being activated by Ras, leading to tumor growth. A similar situation exists with SRGAP3:RAF1 fusion, wherein the kinase portion of C-Raf is joined to SRGAP3 (C–S). A V600E point mutation on B-Raf also has constitutive activity towards MEK. When the p16 checkpoint protein is intact, persistent over-activation of the MAPK pathway can lead to tumor senescence.
Within the cluster of Raf serine-threonine kinases there are 3 isoforms: A-Raf, B-Raf, and C-Raf. Each of these isoforms has the same general principle of an N-terminal regulatory domain that normally inhibits the C-terminal kinase domain. When Ras binds to the N-terminal, this inhibition is released (4). Since each of these kinases is activated by Ras and can in turn phosphorylate MEK, it begs the question as to why mutations in B-Raf are apparently more common compared to A-Raf and C-Raf. B-Raf has only 2 kinase activation sites, whereas A-Raf and C-Raf have 4; this might make it easier to turn B-Raf on with a single point mutation. Furthermore, activated B-Raf is itself a more potent activator of downstream MEK than either A-Raf or C-Raf, and thus has greater oncogenic potential (4). Moreover, as will be discussed below, the BRAF gene on 7q34 has key breakpoints that can produce a constitutively active fusion protein. Whereas comparable tumor-related fusions can occur in RAF1 (encoding C-Raf), they seem less common than in BRAF and, to my knowledge, do not occur in ARAF. The most frequent BRAF alterations in gliomas are gene rearrangement and fusion.
BRAF Fusions
Pilocytic astrocytomas (PAs) are World Health Organization grade I tumors that occur mostly in the posterior fossa in children. When these tumors are in surgically accessible sites, i.e. the cerebellum and more superficial parts of the cerebrum, outcomes tend to be very good, with low rates of recurrence. When tumors are located in deeper midline structures like the diencephalon and brainstem, full resection is rarely achievable and there are higher risks of recurrence and the need for treatment with adjuvant therapy. For decades little was known about the genetic characteristics of these tumors because lower-resolution screening assays showed few consistent abnormalities (5). More recent work on PAs, however, discovered a tandem duplication and rearrangement on 7q34 between BRAF and a gene centromeric to BRAF, KIAA1549, producing a fusion gene (6–9). Although the normal function of KIAA1549 is not known, its participation in the fusion is apparently not uniquely critical because BRAF sometimes fuses with FAM131B on 7q34 (10). Although rather uncommon, RAF1 can also fuse with SRGAP3 on 3p25 (11, 12). The end result in all these variants is to delete the N-terminal Ras-binding regulatory domain, producing constitutive B-Raf (or C-Raf) activity (Fig. 1B).
At first it might seem odd that mutating such a powerful oncogene would produce relatively indolent tumors like PAs, but recall that unmitigated activation of the MAPK pathway is a double-edged sword, capable of triggering differentiation and/or senescence as well as oncogenesis. This phenomenon of oncogene-induced senescence wherein the same pathway that caused the tumor also causes it to “burn out” and stop growing is gradually gaining wider recognition (13). In the case of PAs, constitutive B-Raf activity induces PA-like tumors in grafted mice, but eventually causes senescence unless other genetic lesions are also present (14–17). Specifically, loss of the p16 checkpoint protein produces more aggressive tumors in vivo; this correlates well with the finding that PAs with BRAF fusions have worse outcomes if the p16 gene (CDKN2A) is also deleted and/or its protein expression is absent (Fig. 1B) (14, 18).
So far, I have discussed BRAF fusions exclusively in the context of PAs. That is because these fusions are tightly correlated with PA morphology and PA-like behavior. They are only rarely seen in high-grade pediatric gliomas (19). Overall, roughly 70% of PAs contain BRAF fusions (Table); some now equate BRAF fusion with the diagnosis of PA. There is merit to this because PA morphology can vary markedly and mimic other low-grade gliomas. Perhaps the 15% or so of other low grade gliomas that have been shown to contain fusions, e.g. gangliogliomas and grade II diffuse astrocytomas, are actually PAs in disguise (6–8, 11, 16, 19–22). Consistent with this notion is the finding that the presence of a BRAF fusion has been shown to at least trend towards a relatively favorable prognosis regardless of histologic appearance (16, 18, 22, 23). However, not all studies have shown an association with progression-free survival independent of location (9, 24), and the ability of the neurosurgeon to achieve gross total resection still appears to be the most powerful prognostic variable for determining oval risk of recurrence of pediatric low-grade gliomas (18, 24).
Table 1.
Table Frequencies of KIAA1549:BRAF (B–K) Fusion and BRAF V600E Mutations in Pediatric Gliomas
| BRAF alteration | BRAF location | |||
|---|---|---|---|---|
| Cerebellum | Supratentorial | Non-cerebellar infratentorial | Optic nerve | |
| B–K fusion | 74.2 % | 33.2 % | 50.6 % | 54.2 % |
| V600E | 2.9 % | 17.9 % | 1.6 % | 15.0 % |
| BRAF alteration | BRAF histology | |||||
|---|---|---|---|---|---|---|
| PA | PMA | PXA | GG | DA | HGG | |
| B–K fusion | 66.6 % | 50.0 % | 0.0 % | 21.2 % | 11.9 % | 0.0 % |
| V600E | 5.7 % | 7.7 % | 73.8 % | 19.2 % | 9.7 % | 13.1 % |
At least half of all posterior fossa/infratentorial and optic nerve low-grade gliomas have a B–K fusion, whereas only one third of all supratentorial non-optic nerve tumors have this alteration. In contrast, BRAFV600E mutation is most common in the supratentorium, although still half as frequent as the B–K fusion in this region. BRAF fusions are more associated with PAs and PMAs than any other histotype. Although some studies have identified B–K fusions in non-PA tumors, current thinking is that such tumors may be better classified as a PA or PMA, even if they resemble GGs or DAs. V600E mutations show less restriction in histotype, but are most common in PXAs and GGs. Although not listed in the Table, FAM131B:BRAF, SRGAP3:RAF1, and BRAFins598T mutations occur in 1%–2% of all PAs. These data were extracted and compiled from an aggregate of over 700 pediatric gliomas published by multiple groups (5–11, 15, 17, 20, 22, 33, 34, 45). PA = pilocytic astrocytoma; PMA = pilomyxoid astrocytoma; PXA = pleomorphic xanthoastrocytoma; GG = ganglioglioma; DA = grade II diffuse astrocytoma; HGG = high-grade glioma.
In addition to histologic features, other variables affect the likelihood of a BRAF fusion being present. Nearly 80% of cerebellar PAs have fusions, compared to only 50%–55% of noncerebellar PAs (6–8, 10, 11, 16, 19, 20, 25). The frequency of these fusions also lessens with age, from approximately 80% in the first decade of life to 50% in the second decade, and less than 10% of PAs in patients over 40 years (26). Half of all pilomyxoid astrocytomas (PMAs) contain BRAF fusions (Table) (11, 16, 18). This is consistent with the report of unequivocal PMAs that eventually recurred as equally unequivocal PAs – in other words, PMAs may simply be less mature PAs (27).
Thus, although the diagnostic and prognostic implications of BRAF fusion are not yet completely clear in all situations, in most cases (maybe all) a fusion equates to a PA or PMA, and at the very least it is not an unfavorable prognostic marker. Considering that BRAF fusions in gliomas were first reported only 4 years ago, progress has been remarkable. On the other hand, there is not yet a consensus on the best way(s) of detecting these fusions. This is complicated by the fact that KIAA1549:BRAF fusions occur at multiple sites on both genes. The most common is exon 16 of KIAA1549 joined to exon 9 of BRAF (16-9) in over 60% of fusions, followed by 15-9 in nearly 25% of cases, and 16-11 in 10%–15% of cases (Fig. 2A) (2, 28, 29). Rarer fusions occur between 18-10, 19-9, 16-10, 15-11, and 17-10, as well as the aforementioned FAM131B:BRAF and SRGAP3:RAF1 fusions (23).
Figure 2.
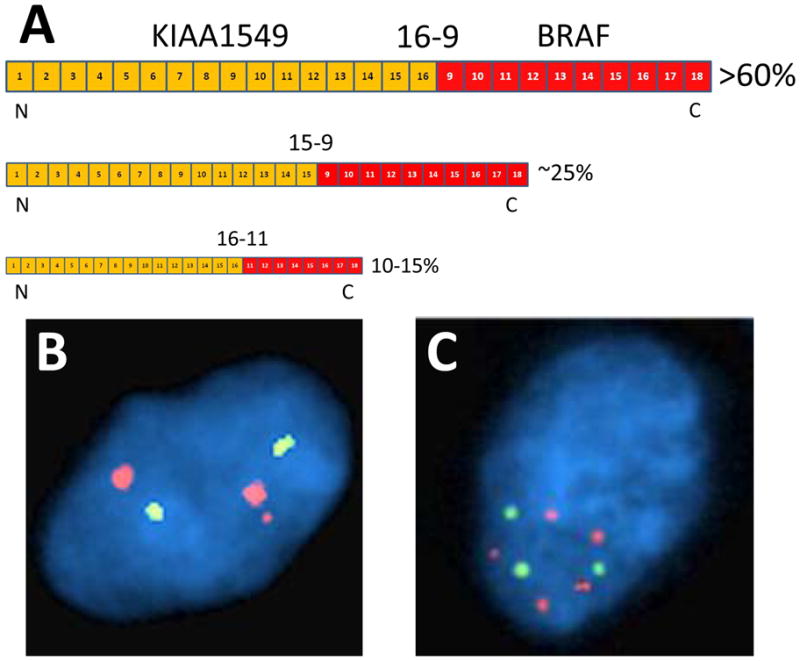
BRAF:KIAA1549 fusions and detection by fluorescence in situ hybridization (FISH). (A) The most common BRAF:KIAA1549 fusion in pediatric low-grade gliomas is between exons 1–16 of KIAA1549 and exons 9–18 of BRAF (16-9), seen in over 60% of tumors. 15-9 and 16-11 fusions are seen in approximately 25% and 15% of tumors, respectively. The common result in all of them is retention of the kinase portion of B-Raf. (B) Using a 3-probe FISH cocktail that encompasses the entire BRAF gene on 7q34, the classic BRAF fusion pattern is 2 large red signals plus a third smaller red signal near one of the larger signals, equating to 2 full copies of BRAF plus the kinase portion of a fusion gene. Green signals = centromeric enumeration probe for chromosome 7. (C) In tumors with high polysomy (frequently seen in pleomorphic xanthoastrocytomas and highly senescent pilocytic astrocytomas), reliable detection of a true BRAF fusion pattern is often difficult. (Probe signal size differences in panels B and C are due to post-image processing variables.)
Therefore, if detection of BRAF fusions is eventually shown to be a predictive marker for response to MAPK-blocking drugs, optimal assays will need to detect as many variations as possible. For widespread utility such methods must also be doable in formalin-fixed, paraffin-embedded (FFPE) tissues. High-resolution copy number assays like oligo array comparative genomic hybridization can detect the extra fusion gene on 7q34 (6, 7), but require high-quality snap-frozen tissues and run the risk of “dilution” by excessive admixed nonneoplastic DNA. A novel, recently published approach is to measure the copy number ratio of BRAF with a BRAF-2 pseudogene on chromosome X via pyrosequencing. This method generated similar results in frozen and FFPE tissues (30).
Fluorescence in situ hybridization (FISH) works on FFPE tissues and can detect abnormal BRAF using a 3-probe cocktail that spans the entire gene (31, 32). A normal cell should show 2 large signals, representing a blending of the signals from the 3 tandem probes. Cells with BRAF fusion will show a third smaller signal near one of the 2 larger signals, corresponding to the additional activation segment and kinase domain (Fig. 2B) (20). This third smaller signal can be difficult to detect, and a consensus has not yet been reached on cutoffs, but this pattern should be clearly seen in at least 10%–15% of tumor cells (20, 33). The reason the cutoff is not higher is because many cells will have an obscured third signal due to close apposition with the normal BRAF signal (thereby impairing resolution), artifactual sectioning loss of the extra signal, or other FISH-specific variables. Also, the reliable detection of this extra small signal is nontrivial if the tumor has high polysomy, as is sometimes the case with pleomorphic xanthoastrocytoma (PXAs) or senescent PAs (Fig. 2C). Still, BRAF FISH can detect all BRAF fusion variants, including the FAM131B form, though, of course, it cannot distinguish between each variant. And FISH retains the critical advantages of needing only 1 unstained cut of tissue, visually targeting specific spots on a slide, and correlating subregions with a matched hematoxylin and eosin section, all of which are most helpful when dealing with sparse or heterogeneous biopsies.
Another popular method is real-time polymerase chain reaction, with probes designed to flank the 16-9 variant that also catch the shorter 16-11 and 15-9 variants, each of which can be readily identified based on the differing lengths of their respective PCR products (22). This approach works in FFPE tissues and has sensitivity and specificity exceeding 90% (32). Of course, this would not identify a FAM131B: BRAF fusion, and none of the assays except array-comparative genomic hybridization would detect SRGAP3:RAF1 fusions unless additional probes were specifically designed to do so.
In the future, the mandated stringency of assay validation and interpretation criteria for BRAF fusion testing will likely be directly proportional to the ability of BRAF to predict targeted therapeutic responsiveness. In other words, a method that seems to work reasonably well for research purposes at this time may not meet future demands for accuracy if BRAF fusions become critical for dictating adjuvant therapy.
BRAF V600E
In gliomas there is another way to turn on B-Raf, i.e. the constitutively active valine-to-glutamate (V600E) point mutation, which activates MEK without first needing upstream Ras phosphorylation. B-Raf V600E exists in diverse tumors, including melanocytic nevi, melanoma, colon cancer, and papillary thyroid cancer. It is present in 10%–15% of grade II–IV diffusely infiltrative pediatric astrocytomas (34–36), but in less than 2% of comparable adult gliomas (35, 37–39). In both children and adults, less than 10% of all PAs have the mutation, and it is present in only 2% of cerebellar PAs (7, 8, 10, 11, 16, 19, 35). Rarely, the mutation can occur in conjunction with a BRAF fusion in the same tumor (16, 18). In adults, either V600E or BRAF fusions even occasionally coexist with IDH1/2 mutations (40). Although sometimes present in tumors with PA-like morphology, V600E is more associated with other tumors in the differential, including 20%–25% of pediatric and adult gangliogliomas and 60%–80% of pleomorphic xanthoastrocytomas in both age groups (8, 16, 35, 39, 41). Thus, in the pediatric population, a V600E result is not quite as helpful as BRAF fusion in differentiating noninfiltrative from infiltrative gliomas, particularly if the differential is between a ganglioglioma and a diffuse glioma overtaking grey matter.
Compared to BRAF fusion, fewer outcome-based studies have been done on V600E-mutant gliomas. When both types of BRAF alteration were compared in a multivariate analysis of pediatric low-grade gliomas, BRAF fusions trended toward longer progression-free survival, whereas B-Raf V600E trended toward shorter progression-free survival (18). Interestingly, in that same cohort neither alteration was more powerful than the presence of p16 deletion, which was a significant independent adverse prognostic marker. This makes sense because loss of p16 inhibits BRAF-induced tumor senescence (3, 14).
Thus, if both fusion and V600E have the same effect, i.e. turning B-Raf on, why do they produce tumors that generally look and act differently? Perhaps this is a clue that the alterations do not have the exact same effect. The KIAA1549, FAM131B, and SRGAP3 components of the BRAF and RAF1 fusions might not be mere passive partners, and they might dictate where the fusion proteins localize, what controls their expression, and/or how easily they are degraded (2). Buttressing this hypothesis is the finding that a truncated BRAF gene mimicking just the kinase component, without any fusion partner, cannot induce tumors in mice (17). Ongoing research will undoubtedly shed more light on these fascinating phenomena.
Unlike the heterogeneity surrounding BRAF fusion testing, V600E detection is quite straightforward because PCR and sequencing work well in FFPE tissues. Of particular excitement is a V600E-specific antibody recently developed by the von Deimling group — the same laboratory that perfected the R132H IDH1 antibody, which is already a mainstay of surgical neuropathology (42, 43). The V600E-specific antibody generates false-negative results in tissues damaged by surgical coagulation or freezing; and staining intensity varies greatly between mutated cases. Nevertheless, it does work in FFPE tissues and is, therefore, likely to become a first-line test not just for certain gliomas, but also for melanomas and other V600E-mutant tumors elsewhere in the body.
Of note, there are also trinucleotide insertions in codon 598 of BRAF that cause constitutive activity similar to the V600E mutation. These are seen in approximately 1%–2% of PAs (10, 12), and would naturally be missed by the V600E antibody unless directly targeted by sequencing.
Therapeutic Relevance of BRAF Alterations
As is often the case with molecular biomarkers, our ability to identify the diagnostic and prognostic value of BRAF alterations in gliomas has outpaced our ability to target it in adjuvant therapies. However, the potential for rapid implementation of targeted therapeutics is higher than typical because there are already many pharmacologic inhibitors that theoretically should work on these tumors (“should” always caution when referring to clinical trials). As of the time of this writing, the MEK inhibitor AZD6244 (selumetinib) is undergoing various phase I and II trials for B-Raf-mutant solid tumors, including pediatric gliomas. Certainly the experience with melanomas and PLX4032 (vemurafenib) is reason for some optimism (44).
But harkening back to the evidence that not all BRAF mutations are equal, a recent study showed that targeted B-Raf inhibition with vemurafenib only worked on V600E glioma cells in vivo, and actually made BRAF-wt xenografts a little worse (36). Furthermore, it is clear that most pediatric low-grade gliomas are driven by the MAPK pathway, if not by B-Raf then by other MAPK components such as C-Raf, K-Ras, or NF1 (10). Perhaps one downstream inhibitor of all these molecules will work equally well on all of them and specific BRAF status will not matter too much. Even if one or more of these drugs proves effective, there is always the possibility of acquired resistance, as has been seen in melanomas (45).
Summary and Recommendations
Our knowledge of the role that B-Raf plays in gliomas has exploded over the last few years. At this point, BRAF testing can be employed in a simple decision tree for pediatric low-grade gliomas (Fig, 3). We may be heading toward a time in which histologic descriptive diagnoses such as “pilocytic astrocytoma” share the spotlight with BRAF, or are even relegated to secondary importance. Depending on the success of current clinical trials, “low-grade glioma with BRAF fusion” or “low-grade glioma with BRAF V600E” could become de rigueur for the pediatric neurooncology world.
Figure 3.

Suggested method of incorporating BRAF testing in pediatric low-grade gliomas. When light microscopy suggests the possibility of a low-grade glioma, the presence of a BRAF fusion suggests either a pilocytic astrocytoma (PA) or its related tumor, pilomyxoid astrocytoma (PMA). Detection of B-Raf V600E, on the other hand, most represents a pleomorphic xanthoastrocytoma (PXA), ganglioglioma (GG), or a diffusely infiltrative astrocytoma (DA). Even a PA might still be in the differential depending on its histologic appearance.
Acknowledgments
Sources of support: Craig Horbinski was supported in part by 1-K08 CA155764-01A1 (NCI), 2P20 RR020171 (NIGMS), and the University of Kentucky College of Medicine Physician Scientist Program.
References
- 1.Keller TH, Pichota A, Yin Z. A practical view of ‘druggability’. Curr Opin Chem Biol. 2006;10:357–61. doi: 10.1016/j.cbpa.2006.06.014. [DOI] [PubMed] [Google Scholar]
- 2.Tatevossian RG, Lawson AR, Forshew T, et al. MAPK pathway activation and the origins of pediatric low-grade astrocytomas. J Cell Physiol. 2010;222:509–14. doi: 10.1002/jcp.21978. [DOI] [PubMed] [Google Scholar]
- 3.Jacob K, Quang-Khuong DA, Jones DT, et al. Genetic aberrations leading to MAPK pathway activation mediate oncogene-induced senescence in sporadic pilocytic astrocytomas. Clin Cancer Res. 2011;17:4650–60. doi: 10.1158/1078-0432.CCR-11-0127. [DOI] [PubMed] [Google Scholar]
- 4.Maurer G, Tarkowski B, Baccarini M. Raf kinases in cancer-roles and therapeutic opportunities. Oncogene. 2011;30:3477–88. doi: 10.1038/onc.2011.160. [DOI] [PubMed] [Google Scholar]
- 5.Sanoudou D, Tingby O, Ferguson-Smith MA, et al. Analysis of pilocytic astrocytoma by comparative genomic hybridization. Brit J Cancer. 2000;82:1218–22. doi: 10.1054/bjoc.1999.1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bar EE, Lin A, Tihan T, et al. Frequent gains at chromosome 7q34 involving BRAF in pilocytic astrocytoma. J Neuropathol Exp Neurol. 2008;67:878–87. doi: 10.1097/NEN.0b013e3181845622. [DOI] [PubMed] [Google Scholar]
- 7.Pfister S. BRAF gene duplication constitutes a mechanism of MAPK pathway activation in low-grade astrocytomas. J Clin Invest. 2008;118:1739–49. doi: 10.1172/JCI33656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sievert AJ, Jackson EM, Gai X, et al. Duplication of 7q34 in pediatric low-grade astrocytomas detected by high-density single-nucleotide polymorphism-based genotype arrays results in a novel BRAF fusion gene. Brain Pathol. 2009;19:449–58. doi: 10.1111/j.1750-3639.2008.00225.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jones DT, Kocialkowski S, Liu L, et al. Tandem duplication producing a novel oncogenic BRAF fusion gene defines the majority of pilocytic astrocytomas. Cancer Res. 2008;68:8673–7. doi: 10.1158/0008-5472.CAN-08-2097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cin H, Meyer C, Herr R, et al. Oncogenic FAM131B-BRAF fusion resulting from 7q34 deletion comprises an alternative mechanism of MAPK pathway activation in pilocytic astrocytoma. Acta Neuropathol. 2011;121:763–74. doi: 10.1007/s00401-011-0817-z. [DOI] [PubMed] [Google Scholar]
- 11.Forshew T, Tatevossian RG, Lawson AR, et al. Activation of the ERK/MAPK pathway: a signature genetic defect in posterior fossa pilocytic astrocytomas. J Pathol. 2009;218:172–81. doi: 10.1002/path.2558. [DOI] [PubMed] [Google Scholar]
- 12.Jones DT. Oncogenic RAF1 rearrangement and a novel BRAF mutation as alternatives to KIAA1549:BRAF fusion in activating the MAPK pathway in pilocytic astrocytoma. Oncogene. 2009;28:2119–23. doi: 10.1038/onc.2009.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dimauro T, David G. Ras-induced senescence and its physiological relevance in cancer. Curr Cancer Drug Targets. 2010;10:869–76. doi: 10.2174/156800910793357998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Raabe EH, Lim KS, Kim JM, et al. BRAF activation induces transformation and then senescence in human neural stem cells: a pilocytic astrocytoma model. Clin Cancer Res. 2011;17:3590–9. doi: 10.1158/1078-0432.CCR-10-3349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jacob K, Quang-Khuong DA, Jones DT, et al. Genetic aberrations leading to MAPK pathway activation mediate oncogene-induced senescence in sporadic pilocytic astrocytomas. Clin Cancer Res. 2011:12. doi: 10.1158/1078-0432.CCR-11-0127. [DOI] [PubMed] [Google Scholar]
- 16.Hawkins C, Walker E, Mohamed N, et al. BRAF-KIAA1549 fusion predicts better clinical outcome in pediatric low-grade astrocytoma. Clin Cancer Res. 2011:5. doi: 10.1158/1078-0432.CCR-11-0034. [DOI] [PubMed] [Google Scholar]
- 17.Gronych J, Korshunov A, Bageritz J, et al. An activated mutant BRAF kinase domain is sufficient to induce pilocytic astrocytoma in mice. J Clin Invest. 2011 Apr;121:1344–8. doi: 10.1172/JCI44656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Horbinski C, Nikiforova MN, Hagenkord JM, et al. Interplay among BRAF, p16, p53, and MIB1 in pediatric low-grade gliomas. Neuro Oncol. 2012:5. doi: 10.1093/neuonc/nos077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jones DT. Tandem duplication producing a novel oncogenic BRAF fusion gene defines the majority of pilocytic astrocytomas. Cancer Res. 2008;68:8673–7. doi: 10.1158/0008-5472.CAN-08-2097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Horbinski C, Hamilton RL, Nikiforov Y, et al. Association of molecular alterations, including BRAF, with biology and outcome in pilocytic astrocytomas. Acta Neuropathol. 2010;119:641–9. doi: 10.1007/s00401-009-0634-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lawson AR, Tatevossian RG, Phipps KP, et al. RAF gene fusions are specific to pilocytic astrocytoma in a broad paediatric brain tumour cohort. Acta Neuropathol. 2010;120:271–3. doi: 10.1007/s00401-010-0693-y. [DOI] [PubMed] [Google Scholar]
- 22.Ida CM, Lambert SR, Rodriguez FJ, et al. BRAF alterations are frequent in cerebellar low-grade astrocytomas with diffuse growth pattern. J Neuropathol Exp Neurol. 2012;71:631–9. doi: 10.1097/NEN.0b013e31825c448a. [DOI] [PubMed] [Google Scholar]
- 23.Lin A, Rodriguez FJ, Karajannis MA, et al. BRAF alterations in primary glial and glioneuronal neoplasms of the central nervous system with identification of 2 novel KIAA1549:BRAF fusion variants. J Neuropathol Exp Neurol. 2012;71:66–72. doi: 10.1097/NEN.0b013e31823f2cb0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tihan T, Ersen A, Qaddoumi I, et al. Pathologic characteristics of pediatric intracranial pilocytic astrocytomas and their impact on outcome in 3 countries: a multi-institutional study. Am J Surg Pathol. 2011:10. doi: 10.1097/PAS.0b013e3182329480. [DOI] [PubMed] [Google Scholar]
- 25.Huang H, Okamoto Y, Yokoo H, et al. Gene expression profiling and subgroup identification of oligodendrogliomas. Oncogene. 2004;23:6012–22. doi: 10.1038/sj.onc.1207781. [DOI] [PubMed] [Google Scholar]
- 26.Hasselblatt M, Riesmeier B, Lechtape B, et al. BRAF-KIAA1549 fusion transcripts are less frequent in pilocytic astrocytomas diagnosed in adults. Neuropathol Appl Neurobiol. 2011:1365–2990. doi: 10.1111/j.1365-2990.2011.01193.x. [DOI] [PubMed] [Google Scholar]
- 27.Johnson MW, Eberhart CG, Perry A, et al. Spectrum of pilomyxoid astrocytomas: intermediate pilomyxoid tumors. Am J Surg Pathol. 2011;34:1783–91. doi: 10.1097/PAS.0b013e3181fd66c3. [DOI] [PubMed] [Google Scholar]
- 28.Hasselblatt M, Riesmeier B, Lechtape B, et al. BRAF-KIAA1549 fusion transcripts are less frequent in pilocytic astrocytomas diagnosed in adults. Neuropathol Appl Neurobiol. 2011;37:803–6. doi: 10.1111/j.1365-2990.2011.01193.x. [DOI] [PubMed] [Google Scholar]
- 29.Dahiya S, Yu J, Kaul A, et al. Novel BRAF Alteration in a sporadic pilocytic astrocytoma. Case Report Med. 2012:418672. doi: 10.1155/2012/418672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Setty P, Gessi M, Waha A, et al. Sensitive determination of BRAF copy number in clinical samples by pyrosequencing. Diagn Mol Pathol. 2011;20:148–57. doi: 10.1097/PDM.0b013e3182143817. [DOI] [PubMed] [Google Scholar]
- 31.Ciampi R, Knauf JA, Kerler R, et al. Oncogenic AKAP9-BRAF fusion is a novel mechanism of MAPK pathway activation in thyroid cancer. J Clin Invest. 2005;115:94–101. doi: 10.1172/JCI23237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tian Y, Rich BE, Vena N, et al. Detection of KIAA1549-BRAF fusion transcripts in formalin-fixed paraffin-embedded pediatric low-grade gliomas. J Mol Diagn. 2011;13:669–77. doi: 10.1016/j.jmoldx.2011.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Korshunov A, Meyer J, Capper D, et al. Combined molecular analysis of BRAF and IDH1 distinguishes pilocytic astrocytoma from diffuse astrocytoma. Acta Neuropathol. 2009;118:401–5. doi: 10.1007/s00401-009-0550-z. [DOI] [PubMed] [Google Scholar]
- 34.Schiffman JD, Hodgson JG, VandenBerg SR, et al. Oncogenic BRAF mutation with CDKN2A inactivation is characteristic of a subset of pediatric malignant astrocytomas. Cancer Res. 2010;70:512–9. doi: 10.1158/0008-5472.CAN-09-1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schindler G, Capper D, Meyer J, et al. Analysis of BRAF V600E mutation in 1,320 nervous system tumors reveals high mutation frequencies in pleomorphic xanthoastrocytoma, ganglioglioma and extra-cerebellar pilocytic astrocytoma. Acta Neuropathol. 2011;121:397–405. doi: 10.1007/s00401-011-0802-6. [DOI] [PubMed] [Google Scholar]
- 36.Nicolaides TP, Li H, Solomon DA, et al. Targeted therapy for BRAFV600E malignant astrocytoma. Clin Cancer Res. 2011;17:7595–604. doi: 10.1158/1078-0432.CCR-11-1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Basto D, Trovisco V, Lopes JM, et al. Mutation analysis of B-RAF gene in human gliomas. Acta Neuropathol. 2005;109:207–10. doi: 10.1007/s00401-004-0936-x. [DOI] [PubMed] [Google Scholar]
- 38.Knobbe CB, Reifenberger J, Reifenberger G. Mutation analysis of the Ras pathway genes NRAS, HRAS, KRAS and BRAF in glioblastomas. Acta Neuropathol. 2004;108:467–70. doi: 10.1007/s00401-004-0929-9. [DOI] [PubMed] [Google Scholar]
- 39.Dias-Santagata D, Lam Q, Vernovsky K, et al. BRAF V600E mutations are common in pleomorphic xanthoastrocytoma: diagnostic and therapeutic implications. PLoS One. 2011;6:17948. doi: 10.1371/journal.pone.0017948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Badiali M, Gleize V, Paris S, et al. KIAA1549-BRAF fusions and IDH mutations can coexist in diffuse gliomas of adults. Brain Pathol. 2012:1750–3639. doi: 10.1111/j.1750-3639.2012.00603.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dougherty MJ, Santi M, Brose MS, et al. Activating mutations in BRAF characterize a spectrum of pediatric low-grade gliomas. Neuro Oncol. 2010:14. doi: 10.1093/neuonc/noq007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Capper D, Preusser M, Habel A, et al. Assessment of BRAF V600E mutation status by immunohistochemistry with a mutation-specific monoclonal antibody. Acta Neuropathol. 2011;122:11–9. doi: 10.1007/s00401-011-0841-z. [DOI] [PubMed] [Google Scholar]
- 43.Capper D, Zentgraf H, Balss J, et al. Monoclonal antibody specific for IDH1 R132H mutation. Acta Neuropathol. 2009;118:599–601. doi: 10.1007/s00401-009-0595-z. [DOI] [PubMed] [Google Scholar]
- 44.Chapman PB, Hauschild A, Robert C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507–16. doi: 10.1056/NEJMoa1103782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Poulikakos PI, Rosen N. Mutant BRAF melanomas--dependence and resistance. Cancer Cell. 2011;19:11–5. doi: 10.1016/j.ccr.2011.01.008. [DOI] [PubMed] [Google Scholar]