

Abstract
We review our experience with unusual ocular pathologies mimicking retinoblastoma that were referred to our institution over the past two decades. After presenting the imaging anatomy of the normal eye, we discuss pertinent clinical and pathological features, and illustrate the ultrasound and magnetic resonance imaging appearance of retinoblastoma, medulloepithelioma, uveal melanoma, persistent fetal vasculature, Coats disease, corneal dermoid, retinal dysplasia and toxocara granuloma. Features useful in discriminating between these entities are emphasized.
Introduction
Leukocoria is an abnormal white reflection from the retina of the eye compared with the normal “red reflex.” From the Greek “leukos” (white) and “kore” (pupil), leukocoria signifies an abnormality located between the lens and the retina, representing a threat to vision and/or life. Adults may have leukocoria as a result of trauma, glaucoma, or other intraocular masses. Leukocoria in children is often the clinical finding of retinoblastoma, but may be due to other ocular diseases. The lengthy differential includes other intraocular malignancies (medulloepithelioma), congenital malformations such as persistent fetal vasculature, Coats disease, corneal dermoid, retinal dysplasia and Norrie disease, and infectious reactions (toxocara granuloma). Retinal detachment and more common etiologies such as ocular trauma or disorganized growth of retinal blood vessels in premature infants (retinopathy of prematurity) can cause leukocoria, but are excluded by eliciting pertinent elements during the history and physical exam. An accurate diagnosis of the other more rare ocular pathologies requires recognition of specific clinical features, imaging and, if needed, histopathologic features. This pictorial essay describes imaging and clinical features of unusual ocular pathologies that were referred to our large retinoblastoma center. An accurate diagnosis was possible using clinical features and imaging although some patients with advanced ocular disease and lack of visual potential at diagnosis underwent enucleation after consultation with the family. We present clinical and pathology findings as well as ultrasound (US) and magnetic resonance imaging (MRI) that illustrate salient features of each disease in addition to an algorithm that may be useful in establishing the correct diagnosis.
Normal Eye Anatomy and Imaging Modalities for Assessing the Eye
An awareness of normal eye anatomy is critical to accurately evaluate lesions presenting with leukocoria and masquerading as retinoblastoma. The US and MRI appearances of the normal eye are reviewed in Figure 1. In general, ultrasound is useful to delineate areas of calcification within intraocular lesions, identify tissue interfaces, and reveal patterns of vascularity using Doppler technology. MRI is less sensitive than US for the detection of calcification but more sensitive to tumor extension into the optic nerve and subarachnoid spaces [1]. Therefore, these modalities are complementary and both are routinely used at our institution. We perform ultrasound with patients awake; a typical examination takes 10 to15 minutes. Only rarely do we find it necessary to perform ultrasound while the patient is sedated for MRI. We use a linear, high-frequency transducer with a small footprint that is placed directly on standard ultrasound gel applied to the closed eyelid. The ultrasound machine’s mechanical index is kept ≤ 0.23 to conform to FDA guidelines and minimize risk of thermal injury to the eye [2,3,4]. Color Doppler is performed routinely to assess the vascularity of the intraocular mass, to assess the relationship of the mass to the optic disc and to help distinguish dense subretinal fluid from a solid mass. MRI requires some level of sedation in order to provide optimal images. Guidelines for standardized MR imaging of the eye were recently published [5]. Currently, at our institution, we use a 32 channel total imaging matrix (TIM) coil to obtain images of the orbits and head during the same scanning session. Imaging of the orbits includes thin (3mm), pre-contrast axial and coronal constructive interference steady state (CISS, heavily T2W) and T1W sequences and post-contrast, fat-saturated T1W sequences. Imaging of the head includes pre-contrast sagittal T1W and axial fat-saturated T2W and diffusion weighted sequences followed by post-contrast, sagittal T1W magnetization prepared rapid gradient echo (MPRAGE) and axial T1W and fluid attenuated inversion recovery (FLAIR) sequences. We do not perform CT for assessment of intraocular tumors due to the inherent exposure of patients to ionizing radiation and the association of radiation induced secondary malignant neoplasms in patients with leukocoria who are ultimately diagnosed with hereditary retinoblastoma. In modern day practice, the combination of US, MRI and fundoscopy obviates the need for CT in the setting of pediatric ocular tumors [5].
Fig. 1.
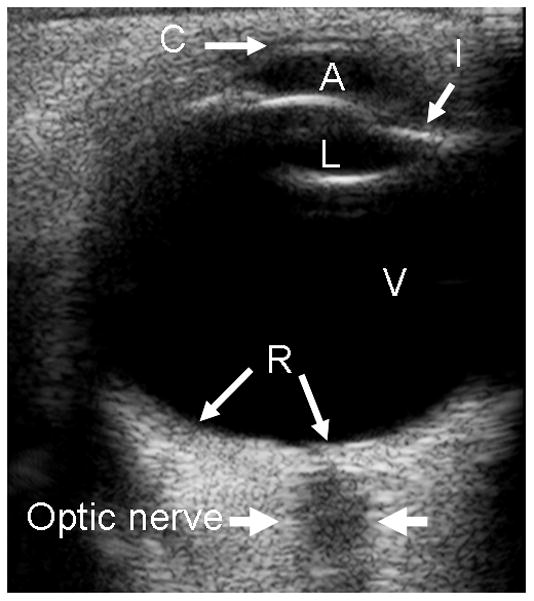
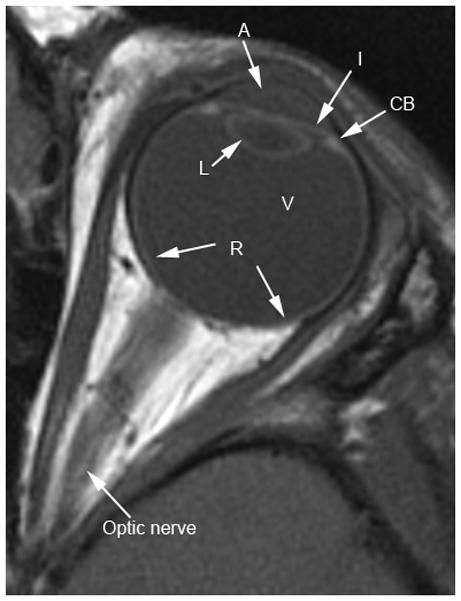
Normal eye anatomy on transverse. a Ultrasound (US). b T1W MR images. C = cornea, L = lens, I = iris, A = anterior chamber, CB = ciliary body, V = vitreous chamber, R = retina
Retinoblastoma
Retinoblastoma (RB) is the most common intraocular malignancy of childhood and the most common cause of childhood leukocoria. While most patients present before four years of age, thirty to forty percent of patients will have a germline mutation in the RB1 gene and present at an earlier age with multifocal, bilateral disease [6]. Patients with the genetic form of retinoblastoma are at an increased risk for developing primary intracranial neuroectodermal tumors in the pineal or suprasellar region, termed “trilateral retinoblastoma”; this form of disease carries a very poor prognosis [7]. Since 3–9% of patients with heritable retinoblastoma will develop trilateral retinoblastoma, neuroimaging every 6–12 months until age 5 has been recommended [6,8]. When diagnosed early and treated with a combination of systemic chemotherapy, focal therapy (i.e. laser phocoagulation, thermotherapy, cryotherapy, plaque radiotherapy), external beam radiation and surgery (enucleation), this malignancy is curable in over 95% of children [6]. When direct ophthalmologic evaluation is limited by cataracts, hyphema or disease progression, imaging with US and MRI provides important disease assessment. Solitary or multiple intraocular masses, often with calcification in older or advanced stage patients, are readily visible by US. Ultrasound can also demonstrate associated retinal detachment and vitreous seeding (Fig. 2a). MRI demonstrates lesions that are hyperintense to vitreous on T1-weighted sequences and hypointense to vitreous on T2-weighted sequences (Figs. 2b and c)[9]. Ultrasound and MRI features of RB have been previously well described [9,10].
Fig. 2.
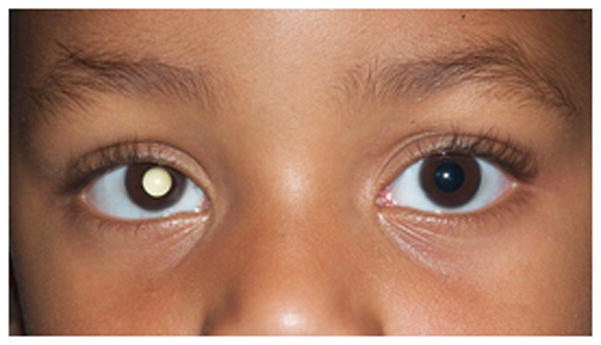
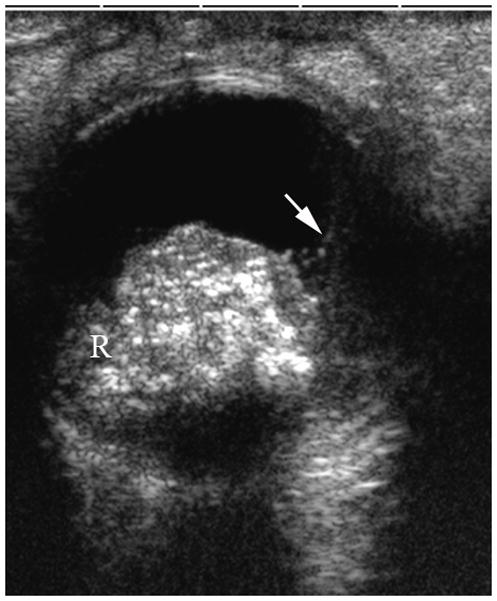
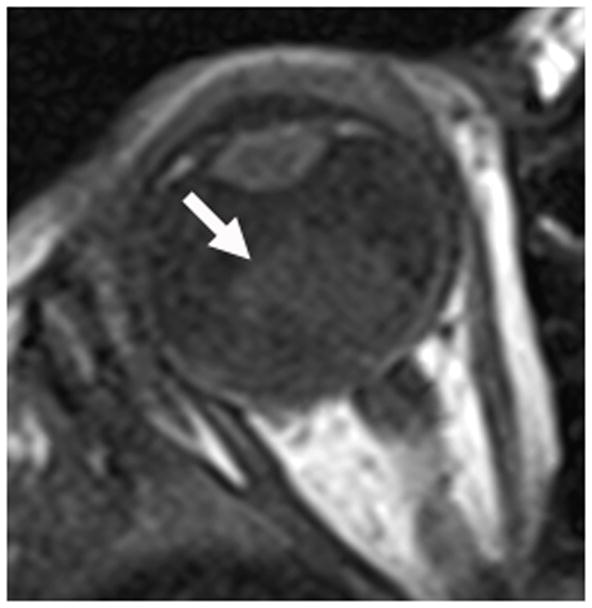
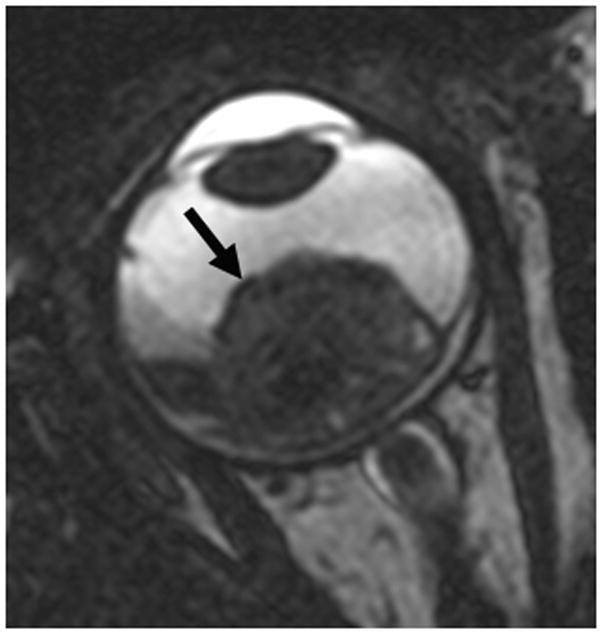

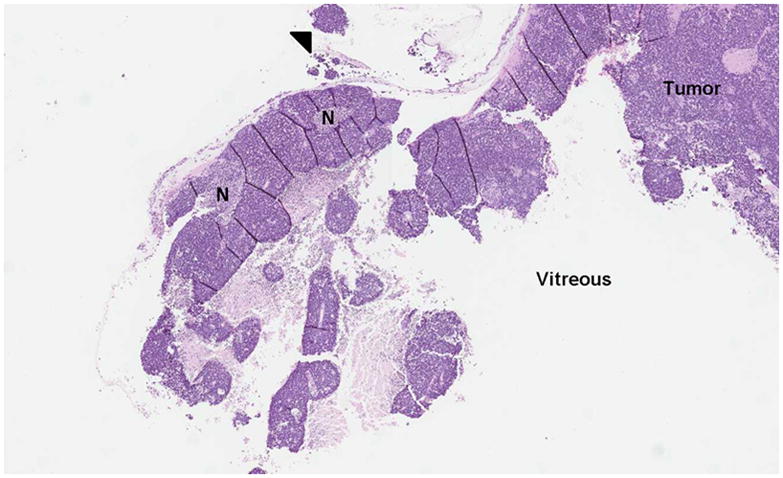
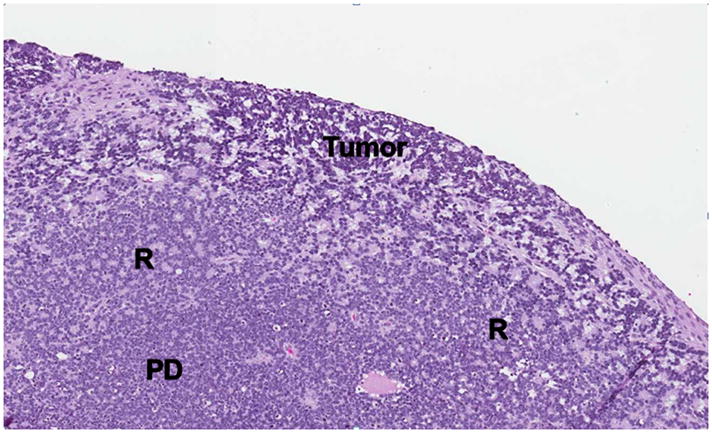
1-year old girl presented with leukocoria of the right eye due to retinoblastoma. a Clinical face photo of leukocoria. b Transverse US image shows the solid retinoblastoma (R) with numerous internal microcalcifications (tiny hyperechoic foci) typical of retinoblastoma. A detached retina (arrow) is also apparent. c T1W axial image of the same eye shows the retinoblastoma (arrow) to be slightly hyperintense to the vitreous. d CISS axial image shows the tumor (arrow) is hypointense to vitreous. These signal characteristics are typical of retinoblastoma. Note that microcalcifications are not apparent on MRI but well visualized by US. Current studies support the use of both US and MRI in the assessment of ocular tumors. Computed tomography adds little information in this setting and is not indicated. e Pupil-optic nerve section at low power showing combined exo-/endo-phytic retinoblastoma. f Retinoblastoma tumor expands into the vitreous, creating vitreous seeds (black arrowhead). Patchy necrosis (N) is present. g Retinoblastoma tumor erodes through retina, distorting the retinal layer. Areas of the tumor form rosettes (R) or are poorly differentiated (PD).
Medulloepithelioma (diktyoma)
Another intraocular malignant tumor, medulloepithelioma, usually occurs in the first decade of life. This non-pigmented, vascular mass is distinctly different in location compared with retinoblastoma; it arises from and is localized to the ciliary body. It is associated with superficial cystic elements, vision loss, a lens notch, and pain due to subluxation of the lens [1,11,12]. Associated retinal detachment is common. Patients may demonstrate rubeosis (neovascularization), leading to secondary neovascular glaucoma [11,12]. With rare presentations of the mass in the optic nerve or retina, especially with dystrophic calcifications, histopathologic examination demonstrating a characteristic pattern of folded cords and sheets (resembling a fisherman’s net, “diktyomatous” pattern) is required to differentiate this lesion from retinoblastoma[1]. Ultrasound or MRI demonstrate a solid mass with cystic components, usually arising from the ciliary body (Fig. 3a–c). The solid component generally enhances markedly on MRI after contrast administration (Fig. 3b)[1]. Although most medulloepitheliomas are cytologically malignant, distant metastasis and mortality is rare [1,11]. Iridocyclectomy may be followed by local recurrence requiring enucleation [11,12].
Fig. 3.
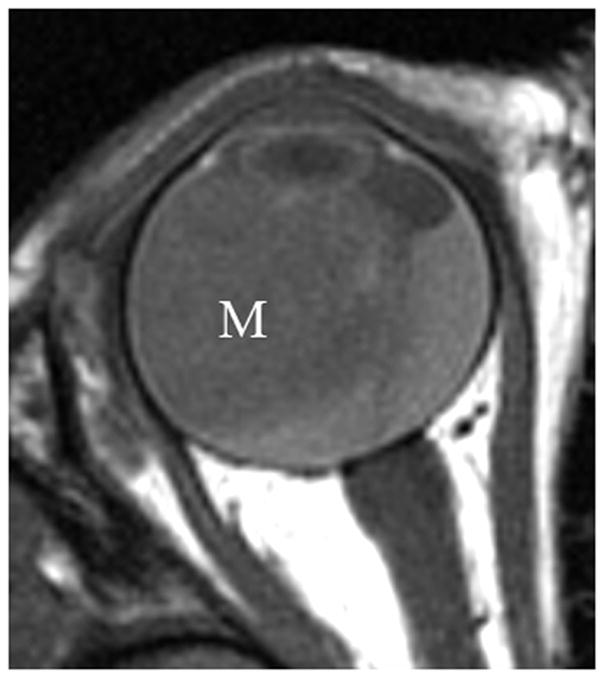
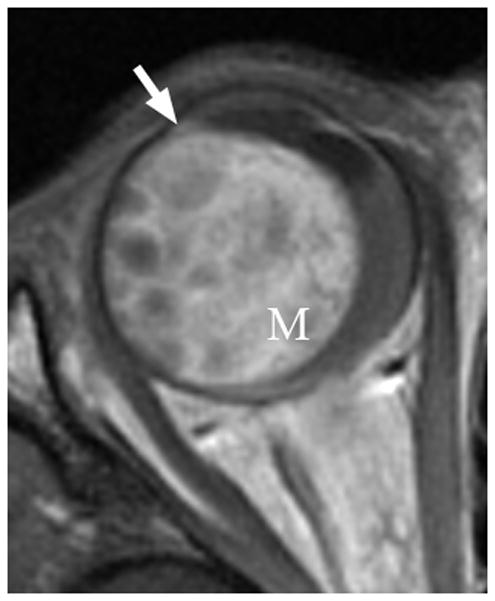
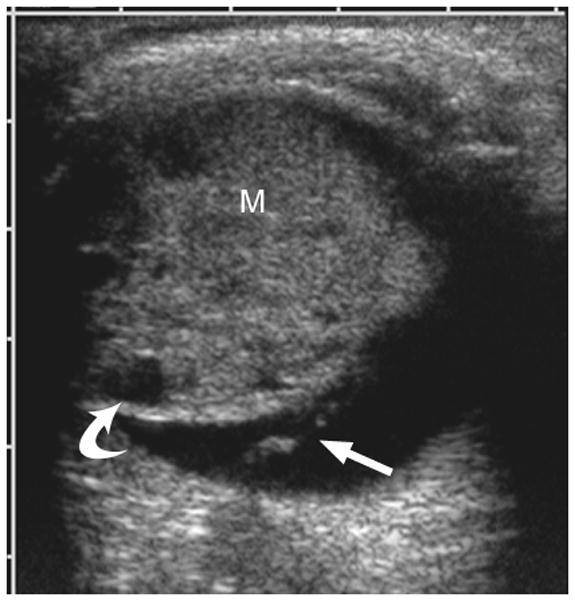

3-year-old boy with medulloepithelioma who presented with 1 week history of exotropia of the right eye. a Pre-contrast-enhanced and b post-contrast-enhanced T1W MRI show marked enhancement of medulloepithelioma (M) arising from lateral ciliary body (arrow). Note small cysts scattered throughout mass. The intense enhancement, tumor location and cysts are features typical of medulloepithelioma. c Transverse US shows medulloepithelioma (M) containing small cysts (curved arrow) arising from lateral ciliary body. There is a possible detached retina (straight arrow), a common complication of this tumor. d Hematoxylin and eosin (H&E) staining of teratoid medulloepithelioma with prominent cyst (straight arrow) and cartilage (curved arrow) formation. The tumor is composed of a proliferation of small round blue cells in nests and cords (fisherman’s net or “diktyomatous” pattern).
Uveal melanoma
Ocular melanoma arises from melanocytes most commonly in the uveal tract (iris, ciliary body and choroid) of the eye. In adults, uveal melanoma is the most common primary ocular malignancy, however, only ~1–2% of uveal melanoma cases are reported in patients less than 20 years of age [13,14]. Most patients are Caucasian and present with unilateral disease [14]. Associated syndromes, such as neurofibromatosis type 1, oculo(dermal) melanocytosis, and dysplastic nevus syndrome [15] and even congenital cases [16] have been reported. In one series, oculo(dermal) melanocytosis (hyperpigmentation of the episclera, uvea, orbit, meninges and periocular skin) was nine times more common in young patients with uveal melanoma than in the general population with uveal melanoma [14]. Local control with radiation or enucleation are the primary treatment modalities. Although short term survival in children (95% at five years) is superior to adults, long-term survival (77%) is similar [14]. The risk of metastasis is life-long and regular monitoring is needed [14]. Uveal melanoma may have one of three configurations; 1) lentiform, 2) mound and 3) mushroom shaped. The mushroom configuration is considered almost pathgnomonic of choroidal melanoma. Serous retinal detachment occurs in about 66% of cases and is a sign of advanced disease [17]. The ultrasound features of uveal melanoma are described in ophthalmology literature and include a solid, hypoechoic mass that lacks calcification (Fig. 4a) [17,18]. The MRI features of uveal melanoma are well described and depend on the amount of melanin within the tumor which can range from absent to strongly pigmented. Approximately 31% of uveal melanomas are amelonotic or minimally pigmented. The remainder exhibit shortening on both T1W and T2W MR images resulting in high signal on T1 and very low signal on T2W images. Uveal melanoma typically show minimal enhancement after contrast administration (Fig. 4b–d) [19].
Fig. 4.
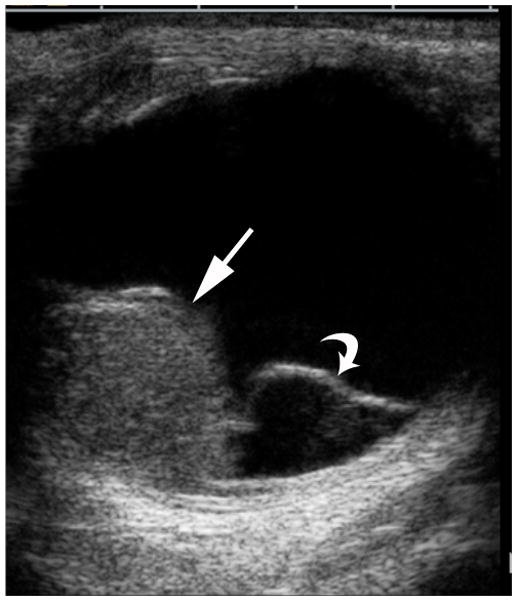
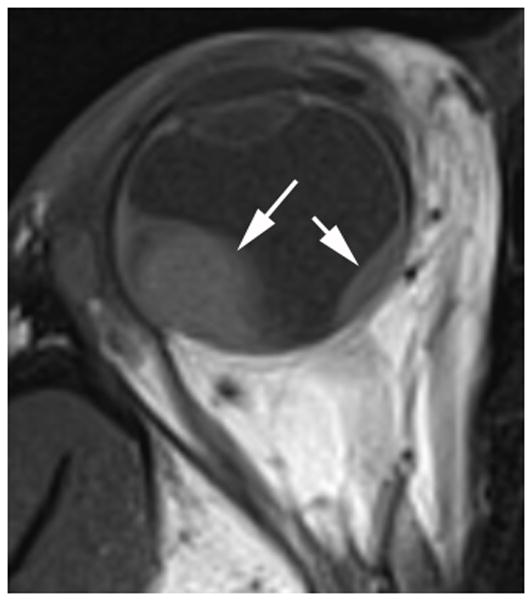
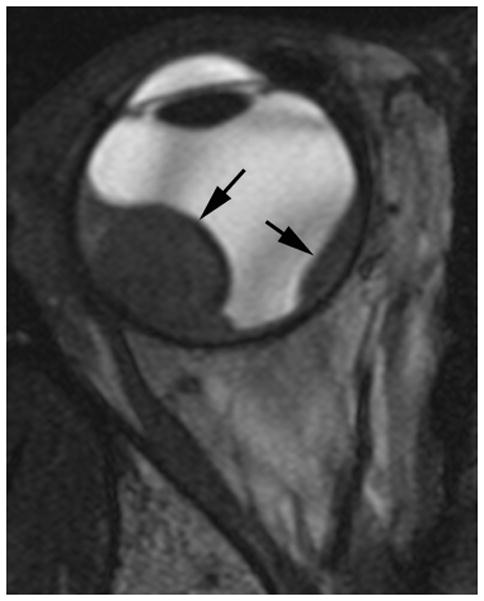
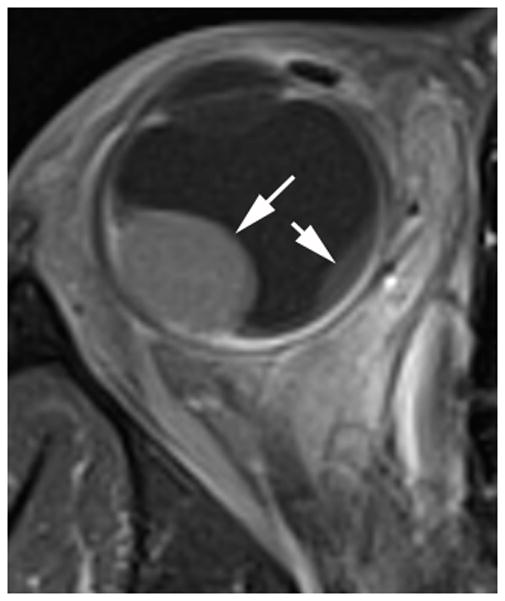



17-year-old boy presented with irritation, redness and blurred vision in right eye. a Transverse US shows mounded appearance of choroidal melanoma (arrows) with associated detached retina (curved arrow). Note lack of calcification in tumor. b Non-contrast enhanced T1W axial MRI shows the choroidal tumor (arrows) to be slightly hyperintense to vitreous. c CISS axial MRI shows the tumor (arrows) to be very low signal intensity. These signal characteristics are typical of most melanomas. d Post-contrast axial TW1 MR shows minimal enhancement of tumor (arrows). The detached retina is difficult to appreciate on these MR images. e Whole eye pathology specimen demonstrates domed shape of the choroidal melanoma as it arises from the choroid with an associated serous retinal detachment (*). The central defect present in the tumor represents artifact from prior fine needle aspiration biopsy (†). f Close up view of the periphery of the tumor where the PAS stain highlights Bruch’s membrane (white arrows). This membrane remains intact giving the tumor its dome shape and provides a barrier from the retina. Disruption of Bruch’s membrane would have produced a classic collar button configuration. g The tumor is composed of uniform sheets of variably pigmented epithelioid cells with prominent nucleoli. Interspersed between the tumor cells are prominent vascular spaces (thin white arrows). Densely pigmented cells with no discernable cytological features are melanophages within the tumor (thick white arrows). Scattered lymphocytes (small round blue cells, *) are invading the tumor.
Persistent fetal vasculature (persistent hyperplastic primary vitreous)
Persistent fetal vasculature (PFV) is the second most common cause of leukocoria [1]. In contrast to retinoblastoma, PFV is characterized by a lack of intraocular calcification and the presence of microophthalmia [1]. PFV is a congenital, non-hereditary, failure of the embryonic primary vitreous to regress, resulting in continued proliferation and formation of a retrolental mass with cataract in the anterior segment [1,20]. This fibrovascular mass may invade the posterior lens capsule where it is characterized by a prominent retinal fold or white opaque membrane [20]. While most cases are sporadic, inherited autosomal dominant or recessive cases have been reported, and this disease can occur with other disorders such as Axenfeld-Rieger Syndrome, neurofibromatosis, and Aicardi syndrome [20]. PVF is unilateral in over 90% of cases, and vitreous hemorrhage and neovascular glaucoma are the most common complications necessitating enucleation [1]. Visualization of a vertical septum (Cloquet canal) between the optic disc and posterior lens is a diagnostic hallmark [1,21,22]. Using Doppler US, a persistent hyaloid artery within the canal is noted in over half of cases (Fig. 4)[1,21].
Coats disease
Coats disease is a congenital, nonhereditary, vascular malformation. Historically, patients with Coats disease underwent enucleation if a diagnosis of retinoblastoma was considered, but this happens less frequently with improved understanding of imaging and clinical features that distinguish these entities [23]. Coats disease is characterized by focal areas of retinal telangiectasia progressing to full breakdown of the blood-retina barrier, allowing blood and blood products to leak into the retina and sub-retinal space. This fluid is comprised of cholesterol crystals and lipid-laden macrophages [24]. Peripheral portions of the retina are affected early, with progressive fluid accumulation leading to thickening and detachment of the retina resulting in vision loss [1]. Coats disease has a male predominance and peak age incidence of 6–8 years (range, 5 months to 71 years) which is older than that of retinoblastoma which has a peak incidence of about 18 months of age [1,23]. Typical presentation (>80%) is unilateral leukocoria, but patients may have strabismus or painful glaucoma [1,23]. Volumetric measurements reveal the involved globe to be smaller than the uninvolved globe in most cases. Magnetic resonance imaging shows lack of enhancement of the subretinal space, absence of calcifications, and high signal intensity on T2W images due to high fat content. Proton MR spectroscopy of the exudate demonstrates a peak at 1–1.6ppm due to lipoproteinaceous material [1]. Ultrasound may demonstrate low-level echoes (cholesterol crystals, lipid-laden macrophages, blood-products) floating in the sub-retinal space, large volume of sub-retinal exudate and detached retina (Fig 5). Calcification is rarely present in Coats disease [1,21].
Fig. 5.

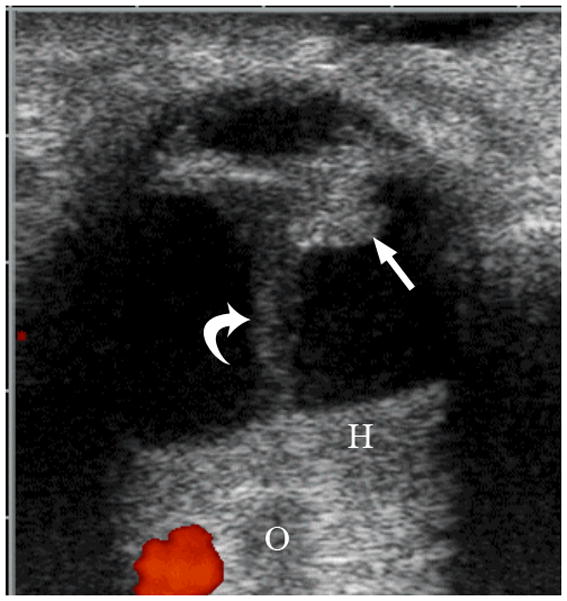
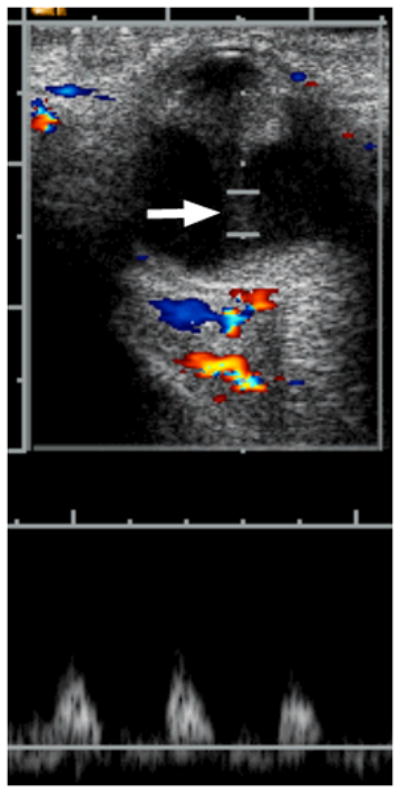
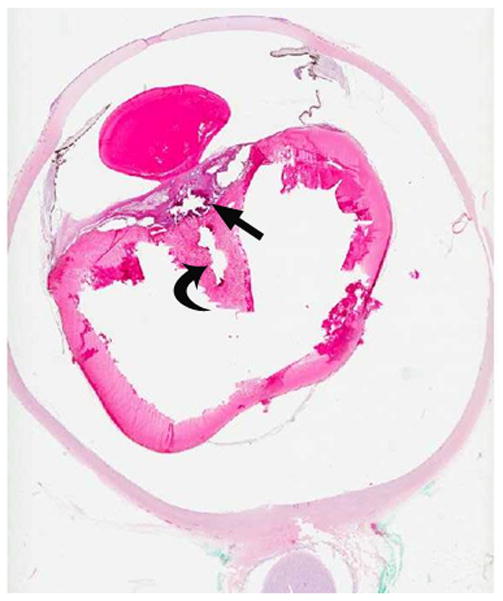
6-week-old girl with persistent fetal vasculature (persistent hyperplastic primary vitreous). The patient’s pediatrician noted an absent light reflex in the right eye at one week of age. a Diagram depicting normal fetal vasculature in the eye [adapted from 7]. b Transverse US image demonstrating echogenic retrolental mass (straight arrow) and hyperechoic band between posterior lens and posterior pole of globe (Cloquet canal) with persistent hyaloid artery (curved arrow). H = layering blood. O = optic nerve. Hemorrhage, Cloquet canal and retrolental mass, as shown here, are features typical of this entity. c Doppler evaluation of Cloquet canal (arrow) shows arterial flow in persistent hyaloid artery. d H&E staining shows partially calcified retrolental mass (straight arrow) and persistent hyaloid artery (curved arrow)
Corneal Dermoid
This congenital malformation of the lens placode results in a hamartomatous corneal lesion at birth (Fig. 6). On the ocular surface, the lesion is round or ovoid in shape, yellowish-white in color, solid and well vascularized [25,26]. It may contain ectodermal derivatives such as hair, sebaceous and sweat glands embedded in connective tissue, covered by epithelium [1]. Imaging features reflect varying degrees of lens malformation (sometimes completely absent) and corneal surface mass.
Fig. 6.
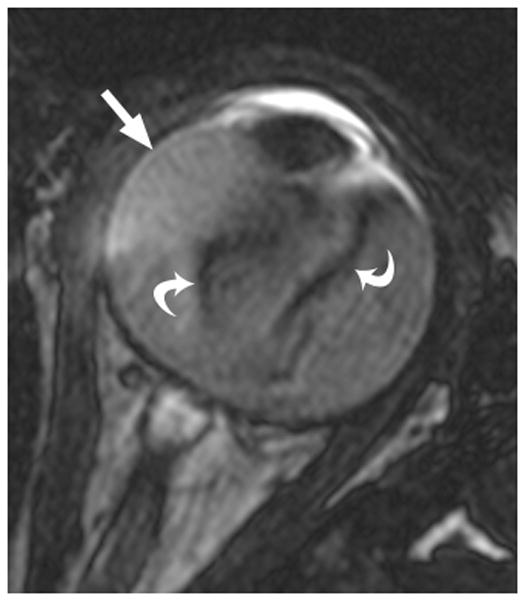
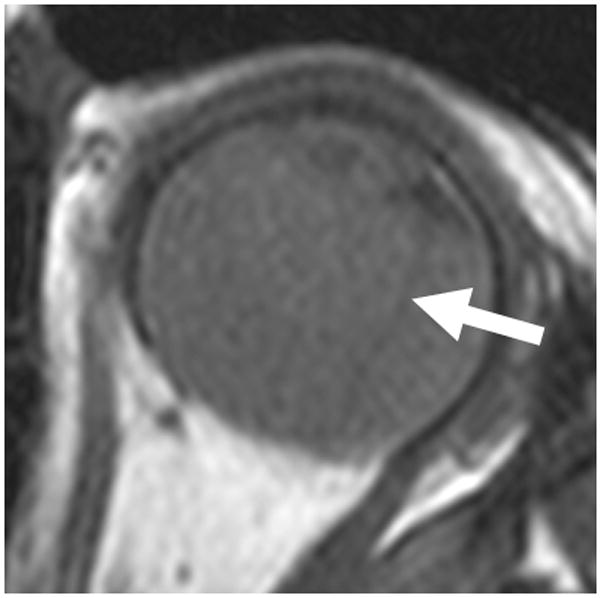
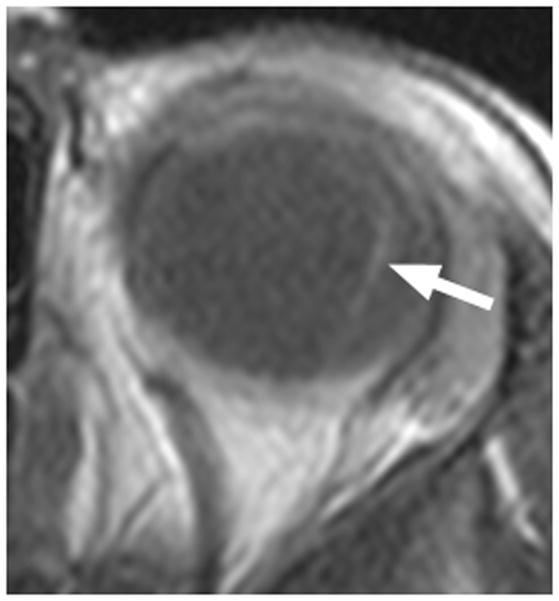
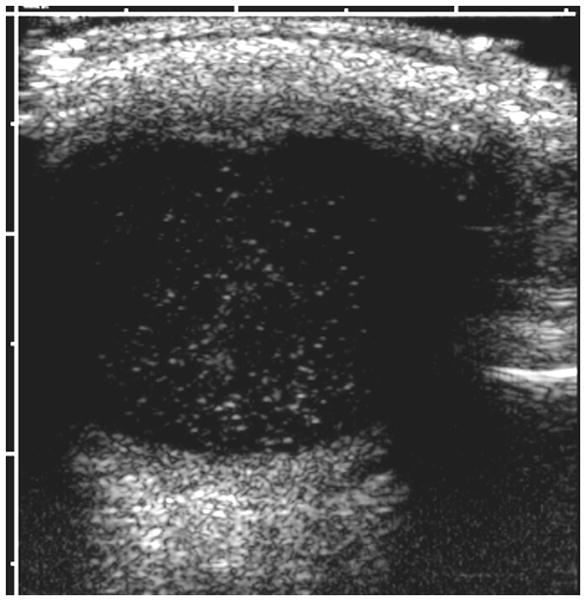
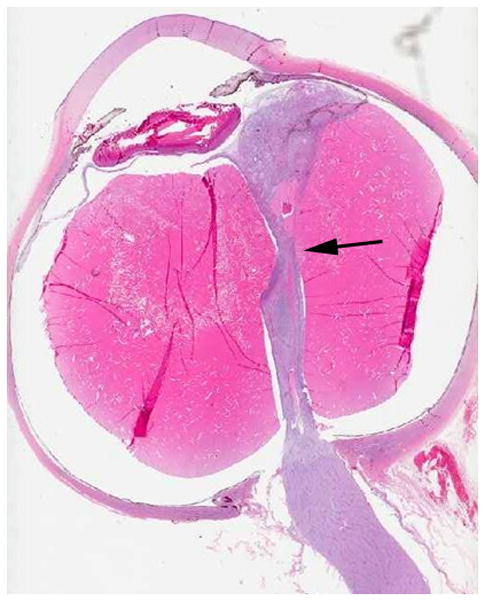
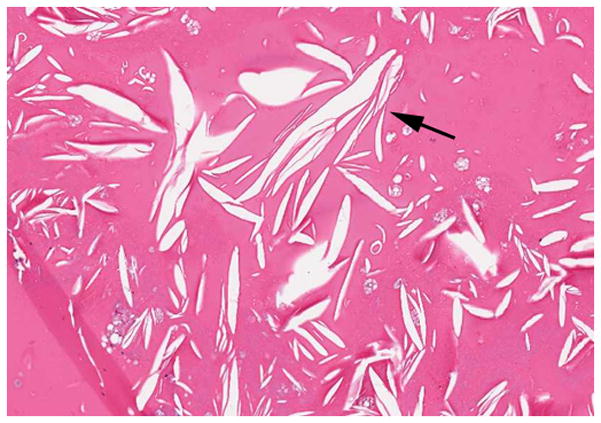
4-year-old boy with Coats disease presented with leukocoria in the left eye and was suspected to have retinoblastoma. a Axial CISS MRI demonstrates exudate with high signal intensity due to cholesterol and lipid-laden subretinal exudate (straight arrow) causing massive retinal detachment (curved arrows). b Axial pre-contrast-enhanced and c post-contrast-enhanced T1W MR image show typical lack of enhancement of subretinal fluid with linear enhancement at the border of exudate and vitreous (arrows). d Transverse US shows tiny echogenic “speckles” likely due to lipoproteinaceous exudate floating in subretinal space. As in this example, microcalcifications are usually absent. e H&E staining of a paraffin section through the optic nerve with subretinal exudate (pink) and severely detached retina (arrow). f H&E staining of exudate shows cholesterol crystals (arrow)
Retinal dysplasia
Retinal dysplasia is a descriptive term for the reduction or complete absence of retinal ganglion cells, nerve fibers, and blood vessels (Fig. 7). On histopathology, retinal malformation, tenting and detachment are combined with rosette formation and optic nerve aplasia. If both eyes are affected, an association with several syndromes has been described (i.e. trisomy 13, or Walker-Warburg syndrome)[27]. If a careful family history and systemic evaluation in a male patient with retinal dysplasia documents blindness shortly after birth, mental retardation and hearing loss, a diagnosis of Norrie disease can be established [28]. Molecular testing to confirm mutations of the NDP gene (Xp11.4) is available and may be recommended with genetic counseling to ascertain carrier status for females [29]. This x-linked recessive disorder may present with a white retrolental membrane (leukocoria) and retinal detachment that eventually leads to phthisis bulbi, a non-functional, shrunken eye. The abnormal vasculature in Norrie disease results in a retinal pseudoglioma with an avascular peripheral space. Children with retained light perception may undergo vitrectomy [30], but no other effective therapies have been established for the ocular manifestation of Norrie disease. On funduscopic examination, retinal dysplasia may range from simple to massive retinal folding. There are often central stalks extending from the posterior lens to optic disc with completely detached retina causing highly disorganized microphthalmic eyes [31]. The intensity of MRI enhancement of dysplastic retinal tissue after contrast administration depends on the number and quality of vessels, but the characteristic linear structure of retinal detachment may be preserved [27]. Proliferation of intraocular tissue may cause leukocoria. Small calcifications may be present [27].
Fig. 7.

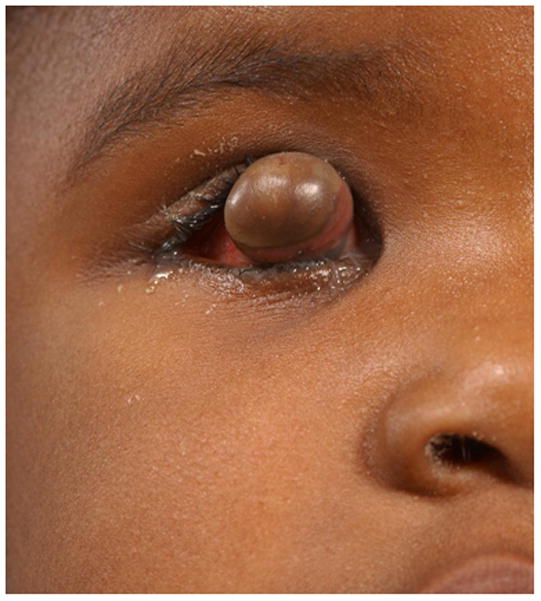
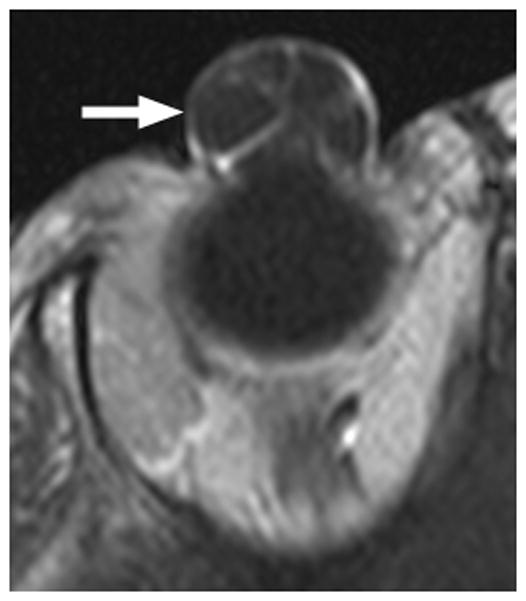
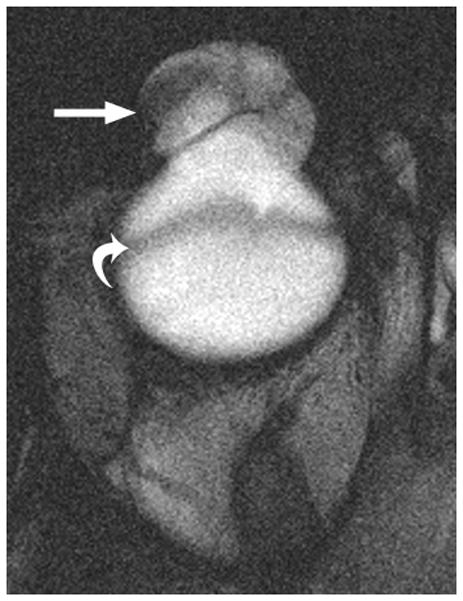
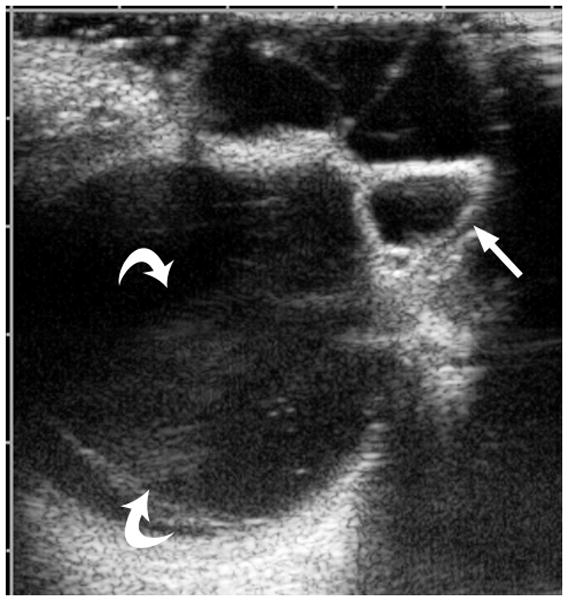


3-year-old girl with corneal dermoid. The mass had been present since birth and grew in proportion with the child. a Diagram showing normal fetal eye development from earliest (left) to most mature (right). During early normal eye development, the surface ectoderm thickens to form a lens placode and invaginates with the underlying neuroepithelium of the optic vesicle (image 2) to form a cup-shaped structure (image 3). The bilayered optic cup will give rise to the neural retina (inner layer) and retinal pigmented epithelium (outer layer, image 4) (adapted from [33]). b Clinical photograph of typical corneal dermoid, a round, yellowish mass arising from the ocular surface c Axial T1W and d CISS MR images show septated, cystic dermoid arising from corneal surface (arrows) and absence of lens. e Transverse US image of corneal dermoid showing anterior chamber cysts (arrow) and absent lens. In this example, vitreous debris is evident on T2W MRI and US images (curved arrows). f Whole eye H&E showing keratinized epithelium in the place of cornea (arrows). The lens is absent suggesting failure to of the embryonic lens placode to invaginate. The neurosensory retina, choroid, sclera and optic nerve appear normal. g Higher power image showing associated dermal elements (straight arrow) and cystic inclusions (curved arrow)
Toxocara endophthalmitis
Resulting from a hypersensitivity response and T-cell granulomatous reaction to infestation by the larval form of the nematode Toxocara canis (in dogs) or Toxocara cati (in felines), toxocara endophthalmitis (Fig. 8) commonly presents with unilateral vision loss and less commonly with strabismus, pain, and redness [1]. Humans become infested by oral-fecal route after exposure to soil or sandboxes frequented by domestic dogs or cats, more commonly puppies or kittens than adult animals [32]. While larvae may reach any organ, ocular involvement (ocular larva migrans) often occurs in the absence of systemic (visceral larva migrans) infestation [1]. Larvae may cause a posterior pole granuloma, peripheral choroidal sclerosing inflammation, or diffuse endophthalmitis with tractional retinal detachment [1]. Secondary sub-retinal exudates may contribute to retinal detachment. Histopathology demonstrates eosinophilic inflammatory changes within the choroid; the small larvae are difficult to detect [1]. When calcification is absent on imaging this condition often mimics Coats disease [1,21,22]. Antihelminthic agents are required for treatment.
Fig. 8.
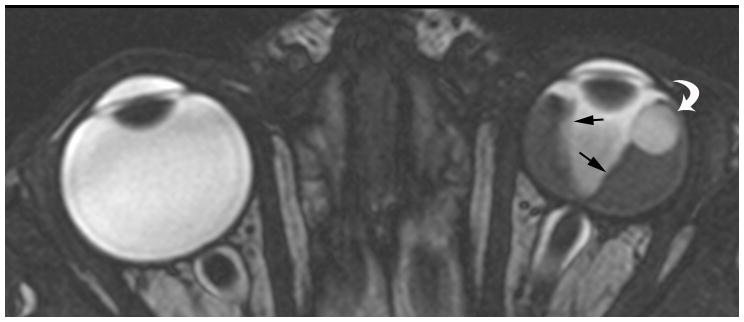
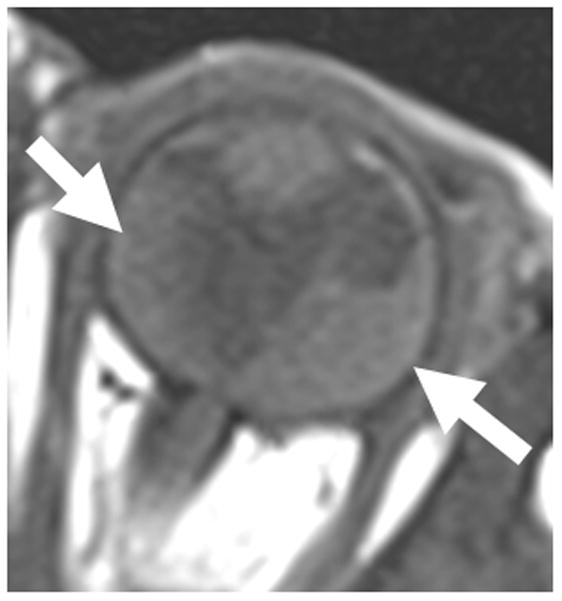
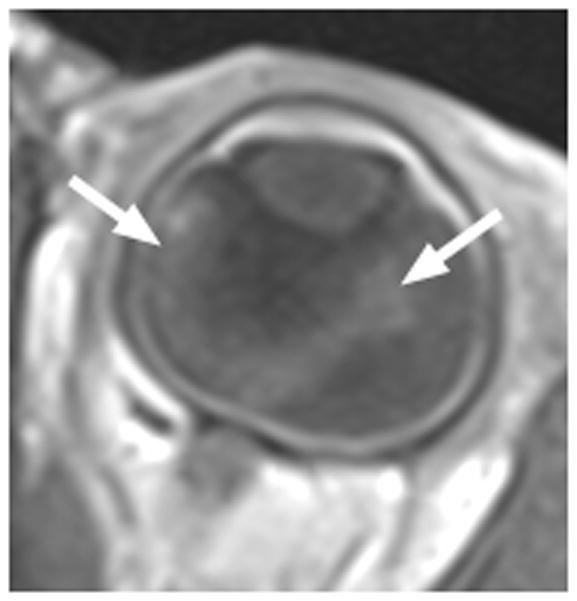
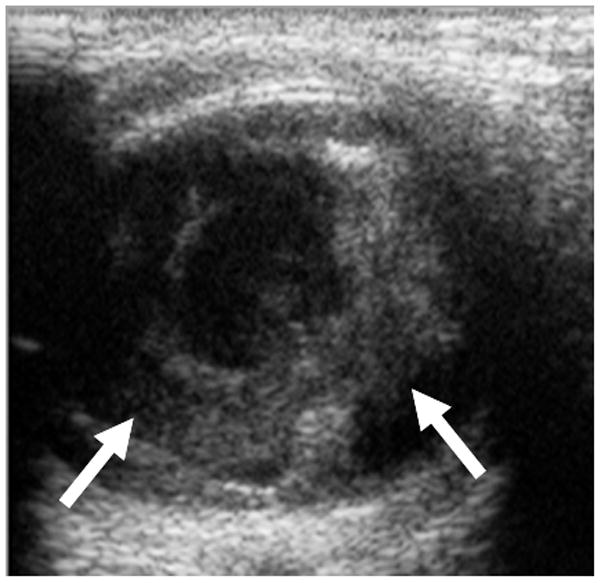
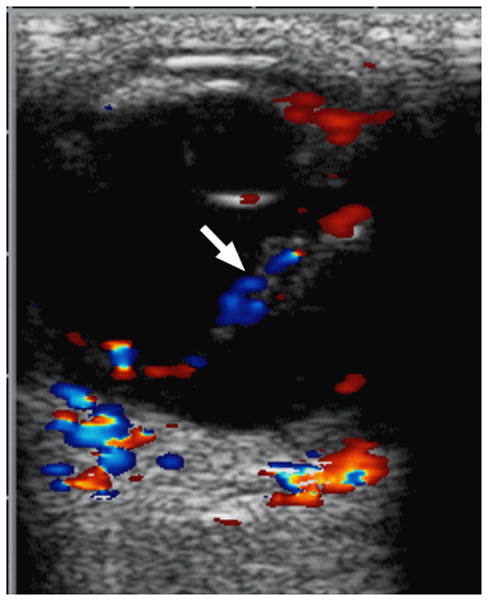
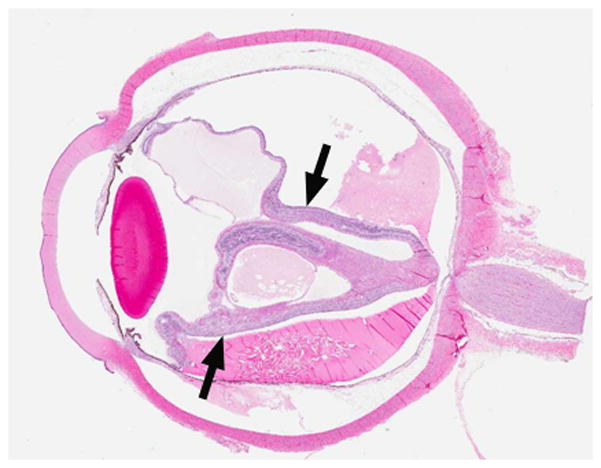
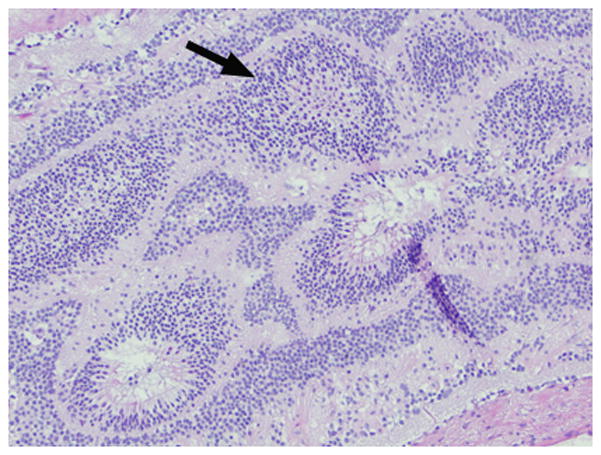
7-month-old boy with retinal dysplasia presented with inability to follow objects with left eye. a Axial CISS MRI shows thickened, detached retina (straight arrows). The vacuole (curved arrow) may be contiguous with vitreous. Note left microphthalmia due to the highly disorganized retina. These features are often seen in retinal dysplasia. b Axial pre-contrast-enhanced T1W MRI showing subretinal exudate (arrows) c Post-contrast-enhanced T1W MRI showing dysplastic thickened retina (arrows) with moderate contrast enhancement. d Transverse ultrasound image shows dysplastic thickened retina (arrows) and subretinal exudate. e Doppler US image shows prominent vascularity of dyplastic retinal tissue (arrow), consistent with degree of enhancement seen on post-contrast MRI in b. Degree of vascularity is variable in dyplastic retina. f Low power H&E staining showing thickened, dysplastic retina (arrows) and subretinal exudate (pink material). g High power H&E staining shows rosettes (arrow) typical of retinal dysplasia
Pediatric ocular masses pose a challenging diagnostic dilemma. While pediatric patients who present with leukocoria (white reflex) require an ophthalmology referral to rule out retinoblastoma, a lengthy differential must be considered by the evaluating physician. A detailed history may identify recent ocular trauma, prematurity, exposure to puppies, or co-existing congenital abnormalities that can distinguish some ocular masses within the differential. However, the more rare ocular pathologies that may mimic retinoblastoma require an ophthalmology examination, imaging, and, if needed, histopathology. The imaging and clinical features described in this pictorial essay illustrate important features to distinguish pediatric ocular masses. We also provide an algorithm (Fig. 10) that may be useful in establishing the correct diagnosis.
Fig. 10.

Fig. 9.
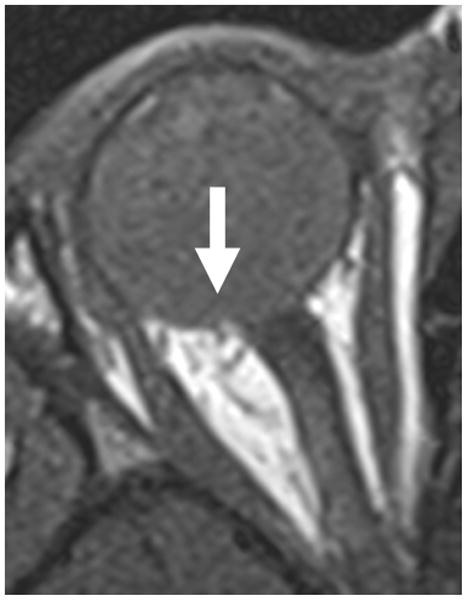
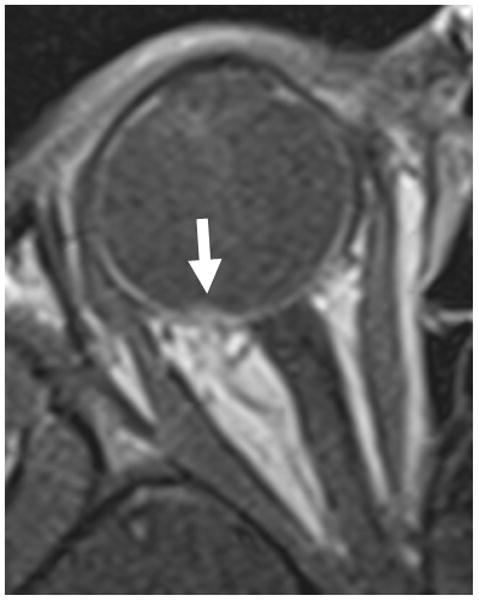
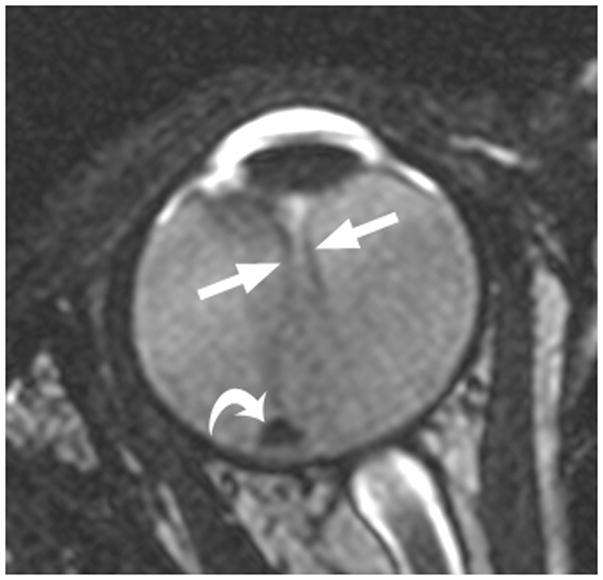
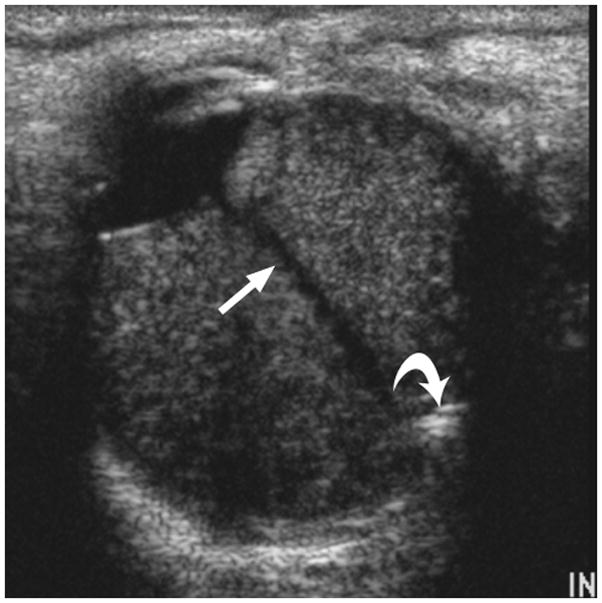
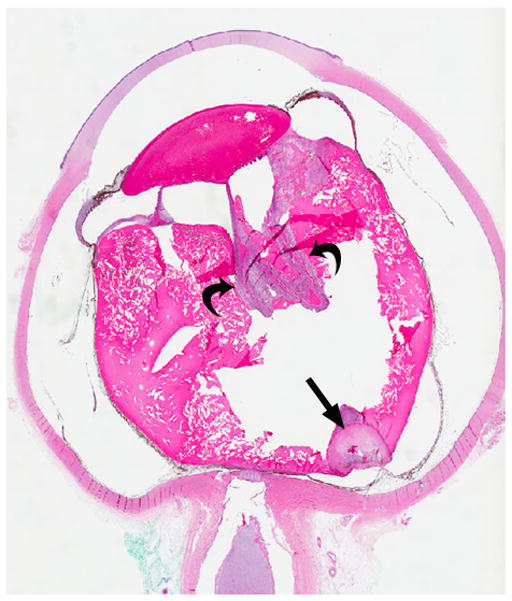
3-year-old boy with toxocara endophthalmitis presented with a one year history of leukocoria and was suspected to have retinoblastoma. a Axial pre-contrast-enhanced and b post-contrast-enhanced T1W MRI show slight enhancement of partially calcified granuloma (arrows) caused by larval invasion of the posterior pole. c Axial CISS MRI shows calcified granuloma in posterior pole (curved arrow) and apposed leaves of severely detached retina (straight arrows) caused by massive sub-retinal exudate. d Longitudinal US images showing apposed leaves of detached retina (straight arrow) and calcified granuloma (curved arrow). Note diffuse low-level echoes due to massive amount of sub-retinal exudate. e H&E staining showing calcified granuloma (straight arrow), exudate (pink material), and detached retina (curved arrows)
Acknowledgments
The authors thank Betsy Williford for providing art work for this project.
References
- 1.Chung EM, Specht CS, Schroeder JW. From the archives of the AFIP: Pediatric orbit tumors and tumorlike lesions: neuroepithelial lesions of the ocular globe and optic nerve. Radiographics. 2007;27:1159–1186. doi: 10.1148/rg.274075014. [DOI] [PubMed] [Google Scholar]
- 2.Nema HV. Ocular tumours--some newer and controversial aspects. Indian J Ophthalmol. 1989;37:162–163. [PubMed] [Google Scholar]
- 3.Ramji FG, Slovis TL, Baker JD. Orbital sonography in children. Pediatr Radiol. 1996;26:245–258. doi: 10.1007/BF01372106. [DOI] [PubMed] [Google Scholar]
- 4.Standard for Real-Time Display of Thermal and Mechanical Acoustic Output Indices on Diagnostic Ultrasound Equipment, rev. 2. NEMA Standards Publication UD 3-2004. 2009 Available via http://www.nema.org/stds/ud3.cfm.
- 5.de Graaf P, Goricke S, Rodjan F, Galluzzi P, Maeder P, et al. Guidelines for imaging retinoblastoma: imaging principles and MRI standardization. Pediatr Radiol. 2011 doi: 10.1007/s00247-011-2201-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shields CL, Shields JA. Diagnosis and management of retinoblastoma. Cancer Control. 2004;11:317–327. doi: 10.1177/107327480401100506. [DOI] [PubMed] [Google Scholar]
- 7.Blach LE, McCormick B, Abramson DH, Ellsworth RM. Trilateral retinoblastoma--incidence and outcome: a decade of experience. Int J Radiat Oncol Biol Phys. 1994;29:729–733. doi: 10.1016/0360-3016(94)90560-6. [DOI] [PubMed] [Google Scholar]
- 8.Kivela T. Trilateral retinoblastoma: a meta-analysis of hereditary retinoblastoma associated with primary ectopic intracranial retinoblastoma. J Clin Oncol. 1999;17:1829–1837. doi: 10.1200/JCO.1999.17.6.1829. [DOI] [PubMed] [Google Scholar]
- 9.Kaste SC, Jenkins JJ, 3rd, Pratt CB, Langston JW, Haik BG. Retinoblastoma: sonographic findings with pathologic correlation in pediatric patients. AJR Am J Roentgenol. 2000;175:495–501. doi: 10.2214/ajr.175.2.1750495. [DOI] [PubMed] [Google Scholar]
- 10.Mahajan A, Crum A, Johnson MH, Materin MA. Ocular neoplastic disease. Semin Ultrasound CT MR. 2011;32:28–37. doi: 10.1053/j.sult.2010.12.001. [DOI] [PubMed] [Google Scholar]
- 11.Canning CR, McCartney AC, Hungerford J. Medulloepithelioma (diktyoma) Br J Ophthalmol. 1988;72:764–767. doi: 10.1136/bjo.72.10.764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shields JA, Eagle RC, Jr, Shields CL, Potter PD. Congenital neoplasms of the nonpigmented ciliary epithelium (medulloepithelioma) Ophthalmology. 1996;103:1998–2006. doi: 10.1016/s0161-6420(96)30394-1. [DOI] [PubMed] [Google Scholar]
- 13.Shields CL, Shields JA, Milite J, De Potter P, Sabbagh R, et al. Uveal melanoma in teenagers and children. A report of 40 cases. Ophthalmology. 1991;98:1662–1666. doi: 10.1016/s0161-6420(91)32071-2. [DOI] [PubMed] [Google Scholar]
- 14.Singh AD, Shields CL, Shields JA, Sato T. Uveal melanoma in young patients. Arch Ophthalmol. 2000;118:918–923. [PubMed] [Google Scholar]
- 15.Pogrzebielski A, Orlowska-Heitzman J, Romanowska-Dixon B. Uveal melanoma in young patients. Graefes Arch Clin Exp Ophthalmol. 2006;244:1646–1649. doi: 10.1007/s00417-006-0347-x. [DOI] [PubMed] [Google Scholar]
- 16.Palazzi MA, Ober MD, Abreu HF, Cardinalli IA, Isaac CR, et al. Congenital uveal malignant melanoma: a case report. Can J Ophthalmol. 2005;40:611–615. doi: 10.1016/s0008-4182(05)80055-9. [DOI] [PubMed] [Google Scholar]
- 17.Bakri SJ, Sculley L, Singh AD. Imaging techniques for uveal melanoma. Int Ophthalmol Clin. 2006;46:1–13. doi: 10.1097/01.iio.0000195859.86805.64. [DOI] [PubMed] [Google Scholar]
- 18.Grabowska A, Abelarias J, Peralta J, Asencio M, Garcia-Cabezas MA, et al. Uveal melanoma in a 19-month-old child. J AAPOS. 2011;15:606–608. doi: 10.1016/j.jaapos.2011.07.018. [DOI] [PubMed] [Google Scholar]
- 19.Lemke AJ, Hosten N, Wiegel T, Prinz RD, Richter M, et al. Intraocular metastases: differential diagnosis from uveal melanomas with high-resolution MRI using a surface coil. Eur Radiol. 2001;11:2593–2601. doi: 10.1007/s003300100936. [DOI] [PubMed] [Google Scholar]
- 20.Shastry BS. Persistent hyperplastic primary vitreous: congenital malformation of the eye. Clin Experiment Ophthalmol. 2009;37:884–890. doi: 10.1111/j.1442-9071.2009.02150.x. [DOI] [PubMed] [Google Scholar]
- 21.Berrocal T, de Orbe A, Prieto C, al-Assir I, Izquierdo C, et al. US and color Doppler imaging of ocular and orbital disease in the pediatric age group. Radiographics. 1996;16:251–272. doi: 10.1148/radiographics.16.2.8966285. [DOI] [PubMed] [Google Scholar]
- 22.Glasier CM, Brodsky MC, Leithiser RE, Jr, Williamson SL, Seibert JJ. High resolution ultrasound with Doppler: a diagnostic adjunct in orbital and ocular lesions in children. Pediatr Radiol. 1992;22:174–178. doi: 10.1007/BF02012488. [DOI] [PubMed] [Google Scholar]
- 23.Shields JA, Shields CL, Honavar SG, Demirci H, Cater J. Classification and management of Coats disease: the 2000 Proctor Lecture. Am J Ophthalmol. 2001;131:572–583. doi: 10.1016/s0002-9394(01)00896-0. [DOI] [PubMed] [Google Scholar]
- 24.Kremer I, Nissenkorn I, Ben-Sira I. Cytologic and biochemical examination of the subretinal fluid in diagnosis of Coats’ disease. Acta Ophthalmol (Copenh) 1989;67:342–346. doi: 10.1111/j.1755-3768.1989.tb01885.x. [DOI] [PubMed] [Google Scholar]
- 25.Kadri RKA, Achar A, Hedge S. Corneal Dermoid. Online J Health Allied Scs. 2011;10:23. [Google Scholar]
- 26.Shields JA, Shields CL. Orbital cysts of childhood--classification, clinical features, and management. Surv Ophthalmol. 2004;49:281–299. doi: 10.1016/j.survophthal.2004.02.001. [DOI] [PubMed] [Google Scholar]
- 27.de Graaf P, van der Valk P, Moll AC, Imhof SM, Schouten-van Meeteren AY, et al. Retinal dysplasia mimicking intraocular tumor: MR imaging findings with histopathologic correlation. AJNR Am J Neuroradiol. 2007;28:1731–1733. doi: 10.3174/ajnr.A0635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Warburg M. Norrie’s Disease. Birth Defects Orig Artic Ser. 1971;7:117–124. [PubMed] [Google Scholar]
- 29.Sims K. NDP-related Retinopathies. In: Pagon RABT, Dolan CR, et al., editors. GeneReviews. Seattle, Washington: 1999. [Google Scholar]
- 30.Walsh MK, Drenser KA, Capone A, Jr, Trese MT. Early vitrectomy effective for Norrie disease. Arch Ophthalmol. 2010;128:456–460. doi: 10.1001/archophthalmol.2009.403. [DOI] [PubMed] [Google Scholar]
- 31.Godel V, Nemet P, Lazar M. Retinal dysplasia. Doc Ophthalmol. 1981;51:277–288. doi: 10.1007/BF00143890. [DOI] [PubMed] [Google Scholar]
- 32.Shields JA. Ocular toxocariasis. A review. Surv Ophthalmol. 1984;28:361–381. doi: 10.1016/0039-6257(84)90242-x. [DOI] [PubMed] [Google Scholar]
- 33.Ali RR, Sowden JC. Regenerative medicine: DIY eye. Nature. 2011;472:42–43. doi: 10.1038/472042a. [DOI] [PubMed] [Google Scholar]