

Abstract
Transthyretin (TTR) binds to the Alzheimer-related peptide beta-amyloid (Aβ), and may protect against Aβ-induced neurotoxicity. In this work, the specific domains on TTR involved with binding to Aβ were probed. An array was constructed of peptides derived from overlapping sequences from TTR. Strong binding of Aβ to TIAALLSPYSYS (residues 106–117) was detected, corresponding to strand G on the inner β-sheet of TTR. Aβ bound weakly to four contiguous peptides spanning residues 59–83, which includes strand E through the E/F helix and loop. To further pinpoint specific residues on TTR involved with Aβ binding, nine alanine mutants were generated: I68A, I73A, K76A, L82A, I84A, S85A, L17A, T106A and L110A. Aβ binding was significantly inhibited only in L82A and L110A, indicating that Aβ binding to TTR is mediated through these bulky hydrophobic leucines. Aβ binding to L17A and S85A was significantly higher than to wild-type TTR. Enhancement of binding in L17A is postulated to arise from reduced steric restriction to the interior L110 site, since these two residues are adjacent in the native protein. The S85A mutation caused a reduction in TTR tetramer stability; increased Aβ binding is postulated to be a direct consequence of the reduced quaternary stability.
Keywords: beta-amyloid, scanning alanine mutagenesis, SPOT peptide arrays, transthyretin
Introduction
Alzheimer's disease (AD) is the most common age-associated neurodegenerative disorder. Characteristic features include extracellular senile plaques, intraneuronal neurofibrillary tangles and extensive neuronal cell death. The plaques are deposits of fibrillar aggregates of the 4 kDa peptide beta-amyloid (Aβ), a proteolytic product of amyloid precursor protein (APP). Aβ spontaneously self-assembles through a multistep process into soluble oligomers and fibrils. Numerous studies suggest that Aβ aggregation is causally linked to toxicity, although the exact mechanism remains unknown. Although it was once widely believed that Aβ fibrils were responsible for neurotoxicity, the current dominant paradigm is that soluble oligomeric intermediates are most toxic (Haass and Selkoe, 2007).
Several recent studies suggest that the transport protein transthyretin (TTR) exerts neuroprotective activity against Aβ toxicity. Johnson and coworkers demonstrated that TTR is upregulated in Tg2576 transgenic mice, which are engineered to overexpress the Swedish mutation of APP (APPSw) (Stein and Johnson, 2002). Administration of anti-TTR antibody led to increased tau phosphorylation and greater neuronal cell death, suggesting that increased TTR expression prevented damage from Aβ deposits (Stein et al., 2004). Increased TTR expression in transgenic AD mice has been confirmed by at least one other group (Li et al., 2011). Buxbaum et al. (2008) demonstrated improved cognition and behavior in progeny from APPSw mice crossed with mice expressing human TTR compared with APPSw alone. TTR was upregulated in AD mice raised in an enriched environment relative to those raised in a control environment; these mice performed better on cognitive tests (Costa et al., 2007). In vitro experiments confirm that TTR associates with Aβ and protects against Aβ toxicity (Li et al., 2011).
TTR is a 55 kDa homotetrameric protein present in both blood (3–7 µM) and cerebrospinal fluid (0.1–0.4 µM). Each monomer contains two four-stranded β-sheets, an ‘inner’ sheet of strands D, A, G and H, and an ‘outer’ sheet of strands C, B, E and F. There is also a short EF α-helix. Monomers assemble into a dimer stabilized by extensive hydrogen bonding between strands F and H, while dimer association into the tetramer is mediated mainly through hydrophobic interactions (Hamilton and Benson, 2001). TTR has two natural ligands, thyroxine and retinol-binding protein (RBP), that bind non-competitively (Raghu and Sivakumar, 2004). Thyroxine binds in the pocket created by tetramer assembly. TTR is a minor thyroxine transporter in plasma, with typically only 15–20% of the binding pocket occupied (Richardson, 2007), but carries up to 80% of thyroxine in the central nervous system (Hamilton and Benson, 2001). TTR also serves as the major transporter of RBP, which binds to TTR residues in the EF loop (Monaco, 2000). Normally, ∼30% of TTR is bound to RBP (Filteau et al., 2000).
Previously, we identified two possible Aβ-binding regions on TTR, strand A and EF helix, via cross linking plus tandem mass spectrometry (Du and Murphy, 2010). With this technique, the native folded structure is retained, but only those domains that are spatially close in the protein complex, and contain lysines, are identified. In the present study, we used two complementary methods to further narrow down and identify the specific residues on TTR that are involved with Aβ binding. First, the SPOT peptide array method was used to systematically screen overlapping peptide sequences derived from TTR for binding to Aβ. SPOT, first developed by Ronald Frank and coworkers (Frank, 1992, 2002), has been widely used to identify antibody epitopes, and is effective at identifying binding domains that involve contiguous residues and are not conformationally dependent. Second, we selected nine residues and constructed single-point mutants in which the targeted residue was replaced with alanine. Binding of Aβ to these alanine mutants was probed. In previous studies we suggested that the stability of TTR tetramers strongly influenced TTR–Aβ binding; further confirmation was obtained in the present work.
Materials and methods
Aβ sample preparation
Aβ was purchased from Anaspec, Inc. (San Jose, CA) as lyophilized powder and used without further purification. All other chemicals were purchased from Fisher (Fair Lawn, NJ) unless otherwise stated. Aβ stock solution was prepared by directly dissolving lyophilized Aβ in 8 M urea as described (Pallitto and Murphy, 2001). Aβ samples were prepared by 20-fold dilution of Aβ stock into phosphate-buffered saline with azide (PBSA: 10 mM Na2HPO4/NaH2PO4, 150 mM NaCl, pH 7.4, with 0.02% w/v NaN3) to a final concentration of 0.8 mg/ml. Aβ samples were used immediately (fresh) or incubated at room temperature overnight (pre-aggregated).
Expression and purification of recombinant TTR
A recombinant plasmid of human transthyretin (pTWIN1-TTR) was constructed as described previously (Liu et al., 2009). The IMPACT-TWIN system (NEB, Iswitch, MA) was chosen because it allows for the expression of protein with fully human sequence with native N- and C-termini and purification by single-step affinity adsorption without the need of proteases. The expression plasmids for TTR mutants were prepared with the QuickChange Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA) using pTWIN1-TTR as the template. Mutations were verified by DNA sequencing. Plasmids were transformed into BL21(DE3)pLysS cells (Promega, Madison, WI). Cells were grown on Luria Bertani medium supplemented with 0.1 mg/ml ampicillin at 37°C until an OD600 of 0.4–0.6 was reached, at which time 0.4 mM isopropyl-β-d-thiogalactopyranoside was added to induce protein expression. After 5 h at 37°C, cells were harvested by centrifugation at 4000 rpm for 15 min at 4°C, resuspended in lysis buffer (20 mM Tris, 500 mM NaCl, 1 mM ethylenediaminetetraacetic acid (EDTA), 20 µM phenylmethylsulfonyl fluoride and 1 mM dithiothreitol (DTT), pH 9.0, containing 8 M urea), and sonicated for 10 min on ice. The suspension was centrifuged for 45 min at 4000 rpm and 4°C to remove cell debris, and the supernatant was collected and diluted in half with lysis buffer without urea. Clarified cell lysate was applied to chitin beads equilibrated with column buffer (20 mM Tris, 500 mM NaCl, 1 mM EDTA, pH 9.0) containing 4 M urea. After loading, the column was washed with column buffer containing 1 mM DTT to remove unbound material, then incubated with cleavage buffer (20 mM Tris, 500 mM NaCl, 1 mM EDTA, pH 6.5) overnight at room temperature. Proteins were collected in elution buffer (20 mM Tris, 500 mM NaCl, 1 mM EDTA, 1 mM DTT, pH 6.5) and dialyzed against PBSA. Protein concentration was determined by absorbance at 280 nm, using an extinction coefficient of 77 600−1 cm−1.
Circular dichroism
Protein stock solutions were dialyzed against phosphate-NaF buffer (10 mM Na2HPO4/NaH2PO4, 150 mM NaF, pH 7.4), diluted to a concentration of 2 µM and transferred into a 1-mm cuvette. Circular dichroism (CD) spectra were collected on an Aviv 202SF CD spectrophotometer from Aviv Biomedical (Lakewood, NJ) at 20°C. Blank solvent spectra were collected and subtracted. Spectra were analyzed using the CONTINLL program provided by CDPro software, using the SMP50 reference set as a basis set.
Tryptophan fluorescence
Protein samples were prepared at 0.1 mg/ml in phosphate buffered saline (PBS). Fluorescence spectra were collected using a QuantaMaster spectrofluorometer (PTI, Birmingham, NJ, USA), with excitation at 290 nm and emission spectra recorded from 300 to 420 nm. Three serial spectra were averaged for each sample and the background signal of solvent was subtracted from the averaged data.
ANS fluorescence
TTR solutions were prepared at 1 μM in PBS and mixed with 29 μM 1-anilinonaphthalene-8-sulfonic acid (ANS, AnaSpec, Fremont, CA, USA). ANS concentration was measured by absorbance at 350 nm using an extinction coefficient of 4950 M−1 cm−1. Fluorescence spectra were collected on a PTI QuantaMaster spectrofluorometer, with excitation at 370 nm and emission spectra recorded from 440 to 500 nm. For each sample, three serial spectra were collected, and the background signal of ANS in PBS was subtracted from the average of the three measurements.
Characterization of quaternary structure by gel electrophoresis
Sodium dodecyl sulphate (SDS) was added to TTR (wildtype (wt) or mutant) samples to a final concentration of 2% (w/v). In some cases, samples were boiled at 95°C for 5 min to denature. Samples were loaded on a precise 4–20% polyacrylamide gradient gel (Pierce, Rockford, IL) along with EZ-Run Protein Ladder (Fisher BioReagents, Fair Lawn, NJ) and electrophoresed using SDS buffer for 45 min at 125 V. Gels were stained with Coomassie blue. To cross-link TTR, 50 μl TTR in PBSA was reacted with 4 μl of 25% glutaraldehyde for 2 min at room temperature. The reaction was terminated by adding 4 μl of 7% (w/v) sodium borohydride in 0.5 M NaOH. Cross-linked protein was denatured by boiling in 2% SDS and analyzed on a precise 4–20% polyacrylamide gradient gel.
Light scattering
Protein samples (0.8 mg/ml in PBSA) were filtered through a 0.02 µm filter directly into a light-scattering cuvette and then placed into a bath of the index-matching solvent decahydronaphthalene with temperature controlled to 20°C. Light-scattering data were collected using a Brookhaven BI-200SM system (Brookhaven Instruments Corp., Holtsville, NY) and an Innova 90C-5 argon laser (Coherent, Santa Clara, CA) operating at 488 nm and 300 mW. The z-averaged hydrodynamic diameter was determined from the autocorrelation function using the method of cumulants. Data were collected for several hours to ensure stability of the samples.
Size exclusion chromatography
Protein samples (∼2 mg/ml in PBSA) were injected onto a BioAssist G3SWXL column (Tosoh, King of Prussia, PA) using a 500 µl sample loop and PBSA as the mobile phase. The mobile phase flow rate was set to 1 ml/min and peaks were detected by absorbance at 280 nm.
SPOT peptide array
The optimal length of peptide in SPOT arrays is 8–16 amino acids (Toepert et al., 2003). A series of peptides with overlapping sequences derived from TTR (Table I) were synthesized onto a cellulose membrane (Sigma-Genosys, St Louis, MO). The SPOT membrane was wetted with a few drops of methanol, rinsed with water and then washed with 10 ml Tris-buffered saline (TBS: 20 mM Tris, 150 mM NaCl, pH 7.6) three times for 10 min each. The membrane was blocked by 10 ml blocking buffer (casein blocking buffer, Thermo Scientific) overnight at room temperature. Aβ was pre-aggregated at room temperature at 0.8 mg/ml as described above. The membrane was washed once with 10 ml TBS and incubated with 10 ml diluted Aβ (pre-aggregated Aβ diluted to 5 μg/ml in blocking buffer) at room temperature overnight. The membrane was washed three times with 10 ml T-TBS (TBS with 0.05% (v/v) Tween-20) for 10 min each, and bound Aβ was transferred onto 0.2 μm poly(vinylidene difluoride) (PVDF) membrane (Millipore Corp, Billerica, MA) at 70 mA three times: for 30, 30 and 60 min, sequentially. The three PVDF membranes were blocked with 10 ml blocking buffer at room temperature overnight and then reacted with monoclonal mouse anti-Aβ antibody 6E10 (Covance, Emeryville, CA) at 1 : 8000 dilution in the blocking buffer for 2 h at room temperature. PVDF membranes were subsequently treated with anti-mouse immunoglobulin/HRP (Pierce) at 1 : 8000 dilution in blocking buffer at room temperature for 2 h. Bound Aβ was visualized by means of ECL™ Western Blotting Analysis System (GE Healthcare, Buckinghamshire, UK). The film was analyzed using ImageJ by measuring the density of the spots and comparing with density of the background.
Table I.
Peptide SPOT number and sequence
| 1; 1–12 | GPTGTGESKCPL | 10; 35–46 | KAADDTWEPFAS | 19; 80–89 | KALGISPFHE |
| 2; 7–18 | ESKCPLMVKVLD | 11; 41–52 | WEPFASGKTSES | 20; 84–93 | ISPFHEHAEV |
| 3; 10–19 | CPLMVKVLDA | 12; 47–58 | GKTSESGELHGL | 21; 88–99 | HEHAEVVFTAND |
| 4; 13–22 | MVKVLDAVRG | 13; 53–64 | GELHGLTTEEEF | 22; 94–105 | VFTANDSGPRRY |
| 5; 15–24 | KVLDAVRGSP | 14; 59–70 | TTEEEFVEGIYK | 23; 100–111 | SGPRRYTIAALL |
| 6; 15–22 | KVLDAVRG | 15; 65–76 | VEGIYKVEIDTK | 24; 106–117 | TIAALLSPYSYS |
| 7; 17–28 | LDAVRGSPAINV | 16; 71–82 | VEIDTKSYWKAL | 25; 112–123 | SPYSYSTTAVVT |
| 8; 23–34 | SPAINVAVHVFR | 17; 74–83 | DTKSYWKALG | 26; 118–127 | TTAVVTNPKE |
| 9; 29–40 | AVHVFRKAADDT | 18; 77–86 | SYWKALGISP |
The number corresponds to the position in the SPOT membrane. The TTR residue numbers are given in italics.
Enzyme-linked immunoassay
Enzyme-linked immunoassay (ELISA) plates (Corning Inc., Corning, NY) were coated with 5 μg/ml of TTR (wt or mutant, 100 μl/well) in coating buffer (10 mM sodium carbonate, 30 mM sodium bicarbonate, 0.05% NaN3, pH 9.6) overnight at room temperature. The plate was washed three times with wash buffer (PBS, 0.05% Tween 20) and incubated with blocking buffer (5% non-fat dry milk in wash buffer) for 2 h at room temperature. For a negative control, TTR was not coated but wells were incubated with blocking buffer. Five replicate wells were prepared at each condition.
Freshly prepared Aβ or pre-aggregated Aβ was diluted to 5 μg/ml in PBS and then immediately added to TTR-coated or negative control wells (50 μl/well). For background, PBS was added instead. The plate was incubated at 37°C for 1 h. After washing the plate, anti-Aβ antibody 6E10 was diluted per manufacturer's instruction in PBS, added to each well (100 μl/well), and incubated at room temperature for 1 h with gently shaking. After washing, anti-mouse HRP antibody (1 : 3000 dilution; Pierce) was added to each well (100 μl/well) and the plate was incubated for 1 h at room temperature with gentle shaking. The plate was washed three times with wash buffer, and then 100 μl 3,3′,5,5′-tetramethylbenzidine substrate solution (Pierce) was added into each well. The plate was incubated at room temperature for 15–30 min; color development was stopped by adding 100 μl 2 M sulfuric acid to each well. Absorbance was measured at 450 nm with EL800 Universal Microplate Reader (Bio-tek Instruments Inc., Winooski, VT). Aβ binding was calculated as the mean of five replicate wells by subtracting the absorbance of the negative control from the sample absorbance; the result was then normalized to Aβ binding to wt TTR.
For experiments with cross-linked TTR, 50 μl wt TTR (0.1 mg/ml) was first mixed with 2 μl of 25% glutaraldehyde solution and incubated at room temperature for 2 min. The cross-linking reaction was terminated by the addition of 2 μl of 7% (w/v) sodium borohydride in 0.5 M sodium hydroxide. Cross-linked TTR was then diluted to 5 μg/ml and coated onto the plate.
Results and discussion
SPOT analysis of Aβ-binding TTR peptide sequence
We previously used cross linking plus tryptic digestion and tandem MS to identify domains in TTR involved with binding to Aβ (Du and Murphy, 2010). Cross links with Aβ were identified that involved K9 and K15, both on or near strand A of TTR, and K76, on the EF helix. The advantage of this technique is that the three-dimensional structure of TTR and Aβ is retained. However, only spatially close domains (cross-linker length = 11.4 Å) containing lysines are identified. We therefore chose to use peptide arrays to test whether specific contiguous sequences of TTR are required for binding of Aβ. Twenty-six overlapping TTR-derived peptides were synthesized onto a cellulose membrane (Table I). Pre-aggregated Aβ was diluted to 5 µg/ml and incubated overnight with the SPOT membrane. Bound Aβ was transferred onto a PVDF membrane three times sequentially, in order to reduce background signal. Binding was probed with antibody 6E10. For the third transfer, as shown in Fig. 1A, an unambiguous dark spot was observed at peptide 24 (TIAALLSPYSYS, residues 106–117 of TTR), which corresponds closely to strand G. No bound Aβ was seen for adjacent peptides 23 (SGPRRYTIAALL) or 25 (SPYSYSTTAVVT). This result at first appears to be inconsistent with our previous identification of residues in strand A by cross-linking/tandem MS (Du and Murphy, 2010). However, in the folded TTR protein, strands A and G are neighboring strands in the inner β-sheet, and strand G contains no lysines. We conclude that Aβ binds to strand G, and was cross linked to lysines in strand A.
Fig. 1.

SPOT membrane images and the corresponding TTR peptide sequences. Table I contains sequences corresponding to spot numbers. (A) Third transfer membrane, with strong binding to spot 24. (B) Second transfer membrane, with weak binding to spots 14–17 in addition to strong binding to spot 24. Density difference between spots 14–17 and background was confirmed by image analysis (not shown).
We also examined the second transferred membrane. Although background staining was higher, weak Aβ binding to spots 14, 15, 16 and 17 was detected (Fig. 1B) and confirmed by quantitative image analysis (not shown). Spots 14–17 correspond to the sequence TTEEEFVEGIYKVEIDTKSYWKALG (residues 59–83), which include strand E as well as the EF helix and loop. This is consistent with our previous identification from cross linking/MS/MS studies that residues near K76 in the EF helix are important in Aβ binding (Du and Murphy, 2010).
Binding of Aβ to TTR alanine mutants
To further test whether the regions identified by SPOT and/or cross linking/MS/MS were involved with Aβ binding, we created nine different TTR mutants where a single residue was mutated to alanine. These mutations included one residue from strand A (L17A), two residues from strand G (T106A and L110A), two residues in strand E (I68A and I73A) and four residues from the EF helix/loop (K76A, L82A, I84A and S85A). L17, T106 and L110 are all hydrophobic residues that point from the inner sheet toward the thyroxine-binding channel. (Residue 108 in wt TTR is an alanine and therefore not a target for mutagenesis.) K76 was identified in the MS/MS study and we considered the possibility that this basic residue could bind to negatively charged residues in the N terminus of Aβ. We chose I68, I73 and L82 as mutation sites because they are hydrophobic residues in the EF helix/loop. I84 and S85, in the EF loop, were selected because they are involved in TTR–RBP complex formation (Berni et al., 1994; Naylor and Newcomer, 1999; Monaco, 2000).
Binding of Aβ (either freshly prepared or pre-aggregated) to alanine TTR mutants was measured by ELISA and compared with wt. The mean and range of two representative data sets are shown in Fig. 2; the experiment was repeated at least 10 times with similar conclusions. Aβ binding was unchanged relative to wt for T106A, I68A, I73A, K76A and I84A. (In a few experiments we observed a modest change in Aβ binding to I68A and I84A but this was not consistently observed.) Binding to L82A and L110A was significantly diminished in all experiments. Aβ binding was increased compared with wt for two alanine mutants, L17A and S85A.
Fig. 2.

Binding of Aβ to TTR alanine mutants. TTR (wt and mutants) were adsorbed to 96-well plates, and binding of Aβ was measured using an ELISA method. There were five replicates in each experiment; shown are mean and range of two repeat experiments. The experiment was repeated at least 10 times with the same residues identified in each case. Residues with absorbances falling to the left (<0.75 × wt) or right (>1.5 × wt) of the double lines are considered to influence Aβ binding.
The positions of the four residues identified in alanine screening as influential in TTR–Aβ binding (L17, L110, L82 and S85) are shown in Fig. 3. To check whether these alanine mutants were correctly folded, we compared CD spectra, Trp and ANS fluorescence spectra, native gel electrophoresis and hydrodynamic size. The collected data are summarized in Table II, Figs 4 and 5. CD spectra were virtually identical for wt, L17A and L82A (Fig. 4A). Quantitative analysis of the spectra agreed with the known secondary structure from crystallographic data (Table II). With L110A there was a slight change in CD spectra that may indicate a very minor loss of α-helix and β-sheet (Table II). Trp fluorescence spectra were all essentially identical for wt and these three mutants (Table II), and consistent with published results (Quintas et al., 2001). With S85A, there was a modest decrease in intensity but no significant change in shape of the CD spectra (Fig. 4A) and at most a very slight loss in helix (Table II). Trp fluorescence was not affected (Table II). These data indicate that the four alanine mutants retain native or near-native secondary and tertiary structure.
Fig. 3.
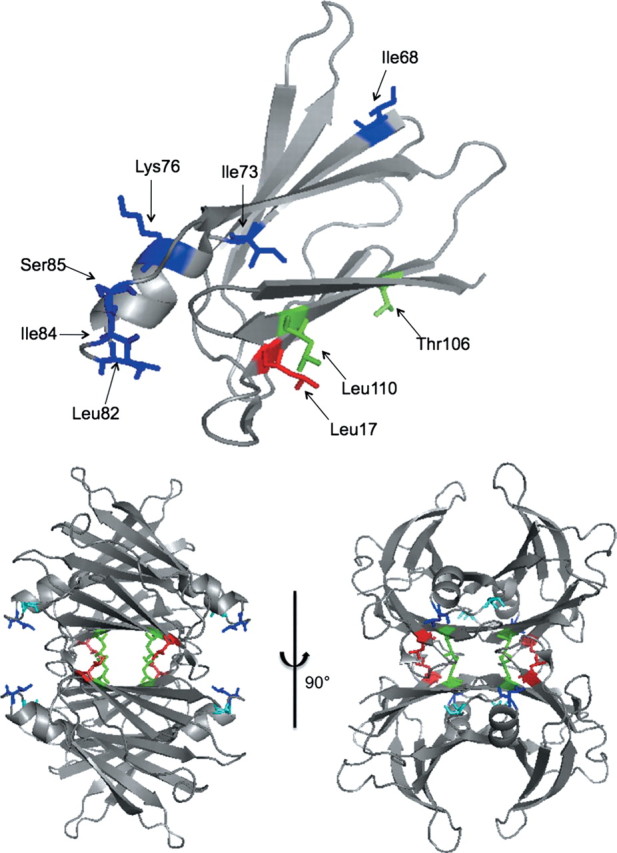
(Top) Ribbon diagram showing location of the residues that were mutated to alanine (one at a time). Only one monomer is shown for clarity. (Bottom) Two views of the TTR tetramer, showing sites of residues identified by alanine screening to affect Aβ binding: L17 (red), L110 (green), L82 (dark blue) and S85 (light blue). Both drawings were generated from Protein Data Bank entry 1DVQ.
Table II.
Physicochemical characterization of TTR mutants
| Secondary structure elements |
Trp λmax (nm) | ANS binding? | Hydrodynamic diameter (nm) | ||||
|---|---|---|---|---|---|---|---|
| β-Sheet | α-Helix | Turn | Disorder | ||||
| Wt | 42 | 8 | 22 | 28 | 337 | Yes | 6.1 ± 0.1 |
| L17A | 41 | 8 | 24 | 27 | 335 | Yes | 6.4 ± 0.2 |
| L82A | 42 | 8 | 23 | 27 | 335 | Yes | 6.6 ± 0.1 |
| L110A | 38 | 6 | 23 | 33 | 336 | Yes | 6.1 ± 0.2 |
| S85A | 42 | 6 | 22 | 30 | 337 | No | 6.2 ± 0.4 |
| L55P | 44 | 9 | 21 | 26 | Partial | 6.4 ± 0.2 | |
| S112I | 43 | 5 | 22 | 30 | No | 5.6 ± 0.3 | |
| F87M/L110M | 41 | 6 | 22 | 31 | No | 5.3 ± 0.3 | |
For comparison, from crystallographic data TTR contains 45% β-sheet and 7% α-helix (Blake et al., 1978).
Fig. 4.
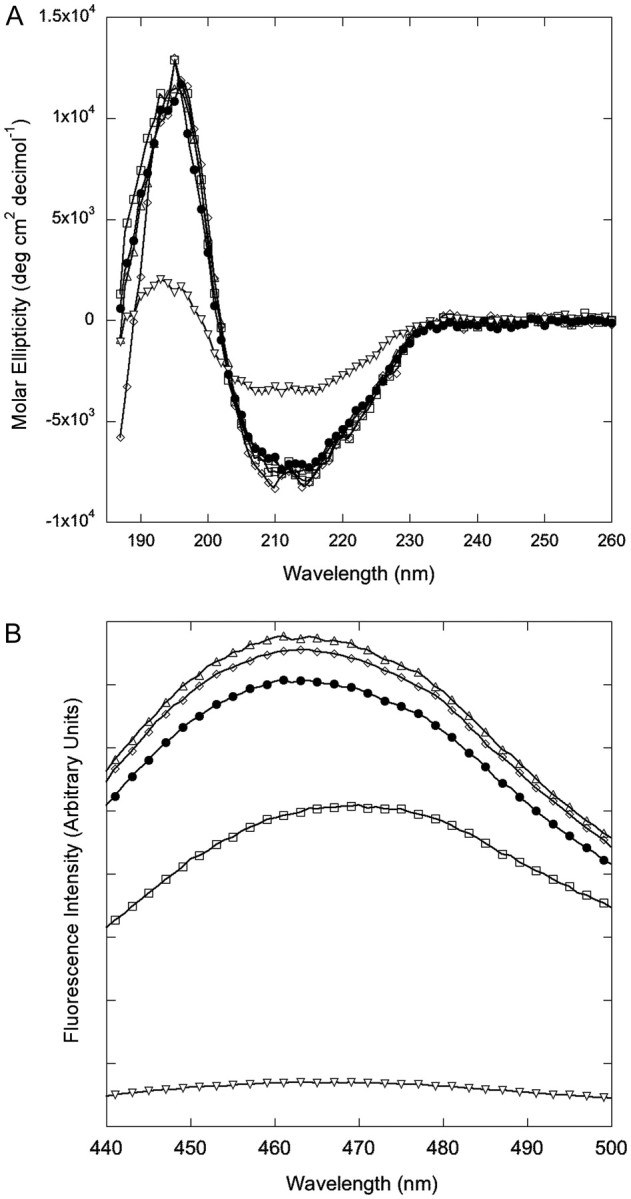
Spectroscopic characterization of secondary and quaternary structure of alanine mutants. (A) Circular dichroism spectra. (B) ANS fluorescence spectra. Open triangle indicates L82A, open diamond indicates L110A, closed circle indicates wt, open square indicates L17A and inverted open triangle indicates S85A.
Fig. 5.

Characterization of tetramer formation in alanine mutants. (A) Size exclusion chromatogram of (reading from bottom up) WT, L17A, L82A, L110A and S85A. Arrows indicate elution times for TTR mutants that form dimers (S112I) and monomers (F87M/L110M). (B) Native gel electrophoresis. Two lanes of each of the indicated proteins were run. Wt TTR tetramers are stable in SDS. (C) Denatured SDS–PAGE. Wt or S85A was cross linked with glutaraldehyde (w/XL) or left uncross linked (w/o XL). Samples were boiled in SDS-containing buffer prior to application to gel.
Wt TTR readily assembles into stable homotetramers; assembly into tetramers is required for formation of the hydrophobic thyroxine-binding channel. ANS is a dye that fluoresces when bound to the thyroxine-binding channel (Cheng et al., 1977); binding causes a large increase in fluorescence intensity and a blue shift in emission maximum to 465–470 nm (Cheng et al., 1977; Liu and Murphy, 2006). ANS spectra were similar for wt, L82A and L110A (Fig. 4B). Fluorescence intensity was reduced for L17A, and the spectrum was red shifted. This is likely due to the fact that the naphthalene ring of ANS normally inserts between K15 and L17 in TTR (Lima et al., 2010), and the change from leucine to alanine at the latter position affects the local environment and therefore fluorescence of this dye.
We used SDS–polyacrylamide gel electrophoresis (PAGE) to test for tetramer formation, because wt TTR tetramers are stable in SDS if the sample is not boiled. Wt, L17A, L82A and L110A all folded into stable tetramers (Fig. 5B). The size exclusion chromatography (SEC) elution profile (Fig. 5A) and the hydrodynamic diameter of wt, L17A, L82A and L110A are all similar (Table II) and consistent with tetramer formation.
In contrast, ANS fluorescence was reduced to near-background levels for S85A (Fig. 4B), indicating loss of the thyroxine-binding channel in this mutant. A mixture of tetramer, dimer and monomer was observed by SEC (Fig. 5A). S85A was monomeric by native gel electrophoresis (Fig. 5B). However, the hydrodynamic radius as measured by dynamic light scattering is consistent with a tetramer (Table II). We interpreted these data to indicate that S85A assembles into a tetramer at moderately high concentrations, but the tetramer is less stable than wt and so dissociates at lower concentrations or in the presence of SDS. To test this, we cross-linked S85A with glutaraldehyde and then analyzed by SDS–PAGE. With cross linking, the tetramer was observed (Fig. 5C), supporting our hypothesis that S85A is able to assemble into tetramers, but these tetramers are less stable than wt.
Taken together, we interpret these data as follows. First, replacing leucines with alanines at positions 82 (EF loop) and 110 (strand G) did not alter the native fold of TTR or its assembly into tetramers, but significantly reduced Aβ binding. We conclude that these two hydrophobic residues, L82 and L110, are required for mediating TTR–Aβ interactions. It should be noted that mutation sites were chosen strategically and were not all-inclusive; thus it is possible that the search strategy missed other TTR residues that are also required for Aβ binding. Second, replacing leucine with alanine at position 17 did not affect native fold, but increased Aβ binding. It is interesting to note that L17 is directly adjacent to L110 in the inner sheet of the TTR tetramer, but closer to the protein's exterior (Fig. 3). We hypothesize that the leucine at position 17 partially blocks access to the L110-binding site, thus explaining the increase in binding to strand G when the bulky leucine residue on strand A is replaced with alanine. T106 is also on strand G but mutation of T106 did not inhibit Aβ binding. This result suggests that Aβ binding is restricted to the C-terminal side of strand G. Alternatively, T106 might participate in Aβ binding but its role could be modest compared with L110 and therefore its replacement with alanine would not significantly reduce the amount of bound Aβ. More detailed measurements of binding kinetics and equilibrium constants are underway which may be able to distinguish between these hypotheses.
Third, we propose that S85A affects TTR–Aβ association indirectly, by rendering TTR tetramers less stable. This essentially increases solvent exposure and access to the interior channel and, hence, to L110. This is consistent with our previous result where we showed that Aβ binds more to an engineered TTR mutant that is stable as a monomer (F87M/L110M; Jiang et al., 2001) than it does to wt (Du and Murphy, 2010). To further test our hypothesis that quaternary structure influences Aβ binding, we generated two additional TTR mutants: L55P, an unstable tetramer responsible for the most aggressive form of familial amyloid polyneuropathy (FAP) (Lashuel et al., 1999); and S112I, a dimeric variant of TTR that is also linked to FAP (Matsubara et al., 2005). We confirmed attainment of the expected secondary and quaternary structure by CD, SEC, native gel electrophoresis and dynamic light scattering (Table II, Fig. 5A, also data not shown). We compared binding of Aβ with wt, L55P, S112I and F87M/L110, and observed significantly greater Aβ binding to all three mutants (Fig. 6). To further test the hypothesis that access to the inner channel facilitates Aβ binding, we cross-linked TTR with glutaraldehyde to restrict access, and then observed a decrease in binding of Aβ to cross-linked TTR compared with uncross-linked TTR (not shown).
Fig. 6.

Aβ binding to TTR mutants L55P (unstable tetramer), S112I (dimer) or F87M/L110M (monomer). Solid bars indicate freshly prepared Aβ and clear bars indicate pre-aggregated Aβ.
Tetramer formation is required for thyroxine binding (Hamilton and Benson, 2001); the binding site includes the hydrophobic patch of L17, T106, A108, L110 and V121 (Hamilton and Benson, 2001). RBP binding to TTR involves residues on the EF loop including L82, I84 and S85 (Berni et al., 1994; Naylor and Newcomer, 1999; Monaco, 2000; Hamilton and Benson, 2001). It is striking that the two residues identified in this study as required for Aβ binding, L110 and L82, are directly involved with binding of the two natural ligands. Both ligands are believed to stabilize TTR's quaternary structure and reduce TTR misfolding and aggregation (White and Kelly, 2001), and there is limited evidence that RBP levels are reduced in AD (Jung et al., 2008). Our results raise the intriguing question as to whether changes in occupancies of the ligand-binding sites could modulate TTR's interaction with Aβ. Experiments are currently underway to test whether Aβ competes with thyroxine and/or RBP for binding to TTR, or more generally whether the presence of either ligand modulates Aβ binding.
TTR, like Aβ, is amyloidogenic. Wt TTR deposits in cardiac tissues cause senile systemic amyloidosis, a disease affecting ∼25% of the elderly (Westermark et al., 1990). Over 80 TTR mutants have been identified that aggregate more aggressively than wt and are associated with FAP and other lethal diseases (Hou et al., 2007). In the generally accepted model of TTR amyloidogenesis, tetramer dissociation to monomers is followed by a small conformational change in the monomer and subsequent reassembly into fibrils (Foss et al., 2005). Disease-associated mutants generally display similar structure but reduced tetramer stability relative to wt (Hornberg et al., 2000; Sorensen et al., 2007; Connelly et al., 2010). The correlation between reduced quaternary stability and increased amyloiodogenicity with TTR is intriguing in light of our observation that Aβ binding increased with reduced tetramer stability. We analyzed the TTR sequence using four different amyloid prediction algorithms and found that strand G, specifically residues 105–110, is repeatedly identified as a putative aggregation site (Fernandez-Escamilla et al., 2004; Galzitskaya et al., 2006; Conchillo-Sole et al., 2007; Tartaglia et al., 2008). The EF helix/loop region was identified in two out of four algorithms. Interestingly, there is evidence that structural perturbation in the EF helix/loop plays a significant role in fibril formation from TTR (Palaninathan et al., 2008; Palmieri et al., 2010). Our data with TTR and Aβ provide a hint that structural similarities between amyloidogenic sites can lead not only to homotypic (self) recognition and aggregation, but also to heterotypic interactions. We speculate that there exists an amyloidogenic regulatory network, where heterotypic interactions (e.g. between TTR and Aβ) alter homotypic interactions (e.g. Aβ–Aβ association and aggregation), in a manner that may change the biological activity of the resulting system.
Acknowledgments
The authors gratefully acknowledge assistance provided by Angelica de Lourdes Rodriguez, Matthew Miklas and Graham Hicken in producing and characterizing alanine mutants, and by Xiaomeng Lu in analyzing TTR using aggregation predictor algorithms. Technical guidance in collection and analysis of CD data was provided by Dr Darrell McCaslin. This work was supported by the National Institutes of Health (R01 AG033493) and the National Science Foundation (CBET-0930102). Circular dichroism spectra were collected at the University of Wisconsin Biophysics Instrumentation Facility, which was established with support from National Science Foundation (BIR9512577) and National Institutes of Health (S10 RR13790).
References
- Berni R., Malpeli G., Folli C., Murrell J.R., Liepnieks J.J., Benson M.D. J. Biol. Chem. 1994;269:23395–23398. [PubMed] [Google Scholar]
- Blake C.C.F., Geisow M.J., Oatley S.J., Rerat B., Rerat C. J. Mol. Biol. 1978;121:339–356. doi: 10.1016/0022-2836(78)90368-6. [DOI] [PubMed] [Google Scholar]
- Buxbaum J.N., Ye Z., Reixach N., et al. Proc. Natl Acad. Sci. USA. 2008;105:2681–2686. doi: 10.1073/pnas.0712197105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng S.Y., Pages R.A., Saroff H.A., Edelhoch H., Robbins J. Biochemistry. 1977;16:3707–3713. doi: 10.1021/bi00635a031. [DOI] [PubMed] [Google Scholar]
- Conchillo-Sole O., De Groot N.S., Aviles F.X., Vendrell J., Daura X., Ventura S. BMC Bioinformatics. 2007;8:65–82. doi: 10.1186/1471-2105-8-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connelly S., Choi S., Johnson S.M., Kelly J.W., Wilson I.A. Curr. Opin. Struct. Biol. 2010;20:54–62. doi: 10.1016/j.sbi.2009.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa D.A., Craechiolo J.R., Bachstetter A.D., Hughes T.F., Bales K.R., Paul S.M., Mervis R.F., Arendash G.W., Potter H. Neurobiol. Aging. 2007;28:831–844. doi: 10.1016/j.neurobiolaging.2006.04.009. [DOI] [PubMed] [Google Scholar]
- Du J.L., Murphy R.M. Biochemistry. 2010;49:8276–8289. doi: 10.1021/bi101280t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Escamilla A.M., Rousseau F., Schymkowitz J., Serrano L. Nat. Biotech. 2004;22:1302–1306. doi: 10.1038/nbt1012. [DOI] [PubMed] [Google Scholar]
- Filteau S.M., Willumsen J.F., Sullivan K., Simmank K., Gamble M. Br. J. Nutr. 2000;83:513–520. [PubMed] [Google Scholar]
- Foss T.R., Wiseman R.L., Kelly J.W. Biochemistry. 2005;44:15525–15533. doi: 10.1021/bi051608t. [DOI] [PubMed] [Google Scholar]
- Frank R. Tetrahedron. 1992;48:9217–9232. [Google Scholar]
- Frank R. J. Immunol. Methods. 2002;267:13–26. doi: 10.1016/s0022-1759(02)00137-0. [DOI] [PubMed] [Google Scholar]
- Galzitskaya O.V., Garbuzynskiy S.O., Lobanov M.Y. Mol. Biol. 2006;40:821–828. [Google Scholar]
- Haass C., Selkoe D.J. Nat. Rev. Mol. Cell Biol. 2007;8:101–112. doi: 10.1038/nrm2101. [DOI] [PubMed] [Google Scholar]
- Hamilton J.A., Benson M.D. Cell. Mol. Life Sci. 2001;58:1491–1521. doi: 10.1007/PL00000791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornberg A., Eneqvist T., Olofsson A., Lundgren E., Sauer-Eriksson A.E. J. Mol. Biol. 2000;302:649–669. doi: 10.1006/jmbi.2000.4078. [DOI] [PubMed] [Google Scholar]
- Hou X., Aguilar M.I., Small D.H. FEBS J. 2007;274:1637–1650. doi: 10.1111/j.1742-4658.2007.05712.x. [DOI] [PubMed] [Google Scholar]
- Jiang X., Smith C.S., Petrassi H.M., Hammarstrom P., White J.T., Sacchettini J.C., Kelly J.W. Biochemistry. 2001;40:11442–11452. doi: 10.1021/bi011194d. [DOI] [PubMed] [Google Scholar]
- Jung S.M., Lee K., Lee J., et al. Neurosci. Lett. 2008;436:153–157. doi: 10.1016/j.neulet.2008.03.010. [DOI] [PubMed] [Google Scholar]
- Lashuel H.A., Wurth C., Woo L., Kelly J.W. Biochemistry. 1999;38:13560–13573. doi: 10.1021/bi991021c. [DOI] [PubMed] [Google Scholar]
- Li X.Y., Masliah E., Reixach N., Buxbaum J.N. J. Neurosci. 2011;31:12483–12490. doi: 10.1523/JNEUROSCI.2417-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lima L.M.T.R., Silva V.D., Palmieri L.D., Oliveira M.C.B.R., Foguel D., Polikarpov I. Bioorg. Med. Chem. 2010;18:100–110. doi: 10.1016/j.bmc.2009.11.025. [DOI] [PubMed] [Google Scholar]
- Liu L., Hou J., Du J.L., Chumanov R.S., Xu Q.G., Ge Y., Johnson J.A., Murphy R.M. Protein Eng. Des. Sel. 2009;22:479–488. doi: 10.1093/protein/gzp025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L., Murphy R.M. Biochemistry. 2006;45:15702–15709. doi: 10.1021/bi0618520. [DOI] [PubMed] [Google Scholar]
- Matsubara K., Mizuguchi M., Igarashi K., et al. Biochemistry. 2005;44:3280–3288. doi: 10.1021/bi048838c. [DOI] [PubMed] [Google Scholar]
- Monaco H.L. Biochim. Biophys. Acta. 2000;1482:65–72. doi: 10.1016/s0167-4838(00)00140-0. [DOI] [PubMed] [Google Scholar]
- Naylor H.M., Newcomer M.E. Biochemistry. 1999;38:2647–2653. doi: 10.1021/bi982291i. [DOI] [PubMed] [Google Scholar]
- Palaninathan S.K., Mohamedmohaideen N.N., Snee W.C., Kelly J.W., Sacchettini J.C. J. Mol. Biol. 2008;382:1157–1167. doi: 10.1016/j.jmb.2008.07.029. [DOI] [PubMed] [Google Scholar]
- Pallitto M.M., Murphy R.M. Biophys. J. 2001;81:1805–1822. doi: 10.1016/S0006-3495(01)75831-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmieri L.D., Lima L.M.T.R., Freire J.B.B., Bleicher L., Polikarpov I., Almeida F.C.L., Foguel D. J. Biol. Chem. 2010;285:31731–31741. doi: 10.1074/jbc.M110.157206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintas A., Vaz D.C., Cardoso I., Saraiva M.J.M., Brito R.M.M. J. Biol. Chem. 2001;276:27207–27213. doi: 10.1074/jbc.M101024200. [DOI] [PubMed] [Google Scholar]
- Raghu P., Sivakumar B. Biochim. Biophys. Acta. 2004;1703:1–9. doi: 10.1016/j.bbapap.2004.09.023. [DOI] [PubMed] [Google Scholar]
- Richardson S.J. Int. Rev. Cytol. 2007;258:137–193. doi: 10.1016/S0074-7696(07)58003-4. [DOI] [PubMed] [Google Scholar]
- Sorensen J., Hamelberg D., Schiott B., McCammon J.A. Biopolymers. 2007;86:73–82. doi: 10.1002/bip.20705. [DOI] [PubMed] [Google Scholar]
- Stein T.D., Anders N.J., DeCarli C., Chan S.L., Mattson M.P., Johnson J.A. J. Neurosci. 2004;24:7707–7717. doi: 10.1523/JNEUROSCI.2211-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein T.D., Johnson J.A. J. Neurosci. 2002;22:7380–7388. doi: 10.1523/JNEUROSCI.22-17-07380.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tartaglia G.G., Pawar A.P., Campioni S., Dobson C.M., Chiti F., Vendruscolo M. J. Mol. Biol. 2008;380:425–436. doi: 10.1016/j.jmb.2008.05.013. [DOI] [PubMed] [Google Scholar]
- Toepert F., Knaute T., Guffler S., Pires J.R., Matzdorf T., Oschkinat H., Schneider-Mergener J. Angew. Chem.-Int. Ed. 2003;42:1136–1140. doi: 10.1002/anie.200390298. [DOI] [PubMed] [Google Scholar]
- Westermark P., Sletten K., Johansson B., Cornwell G.G. Proc. Natl Acad. Sci. USA. 1990;87:2843–2845. doi: 10.1073/pnas.87.7.2843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White J.T., Kelly J.W. Proc. Natl Acad. Sci. USA. 2001;98:13019–13024. doi: 10.1073/pnas.241406698. [DOI] [PMC free article] [PubMed] [Google Scholar]