

Abstract
The construction of expression vector is a basic tool for biotechnology and production of desired proteins, this article summarized the construction of pET-32 α (+) vector techniques which are generally used in research laboratories. The procedures include that acquisition of the exogenous DNA fragment for construction of the vector, subcloning the DNA fragment into pET-32 α (+) expression vector, protein expression in Escherichia coli BL21 (DE3) and protein purification under native conditions in E. coli lysates.
Keywords: Expression vector, Protein expression, Protein purification
Introduction
As a very useful practice in molecular biology lab, recombinant DNA technology (or gene cloning) refer to the transfer of a DNA fragment from one organism to a self-replicating genetic element such as an expression vector. The inserted DNA can then be propagated in a foreign host cells or may be expressed to produce a recombinant protein. A pET-32 α (+) vector series is designed for expression of peptide sequences fused with the 109 aa Trx•Tag™ thioredoxin protein.[1] It belongs to a kind of expression systems which can produce the desired protein when activated after it is inserted into the prokaryotic host cells. Prokaryotic cells such as Escherichia coli are the preferred host for expression of foreign proteins because of their inexpensive, rapid biomass accumulation and simple process scale up.[2]
In this article, we focus on summarizing this useful technique which has been used in our laboratories. Construction of the pET-32 α (+) vector, protein expression and protein purification after expression will be described in detail in this article.
Acquisition of the exogenous DNA fragment for construction of the vector
The DNA to be inserted into the vector could be generally acquired from the multiplying gene using polymerase chain reaction (PCR), it also can be from a gene that artificially synthesized. The methods of getting the interested DNA by PCR are that: First, isolate the total RNA which contains the mRNA of interested DNA fragment and reverse transcript the RNA into cDNA by reverse transcription-polymerase chain reactions (RT-PCR) [Figure 1]. Design the oligonucleotide primers according the published coding sequence (CDs) of the gene on NCBI (http://www.ncbi.nlm.nih.gov/pubmed/), and add the indicated restriction enzyme sites in the primers. Then, amplify the coding region of interested DNA by PCR [Figure 2] and insert the DNA fragment into a cloning vector after purified by gel extraction kit.
Figure 1.

Reverse transcript the RNA into cDNA by RT-PCR
Figure 2.
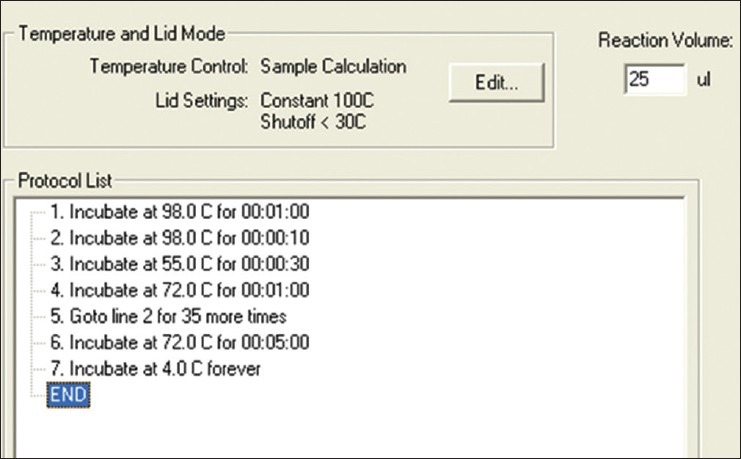
Amplification of the coding region of interested DNA by PCR
Experience
Although getting the DNA fragment from RT-PCR is one of commonly used protocols, many problems may happen and lead to some unexpected results, here are some experiences about how to acquire high quality DNA fragment: In the beginning steps of isolating RNA, insure that all of processing steps should be done in a RNases-free environment and homogenize the sample quickly and sufficiently under room temperature. High purity and integrity of RNA is essential for synthesis of full-length cDNA and latter for high-quality PCR products. Addition to comply with the basic rules of designing primers, the primers designed must be able to extend the template DNA from start codon to stop codon if we wish to express it later. When introducing the restriction sites into the 5’ end of the primer, verify that the DNA fragment amplified does not contain the right restriction sites, and make sure that the restriction sites you choose maintain the DNA fragment in frame with the start codon of the expression vector which allows it be expressed as a functional protein.
Troubleshooting
Low template quality or quantity may happen sometimes and result in low yield or no PCR product, to avoid this, insure to assess the integrity of RNA prior to cDNA synthesis and get rid of the trace amounts of reagents used in RNA purification protocols by washing the RNA pellet totally with 75% ethanol. Poor PCR product may also be caused by the fault primer design and suboptimal thermal cycling conditions, so it is better redesign the primer and optimize the thermal cycling conditions and reaction conditions if this happens. Do not forget to use a high fidelity thermostabile DNA polymerase in your PCR and limit exposure the PCR product to UV to several seconds to avoid a sequence error. Extra bases are required to be added for efficient cleavage before the restriction enzyme sites in the primers, DNA polymerase and other reagents in the DNA product should be removed before digestion, this may lead to an efficient cleavage of the PCR product and easy PCR cloning into the vector.
Subcloning the DNA fragment into pET-32 α (+) expression vector
The gene fragment of coding region which is bounded by a stat codon (ATG) in the 5’ end and a stop codon (TAA, TAG or TGA) in the 3’ end was synthesized by a company with indicated restriction enzyme-cutting site. Then the gene fragment of interest was inserted into pMD18-T vector to construct a recombinant plasmid, which was identified by DNA sequencing. The recombinant pMD18-T plasmid was digested with indicated enzyme, and retrieved DNA fragment of interest was subcloned into the pET-32 α (+) vector by the following protocol to generate a recombinant expression vector [Figure 3].
Figure 3.
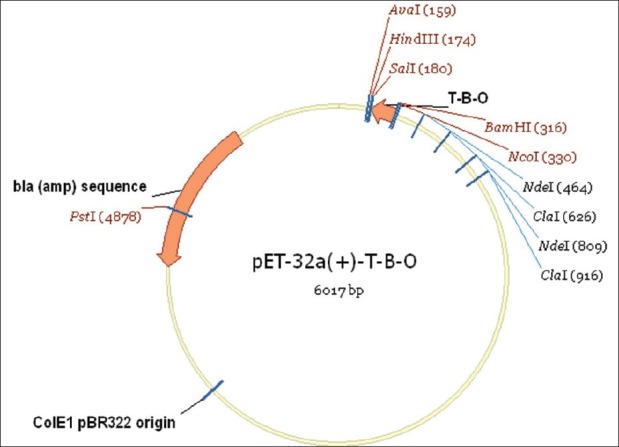
The gene fragment of TBO about 129 bp was inserted into pET-32α (+) vector (5900 bp)
Digest the pET-32 α (+) vector and recombinant pMD18-T vector with the same restriction enzyme at 37°C for 1 hour and then heat inactivate the enzymes by incubating at 65°C for 10 minutes.
Purify the digested pET-32 α (+) vector backbone and the gene fragment cut from pMD18-T vector by gel extraction kit.
Ligate the gene fragment with the pET-32 α (+) vector backbone by T4 DNA ligase at 23°C for 1 hour, the ratio of vector ends and insert ends is 1:3 (fmol).
Mix the 10 μl of the ligation reaction with 100-μl competent E. coli BL21 (DE3) on ice for 10 min, and incubate the mixture at 42°C for 90 seconds. Add 800-μl LB and incubate at 37°C for 40 minutes. Spread on an LB plate containing 100 μg/ml ampicillin. Incubate at 37°C overnight.
Screen for the presence of inserts by colony PCR or prepare DNA and digested with an indicating restriction enzyme [Figure 4].
Figure 4.
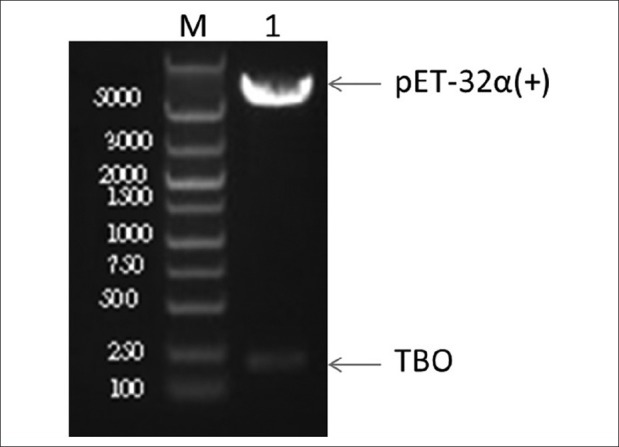
The presence of inserts was identified by restriction enzyme digestion. M: DNA marker, 1: recombinant pET-32α (+) vector after digested with BamHI/SalI
Experience
Sometimes it is still difficult to get the DNA product through PCR because the primers are defined to a limited region with the start codon and stop codon, so artificially synthesizing a gene needed is an alternative choice to get the DNA fragment. For our experience, the expression vectors are large or have a low copy number, so a more efficient way is to clone the DNA fragment into a nonexpression vector first and then subclone it into the expression vector. Insure to set a negative control with no vectors in the competent cells and a positive control with an empty vector alone in the E. coli cells during transformation.
Troubleshooting
1. Low yield or no transformants: The reason may be that the competent E. coli cells have very low transformation efficiency, so check the transformation efficiency with an empty vector DNA before transformation. Excessive amount of ligation mixture used for transformation or excessive amount of ligase used in ligation may also lead to this problem. Before transformation, insure that the DNA fragment contains no contaminants (e.g., excess salts, EDTA, proteins, phenol, etc.) and digested by the correct restriction enzyme properly before ligation. 2. Empty vector with no insert: This may be caused by vector recircluarization or incomplete vector cleavage, try to dephosphorylate the vector and check the cleavage efficiency on an agarose gel may help you solve this problem. 3. Sequence errors in insert: Errors in PCR primers and low fidelity DNA polymerase used in PCR may cause this problem.
Protein expression in E. coli BL21 (DE3)
The recombinant pET-32 α (+) vector could express a desired protein which fused to 109 aa Trx•Tag™ in E. coli BL21 (DE3) after induced by isopropyl β-D-thiogalactoside (IPTG). First, inoculate 10 ml of LB medium containing 100 μg/ml ampicillin with a fresh bacterial colony harboring the recombinant pET-32 α (+) vector at 37°C overnight; Then, add 500-μl overnight culture into 30-ml fresh LB medium containing 100-μg/ml ampicillin, inoculate at 37°C until the OD600 = 0.6. Add IPTG to a final concentration of 1 mM and grow the culture at 32°C for 5 hours in a shaker with 200 rpm [Figure 5], after that, harvest the cells by centrifugation at 4000 g for 15 min. To identify the expression of the fusion protein in the cells, first, collect a part of cells before and after induced by IPTG, centrifuge at 4000 g for 15 min, then, resuspend the pellet with PBS and sonicate them on ice, collect the supernatant and pellet, respectively, and analyze by SDS-PAGE gel [Figure 6].
Figure 5.

Inoculation of E. coli BL21 for protein expression
Figure 6.
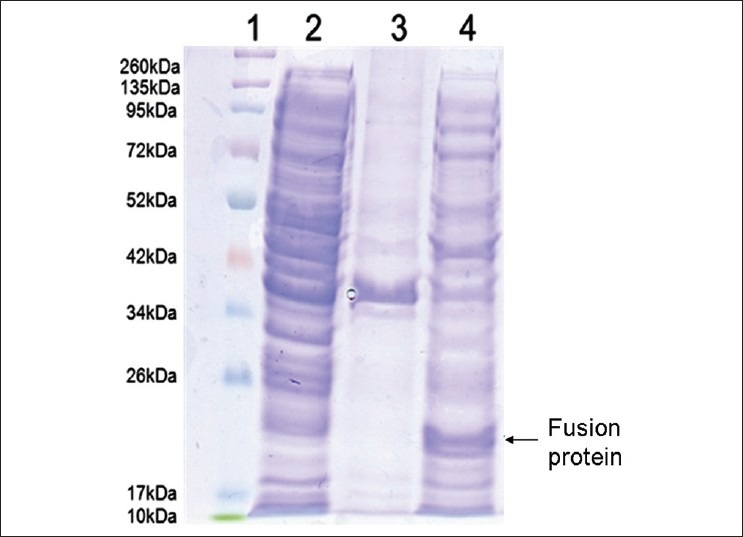
SDS-PAGE analysis of the fusion protein induced by IPTG. Proteins were separated by SDS-PAGE and stained with Coomassie brilliant blue. Lane 1: protein molecular weight marker; Lane 2: supernatant after sonication of uninduced BL-21/pET32 a (+)/pTBO; Lane 3: precipitate after sonication of induced BL-21/ pET 32 a (+)/ pTBO; Lane 4: supernatant after sonication of BL-21/ pET 32 a (+)/ pTBO induced for 5 hours. The molecular weight of pTBO fusion protein is about 22 kDa
Experience
For a heterologous protein expression in a prokaryotic species, first, we need to optimize the codon which is commonly used in E. coli. Second, after adding IPTG in the culture media, allow the culture to grow for different time points such as 3 h, 6 h, 12 h 18 h, and also try different temperature by 37°C, 30°C, 21°C or 18°C, to select the optimized protein expression conditions, this may increase the solubility of expressed protein.
Troubleshooting
To try the lower growth temperature and lower inducer concentration may avoid the formation of inclusion bodies and help with some cases of incorrect disulfide bond formations and hydrophobic proteins. To protect the protein, we may add a proteinase inhibitor to the medium after culturing and break the cells on ice to avoid the degradation.
Protein purification under native conditions in E. coli lysates
Trx•Tag™ is a peptide sequence grafted onto the recombinant protein which could be removed by enterokinase. This protein tag is a sulubilization tag that could assist in the proper folding in proteins and keep them from precipitating. The fusion protein expressed by recombinant pET-32 α (+) vector also contains a His-Tag which is often used for affinity purification, this His-Tag also could be used for identification of fusion protein after purification via Western blotting [Figure 7]. Here is the procedure to purify the protein under native conditions by Ni-NTA spin kit (QIAGEN).
Figure 7.

Western blot analysis of the fusion protein expression. The fusion protein of TBO was expressed in the E.coli BL21 strain, after purification by NI-NTA spin kit, the protein was analyzed by Western blot using anti-His-tag antibody. The Trx. Tag + TBO is 22 KDa
Resuspend a pellet derived from 5-ml culture volume in 630 μl Lysis buffer (NPI-10), sonicate the cell suspension for 10 short burst of 10 second on ice with 30-second intervals.
Centrifuge lysate at 12,000 g for 20 min at 4°C and collect the supernatant.
Equilibrate the Ni-NTA spin column with 600-μl buffer NPI-10, centrifuge for 2 min at 890 g.
Load up to 600-μl lysate containing the 6 × His-tagged protein onto the pre-equilibrated Ni-NTA spin column, centrifuge for 5 min at 270 g and collect the flow-through
Wash the Ni-NTA spin column trice with 600-μl buffer NPI-20, centrifuge for 2 min at 890 g.
Elute the protein twice with 300 μl buffer NPI-500, centrifuge for 2 min at 890 g and collect the eluate.
Experience
All the buffers used in this protocol should be filtered first, in case the solid particles in the buffer block the spin column, and insure the buffers contain the accurate concentration of imidazole. Do not forget to collect the flow-through in each steps, that will allow you to check what kind of proteins eluted in the eluate latter. The NI-NTA spin columns is reusable, that means the spin column can be used for two or three times after equilibrated again by NPI-10.
Troubleshooting
Protein does not bind to the NI-NTA spin column: First, this may be caused by the incorrect binding conditions, such as lower pH value and higher concentration of imidazole in the buffer. Second, the 6× His-tag does not present or is inaccessible in your protein, so you should recheck the expression construction again if the pH value and concentration of imidazole in the buffer are correct.
Protein elutes in the wash buffer: This may be because the wash stringency is too high or wrong buffer conditions, for this problem, to lower the concentration of imidazole or increase the pH value slightly may help sometimes.
Protein precipitates during purification: This will happen when the environment temperature is too low, so performing purification at room temperature may be better than putting it on ice.
In conclusion, as a widely used biotechnique, construction of pET-32 α (+) vector for protein expression and purification in E. coli is fast, inexpensive and scaleable. While as a prokaryotic system, it also has its disadvantage compared with eukaryotic expression system. The property of protein is various and it is difficult to tell if the protein of interest could be expressed well, be soluble, possess activity and crystallize.[3] So the choice of expression strategy including the vector and host should be determined carefully according to the requirement of amount, purity and activity of the protein.
Footnotes
Source of Support: Nil.
Conflict of Interest: None declared.
References
- 1.LaVallie ER, DiBlasio EA, Kovacic S, Grant KL, Schendel PF, McCoy JM. A thioredoxin gene fusion expression system that circumvents inclusion body formation in the E.coli cytoplasm. Biotechnology (N Y) 1993;11:187–93. doi: 10.1038/nbt0293-187. [DOI] [PubMed] [Google Scholar]
- 2.Sahdev S, Khattar SK, Saini KS. Production of active eukaryotic proteins through bacterial expression systems: A review of the existing biotechnology strategies. Mol Cell Biochem. 2008;307:249–64. doi: 10.1007/s11010-007-9603-6. [DOI] [PubMed] [Google Scholar]
- 3.Hartley JL. Cloning technologies for protein expression and purification. Curr Opin Biotechnol. 2006;17:359–66. doi: 10.1016/j.copbio.2006.06.011. [DOI] [PubMed] [Google Scholar]