

Summary
The glyoxylate shunt plays an important role in fatty-acid metabolism, and has been shown to be critical to survival of several pathogens involved in chronic infections. For Mycobacterium tuberculosis (Mtb), a strain with a defective glyoxylate shunt was previously shown to be unable to establish infection in a mouse model. We report the development of novel phenyl-diketo acid (PDKA) inhibitors of malate synthase (GlcB), one of two glyoxylate shunt enzymes, using structure-based methods. PDKA inhibitors were active against Mtb grown on acetate, and over-expression of GlcB ameliorated this inhibition. Crystal structures of complexes of GlcB with PDKA inhibitors were used to guide optimization of potency. A selected PDKA compound demonstrated efficacy in a mouse model of tuberculosis. The discovery of these PDKA derivatives provides chemical validation of GlcB as an attractive target for tuberculosis therapeutics.
Introduction
Despite the availability of good first and second line therapeutics to treat tuberculosis (TB), drug resistance, latency, and persistence render TB a continuing threat to public health and pose an urgent need for the development of novel drugs (World Health Organization, 2011).
The glyoxylate shunt, which plays a central role in fatty-acid metabolism, has long been considered a potential vulnerability of Mtb during infection that could be exploited for developing antitubercular therapeutics (McKinney et al., 2000). The glyoxylate shunt is an anaplerotic bypass of the traditional tricarboxylic acid cycle that allows for incorporation of carbon from acetyl-CoA produced by fatty-acid metabolism. This pathway is utilized in plants, fungi, and prokaryotes, but is absent in mammals. Mtb has been shown to undergo significant metabolic alterations during the course of infection, among them a shift from a reliance on carbohydrates to fatty acids as a principal source of carbon (Bloch and Segal, 1956). The increased reliance on fatty acid β-oxidation and gluconeogenesis in concert with a shift away from glycolysis during infection is supported by analysis of transcriptional profiles (Schnappinger et al., 2003), (Talaat et al., 2004).
The glyoxylate shunt as well as gluconeogenesis have been shown to play a crucial role in Mtb virulence, as both isocitrate lyase and phosphoenolpyruvate carboxykinase, the first committed steps of each pathway, are required for infection in activated macrophages and in animal models (McKinney et al., 2000; Marrero et al., 2010).
The glyoxylate shunt consists of two enzymes: isocitrate lyase (ICL) which hydrolyzes isocitrate into glyoxylate and succinate, and malate synthase (GlcB), which converts glyoxylate into malate using one molecule of acetyl-CoA. The shunt bypasses two CO2-generating steps of the TCA cycle, allowing incorporation of carbon (via acetyl-CoA) and serves to replenish oxaloacetate under carbon-limiting conditions (Kornberg and Krebs, 1957). Icl1 is one of the most highly up-regulated genes in Mtb under conditions that mimic infection (Timm et al., 2003). Further studies demonstrated the essentiality of the glyoxylate shunt for a persistent or chronic infection by showing that Mtb lacking icl1 was unable to persist, and a knockout of both isoforms of icl could not establish an infection in mice and was rapidly cleared (McKinney et al., 2000; Muñoz-Elías and McKinney, 2005). A critical role of the glyoxylate shunt for virulence has been reported for other intracellular and fungal pathogens (Lorenz and Fink, 2001) (Dunn et al., 2009).
Targeting ICL has been a challenge, largely due to its highly polar and small active site that becomes even more constricted during catalysis (Sharma et al., 2000). To date, the most-used in vitro inhibitor of ICL is the succinate analog, 3-nitropropionate which has an IC50 of 3 μM (Muñoz-Elías and McKinney, 2005). In contrast to ICL, GlcB has a much more “druggable” and large active site, consisting of a 20 Å by 7 Å cavity, which normally accommodates the pantothenate tail of the acetyl-CoA. The catalytic Mg2+ is located at the bottom of the cavity (Smith et al., 2003; Anstrom and Remington, 2006). X-ray crystal structures of GlcB bound with substrate glyoxylate or products CoA-SH and malate (Smith et al., 2003) show that the protein conformation is nearly identical regardless of the ligand (r.m.s.d. < 0.5 Å), suggesting that catalysis occurs without significant structural rearrangements. In this paper, we report our structure-based discovery of small molecule inhibitors of Mtb GlcB, and pharmacological validation of GlcB as a drug target. One of the identified GlcB inhibitors with a reasonable potency and favorable toxicity, pharmacokinetic (PK) and pharmacodynamic (PD) profiles, has demonstrated efficacy in a mouse model of TB, and could serve as the basis for a novel class of antituberculars.
Results
Discovery of PDKA, and Crystal Structure of GlcB-inhibitor Complex
A focused library of thirty-five small molecules with a glyoxylate-like substructure were assayed against GlcB and ICL at a single concentration point of 40 μg/ml; of these, nineteen showed activity against GlcB. All of the GlcB-actives were phenyl-diketo acids, exemplified by (Z)-2-hydroxy-4-oxo-4-phenylbut-2-enoic acid (PDKA) (Figure 1A). The parent PDKA exhibited an IC50 of 2.0 μM against GlcB, and was inactive against ICL. Based on these initial findings, approximately one hundred PDKA analogs were synthesized using readily available starting materials and straightforward chemical synthesis (Summa et al., 2004), (Zeng et al., 2008), (Pais et al., 2002), (Tumey et al., 2004), (Adams, 2008). A series of compounds was selected that demonstrated a good balance of enzyme inhibition and whole-cell activity. Aryl diketo acids have also been identified in drug-discovery projects for other Mg2+-dependent enzymes, including HIV-1 integrase and HCV-polymerase, where the keto acid moiety was found to coordinate the catalytic divalent metal cation (Egbertson, 2007).
Figure 1. PDKA inhibitor and its contacts to GlcB.
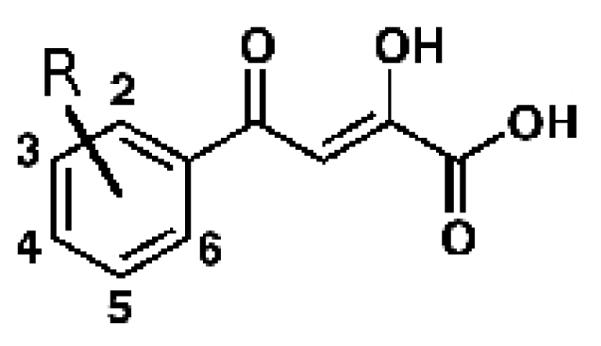

A - PDKA chemical structure drawn here in the enol form most consistent with solution-phase 1H and 13C NMR data; B – diagram of PDKA–GlcB interactions; hydrogen bonds are shown in green. Catalytic Asp633 contacts the face of the phenyl ring. Coordinating interactions between the Mg2+ ion and oxygens of the inhibitor are in purple. Atom colors: black=carbon, red=oxygen, blue=nitrogen, green=magnesium. This figure was made in LigPlot (Laskowski and Swindells, 2011).
The enzyme inhibition activities of these compounds had IC50s against GlcB ranging from 20 nM to >100 μM. However, the minimal inhibitory concentrations (MICs) against cultured mycobacteria were in certain cases in disagreement with the enzyme inhibition level, and had poor reproducibility. We also observed a time-dependent decrease of activity in solutions of inhibitors at room temperature, even during short periods of time. Using UV/Vis spectroscopy and HPLC analysis to assess the stability of our compounds, we found that the parent PDKA was stable in distilled, deionized water and organic solvents (DMSO, MeOH), but unstable (t1/2 ≈ 3 days) in cell growth media or any of several buffer solutions tested. The results from our HPLC-MS analysis were consistent with retro-Claisen decomposition, with acetophenone as a product. Based on previous findings (Egbertson, 2007), we reasoned that the very high degree of conjugation encompassing the PDKA molecule was the driver of the retro-Claisen decomposition. To avoid this decomposition, we hypothesized that the addition of a group to the ortho-position of the PDKA phenyl ring would twist the ring out-of-plane, reducing the degree of conjugation. The structure of the GlcB-PDKA complex showed a 37 degree twist of the phenyl ring of PDKA out-of-plane with the conjugated di-keto moiety. Therefore, we predicted that the twist due to substitution should not significantly impact binding of ortho-substituted PDKAs to the enzyme. Substitution at the ortho-position afforded an approximately ten-fold increase in compound stability to greater than 30 days half-life, and actually improved the IC50 (Table 1). Identical substitutions at the meta- and para-positions had no effect on stability. The extent of conjugation of the ortho-substituted phenyl diketo acids in solution was indeed reduced, as evidenced by the blue shift of their respective UV absorbance peaks (λmax values), when compared to PDKA and meta- and para- substituted compounds (Table 1). Moreover, the magnitude of this spectral shift correlates well with stability.
Table 1.
Stability and activity data for PDKA family inhibitors.
| Compound | R | Half- life (days) |
λmax (nM) |
IC50 (nM) | MIC99 mc2-7000 Mtb acetate (μM) |
MIC99 mc2-7000 Mtb dextrose (μM) |
|---|---|---|---|---|---|---|
| PDKA | Ph | 3 | 340 | 2.0 | 50 | 25 |
| 1 | 2-MePh | 30 | 326 | 1.1 | >200 | 200 |
| 2 | 2-FPh | 12 | 333 | 0.24 | >200 | 200 |
| 3 | 2-ClPh | 35 | 324 | 0.5 | >200 | 200 |
| 4 | 2-BrPh | 18 | 324 | 0.6 | 6.25 | 12.5 |
| 5 | 3-MePh | 11 | 339 | 0.18 | 25 | 50 |
| 6 | 3-ClPh | 7 | 341 | 0.17 | 25 | 25 |
| 7 | 3-BrPh | 7 | 341 | 0.8 | 25 | 25 |
| 8 | 4-MePh | 8 | 340 | 6.0 | 50 | 100 |
| 9 | 4-BrPh | 7 | 345 | 5.7 | 50 | 25 |
| 10 | 2-Cl-6-FPh | >30 | 317 | 2.7 | 50 | 100 |
| 11 | 2-Cl-6-F- 3MePh |
>30 | 321 | 5.5 | 50 | 100 |
All other attempts to stabilize this chemical framework (removing either of the two keto groups, replacing the carbon between them with a nitrogen, or introducing a methyl group to the beta carbon of the diketo acid) resulted in compounds that were inactive against GlcB (data not shown). We evaluated replacement of the PDKA carboxylic acid by proven Mg2+ chelators (catechol, diazole, sulfone, pyridazinone, hydroxylamine), and other bioisosteres such as tetrazole, but all of these failed to afford GlcB inhibitors with promising enzyme activity (IC50 > 100 μM).
We noticed that in the crystals of the glyoxylate-bound Mtb GlcB, Cys619 was often oxidized to cysteine-sulfenic acid, similar to E. coli malate synthase (Anstrom et al., 2003), resulting in a constriction at the entrance to the active site channel. The sulfenic acid is likely to be an artifact resulting from exposure to air during purification, and is not relevant to the metabolic function of GlcB (Quartararo and Blanchard et al., 2011), which should remain reduced in the reducing environment of the cell. We therefore constructed a Cys619Ala Mtb GlcB mutant, which exhibited ~80% of the reaction velocity of the wild-type (kinetic curve shown in Figure S1), and a ten-fold increase in acetyl-CoA KM (from 5 to 50 μM). Examination of the crystal structure of GlcB bound to CoA (1N8W; Smith et al., 2003) shows that the Sγ of Cys619 makes a hydrogen bond with a nitrogen in the pantothenate arm of CoA, which could explain why the C619A mutant enzyme binds the co-factor with less affinity, potentially causing the slight reduction in reaction velocity. A similar Cys619Ser Mtb GlcB mutant has also been described as a suitable model for kinetic studies (Quartararo and Blanchard, 2011). However, with the Cys619Ala mutant, we did not observe the lag in activity reported for the Cys619Ser mutant, and elevating Mg2+ concentrations did not influence the activity (Figure S1). Since the IC50 values of inhibitors measured for the mutated and wild-type enzymes were well-correlated (see Table S1, compare to Table 1), the C619A mutant protein was used in all subsequent crystallographic studies and enzyme assays. Cys619 is located ~5 Å away from the ligand (as shown in Figure S2), and thus does not participate significantly in binding of these inhibitors.
The crystal structure of GlcB (C619A) complexed with PDKA was determined at 1.9 Å resolution (data collection and refinement statistics are presented in Table 2). No significant conformational changes in the protein were observed upon PDKA binding compared to structures with glyoxylate or malate and CoA bound. The backbone RMSD of the superposition between the GlcB:malate complex and the GlcB:PDKA complex is 0.32 Å over 700 Cα atoms, and among 15 active-site residues, the all-atom RMSD is 0.21 Å, excluding Met631, which adopts a different conformer to accommodate the phenyl ring. The diketo acid group of the PDKA coordinates the Mg2+ ion in an edge-on fashion, very similar to glyoxylate (Smith et al., 2003), filling two of the six octahedral coordination sites with one of the carboxylate oxygens and the adjacent ketone oxygen (2.1 Å and 2.2 Å contact distances). Other active site interactions are illustrated in Figure 1B. Carboxylate oxygens of the inhibitor hydrogen-bonded with the backbone nitrogens of Asp462 and Leu461 (dO-N = 3.0 and 3.0 Å, respectively). Both ketone oxygens also form hydrogen bonds (dO-N = 2.9 and 2.9 Å) with the Arg339 side-chain, exhibiting similar contacts as the substrate/product. The catalytic Asp633 (Clark et al., 1988) side-chain oxygen was within hydrogen bonding distance (3.2 Å) to the phenyl ketone oxygen of PDKA. As noted above, the aryl ring of the PDKAs was twisted 37 degrees out of coplanarity with the ketone, and occupied the approximate middle of the active site channel, overlapping the region where the thiol group of CoA normally binds. The aromatic ring forms multiple van der Waals interactions with the Cγ of catalytic Asp633, and the side-chains of Met515, Trp541, and Met631. The carboxylate of the side-chain of Asp633, expected to be deprotonated in view of its catalytic function (Clark et al., 1988), is positioned over the face of the PDKA ring, slightly shifted toward the diketo acid side, with distances from Oδ1 to PDKA atoms C1 and C6 of 4.1 and 3.4 Å, respectively, and from Oδ2 to C1 and C2 – of 3.3 and 4.0 Å.
Table 2. Crystal data collection and refinement statistics.
Data restricted to highest resolution shell are shown in parentheses. All crystals were of the C619A mutant enzyme with His6 tag.
| GlcB complex with PDKA |
GlcB complex with 4 |
GlcB complex with 1 |
GlcB complex with 7 |
GlcB complex with 11 |
|
|---|---|---|---|---|---|
| Data collection | |||||
| Space group | P43212 | P43212 | P43212 | P43212 | P43212 |
| Cell dimensions | |||||
| a, b, c (Å) | 79.33, 79.33, 225.94 |
79.45, 79.45, 226.26 |
77.74, 77.74, 221.98 |
78.22, 78.22, 223.58 |
78.49, 78.49, 224.05 |
| a, b, g (°) | 90, 90, 90 | 90, 90, 90 | 90, 90, 90 | 90, 90, 90 | 90, 90, 90 |
| Resolution (Å) | 50-1.90 (1.93-1.9) |
43.05-1.80 (1.83-1.80) |
44.13-1.82 (1.92-1.82) |
49.57-2.04 (2.13-2.04) |
45.59-2.20 (2.24-2.20) |
| Rsym or Rmerge | 0.109 (0.532) | 0.191 (0.634) | 0.108 (0.554) | 0.099 (0.584) | 0.183 (0.742) |
| I/σI | 13.8 | 14.0 | 6.4 | 6.3 | 9.0 |
| Completeness (%) | 99.9 (99.4) | 97.2 (76.0) | 99.9 (100) | 99.2 (98.6) | 99.9 (100) |
| Redundancy | 13.1 (10.2) | 9.5 (2.6) | 9.7 (9.8) | 15.3 (15.4) | 14.4 (14.8) |
| Refinement | |||||
| Resolution (Å) | 50-1.90 | 43.05-1.8 | 44.13-1.82 | 49.57-2.04 | 45.59-2.2 |
| No. reflections | 57789 | 66563 | 62029 | 45023 | 36507 |
| Rwork/ Rfree | 0.1678/0.2027 | 0.1753/0.2192 | 0.1704/0.2196 | 0.1628/0.2040 | 0.1721/0.2231 |
| No. atoms | |||||
| Protein | 5543 | 5645 | 5562 | 5509 | 5429 |
| Ligand/ion | 14/3 | 15/3 | 15/3 | 15/3 | 17/3 |
| Water | 674 | 859 | 641 | 574 | 400 |
| B-factors | |||||
| Protein | 27.86 | 29.64 | 21.22 | 32.13 | 34.69 |
| Ligand/ion | 30.47/28.14 | 20.7/32.13 | 14.24/27.53 | 30.1/38.4 | 28.87/39.3 |
| Water | 39.65 | 40.55 | 28.79 | 38.58 | 40.78 |
| R.m.s deviations | |||||
| Bond lengths (Å) | 0.013 | 0.007 | 0.007 | 0.007 | 0.008 |
| Bond angles (°) | 1.03 | 1.024 | 1.013 | 1.013 | 1.062 |
The close contact between Asp633 side chain and the aryl ring of the inhibitor is unusual, and resembles anion-π interactions that have recently been reported in small molecules and proteins (Berryman et al., 2007), (Schottel et al., 2008). In most interactions between anions and aromatic groups in biomolecular systems, the anion contacts the aromatic ring on the edge (co-planar) with distances around ~4.5 Å, whereas the contacts of carboxylates over the center of the ring were generally >5.0 Å (Jackson et al., 2007), (Philip et al., 2011). However, face-on anion-π contacts have been observed in inorganic systems, and are enhanced by electron-withdrawing substituents, resulting in lower contact distances and higher binding affinities. This is supported by semi-empirical quantum mechanics calculations for representative model systems that suggests there is a significant energy well for face-on contacts at 3-4 Å (Berryman et al., 2007), (Schottel et al., 2008). All crystal structures of GlcB-inhibitor complexes with PDKA analogs we have solved to date exhibit a close contact between the carboxylate of Asp633 and the face of the aromatic ring of the inhibitor. Among the five structures reported in this paper, the mean distance between the closest arene carbon and either of the Asp633 oxygens is 3.1 Å, with a mean contact angle of 55 degrees relative to the ring plane, consistent with a face-on interaction. A superposition of the ring plane of the Asp633-PDKA pair with the small-molecule crystal structure of tetracyanobenzene (TCB)-NaI complex reported by Berryman et al. (Berryman et al., 2007) (Figure 2) shows a very similar configuration between the two systems. The anion of the TCB-NaI complex (iodine) superimposes on the carboxylate of Asp633 in the GlcB-PDKA structures, and is located over the periphery of the ring (the carbon connecting to the diketo acid moiety), rather than directly over the ring center as suggested by theoretical calculations (Schottel et al., 2008). Presumably this position provides additional localized polarization. Anion positioning over the periphery of the ring rather than over the center has been hypothesized to indicate a partial charge transfer character to the interaction (Berryman et al., 2007). In single ortho-halogen substituted PDKAs (represented in Figure 2 by o-Br-PDKA) bound with GlcB, Asp633 is shifted away from the halogen by 1.7 Å (measured at Oδ2), reflecting the asymmetry created by the electron-withdrawing group, while with symmetric double ortho-halogen-substituted PDKA, the carboxylate of Asp633 is in the same position as with unsubstituted PDKA or o-Me-PDKA (Figure 2).
Figure 2. Anion-π interaction.

PDKA inhibitor (4) with Asp633 superimposed with crystal structure of tetracyanobenzene (TCB) in complex with NaI (606750 entry at Cambridge Crystallographic Data Center as supplementary for small molecule anion-π complex structures (Berryman et al., 2007)) colored by element, TCB ring is slightly off-set for easier visualization. Halide shown as a purple sphere, TCB carbons are colored yellow; Asp633 with gray carbons represent Asp633 position in complex with single ortho-halogen substituted PDKAs (o-Br-PDKA shown), while Asp633 with black carbons represent Asp633 position for all other complexes (with PDKA, 1, 7 and 11). Atom colors: black, gray or yellow=carbon, red=oxygen, blue=nitrogen, white=hydrogen, purple=iodine. Images rendered in CHIMERA (Pettersen et al., 2004).
Given the extra room in the active site around the phenyl ring of the PDKA, we hypothesized that an alternative cyclic structure could be accommodated and might improve affinity. Several replacements for the phenyl ring in PDKA were prepared, including aliphatic moieties/rings, exemplified by cyclohexyl and adamantyl cores, but were found to be inactive against GlcB (data not shown), suggesting a strong requirement for an aromatic moiety. Other aromatic rings such as naphthylene and various heterocycles were evaluated, but were not immediately pursued due to a sub-optimal combination of enzyme inhibition, whole-cell activity against cultured mycobacteria, and pharmacokinetic profile (full SAR data to be published elsewhere). Naphthyl-, indole-, pyrrole-, and thiophene-based diketo acids were active against the enzyme with IC50s ranging from 20 nM to 5 μM, with the first three failing at the level of whole-cell activity, and thiophenes displaying only low whole-cell activity (≥ 50 μM) (attributed to albumin binding) (data not shown). Furan-, quinoline-, benzodioxole-, and benzothiazole-based PDKAs exhibited low enzyme inhibition activity (IC50s ranging from 30 to 100 μM), while thiazole-, pyridine- and pyrimidine-based PDKAs were inactive against the enzyme (data not shown).
Structure-Guided Optimization of PKDA Analogs with High Potency
Structural examination of the GlcB-PDKA complex suggested several opportunities to enhance affinity by making substitutions around the PDKA aromatic ring to optimize interactions within the active site (schematic view of substitution strategy is shown in Figure S3). Because of their importance for stability, we focused on optimizing the potency and whole-cell activity of ortho-substituted PDKAs. Crystal structures of complexes of GlcB with twenty of the synthesized PDKA analogs were determined at resolutions ranging from 1.8 to 2.2 Å (crystallographic statistics for 5 representative data sets are shown in Table 2; example of omit map density is in Figure S2) to evaluate whether binding modes agreed with our predictions, and to guide new ideas. The crystal structure of GlcB in complex with 2-Br-PDKA (4) showed that the Br oriented toward the Val118 side-chain (Figure 3A). Of the ortho-substituted analogs, inhibitor potency was improved compared to unsubstituted PDKA (2.0 μM), the most potent being those with halogens, with a preference for smaller groups: 2-F (0.24 μM) < 2-Cl (0.5 μM) < 2-Br (0.6 μM) < 2-Me (1.1 μM) (Table 1). This was presumably due to the increase of steric clashes with the Val118 side-chain (3.2 Å from Br to the closest Cγ of Val118). There is little space available to accommodate a larger group, and indeed, o-Et-PDKA showed an IC50 of only 35 μM. Crystal structures of ortho-substituted PDKA complexes with GlcB indicated that all the ortho-substituents line up in the same direction, i.e. none are rotated to position 6 (Figure 1A), regardless of their size or nature, as exemplified by the crystal structure of GlcB in complex with the 2-Br derivative (4) (Figure 3). The ring of the ortho-substituted PDKA does not overlap exactly with the position of the parent PDKA ring (Figure 3), where for all of 2-substituted inhibitors it is moved about 0.7 Å away from Val118 to accommodate the group at the 2-position.
Figure 3. Comparing binding of PDKA analogs to GlcB.



Binding of GlcB to inhibitor 4 (A) and inhibitor 11 (B) colored by element, with Cprotein in grey, and Cligand in black. Hydrogen bonds are indicated by solid blue lines, distances from key positions on the phenyl ring to protein residues are marked as dashed lines. (C) - Crystal structure overlay of GlcB complexed with PDKA (in magenta), 1, 4, 7, 11 – represented in ball-and-stick, and CoA represented by a stick model (colored by element), with the Mg atom in green, illustrate the relative positions of the ligands occupying the active site channel (presented by protein surface calculated in CHIMERA (Pettersen et al., 2004). The CoA model and protein surface were made from chain A of the 2GQ3 model.
There was less room in the pocket off the 6-position of the aryl ring compared to the 2-position of the bound PDKA (3.9 Å compared to 4.35 Å to the closest protein atoms). This is likely the reason why the double ortho-substituted compound 2-Cl-6-F (10) (IC50 = 2.7 μM) did not show a better inhibition against the enzyme than PDKA (IC50 = 2.0 μM). However, we cannot rule out the possibility of an unfavorable alteration in the ring’s electron properties compared to a single ortho substituent affecting interaction with Asp633 (Figure 3A,B).
Position 4 (para- to the diketo acid) points directly at the Met631 side-chain (3.6 Å to Met631 Cγ) (Figure 3A,B). Therefore accommodation of a substituent at this position requires the Met631 side-chain to assume a different conformation. And indeed, 4-Me and 4-Br PDKA analogs (8 and 9) showed higher IC50 values than parent PDKA (5.7 and 6.0 μM respectively), likely due to steric interference with the Met631 side-chain.
The structure of the PDKA bound to GlcB showed that position 3 of the aromatic ring was the most promising for extending the PDKA, as any extension should align with the long axis of the channel where the substrate acetyl-CoA binds (Figure 3). This offered the possibility of exploiting the pantothenate binding contacts: e.g. hydrogen bond with the back-bone N of Val119, and van der Waals interactions with the side-chains of Met631, Met515, and the back-bone of Val118-Val119. Indeed, analogs with substitutions at the meta-position showed the largest improvement in potency over PDKA, with the most active compounds being 3-Cl-PDKA (6, IC50 = 0.17 μM) and 3-Me-PDKA (5, IC50 = 0.18 μM). The crystal structure of GlcB in complex with the 3-Br-PDKA (IC50 = 0.8 μM) (7) showed the Br in the van der Waals contact, sandwiched between the side chains of Met515 and Met631, arranged on opposite sides of the channel. It is interesting to note that although sufficient space seemed to exist in the channel to accommodate longer substitutions at the meta- position, alkyl and (CH2)nAr (n = 1, 2; Ar = aryl) substitutions at that position resulted in inhibitors with poor enzyme activity (IC50 > 100 μM). This may be attributed to a sub-optimal angle at which the meta-substituents project off the phenyl ring, potentially resulting in steric clashes with either Met631, or with Val119 and Pro120 on the other side of the channel.
The substituent’s effect on the electronic properties of the aromatic ring of PDKA appeared to be critical for inhibition activity. This is likely due to their influence on ring π-interactions with the carboxylate of catalytic Asp633. For example, 2-Cl-6-F substituted PDKA (10) has an IC50 of 2.7 μM, while 2,6-Me-PDKA which is of similar size and substitution positioning, but not electron-withdrawing, was inactive with an IC50 > 100 μM. Although the activity of compounds with halogens substituted at the 3-position had better enzyme activity than analogs substituted at the 2-position (e.g. 0.5 μM IC50 for 2-Cl-PDKA vs. 0.17 μM for 3-Cl-PDKA), there was a tradeoff with stability, as 2-substituted compounds exhibited longer half-lives, and since stability was important for whole-cell assays and in vivo testing, we chose to pursue compounds with 2-substitutions included. Tri-substituted 2-Cl-6-F-3-Me-PDKA (11) did not result in an additive affinity of each position (IC50 = 5.5 μM). However, 11 proved to be a good candidate for future study, as its methyl ester derivative exhibited a good combination of essential features (reasonable potency, high bioavailability, low toxicity) to modulate mycobacterial growth in an animal model of TB infection.
Anti-mycobacterial Activity of PDKA Inhibitors
We carried out whole-cell testing of growth inhibition by PDKAs on 0.2% acetate-supplemented M9 media to model fatty acid-driven metabolism, and 0.2% dextrose-supplemented 7H9 media to model carbohydrate-oriented metabolism for comparison. Initial testing was conducted with a Biosafety Level 2 (BL2)-approved vaccine strain of Mtb with deletions of the panCD genes and the RD1 region (mc2-7000) (Sambandamurthy et al., 2006). Freshly synthesized inhibitors were solubilized immediately prior to MIC determination (presented in Table 1, 3). Almost all of the PDKA inhibitors (8 of 11 presented in Table 1) with reasonable potency against the enzyme inhibited growth of bacteria on acetate (with MICs from 6.25 μM to 50 μM), and were less potent on carbohydrate (dextrose), with MICs usually 2-4 fold higher than on acetate, indicating that they affect the glyoxylate shunt. In the context of this paper, MICs are reported as MIC99, which refers to the minimum concentration of a compound at which bacteria growth is inhibited by >99%, as assessed by absence of respiration in the rezasurin (AlamarBlue) assay. Testing on other tuberculosis strains and clinical isolates has not yet been carried out.
Table 3.
H37Rv activity, mouse plasma protein binding, and mouse microsomes clearance data for alkyl- and benzyl-ester prodrugs of selected PDKA analogs.
| compound | H37Rv MIC99 on acetate (μM) |
H37Rv MIC99 on dextrose (μM) |
Mouse microsomes clearance (ml/min*gr) |
Mouse plasma protein binding (%) |
|---|---|---|---|---|
| 11 | 16 | 31 | < 0.3 | 70 ± 3.0 |
| Me ester of 11 (12) | 2 | 8 | 0.6 ± 0.2 | 91.9 ± 1.6 |
| Benzyl ester of 11 | < 1 | 2 | > 30 | ND |
| 4 | 16 | 31 | < 0.3 | 67.5 ± 2.6 |
| Me ester of 4 | 8 | 16 | 3.2 ± 0.7 | 87.7 ± 1.6 |
| Benzyl ester of 4 | <1 | 8 | 15.3 ± 6.1 | ND |
| 1 | 16 | 31 | ND | 70.7 ± 1.8 |
| Me ester of 1 | 4 | 16 | 4.6 ± 0.7 | 87.1 ± 1.4 |
| Benzyl ester of 1 | <1 | 16 | >30 | ND |
To improve whole-cell activity, we employed a prodrugging strategy by masking the acid via esterification as a way to enhance cellular uptake. Simple alkyl esters of whole-cell-active PDKA analogs had approximately 8-fold lower MICs compared to their corresponding PDKAs (Table 3). Of particular interest were the ester prodrugs of the ortho-bromo (4), ortho-methyl (5), and 2-chloro-6-fluoro-3-methyl (11) PDKAs, exhibiting MIC values in the ≤ 1 to 8 μM range against H37Rv grown on acetate as a carbon source (Table 3). In agreement with earlier data on the Mtb mc2-7000 strain, PDKA esters inhibited growth of an H37Rv strain on dextrose supplemented media with a 2-4 fold higher MICs. In addition, Caco-2 uptake studies (Yazdanian et al., 1998) demonstrated the enhanced permeability of esters compared to their corresponding acids (from a non-detectable level for acid 11 to a high level of 371 nm/s for the corresponding methyl-ester 12), suggesting they could potentially be orally bioavailable and taken up through the gut. Following the decomposition of the benzyl- and methyl-esters of 11 in whole-cell lysates over time by mass spectrometry demonstrated accumulation of acid 11, and no other product (Table S2). As expected, none of the PDKA esters themselves were directly active against GlcB in the enzyme assay, but upon hydrolysis with longer pre-incubation, each ester decomposed to yield the parent PDKA (free acid) active against the enzyme. Furthermore, PDKA esters inhibited malate synthase activity in mycobacterial cell lysates (Table S1, S2). These compounds achieved IC50s in whole-cell lysates similar to the purified enzyme assay, with no loss of inhibition even after 3 hours of incubation due to potential reactivity or binding to other proteins, arguing that these compounds act on GlcB inside the cell (Table S1).
The methyl ester of 11 (12) was tested using the standard minimal bactericidal concentration (MBC) determination protocol (Motyl et al., 2006), and shown to be bactericidal to mc2-7000 in culture on either acetate or dextrose carbon sources. The MBC was approximately the same as the MIC when grown on acetate, and the MBC was 2-4 fold higher than the MIC on dextrose. Compound 12 was also tested in the Low-Oxygen Recovery Assay (LORA), one of the in vitro models of the non-replicating drug-tolerant state of Mtb (Cho et al., 2007), and was found active against H37Rv grown on dextrose (MIC = 52 μM). The observed 4-to 5-fold shift of MIC in the LORA assay compared to the MABA assay (Microplate Alamar Blue Assay; Franzblau et al., 1998) (MIC = 11 μM) is consistent with what has been observed for other antitubercular drugs with activity against non-replicating bacteria, such as rifampicin (Cho et al., 2007).
To confirm that GlcB is the intracellular target responsible for growth inhibition in Mtb we constructed a GlcB-overexpressing strain in Mtb containing on a plasmid either GlcB under a tetracycline-inducible promoter, or an unrelated Mtb protein (Rv3547) as a control. An 8-fold increase in the MIC (12.5 to 100 μM) was observed after induction of GlcB expression using anhydrotetracycline on M9 media with acetate, and a 4-fold increase in the MIC (25 to 100 μM) on 7H9-dextrose, whereas the strain with the control plasmid (Rv3547) showed no shift in MIC for compound 12. All strains displayed the same MIC for rifampicin (with or without anhydrotetracycline) as the untransformed parental strain. The observed increase in the MIC for compound 12 in response to GlcB overexpression strongly supports on-target activity. In addition, we have tested PDKA inhibitors (4 and 11) at a high concentration of 100 μM on four purified enzymes in the core metabolic pathways from Mtb which react with similar substrates (ICL, phosphoenolpyruvate carboxykinase, isocitrate dehydrogenase, and pyruvate kinase) and observed no inhibition (data not shown), adding to reassurance that inhibition is specific to GlcB.
Inhibition of GlcB in a Murine Model of TB
We evaluated the pharmacologic properties of several of the most potent inhibitors and selected a representative for advancement to a mouse model of TB infection. The most important factors we considered were solubility, chemical stability, plasma protein binding, serum stability, metabolic clearance in microsomes, and pharmacokinetic profile. The ADME guidelines we used were: soluble in PBS at >20 g/ml; stability in human and murine plasma > 70% over 1 h; extent of plasma protein binding known; clearance in mouse microsomes after 30 min < 15 ml/min/kg, and Caco-2 permeability > 10×10−6 cm−1/s. The methyl ester 12 showed the best combination of potency with in vitro pharmacokinetic (PK) properties among the PDKA analogs tested (Table 3), and therefore was selected for further in vivo PK, pharmacodynamic (PD) and toxicity studies. In mice, 12 was orally bioavailable (%F = 92) and attained sufficient blood levels (i.e., at 600 mg/kg dosing: Tmax = 0.25 h, Cmax = 99.5 μg/ml, and AUC8h = 54.3 μg*h/ml). 12 demonstrated good stability in CD1 mouse plasma, in terms of slow hydrolysis of the ester to yield the parent PDKA derivative (t1/2 = 30 min, conversion from 12 to 11 in plasma). The corresponding acid, 11, demonstrated a low rate of clearance in a mouse liver microsome assay (<0.3 ml/min*g), , and a reasonable level of mouse plasma protein binding (70%). (The eventual disposition of 11, whether by metabolism or excretion, was not determined in vivo.) The achieved exposure for 12 in mice, as measured by AUCfree/MICH37Rv/acetate (35 @ 600 mg/kg oral dosing), compared reasonably well to clinical antituberculars such as moxifloxacin (AUCfree/MICH37Rv/acetate = 142 @ 100 mg/kg oral dosing), which is used in treatment of multi-drug resistant tuberculosis (MDR-TB) (Cox et al., 2011). (PK curves for 12 and its activated form 11 are shown in Figure S4.) The PK data show that the peak blood concentration achieved for compound 12 at 300, 500, and 600 mg/kg was approximately 150-200 fold higher than the MIC for H37Rv (2 μM=0.548 μg/ml), and the exposure was maintained above the MIC value for at least 6-7 hours. Assessment in C57BL/6J mice with a single-dose oral administration of 12 formulated in Solutol (30%) and PEG 400 (70%) demonstrated that a dose of 1000 mg/kg (the highest tested) was not lethal, and the maximum tolerated dose (MTD) was determined to be 600 mg/kg. No toxicity effects were observed at 400 mg/kg po twice daily.
Compound 12 was tested in a murine model of acute TB infection (Rullas et al., 2010). C57BL/6J mice were infected intratracheally with 105 CFU and treated for 9 days, followed by determination of bacillary load in the lungs. Several dosing strategies from 300 to 600 mg/kg were tested to determine the best compound exposure above the MIC at or below the MTD established. Treatment with moxifloxacin (30 mg/kg) was used for comparison. At all dosing levels (once daily (uid) and twice daily (bid)), 12 exhibited a statistically significant reduction (p-values < 0.0001) in the Mtb bacterial load compared to the control (Table 4). In fact, at 400 mg/kg bid dosing, 12 reduced the bacterial load by over 100-fold (Δlog10 CFU = 2.12), within an order of magnitude of moxifloxacin (Δlog10 CFU = 3.07). This activity was achieved despite the fact that the dosage, which was limited by the MTD, provided exposure well above the MIC for only 6-8 hours at a time. Thus, inhibition of GlcB resulted in impairment of the ability to establish an acute infection in mice, similar to the results obtained with a Δicl1/2 strain (Muñoz-Elías and McKinney, 2005).
Table 4. Therapeutic efficacy of 12 against Mtb in mice.
Five animals in each testing group were inoculated with 105 colony-forming units (CFU) of Mtb intratracheally, and treatment started one day post-infection. Nine days post-infection the mice were sacrificed and the respective bacterial loads in the lungs were determined. All reductions in CFU were statistically significant over the untreated control group by a T-test (dof=9-10, ‘*’ indicates p<0.0001).
| Compound | Target dose (mg/kg) |
Experimental dose (mg/kg) |
Administration | Decrease in log10 CFU in lungs |
Standard error |
|---|---|---|---|---|---|
| Moxifloxacin | 30 | 36 | Once a day | 3.07* | 0.0639 |
| 12 | 500 | 542 | Once a day | 1.68* | 0.103 |
| 12 | 600 | 718 | Once a day | 1.72* | 0.149 |
| 12 | 300 | 377 | Twice a day | 1.89* | 0.0787 |
| 12 | 400 | 460 | Twice a day | 2.12* | 0.108 |
Discussion
Our studies have shown that malate synthase is essential for Mtb survival both in vitro and in vivo, and this enzyme can be targeted with phenyl-diketo acid (PDKA) inhibitors. Structure-guided design led to the identification of highly potent inhibitors with sub-micromolar IC50s. While the acids in the series displayed difficulty penetrating the cell wall, esters of these compounds acted as prodrugs that could be taken up and hydrolyzed inside cells, leading to potent growth inhibition. Over-expression of the enzyme leads to a ~4-fold increase in MIC for these compounds - evidence that GlcB is the target whose inhibition is responsible for cell death. Furthermore, we observed a correlation in structure-activity relationship, where analogs with different substituents around the PDKA core that inhibit the enzyme (or their corresponding esters) also inhibit whole-cell growth, and analogs with substituents that abrogate activity against the enzyme are also inactive (even in their ester forms) against whole-cells. This correlation of SAR would be highly unlikely if the actual target of these compounds inside the cell were an enzyme other than GlcB. Finally, the compounds are consistently 4-fold more potent in cultures grown on acetate as a carbon source compared to dextrose - conditions where cells rely on the glyoxylate shunt. Based on these observations, we conclude that inhibition of GlcB is the mechanism of action of the PDKAs in vivo.
The observation that inhibitors of GlcB are bactericidal for Mtb grown on carbon sources other than fatty acids, such as carbohydrates like dextrose, was unexpected because inactivation of ICL is tolerated when grown in vitro on carbohydrates (McKinney et al., 2000). Since ICL2 also has partial isocitrate lyase activity (Gould et al., 2006), a Δicl1/icl2 double mutant of Mtb completely lacking a functional glyoxylate shunt was constructed, and was also found to be able to grow on dextrose (Muñoz-Elías and McKinney, 2005). However, the Δicl1/icl2 double mutant grew at a suppressed rate (2-4 day lag), and this growth defect suggests that the glyoxylate shunt might be playing a metabolic role even when Mtb is growing on carbohydrates. Unlike E. coli and other well-studied bacteria, which suppress anaplerosis in the presence of a preferred carbon source like carbohydrates (Fischer and Sauer, 2001), Mtb catabolizes carbohydrates and fatty acids concurrently in vitro with no apparent repression (Carvalho et al., 2010).
Supporting the observation that GlcB is essential in vitro, recent high-density transposon-mutagenesis experiments (analyzed by deep sequencing) have shown that glcB is essential for growth on glycerol (representative of carbohydrates), as well as on cholesterol as a carbon source (Griffin et al., 2011).
One possible explanation for the requirement for GlcB could be to avoid accumulation of glyoxylate, which has been shown to be toxic in other bacteria (Nuñez et al., 2001). However, Mtb is able to grow on glyoxylate as a sole carbon source (our unpublished data), and there are other enzymes (i.e. glycine dehydrogenase or glyoxylate aminotransferase) that can utilize glyoxylate as a substrate (Sakuraba et al., 2008), (Wayne and Lin, 1982). In addition, we found, that when the ICL inhibitor 3-nitropropionate (3-NP) is co-administered at a low sub-MIC concentration (20 μM) with GlcB inhibitors to Mtb cultures grown on 7H9-dextrose media, it causes a decrease in MIC (for example, from 12.5 μM to 1.56 μM for 12). In theory, the presence of 3-NP should reduce glyoxylate accumulation by suppressing flux through the glyoxylate shunt, and would thus be expected to cause an increase in MIC for GlcB inhibitors. The decrease in MIC we observed suggests that the requirement for GlcB in vitro under carbohydrate-supplemented growth conditions might not be limited to a need for glyoxylate detoxification.
There are several alternative reasons why GlcB inhibition under carbohydrate-supplemented growth conditions might result in cell death. Functional GlcB might be required when grown on dextrose to replenish the intermediates on the reductive side of TCA cycle (succinate, malate, and oxaloacetate), which were shown to be maintained at relatively low intracellular concentrations by metabolite tracing (Carvalho et al., 2010). A recent analysis of13 C metabolic flux in Mtb demonstrated a constant flux through the glyoxylate shunt, even with a carbohydrate (glycerol) as a carbon source, and that disruption of icl1 resulted in a loss of viability at a slow growth rate (Beste et al., 2011). This echoes the finding of ICL1 being important to Mtb’s ability to adapt to nutrient-limiting conditions by regulating ATP levels required for entering a non-replicating state (Gengenbacher et al., 2010). These data point to the conclusion that the role of the glyoxylate shunt extends beyond its anaplerotic function in Mtb. Despite the uncertainty about its metabolic role under carbohydrate-supported growth conditions, our results make it clear that inhibition of GlcB is lethal to Mtb grown on multiple carbon sources, and this provides a novel route to antitubercular drug development.
Significance
Novel enzyme targets are needed to drive discovery of new drugs for combating tuberculosis. Because of its role in the glyoxylate shunt, we have investigated malate synthase (GlcB) as an attractive target and identified a series of potent inhibitors with a phenyl-diketo acid (PDKA) scaffold. A selected compound (12) in the PDKA series was shown to have efficacy in a mouse model of infection. While complete sterilization was not achieved, the bacterial load was reduced nearly 100-fold over the course of 9 days. Interestingly, the compound appears to be active during the acute phase of infection, which is consistent with the essential role of GlcB for growth on other carbon sources in addition to fatty acids. Thus, these compounds have the potential to have activity during both acute and chronic phases of infection. It is likely that with further optimization, a more potent compound than 12 (which was chosen for a tradeoff of PK/PD properties) would be able to achieve even higher bacterial clearance in vivo. Nonetheless, the statistically significant reduction of the bacterial load observed indicates that the PDKA compounds could have therapeutic potential, and provides evidence that Mtb GlcB could be a clinically-relevant target. These structural studies will form the foundation for development of better GlcB inhibitors to eventually be used in human clinical trials.
Experimental procedures
Protein overexpression and purification
GlcB with the Cys619 mutated to Ala was cloned into a custom vector p6HisF-11d, expressed in E. coli BL21 cells, and purified by Ni affinity and size exclusion columns as described previously (Smith et al., 2003). As the presence of the His-tag did not change the results of the enzyme assay or crystallization, most of the reported work was done using GlcB with the N-terminal His-tag intact.
DTNB-coupled enzyme assay
For the C619A GlcB mutant enzyme, a DTNB-coupled assay was used to evaluate inhibition activity. A BMG POLARstar OPTIMA plate reader was used to determine the inhibition of GlcB by continuously monitoring the formation of CoA in the forward enzymatic reaction by the increase in absorbance at 412 nm due to 5,5′-dithiobis-(2-nitrobenzoic acid) (DTNB)-CoA adduct formation, over a period of 20 minutes. The 96-well plates contained 100 μl total reaction volume with 13 nM C619A GlcB in the reaction buffer (20 mM Tris pH 7.5 and 5 mM MgCl2). All inhibitors (in 100% DMSO) were added such that the final reaction mixture contained 1% DMSO. Inhibitors were incubated with GlcB in the reaction buffer for 20 min at room temperature before adding 0.6 mM acetyl-CoA. The reaction was initiated by the addition of 1.2 mM glyoxylate and 0.5 mM (final concentration) DTNB. Each data point for the IC50 plots was measured in triplicate. The data reported in this paper reflect the most robust enzyme assay conditions. We noticed that a longer pre-incubation time with GlcB (probed up to 3 hours), and lower Mg2+ concentrations in the assay buffer (1-5 mM, still in vast excess to the KM for this metal ion as a co-factor in the reaction) led to enhanced enzyme inhibition for the PDKA family of inhibitors (relative to control reactions of the enzyme incubated for the same duration, in the same Mg2+ concentrations without inhibitor in each case, which corrects for the slight loss of activity of the enzyme over time due to protein instability). It appears that PDKA inhibitors are slow to reach equilibrium binding, probably because they bind through chelating Mg2+ in the same manner as glyoxylate (Copeland, 2005). However, incubating longer than 30 minutes at room temperature led to a decrease in enzyme velocity and reduced assay reproducibility, and was not done for routine inhibitor testing aiming at building SAR.
Pyruvate dehydrogenase (PDH)-coupled assay
For the wild-type GlcB enzyme, this assay was used because the Cys619 in the active site is sensitive to oxidation by the DTNB in the coupled reaction. Velocity was measured by monitoring the increase of fluorescence (excitation at 340 nm, emission at 480 nm) due to NADH production coupled with the release of CoA-SH. Final concentrations in the reaction mixture were: 3 nM of GlcB, 0.1 UI/ml of PDH, 50 μM glyoxylate, 2 μM acetyl CoA, 500 μM NAD, 500 μM pyruvate, 200 μM thiamine pyrophosphate, 5 mM MgCl2, 0.8 mM EDTA, 50 mM Tricine pH 7.4. EDTA was included in the assay buffer in both assays because it enhances the stability of the enzyme and does not affect the enzyme velocity, though it is over 6-fold lower than the concentration of Mg2+ used. The same assay was used in mycobacterial (BCG) cell lysate (1 mg/ml total protein concentration measured by Bradford assay), with cells harvested at mid-log phase and disrupted by sonication.
Inhibitor synthesis
Chemical syntheses of the PDKA compounds used in this study are described in the Supplemental Information. Chemical structures for compounds 1-12 are shown in Figure S5.
Protein crystallization, data collection, and data analysis
Purified C619A GlcB (with His6 tag) at a concentration of 5 mg/ml in 20 mM Tris-HCl pH 7.5 buffer with 0.1 M NaCl was mixed for crystallization with an equal volume of mother liquor of 18-22% PEG 3350, 0.1 M MgCl2, and 0.1 M Tris-HCl pH 8.0. Crystals were obtained by hanging drop vapor diffusion within 2-3 weeks. Inhibitors were soaked in by transferring pre-formed GlcB crystals into a drop made from mother liquor with 1-5 mM of inhibitor added from a DMSO stock solution such that the final DMSO concentration was below 1%, and incubated for 1-5 days. Prior to data collection crystals were cryo-protected by Fomblin (Sigma), and flash frozen in liquid nitrogen. Data were collected at Argonne National Lab APS synchrotron, beamlines 19- and 23-ID, at 0.98 Å. Diffraction data were indexed, integrated, and scaled in HKL2000 (Otwinowski and Minor, 1997). Data were truncated in CCP4 (1994). 1N8I (Smith et al., 2003)with only the protein atoms included in the refinement was used as the model for the initial rigid body refinement of the isomorphous P43212 crystal in REFMAC (Murshudov et al., 2011). Then iterative runs of inspection and manual modification in COOT (Emsley et al., 2010) and refinement in PHENIX (Adams et al., 2002) with simulated annealing were done to gradually improve the model. The ligand model and dictionary files were created in ELBOW BUILDER from the PHENIX suite, and fitted into the density in COOT. Final refinement was done in BUSTER (Bricogne G, 2010). For data collection and refinement statistics see Table 2. All ligands had strong electron density in the 2Fo-Fc map covering all atoms visible (example shown in Figure S2). After refinement, the halogens in halogen-containing inhibitors displayed negative Fo-Fc peaks over them in the Fourier difference map. This might have been an artifact due to radiation damage, as refinement with data truncated to very low-redundancy reflections showed no negative Fo-Fc density at these sites. For final model refinement, high redundancy sets were used, and individual occupancies were refined for halogen-containing ligands to eliminate disagreement in the Fo-Fc map. Ramachandran statistics are as follows (given in order most favored/additionally favored regions/outliers in percent): GlcB-PDKA - 97.11/2.02/0.87; GlcB-4 – 97.21/1.76/0.73; GlcB-1 – 97.34/2.22/0.44; GlcB-7 – 97.27/2.16/0.58; GlcB-11 – 96.85/3.01/0.14. All of the Ramachandran outliers are on the surface of the protein except Glu273, which hydrogen bonds with a water molecule coordinating the Mg2+. This unusual backbone conformation is well-supported by the electron density in all data sets.
Accession numbers
Crystal structures of GlcB in complex with inhibitors were deposited in the protein data bank under entry IDs 3S9I, 3S9Z, 3SAD, 3SAZ, and 3SB0.
Whole-cell testing
MIC determination was done using the MABA (Franzblau et al., 1998) assay in 96-well plates. For the Mtb mc2-7000 strain (Sambandamurthy et al., 2006), cells were grown in 7H9 media with OADC (Middlebrook) supplement, 0.05% Tyloxapol (Sigma) and 25 μg/ml pantothenate to an OD600 of 1-2. Then cells were diluted into testing media to an OD600 of 0.01 and pipetted into testing plates, 200 μl per well. The two testing media were: 7H9 media with 0.2% dextrose, 0.085% NaCl, 0.05% Tyloxapol, and 25 μg/ml pantothenate or M9 (Sigma) media with 0.25% Na acetate, 2 mM MgSO4, 0.1 mM CaCl2, 0.05% Tyloxapol, and 25 μg/ml pantothenate. Then each compound was added as a 1/2 serial dilution in DMSO (2% DMSO final in a well). 7H9-dextrose plates were incubated for 6 days before staining with resazurin (Sigma), M9-acetate plates were incubated for 3 weeks, and then for additional 2 days after staining at 37 °C with shaking. The lowest concentration where resazurin stayed completely unconverted was recorded as the MIC99 value. MIC99 refers to the minimum concentration at which growth of the experimental strain mc2-7000 is inhibited by >99%, as assessed by absence of respiration in the rezasurin (AlamarBlue) assay. Rifampicin was used as a control: for mc2-7000 Mtb displaying an MIC99 of 0.125 μM in 7H9-dextrose, and an MIC99 of 0.25 μM in M9-acetate. MIC values for rifampicin varied no more than one dilution point from run to run. All MIC99 values reported are overage with 1/2 dilution precision from at least three independent experiments. For H37Rv Mtb strain testing, an inoculum standardized to approximately 1×107 cfu*ml-1 was diluted 1 in 200 in testing media of 7H9, ADC, 0.025% Tween 80, 0.085% NaCl or 7H9, 0.5% albumin, 0.1% Na acetate, 0.025% Tween 80, 0.085% NaCl. Inhibitors were tested as with the mc2-7000 strain. Plates were incubated at 37 °C for six days before and 24-48 hours after staining with resazurin.
Mouse microsome clearance and plasma protein binding
Microsomal intrinsic clearance was measured as described by (Clarke and Jeffrey, 2001). The compound (0.5 μM) was incubated with 0.5 mg/ml microsomal protein, 0.34 mg/mL NADP, 1.56 mg/ml glucose-6-phosphate, and 1.2 units/ml glucose-6-phosphate dehydrogenase, 2.6 mg/ml UDPGA, 0.5% (v/v) methanol in 50 mM potassium phosphate buffer, pH 7.4, at 37 °C. Fifty microliter aliquots of the incubation mixture were withdrawn at various time points over 30 min and added to 100 ml stop solution (80:20:1 (v/v/v) acetonitrile:ethanol:acetic acid) containing internal standard. Samples were snap frozen and stored at −80 °C until analyzed by LC/MS/MS. Prior to analysis, samples were thawed at room temperature, vortexed, then centrifuged, and the supernatant taken for analysis. No cofactor controls were included to assess non-P450 dependent clearance. Clearance was estimated for midazolam in parallel to assure integrity of the microsomal preparations and acceptable interassay variability. The intrinsic clearance was calculated based on the method published by (Obach et al., 1997), using the first-order elimination rate constant for disappearance of the parent compound. This was calculated from the slope of the log-transformed concentration-time curve using SigmaPlot 8.0 (Systat Software Inc). Clearances were expressed in units of ml/min/g liver. The clearance was predicted based on the assumption that the drug concentration (0.5 μM) was most likely well below the KM. The lower limit of quantification was 0.5 ml/min/g liver and this corresponded to <15% decrease in the parent compound in 30 min. The in vitro plasma protein binding of the diketo acids was determined by equilibrium dialysis (3 cells/species) in fresh mouse and human plasma at 0.5 and 5 mg/ml (0.5% (v/v) DMSO final concentration). Spiked plasma samples were mixed gently and triplicate aliquots were collected to verify initial concentrations. Following assembly of dialysis RED devices (Thermo Scientific) (MW cut off limit of 8,000 to 10,000 Da), spiked plasma was placed in the donor compartment of the cell and phosphate buffered saline, pH 7.4, in the receiver compartment. Cells were incubated in a water bath at 37 °C and mixed continuously for 6 h. Triplicate aliquots (volume determined gravimetrically) from donor and receiver compartments were snap frozen and stored at −30 °C prior to analysis. Percent binding was estimated using standard equations which accounted for volume changes due to Donnan effects (Boudinot and Jusko, 1984).
Pharmacokinetic measurement
The compound was assayed as a single oral dose at 20 mg/kg, 100 mg/kg and 600 mg/kg in female C57BL/6J mice, dissolved in PEG400/Solutol 70:30. The sampling scheme post-administration was 15, 30, and 45 minutes, 1, 1.5, 2, 3, 4, and 8 hours; 4 animals per time point. Peripheral blood levels were analysed by LC/MS/MS. Data analysis was performed with WinNonlin 5.2; Non-compartmental analysis (NCA).
Efficacy in mouse model of TB
To assess the therapeutic efficacy of compound 12 against M. tuberculosis in an acute murine model of intratracheal infection (Rullas et al., 2010), mice were infected with 105 CFU and lung homogenates were obtained 9 days after infection (n = 4-5 mice/group for all groups). Compound 12 was administered according to the schedule indicated in Table 4 (either once a day or twice a day) using PEG400/Solutol 70:30 as the vehicle. Moxifloxacin (30 mg/kg) dissolved in Captisol 20% was used as quality control of the assay, and reduced the CFU lung number by 3.07 logs with respect to untreated mice. The average log CFU in the lungs of untreated mice was 7.13.
Supplementary Material
Highlights.
discovery and stabilization of phenyl diketo-acid (PDKA) inhibitors for GlcB
exploring binding interactions and improving potency through structure-based design
PDKAs are bactericidal to Mtb grown on fatty acids and carbohydrates
targeting GlcB with PDKA reduces bacterial load in a mouse model of tuberculosis.
Acknowledgments
The authors would like to thank Dr. Franzblau for testing 12 in the LORA assay, Hongye Li – for cloning GlcB into PDT vector, TAMU chemistry group currently lead by N. Zhou – for synthesizing ~ 400 compounds for this project, Tracey Musa – for comments on the manuscript, and the GSK team: Ortega, F. – for conducting PK experiments, Santos-Villarejo, A. – for plasma protein binding and stability experiments, Trullas, J. – for in vivo efficacy experiments, Alvarez-Gomez D. - for wild type enzyme testing, and Perez-Herranz, E. – for whole-cell H37Rv testing.
This work was funded by grants to Sacchettini, J.C.: by the Welch foundation, grant A-0015, by the NIH grant P01 AIO 68135, and by Global Alliance for TB Drug Development. Data were collected at Argonne National Laboratory, beamlines 19ID and 23ID.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Terwilliger TC. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr D Biol Crystallogr. 2002;58:1948–1954. doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]
- Anstrom DM, Kallio K, Remington SJ. Structure of the Escherichia coli malate synthase G:pyruvate:acetyl-coenzyme A abortive ternary complex at 1.95 A resolution. Protein Sci. 2003;12:1822–1832. doi: 10.1110/ps.03174303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anstrom DM, Remington SJ. The product complex of M. tuberculosis malate synthase revisited. Protein Sci. 2006;15:2002–2007. doi: 10.1110/ps.062300206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berryman OB, Bryantsev VS, Stay DP, Johnson DW, Hay BP. Structural criteria for the design of anion receptors: the interaction of halides with electron-deficient arenes. J Am Chem Soc. 2007;129:48–58. doi: 10.1021/ja063460m. [DOI] [PubMed] [Google Scholar]
- Beste DJV, Bonde B, Hawkins N, Ward JL, Beale MH, Noack S, Nöh K, Kruger NJ, Ratcliffe RG, McFadden J. C Metabolic Flux Analysis Identifies an Unusual Route for Pyruvate Dissimilation in Mycobacteria which Requires Isocitrate Lyase and Carbon Dioxide Fixation. PLoS Pathog. 2011;7:e1002091. doi: 10.1371/journal.ppat.1002091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloch H, Segal W. Biochemical differentiation of Mycobacterium tuberculosis grown in vivo and in vitro. J Bacteriol. 1956;72:132–141. doi: 10.1128/jb.72.2.132-141.1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudinot FD, Jusko WJ. Fluid shifts and other factors affecting plasma protein binding of prednisolone by equilibrium dialysis. J Pharm Sci. 1984;73:774–780. doi: 10.1002/jps.2600730617. [DOI] [PubMed] [Google Scholar]
- Bricogne G,BE. BUSTER. version 2.9 2010.
- Carvalho L.P.S. de, Fischer SM, Marrero J, Nathan C, Ehrt S, Rhee KY. Metabolomics of Mycobacterium tuberculosis reveals compartmentalized co-catabolism of carbon substrates. Chem Biol. 2010;17:1122–1131. doi: 10.1016/j.chembiol.2010.08.009. [DOI] [PubMed] [Google Scholar]
- Cho SH, Warit S, Wan B, Hwang CH, Pauli GF, Franzblau SG. Low-oxygen-recovery assay for high-throughput screening of compounds against nonreplicating Mycobacterium tuberculosis. Antimicrob Agents Chemother. 2007;51:1380–1385. doi: 10.1128/AAC.00055-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke SE, Jeffrey P. Utility of metabolic stability screening: comparison of in vitro and in vivo clearance. Xenobiotica. 2001;31:591–598. doi: 10.1080/00498250110057350. [DOI] [PubMed] [Google Scholar]
- Clark JD, O’Keefe SJ, Knowles JR. Malate synthase: proof of a stepwise Claisen condensation using the double-isotope fractionation test. Biochemistry. 1988;27:5961–5971. doi: 10.1021/bi00416a020. [DOI] [PubMed] [Google Scholar]
- Copeland RA. Evaluation of enzyme inhibitors in drug discovery. A guide for medicinal chemists and pharmacologists. Methods Biochem Anal. 2005;46:1–265. [PubMed] [Google Scholar]
- Cox H, Ford N, Keshavjee S, et al. Rational use of moxifloxacin for tuberculosis treatment. Lancet Infect Dis. 2011;11:259–260. doi: 10.1016/S1473-3099(11)70036-6. [DOI] [PubMed] [Google Scholar]
- Dunn MF, Ramírez-Trujillo JA, Hernández-Lucas I. Major roles of isocitrate lyase and malate synthase in bacterial and fungal pathogenesis. Microbiology. 2009;155:3166–3175. doi: 10.1099/mic.0.030858-0. [DOI] [PubMed] [Google Scholar]
- Egbertson MS. HIV integrase inhibitors: from diketoacids to heterocyclic templates: a history of HIV integrase medicinal chemistry at Merck West Point and Merck Rome (IRBM) Curr Top Med Chem. 2007;7:1251–1272. doi: 10.2174/156802607781212248. [DOI] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer E, Sauer U. A novel metabolic cycle catalyzes glucose oxidation and anaplerosis in hungry Escherichia coli. Journal of Biological Chemistry. 2001;278(47):46446–51. doi: 10.1074/jbc.M307968200. [DOI] [PubMed] [Google Scholar]
- Franzblau SG, Witzig RS, McLaughlin JC, Torres P, Madico G, Hernandez A, Degnan MT, Cook MB, Quenzer VK, Ferguson RM, et al. Rapid, low-technology MIC determination with clinical Mycobacterium tuberculosis isolates by using the microplate Alamar Blue assay. J Clin Microbiol. 1998;36:362–366. doi: 10.1128/jcm.36.2.362-366.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gengenbacher M, Rao SPS, Pethe K, Dick T. Nutrient-starved, non-replicating Mycobacterium tuberculosis requires respiration, ATP synthase and isocitrate lyase for maintenance of ATP homeostasis and viability. Microbiology. 2010;156:81–87. doi: 10.1099/mic.0.033084-0. [DOI] [PubMed] [Google Scholar]
- Gould TA, Langemheen H, van de, Muñoz-Elías EJ, McKinney JD, Sacchettini JC. Dual role of isocitrate lyase 1 in the glyoxylate and methylcitrate cycles in Mycobacterium tuberculosis. Mol Microbiol. 2006;61:940–947. doi: 10.1111/j.1365-2958.2006.05297.x. [DOI] [PubMed] [Google Scholar]
- Griffin JE, Gawronski JD, Dejesus MA, Ioerger TR, Akerley BJ, Sassetti CM. High-resolution phenotypic profiling defines genes essential for mycobacterial growth and cholesterol catabolism. PLoS Pathog. 2011;7:e1002251. doi: 10.1371/journal.ppat.1002251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson MR, Beahm R, Duvvuru S, Narasimhan C, Wu J, Wang H-N, Philip VM, Hinde RJ, Howell EE. A preference for edgewise interactions between aromatic rings and carboxylate anions: the biological relevance of anion-quadrupole interactions. J Phys Chem B. 2007;111:8242–8249. doi: 10.1021/jp0661995. [DOI] [PubMed] [Google Scholar]
- Kornberg HL, Krebs HA. Synthesis of cell constituents from C2-units by a modified tricarboxylic acid cycle. Nature. 1957;179:988–991. doi: 10.1038/179988a0. [DOI] [PubMed] [Google Scholar]
- Laskowski RA, Swindells MB. LigPlot+: Multiple ligand-protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 2011;51:2778–2786. doi: 10.1021/ci200227u. [DOI] [PubMed] [Google Scholar]
- Marrero J, Rhee KY, Schnappinger D, Pethe K, Ehrt S. Gluconeogenic carbon flow of tricarboxylic acid cycle intermediates is critical for Mycobacterium tuberculosis to establish and maintain infection. Proc Natl Acad Sci U S A. 2010;107:9819–9824. doi: 10.1073/pnas.1000715107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinney JD, Bentrup K.H. zu, Muñoz-Elías EJ, Miczak A, Chen B, Chan WT, Swenson D, Sacchettini JC, Jacobs WR, Russell DG. Persistence of Mycobacterium tuberculosis in macrophages and mice requires the glyoxylate shunt enzyme isocitrate lyase. Nature. 2000;406:735–738. doi: 10.1038/35021074. [DOI] [PubMed] [Google Scholar]
- Motyl M, Dorso K, Barrett J, Giacobbe R. Basic Microbiological Techniques Used in Antibacterial Drug Discovery. Current Protocols in Pharmacology. 2006 doi: 10.1002/0471141755.ph13a03s31. [DOI] [PubMed] [Google Scholar]
- Muñoz-Elías EJ, McKinney JD. Mycobacterium tuberculosis isocitrate lyases 1 and 2 are jointly required for in vivo growth and virulence. Nat Med. 2005;11:638–644. doi: 10.1038/nm1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov GN, Skubák P, Lebedev AA, Pannu NS, Steiner RA, Nicholls RA, Winn MD, Long F, Vagin AA. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr D Biol Crystallogr. 2011;67:355–367. doi: 10.1107/S0907444911001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuñez MF, Pellicer MT, Badia J, Aguilar J, Baldoma L. Biochemical characterization of the 2-ketoacid reductases encoded by ycdW and yiaE genes in Escherichia coli. Biochem J. 2001;354:707–715. doi: 10.1042/0264-6021:3540707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obach RS, Baxter JG, Liston TE, Silber BM, Jones BC, MacIntyre F, Rance DJ, Wastall P. The prediction of human pharmacokinetic parameters from preclinical and in vitro metabolism data. J Pharmacol Exp Ther. 1997;283:46–58. [PubMed] [Google Scholar]
- Otwinowski Z, Minor W. Processing of X-ray Diffraction Data Collected in Oscillation Mode. Methods in Enzymology. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- Payne DJ, Gwynn MN, Holmes DJ, Pompliano DL. Drugs for bad bugs: confronting the challenges of antibacterial discovery. Nat Rev Drug Discov. 2007;6:29–40. doi: 10.1038/nrd2201. [DOI] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera–a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Philip V, Harris J, Adams R, Nguyen D, Spiers J, Baudry J, Howell EE, Hinde RJ. A survey of aspartate-phenylalanine and glutamate-phenylalanine interactions in the protein data bank: searching for anion-π pairs. Biochemistry. 2011;50:2939–2950. doi: 10.1021/bi200066k. [DOI] [PubMed] [Google Scholar]
- Quartararo CE, Blanchard JS. Kinetic and chemical mechanism of malate synthase from Mycobacterium tuberculosis. Biochemistry. 2011;50:6879–6887. doi: 10.1021/bi2007299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rullas J, García JI, Beltrán M, Cardona PJ, Cáceres N, García-Bustos JF, Angulo-Barturen I. Fast standardized therapeutic-efficacy assay for drug discovery against tuberculosis. Antimicrob Agents Chemother. 2010 May;54(5):2262–4. doi: 10.1128/AAC.01423-09. 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacchettini JC, Rubin EJ, Freundlich JS. Drugs versus bugs: in pursuit of the persistent predator Mycobacterium tuberculosis. Nat Rev Microbiol. 2008;6:41–52. doi: 10.1038/nrmicro1816. [DOI] [PubMed] [Google Scholar]
- Sakuraba H, Yoneda K, Takeuchi K, Tsuge H, Katunuma N, Ohshima T. Structure of an archaeal alanine:glyoxylate aminotransferase. Acta Crystallogr D Biol Crystallogr. 2008;64:696–699. doi: 10.1107/S0907444908006732. [DOI] [PubMed] [Google Scholar]
- Sambandamurthy VK, Derrick SC, Hsu T, Chen B, Larsen MH, Jalapathy KV, Chen M, Kim J, Porcelli SA, Chan J, et al. Mycobacterium tuberculosis DeltaRD1 DeltapanCD: a safe and limited replicating mutant strain that protects immunocompetent and immunocompromised mice against experimental tuberculosis. Vaccine. 2006;24:6309–6320. doi: 10.1016/j.vaccine.2006.05.097. [DOI] [PubMed] [Google Scholar]
- Schnappinger D, Ehrt S, Voskuil MI, Liu Y, Mangan JA, Monahan IM, Dolganov G, Efron B, Butcher PD, Nathan C, et al. Transcriptional Adaptation of Mycobacterium tuberculosis within Macrophages: Insights into the Phagosomal Environment. J Exp Med. 2003;198:693–704. doi: 10.1084/jem.20030846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schottel BL, Chifotides HT, Dunbar KR. Anion-pi interactions. Chem Soc Rev. 2008;37:68–83. doi: 10.1039/b614208g. [DOI] [PubMed] [Google Scholar]
- Sharma V, Sharma S, Bentrup K.H. zu, McKinney JD, Russell DG, Jacobs WR, Sacchettini JC. Structure of isocitrate lyase, a persistence factor of Mycobacterium tuberculosis. Nat Struct Biol. 2000;7:663–668. doi: 10.1038/77964. [DOI] [PubMed] [Google Scholar]
- Smith CV, Huang C, Miczak A, Russell DG, Sacchettini JC, Bentrup K.H. zu. Biochemical and structural studies of malate synthase from Mycobacterium tuberculosis. J Biol Chem. 2003;278:1735–1743. doi: 10.1074/jbc.M209248200. [DOI] [PubMed] [Google Scholar]
- Talaat AM, Lyons R, Howard ST, Johnston SA. The temporal expression profile of Mycobacterium tuberculosis infection in mice. Proc Natl Acad Sci U S A. 2004;101:4602–4607. doi: 10.1073/pnas.0306023101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timm J, Post FA, Bekker L-G, Walther GB, Wainwright HC, Manganelli R, Chan W-T, Tsenova L, Gold B, Smith I, et al. Differential expression of iron-, carbon-, and oxygen-responsive mycobacterial genes in the lungs of chronically infected mice and tuberculosis patients. Proc Natl Acad Sci U S A. 2003;100:14321–14326. doi: 10.1073/pnas.2436197100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wayne LG, Lin KY. Glyoxylate metabolism and adaptation of Mycobacterium tuberculosis to survival under anaerobic conditions. Infect Immun. 1982;37:1042–1049. doi: 10.1128/iai.37.3.1042-1049.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yazdanian M, Glynn SL, Wright JL, Hawi A. Correlating partitioning and caco-2 cell permeability of structurally diverse small molecular weight compounds. Pharm Res. 1998;15:1490–1494. doi: 10.1023/a:1011930411574. [DOI] [PubMed] [Google Scholar]
- World Health Organization . Global Tuberculosis Control 2011. Switzerland; Geneva: 2011. http://www.who.int/tb/publications/global_report/en/ [Google Scholar]
- Zeng L-F, Jiang X-H, Sanchez T, Zhang H-S, Dayam R, Neamati N, Long Y-Q. Novel dimeric aryldiketo containing inhibitors of HIV-1 integrase: effects of the phenyl substituent and the linker orientation. Bioorg Med Chem. 2008;16:7777–7787. doi: 10.1016/j.bmc.2008.07.008. [DOI] [PubMed] [Google Scholar]
- Collaborative Computational Project, Number 4. Acta Cryst. 1994;D50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.