

Abstract
The neural network controlling breathing must establish rhythmic motor output at a level adequate to sustain life. Reduced respiratory neural activity elicits a novel form of plasticity in circuits driving the diaphragm known as inactivity-induced phrenic motor facilitation (iPMF), a rebound increase in phrenic inspiratory output observed once respiratory neural drive is restored. The mechanisms underlying iPMF are unknown. Here, we demonstrate in anesthetized rats that spinal mechanisms give rise to iPMF and that iPMF consists of at least two mechanistically distinct phases: (1) an early, labile phase that requires atypical PKC (PKCζ and/or PKCι/λ) activity to transition to a (2) late, stable phase. Early (but not late) iPMF is associated with increased interactions between PKCζ/ι and the scaffolding protein ZIP (PKCζ-interacting protein)/p62 in spinal regions associated with the phrenic motor pool. Although PKCζ/ι activity is necessary for iPMF, spinal atypical PKC activity is not necessary for phrenic long-term facilitation (pLTF) following acute intermittent hypoxia, an activity-independent form of spinal respiratory plasticity. Thus, while iPMF and pLTF both manifest as prolonged increases in phrenic burst amplitude, they arise from distinct spinal cellular pathways. Our data are consistent with the hypotheses that (1) local mechanisms sense and respond to reduced respiratory-related activity in the phrenic motor pool and (2) inactivity-induced increases in phrenic inspiratory output require local PKCζ/ι activity to stabilize into a long-lasting iPMF. Although the physiological role of iPMF is unknown, we suspect that iPMF represents a compensatory mechanism, assuring adequate motor output in a physiological system in which prolonged inactivity ends life.
Introduction
To sustain life, neurons controlling breathing must transmit a continuous rhythmic motor output of appropriate magnitude to respiratory muscles, thereby assuring adequate breathing. Since even the briefest pause in breathing threatens life, the respiratory control system may exhibit unique characteristics, such as rapid responses to periods of reduced neural activity. We recently described a novel form of plasticity elicited by reduced central respiratory neural activity (in the absence of hypoxia) known as inactivity-induced phrenic motor facilitation (iPMF) (Baker-Herman and Strey, 2011; Mahamed et al., 2011), a rebound increase in inspiratory motor output to the diaphragm revealed when central respiratory neural activity is restored. iPMF is induced by multiple forms of brainstem-initiated (i.e., central) apnea, including those caused by hyperventilation, increased inhibitory sensory feedback, and anesthesia-induced respiratory depression (Baker-Herman and Strey, 2011; Mahamed et al., 2011). Because of the diverse nature of these initiating stimuli, we suggested that iPMF arises from a common factor: reduced respiratory neural activity (Baker-Herman and Strey, 2011).
Although a central neural apnea results in reduced activity in many respiratory-related neurons throughout the neuraxis, we hypothesized that mechanisms operating within or near the phrenic motor pool give rise to iPMF (Baker-Herman and Strey, 2011). Consistent with this hypothesis, a 4 h disruption in descending synaptic inputs to phrenic motor neurons via unilateral axon conduction block enhances ipsilateral diaphragm EMG activity once axon conduction is restored (Castro-Moure and Goshgarian, 1996), suggesting that mechanisms downstream from brainstem respiratory networks are sufficient to induce plasticity in response to phrenic/diaphragm inactivity. However, it is currently unknown whether inactivity-induced diaphragm EMG facilitation is due to mechanisms operating in the phrenic motor circuit or at the neuromuscular junction. Indeed, both the diaphragm (Mantilla and Sieck, 2009) and phrenic motor pool (Castro-Moure and Goshgarian, 1997) exhibit profound morphological changes in response to prolonged inactivity.
Here, we provide the first evidence that reduced central respiratory neural activity elicits mechanisms of plasticity in or near the phrenic motor pool that manifest as a prolonged enhancement of phrenic burst amplitude once respiratory neural activity is restored (i.e., iPMF). Furthermore, we demonstrate that iPMF requires activity of atypical protein kinase C (aPKC) isoforms, PKCζ and/or PKCι. Specifically, spinal PKCζ/ι activity following a neural apnea is necessary to transition from an early, transient facilitation to a longer-lasting, stable iPMF; however, once formed, continued PKCζ/ι activity is no longer necessary to maintain late iPMF. Finally, early (but not late) iPMF is associated with increased interaction between PKCζ/ι and the scaffolding protein PKCζ-interacting protein (ZIP) (also known as p62) in spinal regions associated with the phrenic motor pool, suggesting activation of PKCζ/ι in the relevant time frame at relevant sites. In contrast, spinal aPKC is not necessary for an activity-independent form of phrenic plasticity, phrenic long-term facilitation (pLTF) following acute intermittent hypoxia (Dale-Nagle et al., 2010), suggesting that these forms of spinal plasticity occur via distinct cellular mechanisms. To our knowledge, this is the first report describing a key role for local PKCζ/ι activity in stabilizing plasticity in any spinal motor output.
Materials and Methods
Animals.
Experiments were performed on 2- to 5-month-old male Sprague Dawley rats (Harlan colony 217). Rats were housed two per cage in a controlled environment (12 h light/dark cycle), with food and water ad libitum. The Institutional Animal Care and Use Committee at the University of Wisconsin–Madison approved all experiments.
Surgical preparation.
Anesthesia was induced with isoflurane in a closed chamber, followed by inhalation of isoflurane through a nose cone (2.5–3.5% in 50% O2, balance N2). Rats were tracheotomized and pump ventilated (tidal volume, 2–2.5 ml; Harvard Apparatus; Rodent Ventilator 683), and isoflurane was continued through the ventilator. End-tidal CO2 was monitored (Respironics Novametrix) and maintained at ∼45 mmHg throughout surgery by adjusting ventilation rate and/or adding CO2 to the inspired gas mix. A bilateral vagotomy was performed to prevent entrainment of respiratory frequency with the ventilator. Tracheal pressure was monitored throughout the surgery to assure rats continued respiratory efforts during surgery. The femoral artery was catheterized for blood pressure measurement and periodic blood gas sampling. Depending on surgeon preference, the tail or femoral vein was catheterized for fluid infusion. C2 laminectomy was performed, and a small hole was cut in the dura to place a soft silicone catheter (2 French; Access Technologies) connected to a Hamilton syringe in the intrathecal space over spinal segment C4. The left phrenic nerve was exposed dorsally, cut distally, desheathed, and submersed in mineral oil. Rats were converted to urethane anesthesia (1.7–1.8 mg/kg, i.v.), and isoflurane was withdrawn. Rats were then paralyzed with pancuronium bromide (2.5 mg/kg, i.v.), and 1–3 ml/h fluid infusion of 1:4 hetastarch (Hespan, 6% hetastarch in 0.9% sodium chloride), and lactated Ringer's solution was started and continued throughout the experiment. Body temperature was maintained at 37°C using a rectal probe and a custom-designed heated table.
Electrophysiological measurements.
The phrenic nerve was placed on a bipolar silver electrode. Phrenic activity was amplified (10,000×), bandpass filtered (0.3–20 kHz), and integrated (time constant, 50 ms). Raw and integrated traces were recorded using either the WinDAQ (DATAQ Instruments) or PowerLab (ADInstruments) data acquisition systems.
End-tidal CO2 was used as an index of arterial PCO2. These values were confirmed by periodically sampling arterial blood at key time points for blood gas analysis (Radiometer Copenhagen; ABL 500). Blood pressure was monitored and used as an indicator of the stability of the preparation and depth of anesthesia by pressor responses to paw pad pinch.
Pharmacological treatments.
Stock solutions of the following compounds were prepared in artificial CSF (aCSF) (in mm: 120 NaCl, 3 KCl, 2 CaCl, 2 MgCl, 23 NaHCO3, 10 glucose bubbled with 95% O2/5% CO2): myristoylated ζ-pseudosubstrate inhibitory peptide (PKCζ-PS) (2 μg/μl; Tocris Bioscience), myristoylated scrambled ζ-pseudosubstrate peptide (scrPKCζ-PS) (2 μg/μl; Tocris Bioscience), (S)-2,6-diamino-N-[(1-(1-oxotridecyl)-2-piperidinyl)methyl]hexanamide (NPC-15437) (2.4 μg/μl; Enzo Life Sciences). Bisindolylmaleimide I (BIM) (Tocris Bioscience) was prepared in 1.7% DMSO (in aCSF; 10 ng/μl) or in 17% DMSO (100 ng/μl). It should be noted that PKCζ-PS is also frequently referred to as “ZIP” (ζ-pseudosubstrate inhibitory peptide) in the literature; however, to avoid confusion with the scaffolding protein ZIP/p62, we refer to this compound as “PKCζ-PS.” All compounds were delivered intrathecally at doses consistent with those reported in the literature that block different forms of spinal plasticity: PKCζ-PS (10 nmol) (Asiedu et al., 2011; Laferrière et al., 2011), scrPKCζ-PS (10 nmol), NPC-15437 (50 nmol) (Laferrière et al., 2011), and BIM (0.24 and 2.4 nmol) (Yashpal et al., 1995; Hua et al., 1999; Ferguson et al., 2008) (M. Devinney and G. Mitchell, personal communication). Vehicle-treated rats received aCSF. For all compounds, a total of 10 μl was delivered in the intrathecal space in 1–2 μl boluses over 2 min.
Electrophysiological protocols.
Approximately 1 h after conversion to urethane anesthesia, CO2 apneic and recruitment thresholds for phrenic activity were determined to set baseline phrenic nerve activity. Apneic threshold was determined by monitoring end-tidal CO2 and slowly increasing ventilator rate and/or lowering inspired CO2 until rhythmic phrenic burst activity ceased. Recruitment threshold was determined by slowly lowering ventilator rate and/or increasing inspired CO2 until phrenic activity resumed. End-tidal CO2 was raised a further 2 mmHg to establish baseline phrenic discharge. After 20–30 min of stable phrenic burst amplitude and frequency, an arterial blood sample was drawn; arterial PCO2 and phrenic burst activity at this time point were considered “baseline” for all subsequent measurements. Rats were then subjected to one of the following three protocols: (1) neural apnea, (2) acute intermittent hypoxia, or (3) time control (see below).
Central (brainstem) respiratory neural activity was eliminated by clamping PaCO2 ∼5 mmHg below the CO2 threshold for breathing. This “neural apnea” was achieved by increasing ventilator rate and/or decreasing inspired CO2 until end-tidal CO2 was just below the apneic threshold and phrenic bursting ceased. Neural apnea was maintained for 30 min, and then respiratory neural activity was resumed by returning arterial PCO2 to baseline values. Since rats were mechanically ventilated, arterial oxygen levels were maintained throughout the neural apnea. Blood gas analysis confirmed that arterial oxygen levels were >150 mmHg throughout the protocol, and that PaCO2 levels returned to within 1.5 mmHg of baseline following restoration of central respiratory neural activity. Rats exposed to a neural apnea received intrathecal injections of the following: (1) aCSF (n = 10), scrPKCζ-PS (n = 8), or PKCζ-PS (n = 8) ∼20 min before neural apnea; (2) scrPKCζ-PS (n = 7), PKCζ-PS (n = 6), NPC-15437 (n = 5), or BIM (n = 3, each dose) 10 min following the resumption of respiratory neural activity; or (3) PKCζ-PS (n = 3) 45 min following the resumption of respiratory neural activity.
Acute intermittent hypoxia (IH) consisted of three 5 min episodes of hypoxia (11%; PaO2 = 36 ± 1 mmHg) separated by a 5 min return to baseline oxygen conditions. Following IH, baseline oxygen conditions were restored. Blood gas analysis confirmed that arterial PO2 was between 35 and 45 mmHg during hypoxia and that arterial CO2 was maintained within 1.5 mmHg of baseline following IH exposure. Rats exposed to IH received intrathecal injections of either (1) scrPKCζ-PS (n = 3) or PKCζ-PS (n = 8) ∼20 min before IH or (2) scrPKCζ-PS (n = 3) or PKCζ-PS (n = 5) 10 min following IH exposures.
To control for any time-dependent changes in phrenic motor output not related to our treatments, results were compared with “time controls” that received similar surgery and experimental duration, but no neural apnea or IH. Time control rats received either intrathecal aCSF (n = 6), PKCζ-PS (n = 7), or NPC-15437 (n = 3).
Phrenic burst amplitude was monitored continuously before, during, and for 60–90 min following the above protocols. Arterial blood samples were analyzed before, during, and at 10–15, 25–30, 60, and 90 min after each treatment protocol to ensure adequate maintenance of baseline arterial PCO2, PO2, SBE (base excess), and pH (note: 90 min blood gas sample was only collected in rats receiving poststimulus drug injections). At the end of each protocol, a maximal CO2 response (90 < PETCO2 < 100) was assessed to ensure that observed responses were not influenced by deterioration of the preparation. To be included in the analysis, rats had to meet the following criteria: arterial PO2 of >150 mmHg (except during hypoxia in IH rats), arterial PCO2 maintained within 1.5 mmHg of baseline (except during neural apnea), and base excess within 3 mEq/L from baseline values.
Tissue preparation for immunoprecipitation.
In separate subgroups of rats, the association of PKCζ and PKCι with ZIP/p62 was measured at different time points following a neural apnea and were compared with a similar surgical duration in time controls. Rats were prepared as described above. At 10 (neural apnea, n = 6; time control, n = 6), 20 (neural apnea, n = 3; time control, n = 3), or 60 (neural apnea, n = 8; time control, n = 4) min following restoration of respiratory neural activity, the C4 spinal segment was harvested and stored en bloc at −80°C for future analysis.
Segments were placed on a freezing microtome, and successive 40 μm sections of the dorsal horn were removed and discarded until the ventral aspect of the central canal was visible. Ventral white matter was removed in the same manner until ventral gray matter was visible. Lateral white matter was removed with a no. 11 scalpel blade under a dissecting microscope. The remaining ventral horn was placed in 200 μl of ice-cold immunoprecipitation (IP) buffer [50 mm Tris-HCl, pH 7.5, 150 mm NaCl, 1 mm EDTA, 1% (v/v) Nonidet P 40, 1× Protease Inhibitor Mixture (Sigma-Aldrich), 1× Halt Phosphatase Inhibitor Mixture (Pierce Biotechnology)] and homogenized in a glass–Teflon Dounce homogenizer with 15–20 strokes of the pestle. Homogenates were centrifuged at 2500 × g for 10 min at 4°C. Protein concentration of the supernatant was measured using the BCA Protein Assay Kit (Pierce Biotechnology) according to the manufacturer's instructions. Supernatants were diluted to 2.21 μg/μl with IP buffer, and 442 μg of total protein was used for immunoprecipitation.
Immunoprecipitation.
Supernatants were incubated with 1 μg of anti-PKCζ polyclonal antibody (C20; Santa Cruz Biotechnology) or 1 μg of preimmune rabbit IgG (Sigma-Aldrich) at 4°C overnight. The substrate–antibody complex was precipitated with protein G-Sepharose (Sigma-Aldrich) at 4°C for 1 h, washed four times in 15× volumes of IP buffer, and eluted by incubation in SDS-PAGE loading buffer [62.5 mm Tris-HCl, pH 6.8, 2% SDS, 20% glycerol, 5% (v/v) β-mercaptoethanol] at 96°C for 5 min.
Immunoblotting.
Samples were resolved by PAGE on 4–15% Tris-HCl gels in Tris/glycine/SDS buffer (Bio-Rad) and transferred to Immobilon polyvinylidene difluoride membranes (Millipore). Blots were blocked in 5% milk in 1× TBST (20 mm Tris, 500 mm NaCl, 0.1% Tween) for 1 h and probed with anti- SQSTM/p62 monoclonal antibody (1:8000; Abcam) overnight at 4°C. Immunoblots were rinsed in 1× TBST and probed with HRP-conjugated horse anti-mouse IgG (1:10,000; Cell Signaling Technology) for 1 h at room temperature. The immunoblot label was expressed using Pierce Super Signal West Dura extended duration chemiluminescent kit (Pierce Biotechnology). Chemiluminescence was detected and digitized using an AutoChemi Imaging system (UVP Bioimaging Systems) with LabWorks 4.6. To ensure equal loading, blots were stripped using Pierce Restore Western blot Stripping Buffer (Pierce Biotechnology) and reprobed with anti-PKCζ polyclonal antibody (1:1000; Santa Cruz Biotechnology) and HRP-conjugated goat anti-rabbit IgG (1:10,000; Cell Signaling Technology).
Immunofluorescence.
In separate subgroups of rats, immunofluorescence was used to assess changes in aPKC levels following a neural apnea and to determine whether aPKC and ZIP/p62 were colocalized in identified phrenic motor neurons. Rats were anesthetized with isoflurane through a nose cone (1–2% isoflurane in 50% O2, balance N2). A 25 μl Hamilton syringe was used to bilaterally inject 10 μl of 5 mg/ml cholera toxin B subunit solution (CTB) (List Biological Laboratories) in the intrapleural space (Mantilla et al., 2009). Rats were allowed to recover for 5 d before electrophysiological experiments described above (neural apnea, n = 3; time control, n = 4). Following electrophysiological measurements, animals were transcardially perfused with 4% paraformaldehyde (PFA) in 1× PBS, pH 7.4.
Spinal segments C3–C6 were collected, postfixed for 24 h in 4% PFA, and cryoprotected by sequential 24 h incubations in 20% sucrose and 30% sucrose in 1× PBS, and 40 μm coronal sections were prepared using a Leica SM200R sliding microtome. Spinal cord sections were incubated with antibodies against CTB (1:10,000 anti-CTB; Calbiochem), aPKC (1:1000 anti-PKCζ/ι C-20; Santa Cruz Biotechnology), and ZIP/p62 (1:500 anti-SQSTM/p62; Abcam) to localize aPKC and ZIP/p62 with CTB-backlabeled phrenic motor neurons. All secondary antibodies were conjugated to Alexa Fluor fluorescent dyes (Invitrogen). Sections were mounted on slides with ProLong Gold (Invitrogen). Immunofluorescence images were obtained as described by Guenther et al. (2012). All images were collected with the same laser and EZ-C1 software settings.
Statistical analysis.
Integrated phrenic burst amplitude was analyzed in 30–60 s bins before (baseline), and 5, 15, 30, 60, and 90 min following neural apnea, intermittent hypoxia, or an equivalent duration in time controls (90 min time point was only analyzed in rats receiving intrathecal compounds after neural apnea). Since there were no differences in phrenic burst amplitude at any time point between time controls receiving intrathecal aCSF or PKCζ-PS, burst amplitude data from these groups were combined for comparison with groups receiving PKCζ-PS or scrPKCζ-PS and neural apnea. Integrated phrenic burst amplitude was expressed as a percentage change from baseline. A two-way repeated-measures ANOVA was used to detect significant differences (Prism 5; GraphPad Software). Individual comparisons were made via Bonferroni's post hoc tests at a significance level of 0.05.
Arterial PCO2, PO2, and pH were analyzed using a two-way repeated-measures ANOVA (Prism 5; GraphPad Software) before (baseline), and 10–15, 25–30, 60, and 90 min following neural apnea, intermittent hypoxia, or an equivalent duration in time controls (90 min time point was only analyzed in rats receiving intrathecal compounds after neural apnea).
ZIP/p62 and PKCζ/ι immunoblot images were analyzed with ImageJ software. Images were converted to an 8 bit format, and thresholds were set manually and masked. ZIP/p62 and PKCζ/ι bands were outlined; the resulting regions were superimposed on the original 8 bit version of the same image, and mean gray value of each region was measured. ZIP/p62 density was normalized to PKCζ/ι density, and ANOVA was used to detect significant differences (Prism 5; GraphPad Software) between groups. Individual differences were determined by Student–Newman–Keuls post hoc tests at a significance level of 0.05. All data are presented as means ± SEM.
Results
To induce a reversible disruption in central respiratory neural activity, rats were hyperventilated just below the CO2 apneic threshold for breathing. This “neural apnea” was maintained for 30 min while continuing arterial oxygen support with the ventilator. Central respiratory neural activity was resumed by restoring arterial PCO2 to baseline levels. Table 1 lists average arterial PCO2, PO2, and pH during baseline and at the end of the protocol; no time-dependent changes in post-neural apnea blood gases were apparent in any rat group.
Table 1.
Arterial levels of PO2, PCO2, and pH in rat groups before and at the end of the protocol
| PO2 | PCO2 | pH | |
|---|---|---|---|
| Time control | |||
| Baseline | 250 ± 7 | 47.4 ± 1.5 | 7.36 ± 0.01 |
| 60 min | 253 ± 3 | 47.4 ± 1.5 | 7.35 ± 0.01 |
| Neural apnea + pre-aCSF | |||
| Baseline | 253 ± 10 | 47.3 ± 1.2 | 7.35 ± 0.01 |
| 60 min | 249 ± 13 | 47.6 ± 1.3 | 7.34 ± 0.01 |
| Neural apnea + pre-PKCζ-PS | |||
| Baseline | 239 ± 5 | 47 ± 1.9 | 7.34 ± 0.01 |
| 60 min | 246 ± 6 | 46.4 ± 1.9 | 7.33 ± 0.02 |
| Neural apnea + pre-scrPKCζ-PS | |||
| Baseline | 240 ± 12 | 49.4 ± 1.5 | 7.34 ± 0.02 |
| 60 min | 241 ± 10 | 49.6 ± 1.6 | 7.33 ± 0.01 |
| IH + pre-PKCζ-PS | |||
| Baseline | 266 ± 12 | 50.3 ± 1.2 | 7.34 ± 0.01 |
| 60 min | 270 ± 11 | 50.6 ± 1 | 7.30 ± 0.01 |
| IH + pre-scrPKCζ-PS | |||
| Baseline | 277 ± 21 | 48.6 ± 2 | 7.37 ± 0.04 |
| 60 min | 270 ± 11 | 48.9 ± 2 | 7.33 ± 0.01 |
| Neural apnea + PKCζ-PS-10 | |||
| Baseline | 276 ± 24 | 51.8 ± 0.9 | 7.34 ± 0.01 |
| 90 min | 297 ± 7 | 51.4 ± 1 | 7.34 ± 0.01 |
| Neural apnea + scrPKCζ-PS-10 | |||
| Baseline | 276 ± 17 | 49.2 ± 1 | 7.33 ± 0.01 |
| 90 min | 281 ± 19 | 49.8 ± 1 | 7.34 ± 0.02 |
| IH + PKCζ-PS-10 | |||
| Baseline | 254 ± 28 | 47.3 ± 2.6 | 7.35 ± 0.02 |
| 90 min | 288 ± 13 | 47.1 ± 2.1 | 7.36 ± 0.01 |
| IH + scrPKCζ-PS-10 | |||
| Baseline | 265 ± 50 | 46.7 ± 1.9 | 7.36 ± 0.01 |
| 90 min | 286 ± 25 | 46.6 ± 1.7 | 7.38 ± 0.01 |
| Neural apnea + PKCζ-PS-45 | |||
| Baseline | 258 ± 4 | 42.3 ± 2 | 7.36 ± 0.02 |
| 60 min | 226 ± 26 | 43 ± 2.3 | 7.37 ± 0.01 |
| Time control + NPC-15437 | |||
| Baseline | 299 ± 20 | 45.4 ± 1.6 | 7.37 ± 0.02 |
| 90 min | 290 ± 6 | 45 ± 1.3 | 7.38 ± 0.01 |
| Neural apnea + NPC-15437 | |||
| Baseline | 282 ± 18 | 42.3 ± 1.3 | 7.38 ± 0.01 |
| 90 min | 303 ± 19 | 42.4 ± 1.4 | 7.40 ± 0.02 |
| Neural apnea + BIM | |||
| Baseline | 275 ± 6 | 43 ± 0.4 | 7.36 ± 0.01 |
| 90 min | 244 ± 15 | 43.1 ± 0.6 | 7.38 ± 0.03 |
No significant differences were observed over time in any group.
iPMF requires spinal aPKC activity
Figure 1A depicts representative compressed phrenic neurograms, illustrating phrenic burst amplitude during baseline, a 30 min neural apnea, and for 60 min following restoration of respiratory neural activity (representative time control traces not shown). Figure 1B represents the average change in phrenic burst amplitude from baseline for 60 min following restoration of respiratory neural activity after neural apnea (or equivalent points in time controls) in all rats. In rats with vehicle (aCSF) injections, phrenic burst amplitude following neural apnea was significantly increased relative to baseline (all time points, p < 0.05) and significantly increased relative to time control rats that experienced the same surgical procedures, but no neural apnea (all time points, p < 0.05; Fig. 1A,B), indicating iPMF. iPMF lasted for at least 60 min following restoration of respiratory neural activity. These data demonstrate that reduced central respiratory neural activity elicits iPMF, consistent with other reports (Baker-Herman and Strey, 2011; Mahamed et al., 2011).
Figure 1.
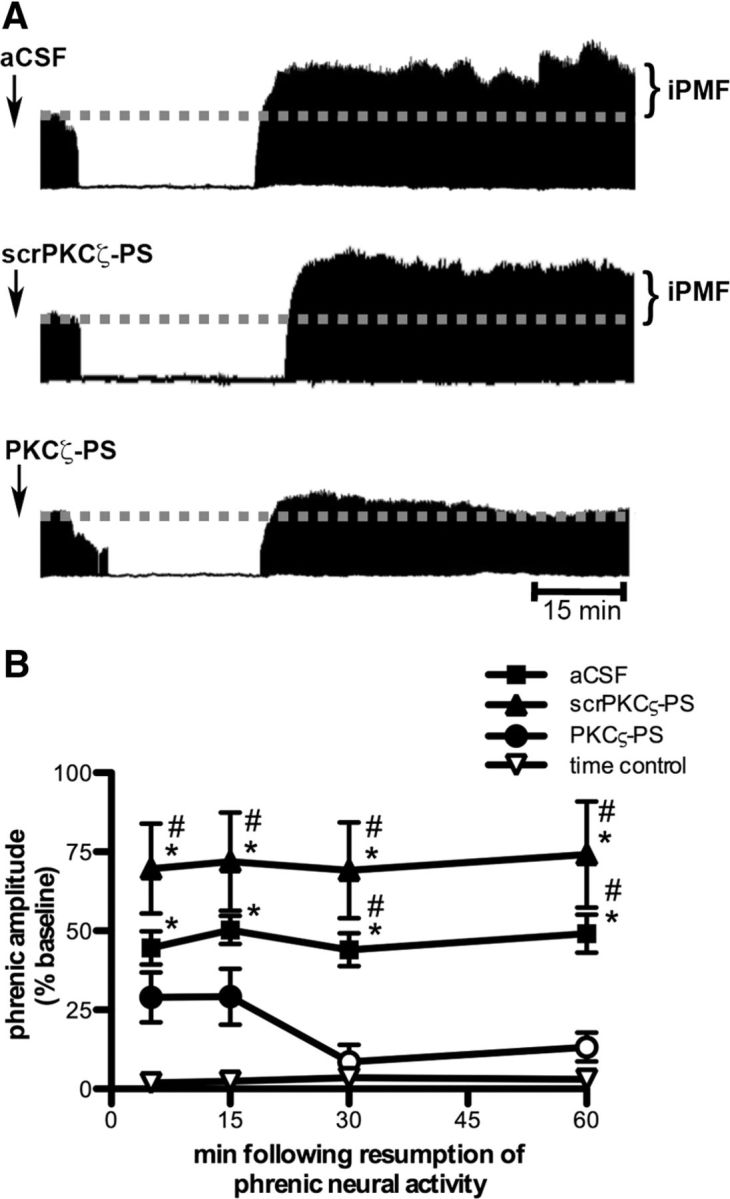
Spinal aPKC activity is necessary for iPMF. A, Representative compressed and integrated phrenic neurograms before, during, and for 60 min following a 30 min neural apnea, illustrating that rats receiving injections (arrows) of intrathecal aCSF (top) or scrPKCζ-PS (middle) before neural apnea expressed a prolonged increase in phrenic burst amplitude following resumption of respiratory neural activity, indicating iPMF. Only modest, transient increases in phrenic burst amplitude after neural apnea were observed rats receiving intrathecal PKCζ-PS (bottom) before a neural apnea, indicating that spinal aPKC inhibition attenuated and shortened iPMF. B, Average change in phrenic burst amplitude from baseline for 60 min following resumption of respiratory neural activity in rats receiving intrathecal aCSF (squares), scrPKCζ-PS (triangles), or PKCζ-PS (circles) 20 min before neural apnea. Prolonged iPMF is expressed in rats receiving control injections of aCSF or scrPKCζ-PS, when compared with baseline or time controls (inverted triangles). Rats receiving intrathecal PKCζ-PS (circles) expressed only transient, modest iPMF since phrenic burst amplitude was significantly increased from baseline only at 5 and 15 min following resumption of respiratory neural activity, which was not significantly different from time controls. No changes in phrenic burst amplitude were observed in time controls receiving similar surgery, but no neural apnea. Mean values ± SEM. The filled symbols indicate significantly different from baseline. *Significantly different from time controls. #Significantly different from PKCζ-PS rats. p < 0.05.
The PKC superfamily is divided into classical, novel, and atypical subfamilies based on structure and requirement for coactivators (Tanaka and Nishizuka, 1994; Reyland, 2009). To determine the role of aPKC activity in iPMF, rats received an intrathecal injection of PKCζ-PS, a cell-permeable pseudosubstrate inhibitory peptide that binds to and inhibits the catalytic domain of all aPKC isoforms. As expected, rats receiving intrathecal scrPKCζ-PS before a neural apnea exhibited significant increases in phrenic burst amplitude relative to baseline and time control rats at all time points following restoration of respiratory neural activity (all p < 0.05; Fig. 1A,B), indicating scrPKCζ-PS does not interfere with iPMF. In contrast, rats pretreated with intrathecal PKCζ-PS expressed significant increases in phrenic burst amplitude relative to baseline shortly after restoration of respiratory neural activity (5 and 15 min; p < 0.05), which was significantly lower than rats receiving scrPKCζ-PS (p < 0.05) and was not significantly different from time controls (p > 0.05; Fig. 1A,B). These data suggest that spinal aPKC inhibition attenuates, but does not eliminate early phases of iPMF. This modest facilitation was only transient since phrenic burst amplitude was no longer significantly different from baseline or time controls by 30 and 60 min following restoration of respiratory neural activity (p > 0.05). Thus, whereas expression of late iPMF requires spinal aPKC activity, early iPMF is only partially dependent on its activity.
Spinal aPKC activity is required to stabilize iPMF
To determine whether continued spinal aPKC activity following resumption of respiratory neural activity is required for late iPMF, rats were exposed to a 30 min neural apnea, and then respiratory neural activity was restored; 10 min following restoration of respiratory neural activity, rats received intrathecal scrPKCζ-PS or PKCζ-PS (i.e., after early iPMF had developed). Figure 2A depicts representative compressed phrenic neurograms, illustrating phrenic burst amplitude during baseline, 30 min neural apnea, and for 90 min following the restoration of respiratory neural activity. Figure 2B illustrates the average change in phrenic burst amplitude from baseline during the first 90 min after neural apnea. Rats receiving intrathecal scrPKCζ-PS or PKCζ-PS 10 min following restoration of respiratory neural activity (scrPKCζ-PS-10 and PKCζ-PS-10, respectively) expressed a similar significant increase in phrenic burst amplitude immediately before intrathecal drug application (both p < 0.05 relative to baseline), indicating that both rat groups began with a similar iPMF magnitude. As expected, intrathecal scrPKCζ-PS-10 did not impair subsequent iPMF expression since phrenic burst amplitude remained significantly increased relative to baseline at all time points following scrPKCζ-PS-10 injections (p < 0.05); these values were not significantly different from phrenic activity before scrPKCζ-PS-10 treatment (p > 0.05). In contrast, phrenic burst amplitude steadily decreased in rats receiving intrathecal PKCζ-PS-10, such that by 30 min after neural apnea, phrenic burst amplitude was significantly reduced relative to that observed before intrathecal PKCζ-PS-10 injections and remained below preinjection levels until the end of the recording (all p < 0.05). In addition, phrenic burst amplitude following intrathecal PKCζ-PS-10 was significantly reduced relative to scrPKCζ-PS-10 at 60 and 90 min after neural apnea (p < 0.05), suggesting that aPKC inhibition during early phases of iPMF attenuates late iPMF.
Figure 2.
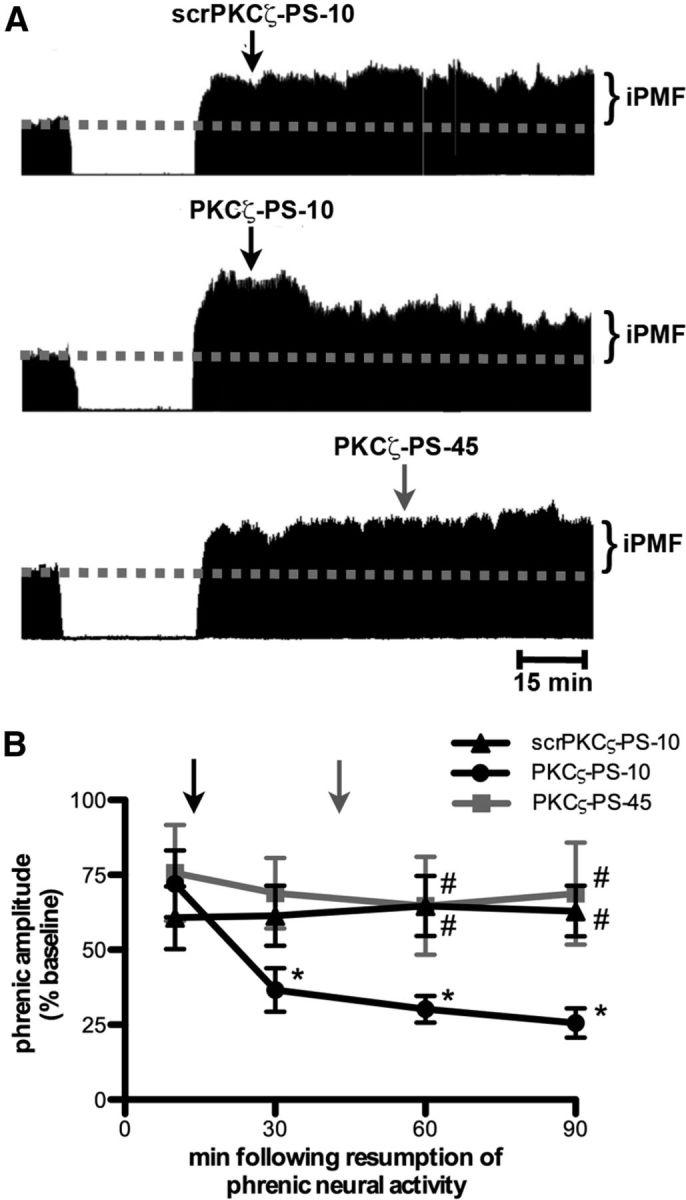
Spinal aPKC activity following a neural apnea stabilizes but does not maintain iPMF. A, Representative compressed and integrated phrenic neurograms before, during, and for 90 min following a 30 min neural apnea, illustrating that a control injection of intrathecal scrPKCζ-PS 10 min (scrPKCζ-PS-10; top) following resumption of respiratory neural activity did not impair neural apnea-induced iPMF. In contrast, intrathecal PKCζ-PS delivered 10 min (PKCζ-PS-10; middle), but not 45 min (PKCζ-PS-45; bottom), following resumption of respiratory neural activity returned phrenic burst amplitude toward baseline. The arrows indicate time points of drug delivery. B, Average change in phrenic burst amplitude from baseline for 90 min following neural apnea in rats receiving intrathecal scrPKCζ-PS-10 (triangles), PKCζ-PS-10 (circles), or PKCζ-PS-45 (gray square) at time points after resuming respiratory neural activity. The arrows indicate time points of drug delivery (black arrow for scrPKCζ-PS-10 and PKCζ-PS-10; gray arrow for PKCζ-PS-45). All rats expressed significant increases in phrenic burst amplitude relative to baseline or time controls (data not shown) immediately before drug injections, indicating iPMF. Significant iPMF was observed in rats receiving intrathecal scrPKCζ-PS-10 for up to 90 min (vs baseline or time controls), whereas iPMF progressively declined following PKCζ-PS-10 injections, such that by 30 min following resumption of respiratory neural activity, iPMF was significantly decreased from the pre-PKCζ-PS-10 injection value. No impairment in iPMF was observed in PKCζ-PS-45 rats. Intrathecal PKCζ-PS-10 was significantly lower than scrPKCζ-PS-10 and PKCζ-PS-45 at 60 and 90 min following neural apnea. Shown are mean values ± SEM. The filled symbols indicate significantly different from baseline. *Significantly different from preinjection time point. #Significantly different from PKCζ-PS-10 rats. p < 0.05.
To determine whether aPKC activity is required to maintain late iPMF, we delivered intrathecal PKCζ-PS 45 min (PKCζ-PS-45) following restoration of respiratory neural activity. As expected, rats expressed a significant iPMF immediately before PKCζ-PS-45 application (p < 0.05; Fig. 2A,B). However, contrary to intrathecal PKCζ-PS-10, intrathecal PKCζ-PS-45 did not interfere with subsequent iPMF expression. Indeed, rats receiving intrathecal PKCζ-PS-45 continued to express significant increases in phrenic burst amplitude from baseline at all measured time points after intrathecal PKCζ-PS-45 (p < 0.05), which was not significantly different from phrenic amplitude before PKCζ-PS-45 injections or scrPKCζ-PS values at any time point (all p > 0.05). Collectively, these data suggest that spinal aPKC activity is necessary to transition from early to late iPMF, but continued aPKC activity is not necessary to maintain late iPMF.
pLTF does not require aPKC activity
To determine whether spinal aPKC activity is a general requirement for phrenic plasticity, we investigated the role of aPKC in a contrasting model, pLTF, following moderate acute IH (Dale-Nagle et al., 2010; Baker-Herman and Strey, 2011). In contrast to iPMF, pLTF is activity independent (MacFarlane and Mitchell, 2009) and is expressed as a progressive ramping of phrenic burst amplitude that is often not significantly increased from baseline until 30 min following IH in anesthetized rats. Figure 3A depicts representative compressed phrenic neurograms, illustrating phrenic burst amplitude before, during, and for 60 min following three hypoxic episodes. Figure 3B illustrates the average change in phrenic burst amplitude from baseline for 60 min after IH. In rats receiving control injections of intrathecal scrPKCζ-PS before IH, phrenic burst amplitude was significantly greater than baseline at all measured time points after IH (p < 0.05) and significantly greater than time controls at 60 min after IH (p < 0.05). Similarly, in rats receiving intrathecal PKCζ-PS before IH, phrenic burst amplitude was significantly increased relative to baseline and time controls at 30 and 60 min (p < 0.05). Importantly, there were no significant differences in phrenic burst amplitude between PKCζ-PS- and scrPKCζ-PS-treated rats at any time point following IH (p > 0.05). These data suggest that spinal pretreatment with an aPKC inhibitor did not impair pLTF following IH.
Figure 3.

Intermittent hypoxia-induced pLTF does not require spinal aPKC activity. A, Representative compressed and integrated phrenic neurograms before, during, and for 60–90 min following three 5 min episodes of hypoxia (11% O2), illustrating the development of pLTF in rats receiving intrathecal scrPKCζ-PS (top) or PKCζ-PS (middle) 20 min before IH, or PKCζ-PS delivered 10 min (PKCζ-PS-10; bottom) following IH. The arrows indicate time points of drug delivery. B, Average change in phrenic burst amplitude from baseline for 60 min following IH in rats receiving intrathecal scrPKCζ-PS (circles) or PKCζ-PS (squares) 20 min before IH. Both rat groups exhibited significantly increased phrenic burst amplitude 60 min following IH relative to baseline. No differences were observed between rats receiving intrathecal scrPKCζ-PS or PKCζ-PS at any point. C, Average change in phrenic burst amplitude from baseline following IH in rats receiving intrathecal scrPKCζ-PS (circles) or PKCζ-PS (squares) 10 min following IH. The arrow indicates time point of drug delivery. Both groups exhibited significant increases in phrenic burst amplitude at 30, 60, and 90 min following IH relative to baseline. No differences were observed between rats receiving intrathecal scrPKCζ-PS-10 or PKCζ-PS-10. Shown are mean values ± SEM. The filled symbols indicate significantly different from baseline. p < 0.05.
A separate group of rats received intrathecal scrPKCζ-PS or PKCζ-PS 10 min after IH exposure (scrPKCζ-PS-10 and PKCζ-PS-10, respectively; Fig. 3A,C; representative trace for scrPKCζ-PS-10 not shown). As expected, scrPKCζ-PS-10 did not impair pLTF; rats receiving intrathecal scrPKCζ-PS-10 expressed significantly increased phrenic burst amplitude relative to baseline and time controls at 30, 60, and 90 min after IH (all p < 0.05). Similarly, rats receiving intrathecal PKCζ-PS-10 expressed significant increases in phrenic burst amplitude relative to baseline and time controls at 30, 60, and 90 min after IH (all p < 0.05). Importantly, increased phrenic burst amplitude following IH was not significantly different between rats receiving intrathecal PKCζ-PS-10 and those receiving intrathecal scrPKCζ-PS-10. Together, these data suggest that, unlike iPMF, pLTF does not require spinal aPKC activity.
iPMF requires PKCζ/ι activity
aPKC isoforms include PKCζ and PKCι/λ (PKCλ is the mouse homolog of PKCι, but for simplicity, we refer to PKCι/λ as PKCι) and a constitutively active form of PKCζ that lacks a regulatory subunit, known as PKMζ (Reyland, 2009). To determine whether iPMF stabilization requires activation of PKCs containing both a regulatory and catalytic subunit, a subgroup of rats received intrathecal NPC-15437 10 min following restoration of respiratory neural activity (Fig. 4). NPC-15437 binds to the regulatory domain of all PKC isoforms and, as such, does not inhibit PKMζ activity (Sullivan et al., 1991). Intrathecal NPC-15437 had no effect on phrenic burst amplitude in time control rats (p > 0.05) (data not shown), suggesting no time-dependent change in phrenic burst amplitude due to NPC-15437 alone. As expected, rats receiving intrathecal NPC-15437 10 min following resumption of respiratory neural activity after neural apnea expressed a significant increase in phrenic burst amplitude relative to baseline and time controls immediately before NPC-15437 injection (p < 0.05), indicating iPMF; however, following intrathecal NPC-15437 delivery, phrenic burst amplitude steadily declined, such that by 15 min after neural apnea, phrenic burst amplitude was significantly lower relative to the 5 min (preinjection) time point and remained at or below these levels until the end of the recording (all p < 0.05). In addition, all measured time points following intrathecal NPC-15437 were not significantly different from baseline or time controls (p > 0.05). These data suggest that an isoform of PKC containing a regulatory subunit is necessary for iPMF expression.
Figure 4.

Stabilization of iPMF requires activity of spinal PKCζ/ι, but not classical or novel PKC isoforms. Average change in phrenic burst amplitude from baseline for 60 min following neural apnea in rats receiving intrathecal NPC-15437 (inverted triangles; inhibits PKC isoforms with a regulatory subunit), 0.24 nmol of BIM (squares; novel and classical PKC inhibitor), or 2.4 nmol of BIM (triangle) 10 min after resumption of respiratory neural activity. The black arrow indicates time point of drug delivery. All rats expressed significant increases in phrenic burst amplitude relative to baseline or time controls (data not shown) immediately before drug injections, indicating iPMF. iPMF progressively declined following NPC-15437 injections; by 15 min following resumption of respiratory neural activity, iPMF was significantly decreased from preinjection value and no longer significantly different from baseline or time controls. No change in iPMF magnitude was observed following intrathecal BIM (0.24 or 2.4 nmol). Rats receiving intrathecal aCSF before neural apnea (from Fig. 1) are shown in gray circles for comparison. Shown are mean values ± SEM. The filled symbols indicate significantly different from baseline. *Significantly different from preinjection time point. p < 0.05.
Since NPC-15437 inhibits full-length PKCs in all subfamilies (i.e., classical, novel, and atypical), we sought to rule out an additional requirement for other, non-atypical PKC isoforms in iPMF. Thus, iPMF was induced by a neural apnea, and then intrathecal BIM (0.24 or 2.4 nmol) was delivered 10 min following restoration of respiratory neural activity (Fig. 4). BIM inhibits classical and novel PKCs with 30–300 times higher affinity than aPKCs (Toullec et al., 1991; Martiny-Baron et al., 1993). The 0.24 nmol dose of BIM is consistent with doses previously used to block other forms of spinal plasticity (Yashpal et al., 1995; Hua et al., 1999; Ferguson et al., 2008), whereas the 2.4 nmol spinal dose of BIM is known to block phrenic motor facilitation induced by intrathecal injections of a classical and novel PKC isoform activator, PMA (M. Devinney and G. Mitchell, personal communication). In contrast to NPC-15437 or PKCζ-PS-10, intrathecal BIM (0.24 or 2.4 nmol) 10 min after neural apnea had no effect on iPMF since phrenic burst amplitude remained elevated from baseline at all time points after drug injection (all p < 0.05). Indeed, phrenic amplitude (and iPMF magnitude) at all time points following intrathecal BIM were not significantly different from pre-BIM levels (0.24 or 2.4 nmol; p > 0.05). Thus, classical and novel PKCs are not necessary to develop late iPMF.
Collectively, our data strongly demonstrate that (1) spinal PKCζ and/or PKCι activity is/are necessary in the transition to late iPMF; (2) once late iPMF is established, neither PKCζ and/or PKCι activity are necessary to maintain iPMF; and (3) neither novel nor classical PKC isoforms are necessary in the transition to late iPMF.
Increased association of PKCζ/ι with p62 during early, but not late iPMF
As previously reported (Guenther et al., 2010), aPKCs were expressed in retrogradely labeled phrenic motor neurons (Fig. 5). aPKC levels in identified phrenic motor neurons did not change 60 min following restoration of respiratory neural activity after a neural apnea (immunofluorescence intensity for time control, 1 + 0.2 A.U.; 60 min after neural apnea, 1 ± 0.1 A.U.; p > 0.05). Confirming these findings, immunoblot analysis indicated that aPKC levels in ventral spinal regions associated with the phrenic motor pool did not change 5 or 60 min following restoration of respiratory neural activity (data not shown).
Figure 5.

aPKC levels in CTB-labeled phrenic motor neurons do not change 60 min following neural apnea. Representative photomicrographs depicting aPKC (green) and CTB (red) in the C4 ventral horn of time controls (top panel) or 60 min following neural apnea (iPMF; bottom panel). The merged image on right demonstrates colocalization of CTB(+) phrenic motor neurons and aPKC. Scale bar, 100 μm (at 20×).
Following activation, PKCζ and PKCι bind to specific binding proteins to assemble a multimeric signaling complex, which confers specificity in aPKC signal transduction (Moscat et al., 2006, 2007). One such binding protein is the scaffolding protein ZIP/p62 [ZIP, ζ-interacting peptide, is the rat homolog of human p62, also known as sequestosome (SQSTM1)]. ZIP/p62 binds to the PB1 domain in the regulatory subunit of PKCζ and PKCι, and, as such, does not interact with PKMζ. Immunofluorescence confirmed that ZIP/p62 and aPKC are colocalized in CTB-labeled phrenic motor neurons (Fig. 6A). ZIP/p62 also labeled large, putative motor neurons in the ventral horn that were not labeled with CTB, suggesting that ZIP/p62 is highly expressed in α motor neurons.
Figure 6.

Increased association between PKCζ/ι and ZIP/p62 in ventral spinal segments containing phrenic motor neurons. A, Representative images depicting CTB (green), aPKC (red), and ZIP/p62 (blue) in the C4 ventral horn. The merged images on right demonstrate colocalization of ZIP/p62 and aPKC in CTB+ phrenic motor neurons. The phrenic motor nucleus is encircled in top panels. Scale bars: Top, 200 μm; bottom, 20 μm. B, C, PKCζ/ι was immunoprecipitated from ventral C4 gray matter homogenates; immunoprecipitates were then immunoblotted for ZIP/p62. B, Representative immunoblots of ZIP/p62 10 or 60 min following resumption of respiratory neural activity after a neural apnea, or equivalent time points in time controls. C, Average change in ZIP/p62 density from time controls illustrating significant increases in ZIP/p62 binding to PKCζ/ι 10 min, but not 20 or 60 min after resumption of respiratory neural activity. *Significantly different from zero, p < 0.05.
To determine whether iPMF is associated with an increase in the interaction between PKCζ/ι and ZIP/p62, spinal segments containing the bulk of the phrenic motor pool (C4) were collected at 10, 20, or 60 min following the restoration of respiratory neural activity or equivalent time points in time controls. aPKC in ventral C4 gray matter homogenates was then immunoprecipitated, and immunoblot analysis for ZIP/p62 was performed on the immunoprecipitates to assess ZIP/p62 binding to endogenous PKCζ/ι (Fig. 6B,C). ZIP/p62 density in rats exposed to neural apnea was expressed relative to time controls. Consistent with our neuropharmacology results, rats exposed to a neural apnea exhibited increased ZIP/p62 binding with PKCζ/ι in spinal segments associated with the phrenic motor nucleus at 10 (p < 0.05), but not 60 (p > 0.05) min following restoration of respiratory neural activity. We observed a trend toward increased ZIP/p62 binding with PKCζ/ι at 20 min following restoration of respiratory neural activity (p = 0.056), but this was not statistically significant. Collectively, these data suggest that early, but not late phases of iPMF are associated with an increased interaction between ZIP/p62 and PKCζ/ι in regions of the spinal cord containing phrenic motor neurons. Collectively, our data indicate that spinal PKCζ and/or PKCι (but not PKMζ) in or near the phrenic motor pool is/are required to transition from early, labile increases in phrenic burst amplitude into longer-lasting, stable iPMF, likely via the formation of a ZIP/p62–PKCζ/ι signaling cassette.
Discussion
We report a key role for spinal PKCζ/ι in iPMF, a novel form of plasticity in phrenic motor output induced by respiratory neural inactivity. iPMF consists of at least two mechanisms: (1) an early, transient phase that is only partially dependent on spinal aPKC activity; and (2) a longer-lasting phase that requires spinal aPKC activity, specifically isoforms PKCζ or PKCι, to develop but not to maintain. We also report that neural apnea induces an early, transient increase in PKCζ/ι binding to ZIP/p62 in spinal regions associated with the phrenic motor pool. We hypothesize that neural apnea leads to formation of a PKCζ/ι–p62/ZIP signaling cassette within the phrenic motor pool, which stabilizes inactivity-induced increases in phrenic burst amplitude and creates long-lasting iPMF. The role of spinal PKCζ/ι activity is unique to iPMF since aPKC inhibition had no effect on pLTF following IH. Overall, our data suggest that spinal mechanisms sense and respond to reduced respiratory-related inputs to phrenic motor neurons. To our knowledge, this is the first report demonstrating a key role for PKCζ/ι in any form of spinal motor plasticity.
PKCζ/ι is required for iPMF but not pLTF
In recent years, multiple, distinct cellular cascades leading to phrenic motor facilitation (pMF) have been described (Zhang et al., 2003; Golder et al., 2008; Dale-Nagle et al., 2010; Hoffman and Mitchell, 2011; MacFarlane et al., 2011; Mahamed et al., 2011; Nichols et al., 2012). The most frequently studied form of pMF is pLTF following acute IH (Dale-Nagle et al., 2010). The working model of pLTF suggests that IH leads to serotonin release near phrenic motor neurons, activating 5-HT2 receptors (Baker-Herman and Mitchell, 2002) and increasing brain-derived neurotrophic factor (BDNF) synthesis; BDNF subsequently activates its high-affinity receptor, TrkB (Baker-Herman et al., 2004), and leads to ERK MAP kinase activation (Wilkerson and Mitchell, 2009). Unlike iPMF, pLTF is activity independent since intermittent spinal serotonin receptor activation elicits pLTF without changes in phrenic activity during 5-HT application and in the absence of hypoxia (MacFarlane and Mitchell, 2009).
Mechanisms giving rise to iPMF were previously unknown. We originally hypothesized that both pLTF and iPMF required spinal aPKC activity since the constitutively active aPKC isoform, PKMζ, plays a prominent role in several models of plasticity (Sacktor et al., 1993; Drier et al., 2002; Ling et al., 2002; Sutton et al., 2004; Pastalkova et al., 2006; Sacktor, 2008; Villareal et al., 2009), including spinal nociceptive plasticity (Laferrière et al., 2011; Zhang et al., 2012). In many studies (including ours), aPKC activity was inhibited with ζ-pseudosubstrate inhibitory peptide (PKCζ-PS), which mimics the autoinhibitory pseudosubstrate sequence in the PKCζ regulatory domain. Although frequently claimed to be a selective inhibitor for PKMζ, PKCζ-PS binds to and inhibits the catalytic domain of all aPKC isoforms (Selbie et al., 1993; Suzuki et al., 2003; Moscat et al., 2006). Thus, it is not possible to identify the specific aPKC isoform using this compound. Contrary to our original hypothesis, rats pretreated with PKCζ-PS exhibited an attenuated, transient iPMF after neural apnea, with no effect on pLTF.
Since late, but not early iPMF was abolished by spinal aPKC inhibition, we sought to determine critical times during iPMF that require spinal aPKC activity. When delivered shortly after early iPMF induction (10 min), PKCζ-PS “erased” the ongoing iPMF; when delivered 45 min after the establishment of iPMF, PKCζ-PS had no effect. Thus, spinal aPKC activity is necessary to transition from a transient to a longer-lasting form of iPMF, suggesting a role for spinal aPKC in stabilization of iPMF. It is unlikely that the relevant isoform for iPMF stabilization is PKMζ since NPC-15437, which inhibits all PKC isoforms via the regulatory domain (Sullivan et al., 1991), also erased the ongoing iPMF. Furthermore, since BIM had no effect on iPMF (a PKC inhibitor with 30–300 times greater affinity for novel/classical vs aPKC isoforms) (Toullec et al., 1991; Martiny-Baron et al., 1993), we suggest that the aPKC isoform(s) necessary for iPMF stabilization is/are PKCζ and/or PKCι.
In contrast to iPMF, spinal aPKC activity is not necessary for pLTF following acute IH since intrathecal PKCζ-PS had no effect on pLTF when delivered either before or after pLTF induction. Thus, the requirement for spinal aPKC activity in phrenic plasticity appears to characterize inactivity-induced phrenic plasticity.
Interactions between PKCζ/ι and ZIP/p62
Scaffolding/adaptor proteins coordinate signaling networks and confer specificity in cell signaling processes by targeting multifunctional kinases to particular sites, and restraining access to unwanted substrates (Mochly-Rosen, 1995; Moscat et al., 2007; Good et al., 2011). One scaffolding/adaptor protein targeting PKCζ/ι is ZIP/p62 (Puls et al., 1997; Sanchez et al., 1998). PKCζ and PKCι interact with ZIP/p62 via the PB1 protein–protein interaction domain unique to the regulatory subunit of aPKCs; thus, PKMζ or classical or novel PKC isoforms are not expected to interact with ZIP/62 (Puls et al., 1997; Sanchez et al., 1998). In response to specific stimuli, PKCζ/ι binds to ZIP/p62, which relocates and anchors the activated kinase to a context-specific signaling complex (Mochly-Rosen, 1995; Samuels et al., 2001). ZIP/p62 is not a substrate, nor does it affect kinase activity of PKCζ or PKCι; rather, ZIP/p62 contains protein binding motifs suggesting a role in shuttling/binding PKCζ/ι to stimulus-specific multimeric signaling complexes (Puls et al., 1997; Sanchez et al., 1998; Suzuki et al., 2003), including complexes associated with AMPA (Jiang et al., 2009), TrkA (Wooten et al., 2001), GABAC (Croci et al., 2003), TNFα (Sanz et al., 1999, 2000), or IL-1 (Sanz et al., 2000) receptors. Thus, ZIP/p62 imparts specificity in substrate selectivity to PKCζ/ι in response to a given stimulus (Moscat et al., 2006, 2007).
Neural apnea induced an early, transient interaction between PKCζ/ι and ZIP/p62 in spinal regions associated with the phrenic motor nucleus, consistent with our neuropharmacology data showing that spinal PKCζ/ι inhibition shortly following restoration of respiratory neural activity impaired subsequent iPMF. We hypothesize that spinal PKCζ/ι stabilizes early, labile increases in phrenic burst amplitude via interactions with ZIP/p62, leading to long-lasting iPMF. In our working model, reduced synaptic inputs to the phrenic motor pool activates PKCζ/ι and leads to formation of a PKCζ/ι–ZIP/p62 signaling cassette, which anchors activated PKCζ/ι at critical subcellular sites to give rise to iPMF. Since phrenic motor neurons richly express both aPKC and ZIP/p62, we speculate that PKCζ/ι–ZIP/p62 complex formation occurs within phrenic motor neurons, although we cannot rule out a similar effect in presynaptic neurons.
Mechanisms whereby neural apnea activates PKCζ/ι and induces ZIP/p62–PKCζ/ι complex formation are not known. TNF receptor 1 (TNF-R1) activation induces ZIP/p62–PKCζ/ι complex formation, a key step in TNFα signaling (Sanz et al., 1999). Our preliminary data suggest that iPMF requires spinal TNFα (our unpublished observations); thus, ZIP/p62 may recruit PKCζ/ι to the TNF-R1 complex during or following phrenic inactivity. ZIP/p62–PKCζ/ι may traffic relevant proteins (Geetha and Wooten, 2003; Jiang et al., 2009) to or from the membrane, thereby increasing synaptic strength between bulbospinal respiratory premotor neurons and phrenic motor neurons. Consistent with this hypothesis, the ZIP/p62–PKCζ/ι cassette recruits AMPA receptors to synapses during early hippocampal LTP (Jiang et al., 2009).
iPMF significance
Although motor neuron pools driving respiratory muscles must be rhythmically active from birth until death, mechanisms enabling respiratory neurons to adapt to prolonged changes in neuronal excitability and/or synaptic inputs throughout life are unknown. iPMF may represent one component in a continuum of “homeostatic plasticity” mechanisms (Turrigiano, 2008) that enable adjustments in neuronal activity during challenges to breathing. In particular, iPMF may preserve inspiratory motor output at times when an animal is at risk from inadequate respiratory stimulation or excessive inhibition (Hengen et al., 2012).
An extreme example of inadequate respiratory stimulation is cervical spinal injury. Traumatic injury induces many changes in the spinal cord, including tissue damage, inflammation, ischemia, and withdrawal of synaptic inputs caudal to injury (Hausmann, 2003). Immediately following a high cervical injury, an initially silent phrenic nerve slowly and spontaneously regains (partial) function over days to weeks after injury (Nantwi et al., 1999; Golder et al., 2001, 2003; Goshgarian, 2003; Golder and Mitchell, 2005; Fuller et al., 2006; Vinit et al., 2007; Lane et al., 2008). Remodeling spinal circuits after injury may restore phrenic motor output ipsilateral to injury by recruiting latent contralateral pathways crossing the midline at the level of the phrenic pool (the “crossed phrenic phenomenon”) (Goshgarian, 2009; Lane et al., 2009; Darlot et al., 2012) or strengthening spared ipsilateral pathways (Vinit et al., 2008; Vinit and Kastner, 2009). Although changes in the spinal microenvironment due to trauma likely initiates a wide variety of cellular/synaptic changes, inactivity may play a prominent role in restoring function by strengthening spared pathways via the same mechanisms responsible for iPMF. Consistent with this hypothesis, increased aPKC enzymatic activity and phosphorylated aPKC expression are observed in spinal segments associated with the phrenic motor pool following cervical spinal injury at times consistent with a role in spontaneous functional recovery (Guenther et al., 2012). While we do not yet know the role for iPMF in compensating for impaired respiratory neural output, understanding mechanisms of iPMF may guide novel therapeutic strategies to treat ventilatory control disorders in patients in whom endogenous plasticity is impaired or insufficient.
Footnotes
This work was supported by National Institutes of Health Grant HL105511. We thank Gordon S. Mitchell for his comments on this manuscript.
The authors declare no competing financial interests.
References
- Asiedu MN, Tillu DV, Melemedjian OK, Shy A, Sanoja R, Bodell B, Ghosh S, Porreca F, Price TJ. Spinal protein kinase M ζ underlies the maintenance mechanism of persistent nociceptive sensitization. J Neurosci. 2011;31:6646–6653. doi: 10.1523/JNEUROSCI.6286-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker-Herman TL, Mitchell GS. Phrenic long-term facilitation requires spinal serotonin receptor activation and protein synthesis. J Neurosci. 2002;22:6239–6246. doi: 10.1523/JNEUROSCI.22-14-06239.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker-Herman TL, Strey KA. Similarities and differences in mechanisms of phrenic and hypoglossal motor facilitation. Respir Physiol Neurobiol. 2011;179:48–56. doi: 10.1016/j.resp.2011.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker-Herman TL, Fuller DD, Bavis RW, Zabka AG, Golder FJ, Doperalski NJ, Johnson RA, Watters JJ, Mitchell GS. BDNF is necessary and sufficient for spinal respiratory plasticity following intermittent hypoxia. Nat Neurosci. 2004;7:48–55. doi: 10.1038/nn1166. [DOI] [PubMed] [Google Scholar]
- Castro-Moure F, Goshgarian HG. Reversible cervical hemispinalization of the rat spinal cord by a cooling device. Exp Neurol. 1996;141:102–112. doi: 10.1006/exnr.1996.0143. [DOI] [PubMed] [Google Scholar]
- Castro-Moure F, Goshgarian HG. Morphological plasticity induced in the phrenic nucleus following cervical cold block of descending respiratory drive. Exp Neurol. 1997;147:299–310. doi: 10.1006/exnr.1997.6615. [DOI] [PubMed] [Google Scholar]
- Croci C, Brändstatter JH, Enz R. ZIP 3, a new splice variant of the PKC-zeta-interacting protein family, binds to GABAC receptors, PKC-zeta, and Kv beta 2. J Biol Chem. 2003;278:6128–6135. doi: 10.1074/jbc.M205162200. [DOI] [PubMed] [Google Scholar]
- Dale-Nagle EA, Hoffman MS, MacFarlane PM, Mitchell GS. Multiple pathways to long-lasting phrenic motor facilitation. Adv Exp Med Biol. 2010;669:225–230. doi: 10.1007/978-1-4419-5692-7_45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darlot F, Cayetanot F, Gauthier P, Matarazzo V, Kastner A. Extensive respiratory plasticity after cervical spinal cord injury in rats: axonal sprouting and rerouting of ventrolateral bulbospinal pathways. Exp Neurol. 2012;236:88–102. doi: 10.1016/j.expneurol.2012.04.004. [DOI] [PubMed] [Google Scholar]
- Drier EA, Tello MK, Cowan M, Wu P, Blace N, Sacktor TC, Yin JC. Memory enhancement and formation by atypical PKM activity in Drosophila melanogaster. Nat Neurosci. 2002;5:316–324. doi: 10.1038/nn820. [DOI] [PubMed] [Google Scholar]
- Ferguson AR, Bolding KA, Huie JR, Hook MA, Santillano DR, Miranda RC, Grau JW. Group I metabotropic glutamate receptors control metaplasticity of spinal cord learning through a protein kinase C-dependent mechanism. J Neurosci. 2008;28:11939–11949. doi: 10.1523/JNEUROSCI.3098-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuller DD, Golder FJ, Olson EB, Jr, Mitchell GS. Recovery of phrenic activity and ventilation after cervical spinal hemisection in rats. J Appl Physiol. 2006;100:800–806. doi: 10.1152/japplphysiol.00960.2005. [DOI] [PubMed] [Google Scholar]
- Geetha T, Wooten MW. Association of the atypical protein kinase C-interacting protein p62/ZIP with nerve growth factor receptor TrkA regulates receptor trafficking and Erk5 signaling. J Biol Chem. 2003;278:4730–4739. doi: 10.1074/jbc.M208468200. [DOI] [PubMed] [Google Scholar]
- Golder FJ, Mitchell GS. Spinal synaptic enhancement with acute intermittent hypoxia improves respiratory function after chronic cervical spinal cord injury. J Neurosci. 2005;25:2925–2932. doi: 10.1523/JNEUROSCI.0148-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golder FJ, Reier PJ, Davenport PW, Bolser DC. Cervical spinal cord injury alters the pattern of breathing in anesthetized rats. J Appl Physiol. 2001;91:2451–2458. doi: 10.1152/jappl.2001.91.6.2451. [DOI] [PubMed] [Google Scholar]
- Golder FJ, Fuller DD, Davenport PW, Johnson RD, Reier PJ, Bolser DC. Respiratory motor recovery after unilateral spinal cord injury: eliminating crossed phrenic activity decreases tidal volume and increases contralateral respiratory motor output. J Neurosci. 2003;23:2494–2501. doi: 10.1523/JNEUROSCI.23-06-02494.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golder FJ, Ranganathan L, Satriotomo I, Hoffman M, Lovett-Barr MR, Watters JJ, Baker-Herman TL, Mitchell GS. Spinal adenosine A2a receptor activation elicits long-lasting phrenic motor facilitation. J Neurosci. 2008;28:2033–2042. doi: 10.1523/JNEUROSCI.3570-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Good MC, Zalatan JG, Lim WA. Scaffold proteins: hubs for controlling the flow of cellular information. Science. 2011;332:680–686. doi: 10.1126/science.1198701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goshgarian HG. The crossed phrenic phenomenon: a model for plasticity in the respiratory pathways following spinal cord injury. J Appl Physiol. 2003;94:795–810. doi: 10.1152/japplphysiol.00847.2002. [DOI] [PubMed] [Google Scholar]
- Goshgarian HG. The crossed phrenic phenomenon and recovery of function following spinal cord injury. Respir Physiol Neurobiol. 2009;169:85–93. doi: 10.1016/j.resp.2009.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guenther CH, Vinit S, Windelborn JA, Behan M, Mitchell GS. Atypical PKC expression in the phrenic motor neurons of the rat. Neuroscience. 2010;169:787–793. doi: 10.1016/j.neuroscience.2010.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guenther CH, Windelborn JA, Tubon TC, Jr, Yin JC, Mitchell GS. Increased atypical PKC expression and activity in the phrenic motor nucleus following cervical spinal injury. Exp Neurol. 2012;234:513–520. doi: 10.1016/j.expneurol.2012.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hausmann ON. Post-traumatic inflammation following spinal cord injury. Spinal Cord. 2003;41:369–378. doi: 10.1038/sj.sc.3101483. [DOI] [PubMed] [Google Scholar]
- Hengen KB, Nelson NR, Stang KM, Johnson SM, Crader SM, Watters JJ, Mitchell GS, Behan M. Increased GABAA receptor ε-subunit expression on ventral respiratory column neurons protects breathing during pregnancy. PLoS One. 2012;7:e30608. doi: 10.1371/journal.pone.0030608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman MS, Mitchell GS. Spinal 5-HT7 receptor activation induces long-lasting phrenic motor facilitation. J Physiol. 2011;589:1397–1407. doi: 10.1113/jphysiol.2010.201657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua XY, Chen P, Yaksh TL. Inhibition of spinal protein kinase C reduces nerve injury-induced tactile allodynia in neuropathic rats. Neurosci Lett. 1999;276:99–102. doi: 10.1016/s0304-3940(99)00818-6. [DOI] [PubMed] [Google Scholar]
- Jiang J, Parameshwaran K, Seibenhener ML, Kang MG, Suppiramaniam V, Huganir RL, Diaz-Meco MT, Wooten MW. AMPA receptor trafficking and synaptic plasticity require SQSTM1/p62. Hippocampus. 2009;19:392–406. doi: 10.1002/hipo.20528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laferrière A, Pitcher MH, Haldane A, Huang Y, Cornea V, Kumar N, Sacktor TC, Cervero F, Coderre TJ. PKMzeta is essential for spinal plasticity underlying the maintenance of persistent pain. Mol Pain. 2011;7:99–109. doi: 10.1186/1744-8069-7-99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane MA, Fuller DD, White TE, Reier PJ. Respiratory neuroplasticity and cervical spinal cord injury: translational perspectives. Trends Neurosci. 2008;31:538–547. doi: 10.1016/j.tins.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane MA, Lee KZ, Fuller DD, Reier PJ. Spinal circuitry and respiratory recovery following spinal cord injury. Respir Physiol Neurobiol. 2009;169:123–132. doi: 10.1016/j.resp.2009.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling DS, Benardo LS, Serrano PA, Blace N, Kelly MT, Crary JF, Sacktor TC. Protein kinase Mzeta is necessary and sufficient for LTP maintenance. Nat Neurosci. 2002;5:295–296. doi: 10.1038/nn829. [DOI] [PubMed] [Google Scholar]
- MacFarlane PM, Mitchell GS. Episodic spinal serotonin receptor activation elicits long-lasting phrenic motor facilitation by an NADPH oxidase-dependent mechanism. J Physiol. 2009;587:5469–5481. doi: 10.1113/jphysiol.2009.176982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacFarlane PM, Vinit S, Mitchell GS. Serotonin 2A and 2B receptor-induced phrenic motor facilitation: differential requirement for spinal NADPH oxidase activity. Neuroscience. 2011;178:45–55. doi: 10.1016/j.neuroscience.2011.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahamed S, Strey KA, Mitchell GS, Baker-Herman TL. Reduced respiratory neural activity elicits phrenic motor facilitation. Respir Physiol Neurobiol. 2011;175:303–309. doi: 10.1016/j.resp.2010.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantilla CB, Sieck GC. Neuromuscular adaptations to respiratory muscle inactivity. Respir Physiol Neurobiol. 2009;169:133–140. doi: 10.1016/j.resp.2009.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantilla CB, Zhan WZ, Sieck GC. Retrograde labeling of phrenic motoneurons by intrapleural injection. J Neurosci Methods. 2009;182:244–249. doi: 10.1016/j.jneumeth.2009.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martiny-Baron G, Kazanietz MG, Mischak H, Blumberg PM, Kochs G, Hug H, Marmé D, Schächtele C. Selective inhibition of protein kinase C isozymes by the indolocarbazole Gö 6976. J Biol Chem. 1993;268:9194–9197. [PubMed] [Google Scholar]
- Mochly-Rosen D. Localization of protein kinases by anchoring proteins: a theme in signal transduction. Science. 1995;268:247–251. doi: 10.1126/science.7716516. [DOI] [PubMed] [Google Scholar]
- Moscat J, Diaz-Meco MT, Albert A, Campuzano S. Cell signaling and function organized by PB1 domain interactions. Mol Cell. 2006;23:631–640. doi: 10.1016/j.molcel.2006.08.002. [DOI] [PubMed] [Google Scholar]
- Moscat J, Diaz-Meco MT, Wooten MW. Signal integration and diversification through the p62 scaffold protein. Trends Biochem Sci. 2007;32:95–100. doi: 10.1016/j.tibs.2006.12.002. [DOI] [PubMed] [Google Scholar]
- Nantwi KD, El-Bohy A, Schrimsher GW, Reier PJ, Goshgarian HG. Spontaneous functional recovery in a paralyzed hemidiaphragm following upper cervical spinal cord injury in adult rats. Neurorehabil Neural Repair. 1999;13:225–234. [Google Scholar]
- Nichols NL, Dale EA, Mitchell GS. Sever acute intermittent hypoxia elicits phrenic long-term facilitation by a novel adenosine-dependent mechanism. J Appl Physiol. 2012;112:1678–1688. doi: 10.1152/japplphysiol.00060.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastalkova E, Serrano P, Pinkhasova D, Wallace E, Fenton AA, Sacktor TC. Storage of spatial information by the maintenance mechanism of LTP. Science. 2006;313:1141–1144. doi: 10.1126/science.1128657. [DOI] [PubMed] [Google Scholar]
- Puls A, Schmidt S, Grawe F, Stabel S. Interaction of protein kinase C zeta with ZIP, a novel protein kinase C-binding protein. Proc Natl Acad Sci U S A. 1997;94:6191–6196. doi: 10.1073/pnas.94.12.6191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyland ME. Protein kinase C isoforms: multi-functional regulators of cell life and death. Front Biosci. 2009;14:2386–2399. doi: 10.2741/3385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacktor TC. PKMzeta, LTP maintenance, and the dynamic molecular biology of memory storage. Prog Brain Res. 2008;169:27–40. doi: 10.1016/S0079-6123(07)00002-7. [DOI] [PubMed] [Google Scholar]
- Sacktor TC, Osten P, Valsamis H, Jiang X, Naik MU, Sublette E. Persistent activation of the zeta isoform of protein kinase C in the maintenance of long-term potentiation. Proc Natl Acad Sci U S A. 1993;90:8342–8346. doi: 10.1073/pnas.90.18.8342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuels IS, Seibenhener ML, Neidigh KB, Wooten MW. Nerve growth factor stimulates the interaction of ZIP/p62 with atypical protein kinase C and target endosomal localization: evidence for regulation of nerve growth factor-induced differentiation. J Cell Biochem. 2001;82:452–466. doi: 10.1002/jcb.1177. [DOI] [PubMed] [Google Scholar]
- Sanchez P, De Carcer G, Sandoval IV, Moscat J, Diaz-Meco MT. Localization of atypical protein kinase C isoforms into lysosome-targeted endosomes through interaction with p62. Mol Cell Biol. 1998;18:3069–3080. doi: 10.1128/mcb.18.5.3069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanz L, Sanchez P, Lallena MJ, Diaz-Meco MT, Moscat J. The interaction of p62 with RIP links the atypical PKCs to NF-kappaB activation. EMBO J. 1999;18:3044–3053. doi: 10.1093/emboj/18.11.3044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanz L, Diaz-Meco MT, Nakano H, Moscat J. The atypical PKC-interacting protein p62 channels NF-kappaB activation by the IL-1-TRAF6 pathway. EMBO J. 2000;19:1576–1586. doi: 10.1093/emboj/19.7.1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selbie LA, Schmitz-Peiffer C, Sheng Y, Biden TJ. Molecular cloning and characterization of PKC iota, an atypical isoform of protein kinase C derived from insulin-secreting cells. J Biol Chem. 1993;268:24296–24302. [PubMed] [Google Scholar]
- Sullivan JP, Connor JR, Tiffany C, Shearer BG, Burch RM. NPC 15437 interacts with the C1 domain of protein kinase C. An analysis using mutant PKC constructs. FEBS Lett. 1991;285:120–123. doi: 10.1016/0014-5793(91)80739-p. [DOI] [PubMed] [Google Scholar]
- Sutton MA, Bagnall MW, Sharma SK, Shobe J, Carew TJ. Intermediate-term memory for site-specific sensitization in Aplysia is maintained by persistent activation of protein kinase C. J Neurosci. 2004;24:3600–3609. doi: 10.1523/JNEUROSCI.1134-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki A, Akimoto K, Ohno S. Protein kinase C lambda/iota (PKClambda.iota): a PKC isotype essential for the development of multicellular organisms. J Biochem. 2003;133:9–16. doi: 10.1093/jb/mvg018. [DOI] [PubMed] [Google Scholar]
- Toullec D, Pianetti P, Coste H, Bellevergue P, Grand-Perret T, Ajakane M, Baudet V, Boissin P, Boursier E, Loriolle F, Duhamel L, Charon D, Kirilovsky J. The bisindolylmaleimide GF 109203X is a potent and selective inhibitor of protein kinase C. J Biol Chem. 1991;266:15771–15781. [PubMed] [Google Scholar]
- Turrigiano GG. The self-tuning neuron: synaptic scaling of excitatory synapses. Cell. 2008;135:422–435. doi: 10.1016/j.cell.2008.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villareal G, Li Q, Cai D, Fink AE, Lim T, Bougie JK, Sossin WS, Glanzman DL. Role of protein kinase C in the induction and maintenance of serotonin-dependent enhancement of the glutamate response in isolated siphon motor neurons of Aplysia californica. J Neurosci. 2009;29:5100–5107. doi: 10.1523/JNEUROSCI.4149-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinit S, Kastner A. Descending bulbospinal pathways and recovery of respiratory motor function following spinal cord injury. Respir Physiol Neurobiol. 2009;169:115–122. doi: 10.1016/j.resp.2009.08.004. [DOI] [PubMed] [Google Scholar]
- Vinit S, Stamegna JC, Boulenguez P, Gauthier P, Kastner A. Restorative respiratory pathways after partial cervical spinal cord injury: role of ipsilateral phrenic afferents. Eur J Neurosci. 2007;25:3551–3560. doi: 10.1111/j.1460-9568.2007.05619.x. [DOI] [PubMed] [Google Scholar]
- Vinit S, Darlot F, Stamegna JC, Sanchez P, Gauthier P, Kastner A. Long-term reorganization of respiratory pathways after partial cervical spinal cord injury. Eur J Neurosci. 2008;27:897–908. doi: 10.1111/j.1460-9568.2008.06072.x. [DOI] [PubMed] [Google Scholar]
- Wilkerson JE, Mitchell Daily intermittent hypoxia augments spinal BDNF levels, ERK phosphorylation and respiratory long-term facilitation. Exp Neurol. 2009;217:116–123. doi: 10.1016/j.expneurol.2009.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wooten MW, Seibenhener ML, Mamidipudi V, Diaz-Meco MT, Barker PA, Moscat J. The atypical protein kinase C-interacting protein p62 is a scaffold for NF-kappaB activation by nerve growth factor. J Biol Chem. 2001;276:7709–7712. doi: 10.1074/jbc.C000869200. [DOI] [PubMed] [Google Scholar]
- Yashpal K, Pitcher GM, Parent A, Quirion R, Coderre TJ. Noxious thermal and chemical stimulation induce increases in 3H-phorbol 12,13-dibutyrate binding in spinal cord dorsal horn as well as persistent pain and hyperalgesia, which is reduced by inhibition of protein kinase C. J Neurosci. 1995;15:3263–3272. doi: 10.1523/JNEUROSCI.15-05-03263.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, McGuire M, White DP, Ling L. Episodic phrenic-inhibitory vagus nerve stimulation paradoxically induces phrenic long-term facilitation in rats. J Physiol. 2003;551:981–991. doi: 10.1113/jphysiol.2003.048157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YH, Kays J, Hodgdon KE, Sacktor TC, Nicol GD. Nerve growth factor enhances the excitability of rat sensory neurons through activation of the atypical protein kinase C isoform, PKMζ. J Neurophysiol. 2012;107:315–335. doi: 10.1152/jn.00030.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]