

Abstract
Following a primary immune response, T cell memory occurs when a subset of antigen specific T cells resist peripheral selection by acquiring resistance to TCR induced death. Recent data have implicated Bim as an essential mediator of the contraction phase of T cell immunity. Herein, we describe that SDF-1α ligation of CXCR4 on activated T cells, promotes two parallel processes which favor survival, phospho-inactivation of Foxo3A as well as BimEL degradation, both in an Akt and Erk dependent manner. Activated primary CD4 T cells treated with SDF-1α therefore become resistant to the pro-apoptotic effects of TCR ligation or IL-2 deprivation and accumulate cells of a memory phenotype. Unlike SDF-1α, gp120 ligation of CXCR4 has the opposite effect as it causes p38 dependent BimEL upregulation. However when activated CD4 T cells are treated with both gp120 and SDF-1α, the SDF-1α driven effects of BimEL degradation and acquired resistance to TCR induced death predominate. These results provide a novel causal link between SDF-1α induced chemotaxis, degradation of BimEL and the development of CD4 T cell memory.
Introduction
An adaptive immune response involves recruitment of T cells to sites of high antigen burden, for example, a site of infection.(1) This is often achieved by inflammatory cells producing chemotactic factors resulting in the homing of T cells.(2) One such chemotactic factor is SDF-1α that initiates chemotaxis of T cells via signaling through CXCR4.(3) After T cells have been recruited, those that encounter cognate antigen proliferate and develop an apoptosis resistant phenotype (2) through an unknown mechanism.
The involvement of SDF-1α/CXCR4 signaling in effector immune responses and T cell development is becoming recognized.(4) For example, in addition to its well described role in coordinating the circulation of mature naïve lymphocytes between blood and secondary lymphoid organs(5), SDF-1α/CXCR4 signaling is critical for enhancing CD34+ thymocyte proliferation and survival through up-regulating Bcl-2 and down-regulating proapoptotic Bax.(6, 7) SDF-1α/CXCR4 is also important for activation, proliferation and survival of effector lymphocytes,(8–10) altogether suggesting that SDF-1α/CXCR4 can contribute to the generation of T cell memory.
SDF-1α/CXCR4 interactions result in Gαi-dependent increases in Ca2+ influx(11, 12), extracellular signal-regulated protein kinase (ERK) 1/2 phosphorylation,(13) activation of phosphatidylinositol 3-kinase (PI3K) and Akt(13, 14) and Gαi-independent activation of mitogen-activated protein kinase (MAPK) p38(15). Among these signals, ERK1/2 and PI3K-Akt promote T cell survival following SDF-1α CXCR4 interaction through posttranslational inactivation of proapoptotic Bad, increased transcription of cell survival-related genes(16) and likely other means.
The pro-apoptotic BH3-only protein, Bcl-2 interacting mediator of death (Bim), is required for apoptosis of T cells during the termination phase of an immune response(17, 18) and consequently contributes to the development of T cell memory(19), a conclusion which is further supported by observations that Bim deficient mice have increased central and effector memory T cells(20–22), and demonstration of a requisite role for Bim in TCR-induced apoptosis of activated T cells(23).
There are three major Bim isoforms: Bim extra-long (BimEL), Bim long (BimL) and Bim short (BimS,), generated by alternative splicing of the Bim gene,(24) which differ in their proapoptotic potency.(25) BimEL contains the six proline-directed S-P or T-P ERK1/2 phosphorylation sites, which regulate the proteosomal turnover of BimEL(25–27). Both BimEL and BimL contain a dynein light chain 1 (DLC1) interaction domain allowing their sequestration in viable cells(25), yet BimL lacks DEF/FXF-type ERK1/2 docking domain(25) and the three ERK1/2 phosphorylation sites including S65 that influence protein turnover(26, 28). BimS contains only the pro-death BH3-only domain and C-terminal hydrophobic tail, required for insertion into the outer mitochondrial membrane.
Death stimuli including cytokine withdrawal cause activation of the forkhead-like transcription factor Foxo3A(29, 30), which promotes apoptosis by upregulating proapoptotic gene products including Bim(29, 30), and concomitantly down regulating antiapoptotic factors, such as FLIP.
In the case of Bim, this events are counteracted by prosurvival stimuli which promote Bim retention in the cytoplasm through Akt-mediated phosphorylation BimEL at Ser(31), which causes it to associate with 14-3-3 proteins where it cannot induce death as it cannot interact with Bax and or Bak.(32) Other survival factors such as nerve growth factor or interleukin-3 (IL-3) induce ERK-dependent phosphorylation, which causes BimEL to be degraded by the proteosome(25–27).
Since SDF-1α ligation of CXCR4 is known to activate both AKT and Erk, we sought to determine whether these events would affect either Foxo3A or BIM in a manner which altered the survival of cells treated in such a way. In addition, since HIV gp120 also signals through CXCR4 we questioned whether gp120 would have the same effects on cell survival as SDF-1α.
Materials and Methods
Cell Culture and Reagents
CD4+ T cells were isolated from the blood of healthy volunteer blood donors by using RossetteSep CD4 enrichment cocktail in accordance with the manufacture’s protocol (StemCell Technologies, Vancouver, British Columbia, Canada). The remaining cell population was repeatedly found to be 98% CD4+ T cells as determined by flow cytometry. CD4+ T were maintained in RPMI 1640 supplemented with 10% fetal bovine serum (Invitrogen, Carlsbad, CA), 2 mM L-glutamine, and antibiotics (penicillin 100 U/ml, streptomycin 100 μg/ml) at 0.5×106 cells/ml. Naïve and memory CD4+ T cells were isolated by negative selection from the blood of healthy volunteer blood donors by using EasySep human naive or memory enrichment CD4+ T cell kit in accordance with the manufacturer’s protocol (StemCellTechnologies, Vancouver, British Columbia, Canada). The fresh CD4+ T cells used in the various experiments were stimulated with PHA (1 μg/ml) for twenty four hours, and then cells were washed twice with RPMI 1640 and maintained in media supplemented with 50U/ml of IL-2 for 24–48 hours. The expression of activation markers as CD69, CD25 and HLA-DR on resting and activated CD4 T cells were determined by Flow Cytometry. Jurkat and Sup T1 T cells were maintained in RPMI 1640 supplemented with 10% fetal bovine serum (Invitrogen, Carlsbad, CA), 2 mM L-glutamine, and antibiotics (penicillin 100 U/ml, streptomycin 100 μg/ml) at 0.5×106 cells/ml. CD4 T cells were incubated with HIV-1 X4 gp120IIIB (Immuno Diagnostics, Inc. Woburn, MA) or gp120 IIIB pretreated with soluble CD4 (1:3 ratio) (Immuno Diagnostics, Inc. Woburn, MA) at concentrations of 1 μg/ml/2×106 cells for indicated time at 37°C. For Bim half-life measurements resting and activated primary human CD4 T cells were treated with cycloheximide (100 μM) and followed by western blot, over time.
Sources of antibodies were as follows: anti-CXCR4 12G5 and SDF-1α (R&D Systems, Minneapolis, MN); anti-Bim antibodies (Chemicon, Australia); anti-Bcl-2 (CalBiochem, La Jolla, CA (Ab-1)); Anti-Fas (CH-11) (Upstate Cell Signaling solutions, Charlottesville, VA); Anti-phospho-Erk (T280/Y282), anti-Erk1/2, Foxo3A antibodies (Santa Cruz Biotechnology).
Neutralizing F (ab′)2-huFas M3 antibodies were obtained from Immunex Corporation. Foxo3A (S253), and phospho-Foxo3A (t32), anti-phospho-p38 (T180/Y182), anti-GSK-3β, anti-phospho- Akt (Ser473), and anti-Akt antibodies were purchased from Millipore. Anti-CXCR4-PE, anti-CD4-PE, anti-CD25-FITC, anti-CD127-PE, anti-CD4PerCP, anti-CD62-PE, anti-CD45RO-FITC, anti-CD45RA-PE-Cy-7, anti-HLA-DR-PE, anti-CD127, AnnexinV-Cy-5, AnnexinV-APC, IgG1κ-PE-Cy7, IgG2a-FITC, IgG1κ-PE, and propidium iodine, were purchased from BD Biosciences.
Plasmids and Transfection
The human BimEL cDNA was a gift from Dr. Scott Kaufmann (Mayo Foundation, Rochester, MN). The N-terminus BimL construct was PCR amplified using a sense primer with BamH1 site (5′-CGGATCCATGGCAAAGCAA) coupled with antisense spliced primer (5′-CCTGTCTTGTGGCTCTGT) and C-terminus BimL was PCR amplified using a antisense primer with EcoR1 site (5′-CGAATTCTCAATGCATTCT) coupled with sense splicing primer (5′-ACAGAGCCACAAGACAGGAGCCCA). The N-terminus BimS construct was PCR amplified using a sense primer with BamH1 site (5′-CGGATCCATGGCAAAGCAA) coupled with antisense splicing primer (5′-GGAAGCTTGTGGCTC) and C-terminus BimS was PCR amplified using a antisense primer with EcoR1 site (5′-CGAATTCTCAATGCATTCT) coupled with sense splicing primer (5′-CCACAAGCTTCCATGAGG). The BimEL, BimL and BimS were cloned into either mammalian expression vector pEF1 (Invitrogen) to express full size Bim proteins or into GFP-pCDNA3 (Invitrogen) to express GFP-tagged Bim proteins. All Bim constructs were confirmed by DNA sequence analysis and tested for expression prior to experimental use. The kinase dead p38 cDNA was a gift from Dr. M. Karin (University of California, San Diego)(33).
Jurkat T cells were transfected with plasmid of interest using an Electro Square Porator T820 (BTX, San Diego, CA) at 325volts for 10 msec. Activated primary human CD4+ T cells were nucleofected using an Amaxa electroporator (Amaxa Inc., Koeln, Germany) with programmed routine T-23 as per manufacturer’s recommendations. SupT1 T cells were stably transfected with control vector pEF1 (Invitrogen) or kinase dead form of Ha-tagged p38 (T180A/Y182F), at 300V for 10 msec.
Cell Extract Preparation, Immunoblotting and Immunoprecipitation
To obtain total cellular proteins, cells were lysed in [40 mM Tris-HCl (pH=8), 0.25 M NaCl, 1% Triton X–100, 6 mM EDTA, 6 mM EGTA, 10 mM para-nitrophenyl phosphate, 10 mM b-glycerophosphate, 300 μM sodium orthovanadate, 1 mM DDT, 2 μM phenylmethylsulfonyl fluoride (PMSF), aprotinin at 10 μg/ml, leupeptin at 1 μg/ml, pepstatin 1 μg/ml] and centrifuged at 1200 × g for 15 min at 4°C. The amount of cellular protein present in the clarified supernatant was calculated by using the Bio-Rad (Hercules, CA) protein assay, and equal amounts separated by SDS-polyacrylamide gel electrophoresis (PAGE) and transferred to Immobilon-P membranes (Millipore, Bedford, MA). Immunoblotting was performed with specific antibodies and visualized by using the ECL Western blotting detection kit (Amersham, Buckinghamshire, England).
Chemotaxis of primary human CD4 T cells was performed by using a 24-well transmigration chamber plates (5μM pore size, Transwell system (Costar, Cambridge, MA). A total of 40nM of SDF-1α in 600μl of media was added to the lower chamber.
Cell Death Analysis and Flow Cytometry
Cell death was analyzed by staining with AnnexinV-Cy-5 and propidium iodine following the manufacturer’s instructions (BD Biosciences). T cell phenotyping studies were performed by using six-color Flow analysis on FACSCantoII cytometer and using FACSDiva 6.0 software. Briefly, 2×106 cells were resuspended in 200μl of PBS+0.5%BSA, stained with the indicated primary conjugated antibodies for 20 min (anti-CD4PerCP, anti-CD62L-PE, anti-CD45RO-FITC, anti-CD45RA-PE-Cy-7, anti-CD44-APC-Cy7, anti-CD27 APC), washed and analyzed immediately. For analysis of cell death, cells were stained in binding buffer (140 mM NaCl, 10 mM HEPES/NaOH (pH 7.4), and 2.5 mM CaCl2) as described above except that anti-CD27-APC was substituted with AnnexinV-APC.
Results
Resting and activated primary human CD4 T cells have different levels of Bim expression and different half-life of BimEL
As our purpose was to determine the effect of CXCR4 signaling on Bim and Foxo3A, we isolated primary human CD4 T cells and either activated them with PHA/IL-2, or left them untreated, and characterized cells for CXCR4 and activation markers expression (CD25, CD69 and HLA-DR) (Fig. 1A). Resting (CD4+ CD25- CD69- HLA-DR-) and activated (CD4+ CD25+ CD69+HLA-DR+) T cells were harvested and Bim expression evaluated. As shown in Figure 1B and C, 23KD BimEL levels are lower in activated CD4 T cells compared with resting cells as determined by densitometry (RLU, lower panel). The reduced levels of BimEL in activated CD4 T cells are also associated with increased phosphorylation of Foxo3A (Fig. 1B). By contrast, the levels of both BimL and BimS were not decreased in activated compared to resting CD4 T cells (Fig. 1B) even following in vitro activation with PHA/IL-2 (Fig. 1C). These three Bim species seen were confirmed to be BimEL, BimL and BimS, by individually overexpressing BimEL, BimL and BimS in Jurkat T cells (Fig. 1D). Finally, the half-life of BimEL was compared between resting and activated CD4 T cells (Fig. 1E), demonstrating a shorter half-life of BimEL in activated CD4 T cells. Therefore, in activated cells there is a reduced level of BimEL, which is associated with phosphorylated Foxo3A, and a shorter protein half-life.
Figure 1. Different pattern of Bim isoform expression in resting and activated primary human CD4 T cells.
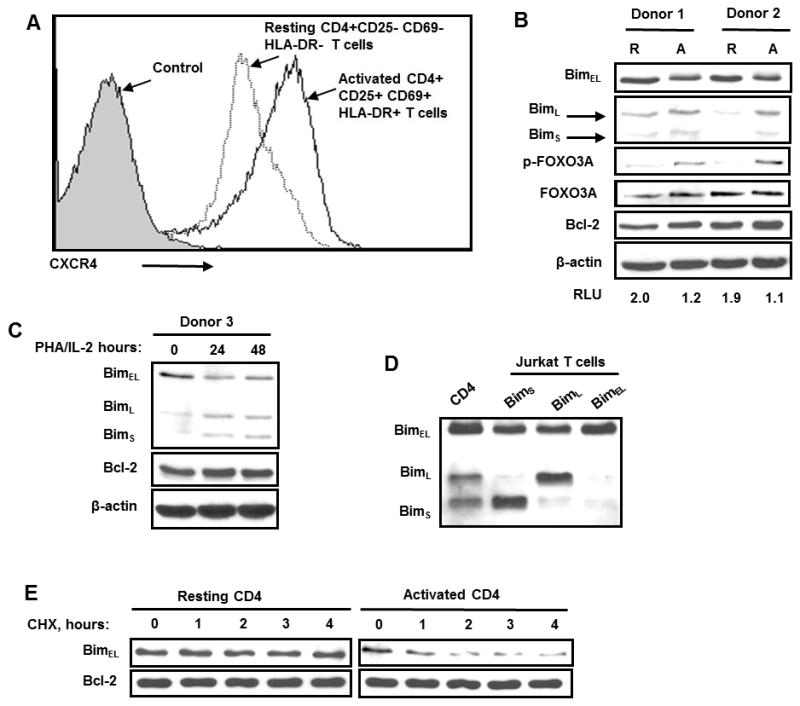
(A) Activated and resting primary human CD4 T cells were stained with anti-CXCR4-PE antibodies and then CXCR4 expression were analyzed by Flow Cytometry (B) CD4T cells were isolated from three healthy volunteer blood donors (Donor 1 and Donor 2), and left untreated for 36–48 hours, and the Bim protein levels analyzed. Equal loading of cell lysates were confirmed by immunoblotting with anti-β-actin antibodies. (C) Isolated primary human CD4 T cells were stimulated with PHA+IL-2 over time course or left untreated and then Bim levels were determined by immunoblotting. (D) Cell lysates from activated CD4 T cells were compared with Jurkat T cells electroporated with 12 μg of pEF1- BimS, pEF1- BimL, pEF1- BimEL were analyzed for Bim. (E) Resting and activated primary human CD4 T cells were treated with cycloheximide (100 μM) over time and the Bim protein levels were visualized by immunoblotting.
SDF-1α ligation of CXCR4 accelerates BimEL degradation in activated primary human CD4 T cells
CXCR4/SDF-1α signaling can activate both Erk and Akt, which are known to phosphorylate Foxo3A, which has been reported to phosphorylate Bim. In order to test if this signaling pathway is operational in T cells, we assess whether SDF-1α would phosphorylate Foxo3A and phosphorylate and degrade Bim. In activated CD4 T cells SDF-1α induced both Foxo3A phosphorylation and BimEL phosphorylation and subsequent degradation (Fig. 2A). The phosphorylation of Foxo3A and of BimEL occurs in concert with Akt and Erk phosphorylation, (Fig. 2A); p38 phosphorylation is not sufficient to induce BimEL phosphorylation and degradation as demonstrated in experiments that induce p38 phosphorylation without Akt or Erk phosphorylation by treating with 12G5 antibodies (Fig. 2B). Next we questioned whether inducing Erk and Akt phosphorylation independent of CXCR4 ligation by SDF-1α would be sufficient to cause BimEL degradation. Treatment of CD4 T cells with PMA and ionomycin caused phosphorylation of Erk and Akt, which, as predicted, lead to the phosphorylation and degradation of BimEL (Fig. 2C).
Figure 2. SDF-1αinduces BimEL degradation in activated CD4 T cells.
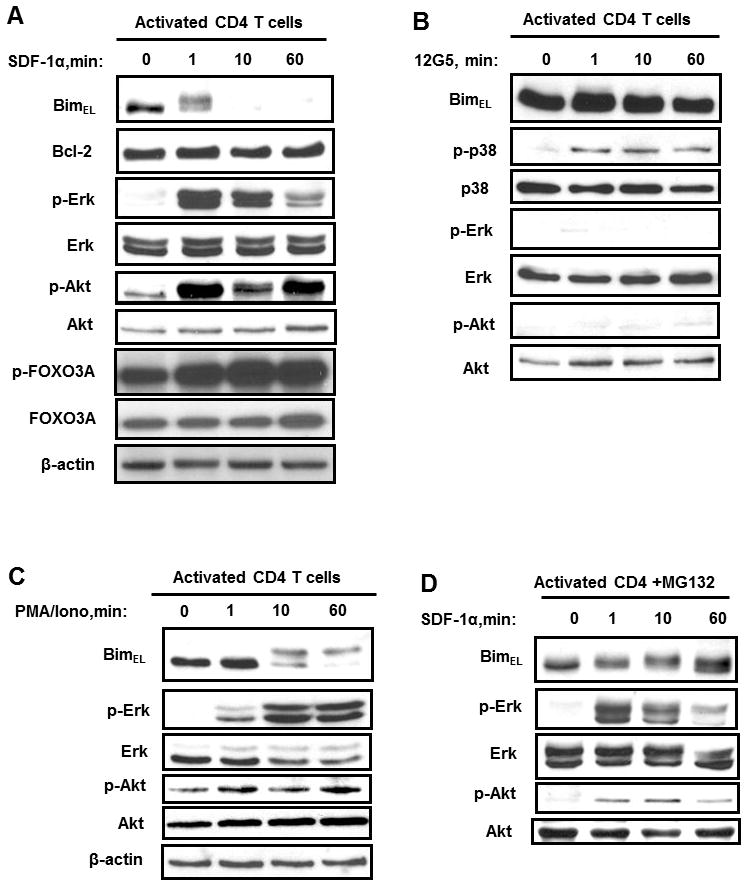
(A) Activated primary human CD4 T cells were incubated with SDF-1α(125 nM) or BSA over time course and then Bim levels as well as phosphorylation of Akt, Erk and GSK-3β were analyzed by immunoblotting. Total amount of Akt, Erk and GSK-3β were also detected. This experiment is representative of three other experiments performed with CD4 T cells from different subjects. (B) Activated primary human CD4 T cells were treated 12G5 (anti-CXCR4) or control and activation of p38, Erk, and Akt were assessed. This experiment represents one of three experiments. (C) Activated primary CD4+ T cells were stimulated with PMA (5 ng/ml) and ionomycin (1μM) over time. Activation of Akt and Erk was analyzed by immunoblotting as described above. (D) Activated primary human CD4 T cells were pre-treated with proteasome inhibitor MG132 (25μM) for 30 minutes and then stimulated with SDF-1α, and the Bim protein levels were visualized. This experiment was repeated at least three times.
BimEL contains six proline-directed S-P or T-P phosphorylation motifs for Erk(27, 28) and one phosphorylation site for Akt, Ser87,(31) which targets Bim for proteosomal degradation. To test the hypothesis that SDF-1α induced activation of Akt and/or Erk results in BimEL phosphorylation which causes proteosomal degradation, we pretreated activated CD4 T cells with the proteosome inhibitor MG132. Following SDF-1α treatment in the presence of MG132, BimEL degradation was blocked (Fig. 2D). Altogether these data suggest that Akt and Erk activity in activated CD4 T cells lead to BimEL phosphorylation leading to proteosomal degradation of BimEL.
Akt and Erk are required for SDF-1α induced BimEL degradation
Having demonstrated that Akt and Erk are associated with SDF-1α/CXCR4 induced Bim degradation; we used specific inhibitors to assess whether Akt and Erk were necessary. Primary CD4 T cells were pretreated with Gαi -protein inhibitor, pertussis toxin (PT)(34), Akt inhibitor VIII(35) and the Erk inhibitor PD98059(36), and then stimulated with SDF-1α (Fig. 3). As expected the Gαi-protein inhibition by PT completely blocked phosphorylation of Akt and Erk and consequently blocked the degradation of BimEL. Inhibiting Erk by PD98059 or inhibiting Akt with Akt inhibitor VIII also blocks the phosphorylation and degradation of BimEL, demonstrating that both Akt and Erk are required.
Figure 3. Pharmocological inhibition of G-proteins, Akt and Erk block SDF-1α induced BimEL degradation.
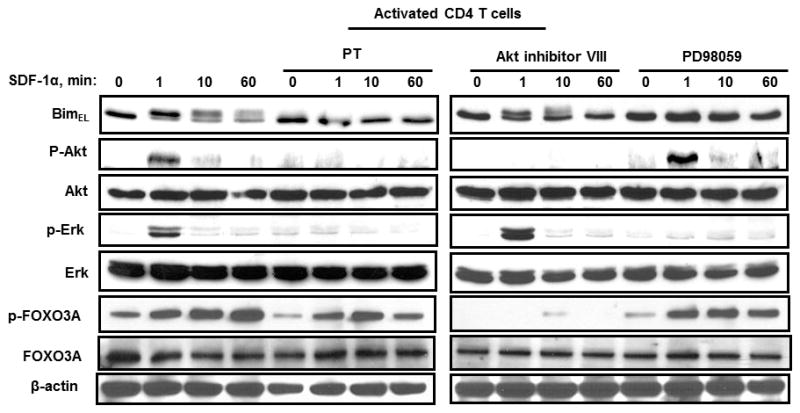
Activated primary human CD4 T cells left untreated or were pre-treated with either Pertussis toxin (0.5 μg/ml), Akt inhibitor VIII (2μM), or PD98059 (20μM) for 30 minutes and then stimulated with SDF-1α (125 nM) or with BSA control (1 μg/ml). Bim levels and Akt, Erk, and Foxo3A phosphorylation were analyzed. All experiments were performed at least three times.
Activated primary CD4 T cells treated with SDF-1α develop resistance to Bim mediated apoptosis
Because Bim mediates CD4 T cell death at the end of the expansion phase of an immune response (20–23), we next assessed whether activated CD4 T cells treated with SDF-1α become resistant to Bim dependent apoptosis induced by either IL-2 deprivation (37) or TCR ligation (38). First, we observed that while IL-2 withdrawal induces death of activated CD4 T cells (Fig. 4A), SDF-1α treatment of these cells induces Bim degradation (Fig. 4B) which reduces the amount of IL-2 withdrawal-induced death. Second, cross-linking of primary human activated CD4 T cells with OKT3 was not blocked by neutralizing anti-Fas antibodies (Fig. 4C); consistent with recent reports demonstrating that TCR mediated contraction of the immune response can occur in a Fas independent Bim dependent manner (17,18, 20–23). Pretreatment of the same primary human activated CD4 T cells with SDF-1α blocked this TCR induced apoptosis (Fig. 4D) and this protective effect is partially reversed by Pertussis toxin, Akt inhibitor VIII, and by PD98059 (Fig. 4D). Finally, activated CD4 T cells treated with SDF-1α become resistant to Bim-dependent apoptosis induced by ionomycin treatment (37), not Bim independent death induced by Fas ligation. Pretreatment of CD4 T cells with Pertussis toxin, Akt inhibitor VIII, or PD98059, reduced the SDF-1α protection against ionomycin induced death (Fig. 4E). Altogether, these results indicated that SDF-1α-induced BimEL degradation renders activated CD4 T cells resistant to Bim-dependent apoptosis induced by IL-2 deprivation, TCR- and ionomycin stimulation.
Figure 4. Pharmocological inhibition of G-proteins, Akt and Erk reverses SDF-1α protective effect on Bim mediated CD4 T cells death.

(A) Activated primary CD4 T were washed for three times and cultured for six hours in IL-2 free media or in the media supplemented with 50u/ml of IL-2. Then cells were incubated with SDF-1α(125 nM) or BSA. Forty eight hours later, the apoptosis were measured as increase (%) of AnnexinV positive CD4 T cells above control treated cells. This experiment is performed in triplicates from three different donors (n=3). P values were determined by Student’s paired t test. (B) Activated primary CD4 T cells were washed for three times and cultured for 6 hours in IL-2-free media or in the media supplemented with 50u/ml of IL-2. Then cells were incubated with SDF-1α(125 nM) or BSA over time course and then Bim levels as well as phosphorylation of Akt and Erk were analyzed by immunoblotting. Total amount of Akt and Erk were also detected. This experiment is representative of three other experiments performed with CD4 T cells from different subjects. (C) Activated primary human CD4 T cells were incubated with OKT3 (1 μg/ml) or with isotype control IgG in the presence or absence of neutralazing anti-Fas antibodies M3 for 18 hours and cell death was assessed by measuring Annexin V positive CD4 T cells. The data is representative of three independent experiments. (D) Activated primary human CD4 T cells left untreated or were pre-treated with either Pertussis toxin, Akt inhibitor VIII or PD98059 as described above and then treated with SDF-1α or with BSA for one hour followed by OKT3 (1μg/ml) or with isotype control IgG for 18 hours, then cell death was assessed by measuring AnnexinV positive CD4 T cells. Cell death expressed as percent increase in AnnexinV positivity of CD4 T cells. The data is representative of three independent experiments. P values were determined by Student’s paired t test. (E) Activated primary human CD4 T cells left untreated or were pre-treated with either Pertussis toxin, Akt inhibitor or PD98059 as described above and then treated with SDF-1α (125 nM) or BSA (1 μg/ml, control) for one hour and then cells were divided into two groups. One group of CD4 T cells were treated with either ionomycin (1μM) or vehicle control (DMSO) for 18 hours. Other groups of CD4 T cells were treated with either the anti-Fas agonist anti-CD95 (CH11, 1μg/ml) or isotype control. Apoptosis was measured as increase (%) of AnnexinV positive CD4 T cells above control treated cells.
SDF-1α ligation of activated cells increases the proportion of memory cells
The biological function of SDF-1α is to induce chemotaxis of T cells to areas of inflammation in order to mediate a primary adaptive immune response. Since a central purpose of the adaptive immune response is to develop immunologic memory of antigen specific cells, we hypothesized that those T cells which undergo chemotaxis in response to SDF-1α should be favored to resist cell death induced by TCR stimulation or by IL-2 deprivation and develop a memory phenotype. First we tested this hypothesis by incubating activated CD4 T cells in the presence or absence of SDF-1α and found that SDF-1α significantly increases the fraction of cells which are of effector memory type (CD4+/CD25+/CD127low/CD45RO+/CD45RA, Fig. 5A). Next we isolated by negative selection naïve and memory CD4 T cells (Fig. 5B), and analyzed Bim content in the freshly isolated CD4 T cells before and after SDF-1α stimulation. Memory CD4 T cells have less BimEL (Fig. 5B), and BimEL is degraded following SDF-1α stimulation in memory CD4 T cells (Fig. 5C). We therefore predicted that the differences in Bim would inhibit apoptosis of activated memory CD4 T cells following IL-2 withdrawal (Fig. 5E). Indeed, SDF-1α treatment significantly decreases apoptosis in activated memory, but not activated naive CD4 T cells following IL-2 withdrawal (Fig. 5D and E).
Figure 5. SDF-1α stimulation degrades BimEL and decreases apoptosis of memory CD4 T cells.

(A) CD4+ T cells were isolated from healthy volunteer donors (n=4) and stimulated with OKT-3 (1μg/ml) and cultured for 48 hours in the presence or absence of 40nM of SDF-1α. Percent of effector memory CD4+/CD25+/CD127low/CD45RO+/CD45RA− cells were measured in duplicates by multi-color Flow Cytometry analysis. (B) Naïve and memory CD4+ T cells were isolated from healthy volunteer blood donors (n=3), and immediately analyzed for Bim EL content by Western blot. (C) Freshly isolated naive and memory CD4+ T cells were stimulated with SDF-1α (40 nM) for the indicated times and Bim and β-actin levels analyzed by immunoblotting. (D and E) Freshly isolated naive and memory CD4+ T cells were stimulated with PHA (1 ug/ml) and cultured for 48 hours in the media with 50u/ml of IL-2, then cultured for 48 hours in the presence or absence of SDF-1α (40 nM) plus or minus 50u/ml of IL-2. Cell death was assessed by measuring of AnnexinV positive CD4+/CD45RO+/CD45RA- and CD4+/CD45RO-/CD45RA+ T cells. This experiment is representative of three other experiments performed with naive and memory CD4+ T cells from different subjects.
Altogether, these results indicate that memory CD4 T cells have less BimEL than naive CD4 T cells, and memory CD4 T cells degrade BimEL in response to SDF-1α, which confers an acquired resistance to cell death following IL-2 withdrawal, resulting in an accumulation of cells expressing a T cell memory phenotype.
Co-ligation of SDF-1α and HIV-1 gp120 to CXCR4 degrades BimEL
Having shown that SDF-1α ligation of CXCR4 results in BimEL degradation in an Akt and Erk dependent manner, we next assessed what effect gp120 ligation of CXCR4 had on Bim. Having previously demonstrated that gp120/CXCR4 ligation activates p38 but not Akt and Erk (51), it was not surprising that gp120 binding to CXCR4 did not induce BimEL degradation, but rather gp120 increases BimEL, whether gp120 is used alone (Fig. 6A) or in the presence of soluble CD4 which targets gp120 to signal exclusively through the CXCR4 receptor (Fig. 6B), consistent with the known pro-apoptotic effects of gp120 on T cell survival (52, 53). The increase in BimEL induced by gp120 is p38 dependent since SupT1 cells stably expressing kinase dead p38 fail to upregulate BimEL in response to gp120 treatment, whereas the same cells stably expressing the vector control efficiently upregulate BimEL following gp120 treatment (Fig. 6C).
Figure 6. SDF-1α degrades BimEL in the presence of HIV-1 gp120.

(A and B) Activated primary human CD4+ T cells were incubated with HIV-1 X4 gp120 IIIB or gp120 IIIB pretreated with soluble CD4 (3:1 ratio) (gp120 IIIB + sCD4) at concentrations of 5 μg/ml/2×106 cells for indicated time at 37°C. (C) Stably transfected SupT1 T cells with either control vector SFFV2 or kinase dead form of p38 were treated with gp120 IIIB + sCD4 for four hours. The BimEL, β-actin levels were visualized by immunoblotting. The expression of HA- tagged p38 kinase dead were confirmed by immunoblotting with HA antibodies. (D) Activated primary human CD4+ T cells were incubated with gp120IIIB (40nM), SDF-1α (40nM) or both (1:1 molar ratio). The BimEL and β-actin levels as well as Akt and Erk phosphorylation were analyzed as described above. (E) Activated primary human CD4+ T cells were stimulated with SDF-1α (40nM) in the presence of increased concentration of gp120 (10, 50 and 100 nM). The BimEL and β-actin levels were analyzed as described above.
Since gp120 and SDF-1α have opposing effects on BimEL levels, we questioned which effect would predominate in a circumstance where both stimuli were present. Such a situation would likely occur in an HIV infected viremic patient, in whom SDF-1α is produced, for example at a site of tissue injury. Treatment of cells with both SDF-1α and gp120 results in BimEL phosphorylation and subsequent degradation similar to what occurs in the cells treated with SDF-1α alone (Fig. 6E). The physiologic levels of gp120 in a patient are likely between 500 ng/ml and 5 μg/ml(39); these doses of gp120 as well as much higher doses were unable to reverse the SDF-1α induced degradation of BimEL.
Discussion
While mechanisms explaining how activated CD4 T cells die during an immune response have been intensively investigated, the molecular events of how activated CD4 T cells survive and become memory T cells are still not completely understood. Although Bim is a key mediator of activated T cell death(17, 18), it is not known whether the signals that control survival of activated T cells also impact Bim. In the current report we have shown that activated primary CD4 T cells have less BimEL, due to a shorter BimEL half-life in these cells than in resting cells. This is in agreement with previously published observation that BimEL levels in primary CD4 T cells depends on phoshorylation and subsequent inactivation of FOXO3A by PI3K/Akt signaling pathway(30). While others groups observed the increase of Bim protein in activated T cells(40–42), this apparent discrepancy is probably due to long-term culture (6–14 days) of activated T cells that associated with decrease of PI3K/Akt activity(41, 42) or restimulation of activated T cells that results in Bim upregulation(23, 41). Moreover SDF-1α treatment induces phosphorylation and inactivation of Foxo3A, as well as phosphorylation and degradation of BimEL due to Akt/Erk, the net effect of which renders these cells resistant to apoptosis induced by IL-2 deprivation or TCR ligation, and thus SDF-1α maintains memory cells by downregulating Bim.
Our findings are consistent with the following previously published observations: First, SDF-1α promotes survival of serum-deprived T cells through increased transcription of cell survival-related genes and posttranslational inactivation of Bad(16), in an Akt and MAPK dependent manner. Second, absence of Bim increases the number of effector T cells which become memory(19). Third, central memory CD4 T cells have low levels of Bim(43), increased Akt activity and inactive Foxo3A., which agrees with our observation that memory CD4 T cells have less BimEL than naïve CD4 T cells. Fourth, IL-2 and IL-15 both inactivate Bim, and promote the survival of activated CD4 T cells(30, 44, 45). Similarly, Akt activation (which is known to degrade Bim) by OX40(46) enhances survival of CD4 T cells. Altogether these observations coupled with our current data suggest that factors which activate Akt, including SDF-1α/CXCR4 ligation, promote T cell survival via Bim degradation.
We demonstrate that recruitment of CD4 T cells by SDF-1α decreases BimEL and confers resistance to apoptosis induced by IL-2 deprivation or TCR stimulation which, in turn, promotes accumulation of T cells with a memory phenotype. These observations provide novel insights of the role of SDF-1α/CXCR4 signaling and Bim in the contraction of the effector T cell pool. Since both Bim and Fas cooperate during contraction of immune response(20–22), the role of SDF-1α/CXCR4 is to promote survival of a subset of effector CD4 T cells which are destined to die in a Bim dependent manner. Indeed, the SDF-1α/CXCR4 does not affect Fas mediated death of activated CD4 T cells (Figure 5A). Taking into consideration that chronically re-stimulated CD4 T cells die preferentially in Fas dependent manner(47), it is plausible that SDF-1α/CXCR4 promotes the survival of acutely re-stimulated CD4 T cells, which preferentially die in a Bim dependent manner. Therefore, the strength and duration of antigen stimulation may be crucial for the effect of SDF-1α/CXCR4 on T cell survival.
This is consistent with the observation that integrin linked kinase (ILK) deficiency impairs the chemotactic response to SDF-1α, which increases peripheral T cell apoptosis and decreases 20-fold the number of lymphoid cells in peripheral lymphoid organs.(48) In contrast, enhanced chemotaxis of T cells in response to SDF-1α results in an increased number of effector memory T cells in patients with WHIM (warts, hypogammaglobulinemia, infections, and myelokathexis) syndrome(49). Therefore, in these two conditions, the ability of T cells to mount a chemotactic response to SDF-1α correlates directly with the ability to form memory T cells.
The normal function of Bim is to bind and sequester the antiapoptotic protein Bcl2(50). Therefore we propose a model whereby SDF-1α induced decreases in BimEL allows for more Bcl2 to render the cell resistant to apoptosis. Therefore, in addition to previously described transcriptional upregulation of survival genes, SDF-1α/CXCR4 signaling imposes an antiapoptotic effect in two ways: through BimEL degradation; and upregulation of other antiapoptotic regulatory proteins
The enigma of HIV infection is that despite effective therapies which almost completely suppress viral replication, a viral reservoir persists, even after years of effective antiretroviral therapy (reviewed in (51). The principal cell type that HIV infects is activated, antigen specific CD4 T cells, yet the cell type which harbors the majority of latent virus in resting central memory CD4 T cells(52), Therefore some event must occur which stimulates a subset of the activated and infected CD4 T cells to change to a latently infected CD4 central memory cell. The survival of these cells appears to be critically dependent upon phosphorylation of Foxo3A(43), although the signals which drive such phosphorylation are incompletely understood. The critical importance of Foxo3A phosphorylation is demonstrated by the existence of very high levels of phospho Foxo3A in cells from HIV infected elite controllers, who are endowed with the capacity to spontaneously control viral replication(53). Further experiments from this group demonstrate that Silencing Foxo3A by siRNA or by a dominant-negative form (Foxo3A Nt) prolongs the survival of central memory CD4 T cells, arguing a causal role of Foxo3A inactivation in maintenance of HIV persistence. Consequently, it is of extreme importance to understand the events which promote Foxo3A phospho-inactivation, as well as the downstream effects of this event on cell survival. In that regard, we now demonstrate, for the first time, that SDF-1α treatment of activated T cells results in both Akt and Erk phosphorylation, which are known regulators of Foxo3A activity. Moreover we further demonstrate that this signaling pathway results in BimEL degradation and an acquired resistance to cell death induced by TCR ligation or IL-2 deprivation, which promotes accumulation of cells expressing a memory phenotype. Altogether, our results identify BimEL as a downstream target for SDF-1α/CXCR4 signaling and therefore provide novel insight into the molecular mechanism that regulates T cells survival, HIV persistence, and the development of CD4 T cell memory.
Acknowledgments
Grant Support: This work was supported by the National Institutes of Health (R01 AI62261) (A.D.B.).
The authors gratefully acknowledge the secretarial expertise of Ms. Tammy Engel.
Footnotes
Authorship
Contribution: S.A.T., A.A.C, G.D.B, and X.D. performed experiments; S.A.T., S.A.R., R.S.A. and A.D.B analyzed results; S.A.T. and A.D.B. made the figures; S.A.T., S.A.R. and A.D.B. designed the research; and S.A.T. and A.D.B. wrote the paper.
Conflict of Interest Disclosure
The authors declare no competing financial interests.
References
- 1.Roman E, Miller E, Harmsen A, Wiley J, Von Andrian UH, Huston G, Swain SL. CD4 effector T cell subsets in the response to influenza: heterogeneity, migration, and function. J Exp Med. 2002;196:957–968. doi: 10.1084/jem.20021052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ebert LM, Schaerli P, Moser B. Chemokine-mediated control of T cell traffic in lymphoid and peripheral tissues. Mol Immunol. 2005;42:799–809. doi: 10.1016/j.molimm.2004.06.040. [DOI] [PubMed] [Google Scholar]
- 3.Campbell DJ, Kim CH, Butcher EC. Chemokines in the systemic organization of immunity. Immunol Rev. 2003;195:58–71. doi: 10.1034/j.1600-065x.2003.00067.x. [DOI] [PubMed] [Google Scholar]
- 4.Klein RS, Rubin JB. Immune and nervous system CXCL12 and CXCR4: parallel roles in patterning and plasticity. Trends Immunol. 2004;25:306–314. doi: 10.1016/j.it.2004.04.002. [DOI] [PubMed] [Google Scholar]
- 5.Kim CH. SLC/exodus2/6Ckine/TCA4 induces chemotaxis of hematopoietic progenitor cells: differential activity of ligands of CCR7, CXCR3, or CXCR4 in chemotaxis vs. suppression of progenitor proliferation. J Leukoc Biol. 1999:3. doi: 10.1002/jlb.66.3.455. [DOI] [PubMed] [Google Scholar]
- 6.Hernandez-Lopez C, Varas A, Sacedon R, Jimenez E, Munoz JJ, Zapata AG, Vicente A. Stromal cell-derived factor 1/CXCR4 signaling is critical for early human T-cell development. Blood. 2002;99:546–554. doi: 10.1182/blood.v99.2.546. [DOI] [PubMed] [Google Scholar]
- 7.Ara T, Itoi M, Kawabata K, Egawa T, Tokoyoda K, Sugiyama T, Fujii N, Amagai T, Nagasawa T. A role of CXC chemokine ligand 12/stromal cell-derived factor-1/pre-B cell growth stimulating factor and its receptor CXCR4 in fetal and adult T cell development in vivo. J Immunol. 2003;170:4649–4655. doi: 10.4049/jimmunol.170.9.4649. [DOI] [PubMed] [Google Scholar]
- 8.Foussat A, Balabanian K, Amara A, Bouchet-Delbos L, Durand-Gasselin I, Baleux F, Couderc J, Galanaud P, Emilie D. Production of stromal cell-derived factor 1 by mesothelial cells and effects of this chemokine on peritoneal B lymphocytes. Eur J Immunol. 2001;31:350–359. doi: 10.1002/1521-4141(200102)31:2<350::aid-immu350>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 9.Yonezawa A, Hori T, Sakaida H, Uchiyama T. SDF-1A has costimulatory effects on human T cells: possible involvement of MAPK (ERK2) activation. Microbiol Immunol. 2000;44:135–141. doi: 10.1111/j.1348-0421.2000.tb01256.x. [DOI] [PubMed] [Google Scholar]
- 10.Nanki T, Hayashida K, El-Gabalawy HS, Suson S, Shi K, Girschick HJ, Yavuz S, Lipsky PE. Stromal cell-derived factor-1-CXC chemokine receptor 4 interactions play a central role in CD4+ T cell accumulation in rheumatoid arthritis synovium. J Immunol. 2000;165:6590–6598. doi: 10.4049/jimmunol.165.11.6590. [DOI] [PubMed] [Google Scholar]
- 11.Oberlin E, Amara A, Bachelerie F, Bessia C, Virelizier JL, Arenzana-Seisdedos F, Schwartz O, Heard JM, Clark-Lewis I, Legler DF, Loetscher M, Baggiolini M, Moser B. The CXC chemokine SDF-1A is the ligand for LESTR/fusin and prevents infection by T-cell-line-adapted HIV-1. Nature. 1996;382:833–835. doi: 10.1038/382833a0. [DOI] [PubMed] [Google Scholar]
- 12.Bleul CC, Farzan M, Choe H, Parolin C, Clark-Lewis I, Sodroski J, Springer TA. The lymphocyte chemoattractant SDF-1A is a ligand for LESTR/fusin and blocks HIV-1 entry. Nature. 1996;382:829–833. doi: 10.1038/382829a0. [DOI] [PubMed] [Google Scholar]
- 13.Ganju RK, Brubaker SA, Meyer J, Dutt P, Yang Y, Qin S, Newman W, Groopman JE. The alpha-chemokine, stromal cell-derived factor-1alpha, binds to the transmembrane G-protein-coupled CXCR-4 receptor and activates multiple signal transduction pathways. J Biol Chem. 1998;273:23169–23175. doi: 10.1074/jbc.273.36.23169. [DOI] [PubMed] [Google Scholar]
- 14.Sotsios Y, Whittaker GC, Westwick J, Ward SG. The CXC chemokine stromal cell-derived factor activates a Gi-coupled phosphoinositide 3-kinase in T lymphocytes. J Immunol. 1999;163:5954–5963. [PubMed] [Google Scholar]
- 15.Vlahakis SR, Villasis-Keever A, Gomez T, Vanegas M, Vlahakis N, Paya CV. G protein-coupled chemokine receptors induce both survival and apoptotic signaling pathways. J Immunol. 2002;169:5546–5554. doi: 10.4049/jimmunol.169.10.5546. [DOI] [PubMed] [Google Scholar]
- 16.Suzuki Y, Rahman M, Mitsuya H. Diverse transcriptional response of CD4(+) T cells to stromal cell-derived factor (SDF)-1: cell survival promotion and priming effects of SDF-1A on CD4(+) T cells. J Immunol. 2001;167:3064–3073. doi: 10.4049/jimmunol.167.6.3064. [DOI] [PubMed] [Google Scholar]
- 17.Hildeman DA, Zhu Y, Mitchell TC, Bouillet P, Strasser A, Kappler J, Marrack P. Activated T cell death in vivo mediated by proapoptotic bcl-2 family member bim. Immunity. 2002;16:759–767. doi: 10.1016/s1074-7613(02)00322-9. [DOI] [PubMed] [Google Scholar]
- 18.Pellegrini M, Belz G, Bouillet P, Strasser A. Shutdown of an acute T cell immune response to viral infection is mediated by the proapoptotic Bcl-2 homology 3-only protein Bim. Proc Natl Acad Sci U S A. 2003;100:14175–14180. doi: 10.1073/pnas.2336198100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wojciechowski S, Jordan MB, Zhu Y, White J, Zajac AJ, Hildeman DA. Bim mediates apoptosis of CD127(lo) effector T cells and limits T cell memory. Eur J Immunol. 2006;36:1694–1706. doi: 10.1002/eji.200635897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hughes PD, Belz GT, Fortner KA, Budd RC, Strasser A, Bouillet P. Apoptosis regulators Fas and Bim cooperate in shutdown of chronic immune responses and prevention of autoimmunity. Immunity. 2008;28:197–205. doi: 10.1016/j.immuni.2007.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hutcheson J, Scatizzi JC, Siddiqui AM, Haines GK, 3rd, Wu T, Li QZ, Davis LS, Mohan C, Perlman H. Combined deficiency of proapoptotic regulators Bim and Fas results in the early onset of systemic autoimmunity. Immunity. 2008;28:206–217. doi: 10.1016/j.immuni.2007.12.015. [DOI] [PubMed] [Google Scholar]
- 22.Weant AE, Michalek RD, Khan IU, Holbrook BC, Willingham MC, Grayson JM. Apoptosis regulators Bim and Fas function concurrently to control autoimmunity and CD8+ T cell contraction. Immunity. 2008;28:218–230. doi: 10.1016/j.immuni.2007.12.014. [DOI] [PubMed] [Google Scholar]
- 23.Snow AL, Oliveira JB, Zheng L, Dale JK, Fleisher TA, Lenardo MJ. Critical role for BIM in T cell receptor restimulation-induced death. Biol Direct. 2008;3:34. doi: 10.1186/1745-6150-3-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.O’Connor L, Strasser A, O’Reilly LA, Hausmann G, Adams JM, Cory S, Huang DC. Bim: a novel member of the Bcl-2 family that promotes apoptosis. Embo J. 1998;17:384–395. doi: 10.1093/emboj/17.2.384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ley R, Balmanno K, Hadfield K, Weston C, Cook SJ. Activation of the ERK1/2 signaling pathway promotes phosphorylation and proteasome-dependent degradation of the BH3-only protein, Bim. J Biol Chem. 2003;278:18811–18816. doi: 10.1074/jbc.M301010200. [DOI] [PubMed] [Google Scholar]
- 26.Akiyama T, Bouillet P, Miyazaki T, Kadono Y, Chikuda H, Chung UI, Fukuda A, Hikita A, Seto H, Okada T, Inaba T, Sanjay A, Baron R, Kawaguchi H, Oda H, Nakamura K, Strasser A, Tanaka S. Regulation of osteoclast apoptosis by ubiquitylation of proapoptotic BH3-only Bcl-2 family member Bim. Embo J. 2003;22:6653–6664. doi: 10.1093/emboj/cdg635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Luciano F, Jacquel A, Colosetti P, Herrant M, Cagnol S, Pages G, Auberger P. Phosphorylation of Bim-EL by Erk1/2 on serine 69 promotes its degradation via the proteasome pathway and regulates its proapoptotic function. Oncogene. 2003;22:6785–6793. doi: 10.1038/sj.onc.1206792. [DOI] [PubMed] [Google Scholar]
- 28.Ley R, Ewings KE, Hadfield K, Howes E, Balmanno K, Cook SJ. Extracellular signal-regulated kinases 1/2 are serum-stimulated “Bim(EL) kinases” that bind to the BH3-only protein Bim(EL) causing its phosphorylation and turnover. J Biol Chem. 2004;279:8837–8847. doi: 10.1074/jbc.M311578200. [DOI] [PubMed] [Google Scholar]
- 29.Dijkers PF, Medema RH, Lammers JW, Koenderman L, Coffer PJ. Expression of the pro-apoptotic Bcl-2 family member Bim is regulated by the forkhead transcription factor FKHR-L1. Curr Biol. 2000;10:1201–1204. doi: 10.1016/s0960-9822(00)00728-4. [DOI] [PubMed] [Google Scholar]
- 30.Stahl M, Dijkers PF, Kops GJ, Lens SM, Coffer PJ, Burgering BM, Medema RH. The forkhead transcription factor FoxO regulates transcription of p27Kip1 and Bim in response to IL-2. J Immunol. 2002;168:5024–5031. doi: 10.4049/jimmunol.168.10.5024. [DOI] [PubMed] [Google Scholar]
- 31.Qi XJ, Wildey GM, Howe PH. Evidence that Ser87 of BimEL is phosphorylated by Akt and regulates BimEL apoptotic function. J Biol Chem. 2006;281:813–823. doi: 10.1074/jbc.M505546200. [DOI] [PubMed] [Google Scholar]
- 32.Harada H, Quearry B, Ruiz-Vela A, Korsmeyer SJ. Survival factor-induced extracellular signal-regulated kinase phosphorylates BIM, inhibiting its association with BAX and proapoptotic activity. Proc Natl Acad Sci U S A. 2004;101:15313–15317. doi: 10.1073/pnas.0406837101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lin A, Minden A, Martinetto H, Claret FX, Lange-Carter C, Mercurio F, Johnson GL, Karin M. Identification of a dual specificity kinase that activates the Jun kinases and p38-Mpk2. Science. 1995;268:286–290. doi: 10.1126/science.7716521. [DOI] [PubMed] [Google Scholar]
- 34.Kaslow HR, Burns DL. Pertussis toxin and target eukaryotic cells: binding, entry, and activation. Faseb J. 1992;6:2684–2690. doi: 10.1096/fasebj.6.9.1612292. [DOI] [PubMed] [Google Scholar]
- 35.Barnett SF, Defeo-Jones D, Fu S, Hancock PJ, Haskell KM, Jones RE, Kahana JA, Kral AM, Leander K, Lee LL, Malinowski J, McAvoy EM, Nahas DD, Robinson RG, Huber HE. Identification and characterization of pleckstrin-homology-domain-dependent and isoenzyme-specific Akt inhibitors. Biochem J. 2005;385:399–408. doi: 10.1042/BJ20041140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dudley DT, Pang L, Decker SJ, Bridges AJ, Saltiel AR. A synthetic inhibitor of the mitogen-activated protein kinase cascade. Proc Natl Acad Sci U S A. 1995;92:7686–7689. doi: 10.1073/pnas.92.17.7686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bouillet P, Metcalf D, Huang DC, Tarlinton DM, Kay TW, Kontgen F, Adams JM, Strasser A. Proapoptotic Bcl-2 relative Bim required for certain apoptotic responses, leukocyte homeostasis, and to preclude autoimmunity. Science. 1999;286:1735–1738. doi: 10.1126/science.286.5445.1735. [DOI] [PubMed] [Google Scholar]
- 38.Bouillet P, Purton JF, Godfrey DI, Zhang LC, Coultas L, Puthalakath H, Pellegrini M, Cory S, Adams JM, Strasser A. BH3-only Bcl-2 family member Bim is required for apoptosis of autoreactive thymocytes. Nature. 2002;415:922–926. doi: 10.1038/415922a. [DOI] [PubMed] [Google Scholar]
- 39.Cummins NW, Rizza SA, Badley AD. How much gp120 is there? J Infect Dis. 201:1273–1274. doi: 10.1086/651434. author reply 1274–1275. [DOI] [PubMed] [Google Scholar]
- 40.Bosque A, Pardo J, Martinez-Lorenzo MJ, Iturralde M, Marzo I, Pineiro A, Alava MA, Naval J, Anel A. Down-regulation of normal human T cell blast activation: roles of APO2L/TRAIL, FasL, and c- FLIP, Bim, or Bcl-x isoform expression. J Leukoc Biol. 2005;77:568–578. doi: 10.1189/jlb.0904514. [DOI] [PubMed] [Google Scholar]
- 41.Sandalova E, Wei CH, Masucci MG, Levitsky V. Regulation of expression of Bcl-2 protein family member Bim by T cell receptor triggering. Proc Natl Acad Sci U S A. 2004;101:3011–3016. doi: 10.1073/pnas.0400005101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bosque A, Aguilo JI, Alava MA, Paz-Artal E, Naval J, Allende LM, Anel A. The induction of Bim expression in human T-cell blasts is dependent on nonapoptotic Fas/CD95 signaling. Blood. 2007;109:1627–1635. doi: 10.1182/blood-2006-05-022319. [DOI] [PubMed] [Google Scholar]
- 43.Riou C, Yassine-Diab B, Van grevenynghe J, Somogyi R, Greller LD, Gagnon D, Gimmig S, Wilkinson P, Shi Y, Cameron MJ, Campos-Gonzalez R, Balderas RS, Kelvin D, Sekaly RP, Haddad EK. Convergence of TCR and cytokine signaling leads to FOXO3a phosphorylation and drives the survival of CD4+ central memory T cells. J Exp Med. 2007;204:79–91. doi: 10.1084/jem.20061681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Benczik M, Gaffen SL. The interleukin (IL)-2 family cytokines: survival and proliferation signaling pathways in T lymphocytes. Immunol Invest. 2004;33:109–142. doi: 10.1081/imm-120030732. [DOI] [PubMed] [Google Scholar]
- 45.Boyman O, Purton JF, Surh CD, Sprent J. Cytokines and T-cell homeostasis. Curr Opin Immunol. 2007;19:320–326. doi: 10.1016/j.coi.2007.04.015. [DOI] [PubMed] [Google Scholar]
- 46.Song J. The costimulation-regulated duration of PKB activation controls T cell longevity. Nat Immunol. 2004:5. doi: 10.1038/ni1030. [DOI] [PubMed] [Google Scholar]
- 47.Green DR. Fas Bim boom! Immunity. 2008;28:141–143. doi: 10.1016/j.immuni.2008.01.004. [DOI] [PubMed] [Google Scholar]
- 48.Liu E, Sinha S, Williams C, Cyrille M, Heller E, Snapper SB, Georgopoulos K, St-Arnaud R, Force T, Dedhar S, Gerszten RE. Targeted deletion of integrin-linked kinase reveals a role in T-cell chemotaxis and survival. Mol Cell Biol. 2005;25:11145–11155. doi: 10.1128/MCB.25.24.11145-11155.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gulino AV, Moratto D, Sozzani S, Cavadini P, Otero K, Tassone L, Imberti L, Pirovano S, Notarangelo LD, Soresina R, Mazzolari E, Nelson DL, Notarangelo LD, Badolato R. Altered leukocyte response to CXCL12 in patients with warts hypogammaglobulinemia, infections, myelokathexis (WHIM) syndrome. Blood. 2004;104:444–452. doi: 10.1182/blood-2003-10-3532. [DOI] [PubMed] [Google Scholar]
- 50.Zhu Y, Swanson BJ, Wang M, Hildeman DA, Schaefer BC, Liu X, Suzuki H, Mihara K, Kappler J, Marrack P. Constitutive association of the proapoptotic protein Bim with Bcl-2-related proteins on mitochondria in T cells. Proc Natl Acad Sci U S A. 2004;101:7681–7686. doi: 10.1073/pnas.0402293101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chomont N, Dafonseca S, Vandergeeten C, Ancuta P, Sekaly RP. Maintenance of CD4+ T-cell memory and HIV persistence: keeping memory, keeping HIV. Curr Opin HIV AIDS. 2011;6:30–36. doi: 10.1097/COH.0b013e3283413775. [DOI] [PubMed] [Google Scholar]
- 52.Chomont N, El-Far M, Ancuta P, Trautmann L, Procopio FA, Yassine-Diab B, Boucher G, Boulassel MR, Ghattas G, Brenchley JM, Schacker TW, Hill BJ, Douek DC, Routy JP, Haddad EK, Sekaly RP. HIV reservoir size and persistence are driven by T cell survival and homeostatic proliferation. Nat Med. 2009;15:893–900. doi: 10.1038/nm.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.van Grevenynghe J, Procopio FA, He Z, Chomont N, Riou C, Zhang Y, Gimmig S, Boucher G, Wilkinson P, Shi Y, Yassine-Diab B, Said EA, Trautmann L, El Far M, Balderas RS, Boulassel MR, Routy JP, Haddad EK, Sekaly RP. Transcription factor FOXO3a controls the persistence of memory CD4(+) T cells during HIV infection. Nat Med. 2008;14:266–274. doi: 10.1038/nm1728. [DOI] [PubMed] [Google Scholar]