

Abstract
Amyotrophic lateral sclerosis (ALS) is a terminal disease involving the progressive degeneration of motor neurons within the motor cortex, brainstem and spinal cord. Most cases are sporadic (sALS) with unknown causes suggesting that the etiology of sALS may not be limited to the genotype of patients, but may be influenced by exposure to environmental factors. Alterations in epigenetic modifications are likely to play a role in disease onset and progression in ALS, as aberrant epigenetic patterns may be acquired throughout life. The aim of this study was to identify epigenetic marks associated with sALS. We hypothesize that epigenetic modifications may alter the expression of pathogenesis-related genes leading to the onset and progression of sALS. Using ELISA assays, we observed alterations in global methylation (5 mC) and hydroxymethylation (5 HmC) in postmortem sALS spinal cord but not in whole blood. Loci-specific differentially methylated and expressed genes in sALS spinal cord were identified by genome-wide 5mC and expression profiling using high-throughput microarrays. Concordant direction, hyper- or hypo-5mC with parallel changes in gene expression (under- or over-expression), was observed in 112 genes highly associated with biological functions related to immune and inflammation response. Furthermore, literature-based analysis identified potential associations among the epigenes. Integration of methylomics and transcriptomics data successfully revealed methylation changes in sALS spinal cord. This study represents an initial identification of epigenetic regulatory mechanisms in sALS which may improve our understanding of sALS pathogenesis for the identification of biomarkers and new therapeutic targets.
Introduction
Amyotrophic lateral sclerosis (ALS) is a progressive and terminal neurodegenerative disease characterized by the selective degeneration of motor neurons within the motor cortex, brainstem and spinal cord [1]. In the United States, approximately 14 cases of ALS are diagnosed each day and 30,000 people are living with the disease. The average time from disease onset to death is 3 years and no treatment that substantially improves the clinical course of the disease is currently available [1].
Proposed pathogenic mechanisms of ALS include oxidative stress, glutamate excitotoxicity, impaired axonal transport, neurotrophic deprivation, neuroinflammation, apoptosis, altered protein turnover, and mitochondrial dysfunction [1], [2]. Moreover, influences from astrocytes and microglia in the motor neuron microenvironment contribute to pathogenesis [3]. In the last 20 years, a search for genetic factors has identified several genes associated with familial ALS (fALS) and a few with sporadic ALS (sALS) [4]–[6]. Because fALS only accounts for 5–10% of all cases of ALS, the causes leading to the vast majority of ALS (sALS) are poorly understood [1].
Environmental exposure to toxins, excessive physical activity, dietary factors, and changes in immunity increase the risk of developing sALS [7]. These factors may drive epigenetic changes, which are well suited to explain disease onset and progression in sALS, as they may be acquired throughout life. Epigenetic modifications, including covalent modifications of DNA and histones as well as RNA editing, dynamically regulate gene expression without altering the genetic code [8], [9]. These modifications are important in chromosome integrity, cellular differentiation, development, and aging [8], [10]. Two such modifications, 5-methylcytosine (5 mC) and 5-hydroxymethylcytosine (5 HmC) are associated with repression or activation of gene expression, respectively, in response to environmental and developmental factors linked to age-related diseases [11].
5mC at CpG (cytosine nucleotide separated by a phosphate from a guanine nucleotide) sites is a reversible mechanism facilitated by DNA (cytosine-5)-methyltransferases (DNMTs). Conversely, the Fe(II) and α-ketoglutarate (α-KG)-dependent ten-eleven translocation (TET) family of proteins catalyze oxidation and decarboxylation reactions of 5mC leading to 5-hydroxymethylcytosine (5 HmC), 5-formylcytosine (5 fC) and 5-carboxylcytosine (5 caC) [12], [13]. 5 HmC may be an intermediate for passive (during DNA replication) and active demethylation and/or serve as a docking site for proteins with high affinity for 5 HmC, thereby dissociating interactions between the transcriptional repression machinery and 5 mC [14].
In addition to the identification of alterations in global 5 HmC associated with sALS, this study represents one of the first methylation assessments of sALS by integrating methylome and transcriptome profiles of postmortem frozen human spinal cord samples. We identified differentially methylated sALS spinal cord genes exhibiting concordant mRNA expression overrepresented in functional categories implicated in sALS. These data support a role for epigenetic regulation in sALS and it may provide a better understanding of disease pathogenesis and facilitate the discovery of new therapeutic targets.
Results
A workflow of our data analysis is provided in Fig. 1.
Figure 1. Flow chart for genome-wide epigenetic and expression analysis.
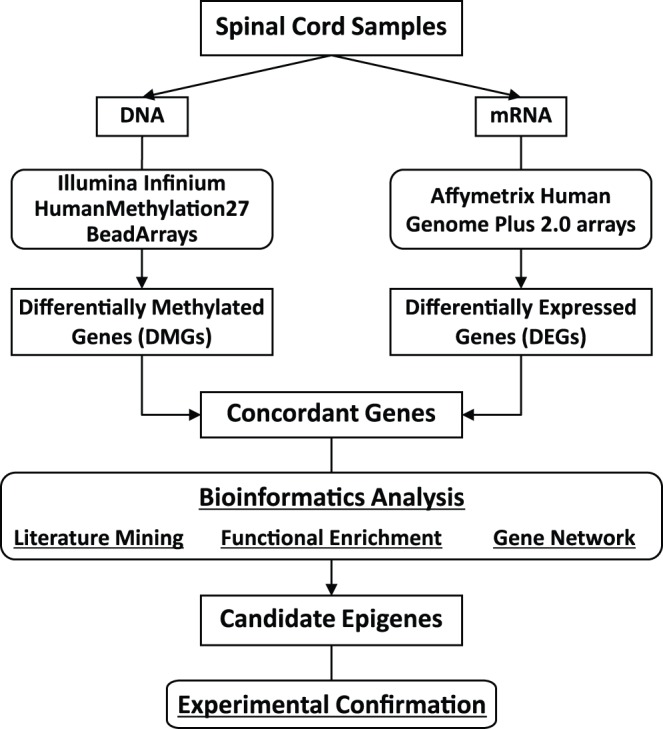
DNA and RNA from postmortem human spinal cord samples were subjected to high-throughput epigenetic and gene expression screening, respectively. DMGs and DEGs representing concordant direction were further analyzed by bioinformatics analyses. Finally, identified candidate genes were experimentally confirmed in the spinal cord.
Global 5mC is Increased in sALS Spinal Cord
Chestnut et al. recently reported an increase in DNMTs and 5mC immunoreactivity in ALS brain and spinal cord, suggesting that a global increase in 5mC is associated with the pathogenesis of ALS [15]. We assessed global 5mC of genomic DNA extracted from postmortem human spinal cord samples (sALS, n = 11; matching controls, n = 11; Tables 1, S1) using a colorimetric ELISA approach. We observed a modest but significant 1.4-fold increase in global 5mC in sALS (3.58±0.18) compared to controls (2.56±0.18) (p = 0.0006, Fig. 2), confirming previous observations [15].
Table 1. Demographics of the subjects for methylation and expression profiling of postmortem spinal cord.
| Characteristics | sALS group | Control group | p-value | |
| Number of subjects | 12 | 11 | – | |
| Age (years)(a) | 55 (35–71) | 55 (36–73) | NS | |
| Gender | Male | 9 | 9 | NS |
| Female | 3 | 2 | – | |
| Spinal cord location | Cervical | 9 | 7 | NS |
| Thoracic | 3 | 4 | NS | |
| Onset | Bulbar | 1 | 0 | – |
| Limb | 6 | 0 | – | |
| NA | 4 | 0 | – | |
| Postmortem interval (hours)(b) | 14.5±7.0 | 15.2±6.2 | NS | |
| Cause of death | ALS | 11 | 0 | <0.001 |
| Accident | 0 | 2 | – | |
| PC | 0 | 2 | – | |
| ASCVD | 0 | 5 | – | |
| Cancer | 0 | 1 | – | |
| Cardiac arrest | 0 | 1 | – |
(a) median (range); (b) mean ± standard deviation; PC, pulmonary complications; ASCVD, atherosclerotic cardiovascular disease; NS, not significant; NA, no data available.
Figure 2. Global 5mC is increased in spinal cord of sALS.

Genomic DNA extracted from control (open bar) or sALS (filled bar) postmortem human spinal cord was analyzed with an ELISA colorimetric assay for methylation, (5mC; Ctrl = 11, sALS = 11). Outliers, outside of median ±1.5 x Inter-Quartile-Range, were excluded. The data is presented as mean ± SEM of percent (%) 5mC using box and whiskers vertical bars plotting minimum to maximum values. A two-sample equal variance t-test used; ***p<0.001 compared to control group (Ctrl) with an achieved power of 99%.
Genome-wide 5mC Profiling in sALS Spinal Cord
To identify loci-specific differentially methylated genes (DMGs) in sALS, postmortem spinal cord tissue (Tables 1, S1) was subjected to an unbiased genome-wide methylation screen using the Infinium Human Methylation27 DNA BeadChip (HM27K) array. We identified 4,261 significant differentially methylated autosomal CpG sites, representing 3,574 genes. Functional enrichment analysis identified biological categories of extracellular region (p = 3.5E-30), defense response (p = 2.3E-18), cytokine activity (p = 2.7E-16), immune response (p = 6.9E-15), JAK-STAT signaling pathway (p = 2.2E-7), steroid hormone biosynthesis (p = 3.6E-7), and drug metabolism (p = 3.1E-3) were over-represented in spinal cord DMGs (Fig. 3). This suggests a role for epigenetic regulation in molecular mechanisms known to drive sALS pathogenesis [16]–[18]. Furthermore, the identified DMGs included previously reported epigenetically regulated genes such as runt-related transcription factor 3 (RUNX3), TNF-related apoptosis-inducing ligand (TRAIL/TNFSF10), H19, neuritin 1 (NRN1) and signal transducer and activator of transcription 5A (STAT5A) [19]–[23], validating our approach. The ALS-dependent differential methylation of three genes encoding the chemokines CKLF-like MARVEL transmembrane domain-containing proteins 2 and 3 (CMTM2 and CMTM3) and the chemokine (C-X-C motif) ligand 12 (CXCL12), as well as the two transcription factors, STAT5A and the CCAAT/enhancer binding protein beta (C/EBPB) was assessed by pyrosequencing (Fig. S1). These results indicate parallel direction of differential methylation between the two assays.
Figure 3. Biological functions of DMGs, DEGs, and concordant epigenes.

Overrepresented biological functions were identified using DAVID. The top 20 biological functions ordered by p-value were collected from each gene set, and redundant terms were combined. The values in the table correspond to -log10 (DAVID p-value), ranging from 0 (white) to 30 (bright red). The values are not normalized across different gene sets with variable numbers of genes. The order of biological terms was based on the log-transformed p-values of DMGs and DEGs.
Genome-wide Expression Profiling in sALS Spinal Cord
An increase in promoter 5mC (hyper-methylation) is associated with gene silencing, while a decrease (hypo-methylation) reflects up-regulation of gene expression [8]. To identify functionally relevant epigenes, we performed genome-wide gene expression profiling of total RNA from sALS (n = 12) and control (n = 10) spinal cord (Tables 1, S1). We found 1,182 DEGs in sALS enriched with immune response-related biological categories such as defense response (p = 2.3E-20), inflammatory response (p = 1.3E-18), T-cell activation (p = 3.0E-9), leukocyte activation (p = 1.6E-8), cytokine binding (p = 1.7E-7) as well as carbohydrate binding (p = 7.2E-6), transmembrane receptor protein tyrosine kinase signaling (p = 2.6E-5), cell death (p = 1.5E-3), and JAK-STAT signaling (p = 3.6E-2) (Fig. 3). Several of these biological categories overlap with the 5mC profile, suggesting methylation may play a role in the regulation of these DEGs.
Comparison between DMGs and DEGs in sALS Spinal Cord
DMGs and DEGs were analyzed for their direction of change to select genes demonstrating concordant and potentially true regulation of gene expression by methylation. Between 3,574 DMGs and 1,182 DEGs, 251 genes were common (Table S2), of which approximately 70% were hyper-methylated as observed with global 5mC. Among these common genes, 112 epigenes demonstrated concordant direction of methylation and expression changes (hypo-methylation/up-regulation (51 genes; Table 2) or hyper-methylation/down-regulation (61 genes; Table 3) (Fig. 4). These concordant epigenes were enriched in biological categories including immune response (p = 7.1E-5), plasma membrane part (p = 9.6E-5), defense response (p = 0.03), and neuron adhesion (p = 0.05) (Fig. 3).
Table 2. Hypo-methylated and up-regulated concordant epigenes.
| GeneID | Description | Symbol | Δ5 mc (%) | DS | FC | SM | ALSoD& OS |
| 58475 | membrane-spanning 4-domains, subfamily A, member 7 | MS4A7 | 6 | −19.0 | 4.6 | ||
| 1439 | colony stimulating factor 2 receptor, beta, low-affinity | CSF2RB | 6.6 | −35.4 | 3.8 | ||
| 9056 | solute carrier family 7 (amino acid transporter light chain, y+L system), member 7 | SLC7A7 | 6.6 | −18.6 | 3.5 | ||
| 822 | capping protein (actin filament), gelsolin-like | CAPG | 5.8 | −13.9 | 3.4 | ||
| 6556 | solute carrier family 11 (proton-coupled divalent metal ion transporters), member 1 | SLC11A1 | 8.6 | −34.4 | 3.1 | ||
| 54209 | triggering receptor expressed on myeloid cells 2 | TREM2 | 7.1 | −26.0 | 2.9 | ||
| 912 | CD1d molecule | CD1D | 5.6 | −19.7 | 2.8 | ✓ | |
| 10457 | glycoprotein (transmembrane) nmb | GPNMB | 10.1 | −43.1 | 2.8 | ✓ | |
| 2203 | fructose-1,6-bisphosphatase 1 | FBP1 | 7.7 | −96.5 | 2.7 | ||
| 11006 | leukocyte immunoglobulin-like receptor, subfamily B, member 4 | LILRB4 | 4.6 | −14.0 | 2.7 | ||
| 27036 | sialic acid binding Ig-like lectin 7 | SIGLEC7 | 5.3 | −15.9 | 2.7 | ✓ | |
| 4046 | lymphocyte-specific protein 1 | LSP1 | 6.4 | −20.1 | 2.7 | ||
| 864 | runt-related transcription factor 3 | RUNX3 | 7.5 | −32.8 | 2.6 | ||
| 11025 | leukocyte immunoglobulin-like receptor, subfamily B, member 3 | LILRB3 | 5.9 | −14.0 | 2.5 | ||
| 2207 | Fc fragment of IgE, high affinity I, receptor for; gamma polypeptide | FCER1G | 5.5 | −20.2 | 2.4 | ||
| 10261 | immunoglobulin superfamily, member 6 | IGSF6 | 10.2 | −50.2 | 2.3 | ||
| 286256 | lipocalin 12 | LCN12 | 5.2 | −18.4 | 2.3 | ||
| 1230 | chemokine (C-C motif) receptor 1 | CCR1 | 7.3 | −25.2 | 2.3 | ||
| 51225 | ABI family, member 3 | ABI3 | 6.3 | -19.2 | 2.3 | ||
| 1522 | cathepsin Z | CTSZ | 5.8 | −39.3 | 2.2 | ✓ | |
| 26157 | GTPase, IMAP family member 2 | GIMAP2 | 7.2 | −35.0 | 2.2 | ||
| 10023 | frequently rearranged in advanced T-cell lymphomas | FRAT1 | 5.2 | −46.0 | 2.2 | ||
| 1117 | chitinase 3-like 2 | CHI3L2 | 7.1 | −22.6 | 2.1 | ||
| 64344 | hypoxia inducible factor 3, alpha subunit | HIF3A | 5.8 | −48.9 | 2.1 | ||
| 10320 | IKAROS family zinc finger 1 (Ikaros) | IKZF1 | 6.8 | −22.2 | 2.1 | ||
| 1051 | CCAAT/enhancer binding protein (C/EBP), beta | C/EBPB | 4.9 | −54.7 | 2.1 | ||
| 1601 | disabled homolog 2, mitogen-responsive phosphoprotein (Drosophila) | DAB2 | 3.9 | −13.7 | 2.0 | ||
| 84898 | plexin domain containing 2 | PLXDC2 | 3 | −23.7 | 2.0 | ||
| 10990 | leukocyte immunoglobulin-like receptor, subfamily B, member 5 | LILRB5 | 7.5 | −36.6 | 2.0 | ||
| 254295 | phytanoyl-CoA dioxygenase domain containing 1 | PHYHD1 | 3.1 | −13.6 | 1.9 | ||
| 4488 | msh homeobox 2 | MSX2 | 5.4 | −51.9 | 1.9 | ||
| 558 | AXL receptor tyrosine kinase | AXL | 3.3 | −14.5 | 1.8 | ||
| 5359 | phospholipid scramblase 1 | PLSCR1 | 7.3 | −82.6 | 1.8 | ||
| 23526 | histocompatibility (minor) HA-1 | HMHA1 | 0 | −33.5 | 1.8 | ||
| 50856 | C-type lectin domain family 4, member A | CLEC4A | 9.5 | −45.3 | 1.7 | ||
| 116843 | chromosome 6 open reading frame 192 | C6orf192 | 3.4 | −16.5 | 1.7 | ||
| 55544 | RNA binding motif protein 38 | RBM38 | 4.9 | −20.8 | 1.7 | ||
| 23209 | megalencephalic leukoencephalopathy with subcortical cysts 1 | MLC1 | 7.4 | −29.0 | 1.7 | ||
| 23418 | crumbs homolog 1 (Drosophila) | CRB1 | 6.2 | −15.2 | 1.7 | ||
| 1501 | catenin (cadherin-associated protein), delta 2 | CTNND2 | 2.9 | −18.0 | 1.6 | ||
| 5045 | furin (paired basic amino acid cleaving enzyme) | FURIN | 5.9 | −19.7 | 1.6 | ✓ | ✓ |
| 1317 | solute carrier family 31 (copper transporters), member 1 | SLC31A1 | 5.3 | −19.4 | 1.6 | ✓ | |
| 123920 | CKLF-like MARVEL transmembrane domain containing 3 | CMTM3 | 3.9 | −16.6 | 1.6 | ||
| 6776 | signal transducer and activator of transcription 5A | STAT5A | 4.2 | −16.6 | 1.6 | ||
| 57522 | SLIT-ROBO Rho GTPase activating protein 1 | SRGAP1 | 0.3 | −43.9 | 1.5 | ||
| 9404 | leupaxin | LPXN | 4 | −13.9 | 1.5 | ||
| 122618 | phospholipase D family, member 4 | PLD4 | 5.5 | −18.0 | 1.5 | ||
| 64859 | oligonucleotide/oligosaccharide-binding fold containing 2A | OBFC2A | 7.2 | −99.8 | 1.5 | ||
| 10628 | thioredoxin interacting protein | TXNIP | 4.3 | −26.1 | 1.4 | ||
| 79939 | solute carrier family 35, member E1 | SLC35E1 | 0 | −46.6 | 1.3 | ||
| 5660 | prosaposin | PSAP | 0 | −33.5 | 1.3 |
Genes are ordered by expression fold change (FC). Percent methylation change (Δ5 mc (%)) is calculated by |BetaALS – Betacontrol|/Betacontrol. DiffScore (DF), Differential methylation (a transformation of the p-value; p = 0.05, p = 0.01, p = 0.001 are equivalent to DiffScore = ±13, ±20, ±30, respectively). Fold change (FC) in gene expression ‘✓’ indicates that the corresponding gene was identified as an ALS-associated gene by SciMiner (SM), the ALSoD database, and other high-throughput microarray studies (OS) on human ALS spinal cord samples, respectively. See Table S2 more details.
Table 3. Hyper-methylated and down-regulated concordant epigenes.
| GeneID | Description | Symbol | Δ5 mc (%) | DS | FC | SM | ALSoD& OS |
| 7306 | tyrosinase-related protein 1 | TYRP1 | 9 | 50 | −7.2 | ✓ | |
| 2706 | gap junction protein, beta 2, 26 kDa | GJB2 | 16.3 | 18.4 | −4.7 | ||
| 26266 | solute carrier family 13 (sodium/sulfate symporters), member 4 | SLC13A4 | 4.6 | 26.2 | −4.2 | ||
| 4060 | lumican | LUM | 8.6 | 37 | −4.1 | ✓ | |
| 90523 | muscular LMNA-interacting protein | C6orf142 | 6.7 | 343.9 | −4.1 | ||
| 149461 | claudin 19 | CLDN19 | 5.6 | 31.6 | −4.1 | ||
| 112464 | protein kinase C, delta binding protein | PRKCDBP | 6.3 | 28.6 | −3.8 | ||
| 195814 | short chain dehydrogenase/reductase family 16C, member 5 | SDR16C5 | 8.1 | 16.7 | −3.5 | ||
| 64073 | chromosome 19 open reading frame 33 | C19orf33 | 5.3 | 29.2 | −3.4 | ||
| 158038 | leucine rich repeat and Ig domain containing 2 | LINGO2 | 2.4 | 42.7 | −3.3 | ✓ | |
| 51299 | neuritin 1 | NRN1 | 3.9 | 18.1 | −2.5 | ✓ | |
| 8727 | catenin (cadherin-associated protein), alpha-like 1 | CTNNAL1 | 6.4 | 17.1 | −2.5 | ✓ | |
| 51200 | carboxypeptidase A4 | CPA4 | 7.3 | 47.3 | −2.4 | ||
| 163782 | KN motif and ankyrin repeat domains 4 | KANK4 | 15.1 | 100.3 | −2.4 | ||
| 57664 | pleckstrin homology domain containing, family A, member 4 | PLEKHA4 | 11.9 | 26.4 | −2.4 | ||
| 283120 | H19, imprinted maternally expressed transcript (non-protein coding) | H19 | 5.6 | 48.2 | −2.3 | ||
| 1755 | deleted in malignant brain tumors 1 | DMBT1 | 9.6 | 36.3 | −2.3 | ||
| 3045 | hemoglobin, delta | HBD | 7.8 | 77.9 | −2.3 | ||
| 8743 | tumor necrosis factor (ligand) superfamily, member 10 | TNFSF10 | 7.2 | 33 | −2.2 | ✓ | |
| 4826 | neuronatin | NNAT | 5.8 | 21.1 | −2.2 | ||
| 2676 | GDNF family receptor alpha 3 | GFRA3 | 4.9 | 19.3 | −2 | ||
| 5121 | Purkinje cell protein 4 | PCP4 | 1.3 | 15.9 | −2 | ✓ | |
| 90139 | tetraspanin 18 | TSPAN18 | 5.2 | 24.8 | −2 | ||
| 5858 | pregnancy-zone protein | PZP | 9.5 | 48.7 | −2 | ||
| 10586 | mab-21-like 2 (C. elegans) | MAB21L2 | 3.9 | 19.4 | −1.9 | ✓ | |
| 64288 | zinc finger protein 323 | ZNF323 | 8.6 | 32.6 | −1.9 | ||
| 84708 | ligand of numb-protein X 1 | LNX1 | 9.4 | 30.5 | −1.9 | ||
| 144347 | family with sequence similarity 101, member A | FAM101A | 6.9 | 33.9 | −1.8 | ||
| 25960 | G protein-coupled receptor 124 | GPR124 | 6.6 | 18.2 | −1.8 | ||
| 6335 | sodium channel, voltage-gated, type IX, alpha subunit | SCN9A | 6.5 | 47.3 | −1.8 | ✓ | |
| 23089 | paternally expressed 10 | PEG10 | 10.1 | 21.2 | −1.8 | ||
| 254228 | family with sequence similarity 26, member E | FAM26E | 5.8 | 97 | −1.8 | ||
| 5332 | phospholipase C, beta 4 | PLCB4 | 6.7 | 49.5 | −1.8 | ||
| 168537 | GTPase, IMAP family member 7 | GIMAP7 | 7.8 | 31.1 | −1.7 | ||
| 4199 | malic enzyme 1, NADP(+)-dependent, cytosolic | ME1 | 7.4 | 16.9 | −1.7 | ✓ | |
| 135932 | transmembrane protein 139 | TMEM139 | 8.6 | 26.9 | −1.7 | ||
| 6932 | transcription factor 7 (T-cell specific, HMG-box) | TCF7 | 5.3 | 87.4 | −1.7 | ||
| 79827 | CXADR-like membrane protein | ASAM | 9.7 | 87.6 | −1.6 | ||
| 152330 | contactin 4 | CNTN4 | 0.6 | 48.8 | −1.6 | ✓ | |
| 55228 | PNMA-like 1 | PNMAL1 | 8.8 | 30.6 | −1.6 | ||
| 150350 | ENTH domain containing 1 | ENTHD1 | 4.8 | 81.9 | −1.5 | ||
| 27237 | Rho guanine nucleotide exchange factor (GEF) 16 | ARHGEF16 | 7.5 | 101.6 | −1.5 | ||
| 57415 | chromosome 3 open reading frame 14 | C3orf14 | 7.9 | 78.2 | −1.5 | ||
| 84909 | chromosome 9 open reading frame 3 | C9orf3 | 0.1 | 40.4 | −1.5 | ✓ | |
| 2326 | flavin containing monooxygenase 1 | FMO1 | 9.4 | 61.1 | −1.5 | ✓ | ✓ |
| 5172 | solute carrier family 26, member 4 | SLC26A4 | 9.4 | 23.5 | −1.5 | ✓ | |
| 9467 | SH3-domain binding protein 5 (BTK-associated) | SH3BP5 | 7 | 50.6 | −1.5 | ||
| 166 | amino-terminal enhancer of split | AES | 5.8 | 42.7 | −1.4 | ||
| 3768 | potassium inwardly-rectifying channel, subfamily J, member 12 | KCNJ12 | 14.5 | 343.9 | −1.4 | ||
| 22927 | hyaluronan binding protein 4 | HABP4 | 12.2 | 45.1 | −1.4 | ||
| 8537 | breast carcinoma amplified sequence 1 | BCAS1 | 8.7 | 59.2 | −1.4 | ||
| 8817 | fibroblast growth factor 18 | FGF18 | 7.8 | 15.8 | −1.4 | ✓ | |
| 5213 | phosphofructokinase, muscle | PFKM | 5.9 | 29.3 | −1.4 | ||
| 85453 | TSPY-like 5 | TSPYL5 | 11.7 | 32 | −1.4 | ||
| 4233 | met proto-oncogene (hepatocyte growth factor receptor) | MET | 6 | 22.9 | −1.4 | ||
| 135250 | retinoic acid early transcript 1E | RAET1E | 9.6 | 89.7 | −1.3 | ||
| 2260 | fibroblast growth factor receptor 1 | FGFR1 | 5.1 | 18.7 | −1.3 | ✓ | |
| 90135 | BTB (POZ) domain containing 6 | BTBD6 | 9.3 | 23.5 | −1.3 | ||
| 744 | metallophosphoesterase domain containing 2 | MPPED2 | 7.3 | 41.8 | −1.3 | ||
| 3749 | potassium voltage-gated channel, Shaw-related subfamily, member 4 | KCNC4 | 3.2 | 28.2 | −1.3 | ✓ | |
| 91977 | myozenin 3 | MYOZ3 | 5 | 15.3 | −1.3 |
Genes are ordered by expression fold change (FC). Percent methylation change (Δ5 mc (%)) is calculated by |BetaALS – Betacontrol|/Betacontrol. DiffScore (DF), Differential methylation (a transformation of the p-value; p = 0.05, p = 0.01, p = 0.001 are equivalent to DiffScore = ±13, ±20, ±30, respectively). Fold change (FC) in gene expression ‘✓’ indicates that the corresponding gene was identified as an ALS-associated gene by SciMiner (SM), the ALSoD database, and other high-throughput microarray studies (OS) on human ALS spinal cord samples, respectively. See Table S2 more details.
Figure 4. Overlapping of DMGs and DEGs.

Identified DMGs and DEGs were compared for overlapping. Among 3,574 DMGs and 1,182 DEGs, 251 genes were common. Among these shared genes, 112 genes had concordant direction of change in methylation and expression (hyper-methylation with down-regulation or hypo-methylation with up-regulation) (shaded circle).
Gene co-citation network analysis revealed potential associations between some of the concordant genes identified in our screens. Among the 112 concordant epigenes, 53 genes were co-cited at least once in PubMed abstracts at the sentence or abstract-level. Within the co-citation network, the epigenes were classified into representative biological functions such as immune response, antigen presentation, tumor/suppressor related, and extracellular matrix repair (Fig. 5). In this network, the transcription factors C/EBPB and STAT5A were highly connected, suggesting a role in sALS pathogenesis. Moreover, some of these connections were directly or indirectly associated with concordant genes previously related to neuronal development such as muscle segment homeobox (msh) 2 (MSX2), megalencephalic leukoencephalopathy with subcortical cysts 1 (MLC1), RUNX3, catenin delta-2 (CTNND2), receptor tyrosine kinase (AXL), neuronatin (NNAT), and NRN1 [24]–[30] (Fig. 5).
Figure 5. Literature-based association network of concordant epigenes.

Literature-derived information for potential associations among the concordant epigenes was obtained using Genomatix Pathway System (GePS). Among the 112 concordant epigenes, 53 genes were co-cited at least once in PubMed abstracts at sentence-level (solid lines) or abstract-level (dashed lines). These epigenes were grouped by their representative biological role: immune response (dark pink), antigen presentation (light pink), tumor/suppressor related (green), extracellular matrix repair (yellow), and others (gray). Neuronal development-related genes are enclosed by a square. The epigenes with more than 5 connections to other genes are enlarged. A diamond-shape represents a transcription factor.
SciMiner identified 4,128 genes from ALS-related publications (as of 7/23/2012), which were compared to our 112 concordant epigenes. Fourteen genes were identified in two or more ALS-related publications with frequencies that were significantly different from those in over 20 million abstracts in PubMed (p<0.05). Fifty-one genes demonstrated ≥2-fold altered expression, including the chitinase 3-like protein 2 (CHI3L2), the triggering receptor expressed on myeloid cells-2 (TREM2), cathepsin Z (CTSZ), the lumican precursor protein (LUM), H19, and TRAIL/TNFSF10 (Tables 2, 3). Thus, bioinformatics evaluation of the concordant epigenes identified by integrating methylomic and transcriptomic analyses detected both novel and previously known ALS-related genes.
Experimental Confirmation using Real-time Polymerase Chain Reaction (RT-PCR)
Expression of 14 concordant epigenes selected either from the ALS-related literature or from the expression array data was confirmed by RT-PCR (Figs. 6 and S2, Table 4). Of the genes previously related to the ALS literature, NRN1, FMO1, and the lumican precursor protein (LUM) were under-expressed, while the lysosomal protease CTSZ was over-expressed in sALS. No significant difference between sALS and control subjects was observed for FES-upstream region (FURIN). Novel sALS-associated epigenes such as STAT5A, TREM2, the high-affinity IgE receptor (FCER1G), CHI3L2, and the proton-couple divalent metal ion transporter solute carrier family 11 member 1A (SLC11A1) were over-expressed in sALS. SLC11A1 presented the highest increase by 12.7-fold. Down-regulation was validated for gap junction ß-2 (GJB2)/Connexin-26 as well as imprinted genes such as H19, NNAT, and the paternally expressed 10 (PEG10). In summary, the RT-PCR expression data indicate high concordance with the microarray expression data, validating our results.
Figure 6. RT-PCR confirmation of concordant epigenes in spinal cord.

RNA was extracted from the postmortem human spinal cord tissue that was used for the methylation analysis from sALS (n = 8–11) subjects and controls (n = 8–11) and subjected to RT-PCR. Results were normalized to glyceraldehyde-3- phosphate dehydrogenase (GAPDH) except for STAT5A which was normalized to TATA box binding protein (Tbp) and presented as fold changes calculated by the 2−ΔΔC T method. Similar results were obtained when using different housekeeping genes (Fig. S2); *p<0.05, **p<0.01, ***p<0.001 compared to the control group (Ctrl). Data mean ± SEM is plotted using box and whiskers vertical bars plotting minimum to maximum values.
Table 4. Confirmation of microarray differential expression in spinal cord using RT-PCR.
| Symbol | Control | sALS | <p-value | Fold-Change | Microarray Fold-Change | Confirmed |
| CTSZ | 1.10±0.07 | 2.06±0.23 | 0.001 | 1.9 | 2.2 | Yes |
| STAT5A | 1.10±0.12 | 1.58±0.20 | 0.05 | 1.4 | 1.6 | Yes |
| TREM2 | 1.20±0.17 | 3.18±0.72 | 0.05 | 2.6 | 2.9 | Yes |
| FCER1G | 1.10±0.11 | 2.30±0.19 | 0.001 | 2.1 | 2.4 | Yes |
| CHI3L2 | 1.32±0.29 | 3.13±0.48 | 0.01 | 1.6 | 2.1 | Yes |
| SLC11A1 | 1.34 0.23 | 17.00±3.38 | 0.001 | 12.7 | 3.1 | Yes |
| NRN1 | 1.19±0.18 | 0.49±0.05 | 0.001 | −2.4 | −2.5 | Yes |
| NNAT | 1.14±0.20 | 0.52±0.06 | 0.05 | −2.2 | −2.2 | Yes |
| FMO1 | 1.41±0.40 | 0.38±0.07 | 0.001 | −3.7 | −1.5 | Yes |
| GJB2 | 1.38±0.27 | 0.35±0.07 | 0.001 | −3.9 | −4.7 | Yes |
| FURIN | 1.11±0.16 | 0.83±0.13 | 0.17 | −1.3 | 1.6 | No |
| H19 | 1.28±0.15 | 0.68±0.20 | 0.05 | −1.9 | −2.3 | Yes |
| PEG10 | 1.08±0.08 | 0.79±0.07 | 0.01 | −1.4 | −1.8 | Yes |
| LUM | 1.40±0.29 | 0.32±0.13 | 0.01 | −4.4 | −4.1 | Yes |
‘Yes’ indicates the differential expression of the corresponding gene is statistically significant and demonstrated the same direction of change as in the microarray data.
Global 5 HmC Increases in sALS Spinal Cord
5 HmC, an alternate epigenetic modification of DNA, is increased in brain compared to other human tissues, and alterations in global 5 HmC are associated with age-related neurodegenerative disorders, suggesting an important role of 5 HmC in neuronal the function [31]–[34]. We measured global 5 HmC for sALS and control spinal cord samples previously analyzed for global 5mC. We observed an approximately 3.0-fold increase in global 5 HmC in sALS (0.31±0.02) compared to controls (0.11±0.03) (p<0.0001) (Fig. 7). This is the first report of aberrant levels of global 5 HmC associated with the pathogenesis of sALS.
Figure 7. Global 5 HmC is increased in spinal cord of sALS.

Genomic DNA extracted from control or sALS postmortem human spinal cord was analyzed with an ELISA colorimetric assay for hydroxymethylation (5 HmC; Ctrl = 9, sALS = 8). The data is presented as mean ± SEM of percent (%)5 HmC using a two-sample equal variance t-test and plotted using box and whiskers vertical bars with minimum to maximum values; ****p<0.0001 compared to the control group (Ctrl).
Global 5mC and 5 HmC in sALS Whole Blood
High correlation of epigenetic marks in spinal cord and blood may be useful for diagnostic and therapeutic application in ALS. We investigated whether global 5mC and 5 HmC would be altered in sALS whole blood similarly to spinal cord. Whole blood genomic DNA from a different cohort (Tables 5, S1) was subjected to global 5mC and global 5 HmC by ELISA. The levels of percent global 5mC and 5 HmC in whole blood were 10 fold lower compared to spinal cord, in agreement with recent reports [33], [35]. Contrary to spinal cord, no differential percent global 5mC (controls, 0.408±0.300; ALS, 0.405±0.027 p = 0.941) and global 5 HmC (controls, 0.033±0.005; ALS, 0.032±0.004; p = 0.401) (Fig. 8) were observed in whole blood.
Table 5. Characteristics of sALS and control subjects used for global 5mC and 5 HmC in whole blood.
| Characteristics | sALS group | Control group | p-value | |
| Number of subjects | 11 | 12 | – | |
| Age (years)(a) | 60 (48–68) | 59 (48–70) | NS | |
| Gender | Male | 7 | 7 | NS |
| Female | 4 | 5 | NS | |
| Onset | Bulbar | 3 | 0 | – |
| Limb | 8 | 0 | – | |
| Disease duration (months)(a)(b) | 45 (16–64) | – | – | |
| Health condition of controls(c) | Anxiety | 0 | 1 | – |
| Asthma | 0 | 2 | – | |
| Cancer | 0 | 3 | – | |
| Healthy | 0 | 5 | – | |
| Hearing loss | 0 | 1 | – | |
| Hypercholesterolemia | 0 | 1 | – | |
| Hypothyroidism | 0 | 2 | – | |
| Hypertension | 0 | 1 | – | |
median (range);
mean ± standard deviation;
some patients presented more than one condition; NS, not significant; ‘–‘, value not available.
Figure 8. Changes in global 5 HmC and 5mC are not detected in ALS whole blood.

Genomic DNA extracted from control or sALS human whole blood was analyzed for 5mC (Ctrl n = 11, ALS n = 11; p = 0.94) and 5 HmC (Ctrl n = 11, sALS n = 11; p = 0.40). Percent (%) 5mC and 5 HmC is presented as mean ± SEM using a two-sample equal variance t-test and graphed using box and whiskers vertical bars plotting minimum to maximum values.
Discussion
Although several genes have been implicated in the pathogenesis of ALS, the causes leading to most cases remain unknown. Environmental factors may be associated with the onset and development of sALS by altering epigenetic regulation [7], [8]. The aim of this study was to identify sALS-associated epigenetic marks resulting in aberrant gene expression. Abnormal 5mC patterns of repetitive elements such as Alu and LINE1, as well as altered function of methylation regulators such as the DNMTs, lead to changes in global 5mC or 5 HmC associated with neurodegeneration [15], [36]. We demonstrate increased global methylation in sALS spinal cord, perhaps due to an increase in DNMT activity [15]. Furthermore, we report for the first time an increase in global 5 HmC in sALS spinal cord. Increased 5mC and 5 HmC may be due to 5mC providing more substrate for the TET proteins [10], TETs are not differentially expressed in spinal cord sALS according to our microarray data (data not shown). TET should decrease the amount of 5mC only if 5mC is not increasing at a faster rate than the oxidation reaction. Although normal aging leads to increased global 5 HmC in mouse hippocampal DNA independently of increased levels of oxidative stress [31], in ALS, increased oxidative DNA damage and free radicals may contribute to global 5 HmC dysregulation. The base excision repair (BER) pathway responsible for oxidative DNA damage restoration and one of the active demethylation pathways, is deficient in ALS [14], [37], [38].
Methylomics and transcriptomics analyses identified potential biologically relevant epigenes in postmortem sALS spinal cord. These epigenes were enriched with biological functions related to inflammation and the immune responses, previously linked to ALS [39]–[41]. Our data suggest that alterations in gene expression of immune-related genes in sALS may be regulated by methylation. Immune-related concordant epigenes including TREM2, chemokine (C-C motif) receptor 1/RANTES receptor (CCR1), SLC11A1, the transmembrane receptor C-type lectin domain family 4 member A isoform 1 (CLEC4A), and the IgE receptor (FCER1G) were found to be over-expressed in sALS. Our findings suggest an infiltration of myeloid cells, mast cells, or natural killer cells to the damaged area and/or activation of resident microglia [42]–[44]. Supporting our observations, neuro-inflammation was recently associated with systemic macrophage activation independent of T-cell activation and the recruitment of activated inflammatory monocytes to the spinal cord in ALS [45], [46]. Although immunosuppressive and anti-inflammatory therapies have shown to delay disease onset in ALS animal models, clinical trials have not revealed a major effect on disease progression or survival [46]–[50]. This suggests that continuous activation of microglia leading to neuronal damage surpasses the capacity of the nervous system to respond to immunosuppressive and anti-inflammatory therapies at later stages of ALS, implicating a need for biomarkers identifying early immune-related changes in sALS.
Co-citation network and literature mining approaches identified connections among novel and previously implicated ALS-related epigenes and pathways [51], [52]. The transcription factors STAT5A and C/EBPB are highly connected in our co-citation network and their interplay promote activation of various genes including interleukin-6 (IL-6) [53]. Moreover, recent reports implicate C/EBPB and STAT5A in ALS pathogenesis and neurodegeneration. For instance, expression of C/EBPB in ALS microglia from spinal cord suggests an important role of C/EBPB in the regulation of neurotoxic genes in the ALS neuronal microenvironment [45], [54]. Furthermore, changes in STAT5A expression may reflect an altered inflammatory response contributing to the pathogenesis of ALS. Over-expression of STAT5A reduces neuronal degeneration associated with spinal muscular atrophy, a neurodegenerative disease with similar pathogenesis as ALS, and it provides oligodendrocyte protection, which in turn favors neuronal environment preservation [55]–[57]. Whether positive regulation of STAT5A in sALS is due to an anti-apoptotic response to compensate for the degeneration of the nervous system, or its over-expression is responsible, in part, for the pathogenesis of the disease remains to be determined. Interestingly, we observed potential transcription factor binding sites (TFBSs) for STAT5A and C/EBPB in 40% and 48% of the promoters of our identified DEGs, respectively; the binding sites for STAT5A and C/EBPB are 1.2 (p = 4.1E-12) and 1.3 (p = 3.8E-13) times more frequent in the DEGs than in the vertebrate promoters, respectively. Our observations suggest epigenetic mechanisms, in part, drive the expression of central regulators of downstream targets in sALS.
Our study identified ALS-dependent methylation dysregulation of several genes previously implicated in neuronal development, differentiation, and proliferation, including Slit-Robo Rho GTPase activating protein 1 (SRGAP1), crumbs homolog 1 (CRB1), MSX2, MLC1, CTNND2, AXL, RUNX3, NNAT, and NRN1 [24]–[30], [58], [59]. Altered expression and/or mutations in CRB1, MLC1, and CTNND2 are associated with mental retardation and neurodegeneration [26], [28], [60], [61]. Moreover, we found CRB1 to be hypo-methylated and up-regulated in sALS, in agreement with overexpression observed by Kudo et al. in spinal cord from a mouse model of familial ALS (SOD1 G93A) as well as human sALS [61]. Interestingly, most of these genes were identified by our literature-based association network of concordant epigenes and were connected to C/EBPB and STAT5A. Analysis of the promoter region of these genes indicates a high incidence of potential TFBSs for these two transcription factors, suggesting a potential role of STAT5A and C/EBPB in the regulation of neuronal genes in the pathogenesis of sALS. Our observations suggest sALS-related alterations in methylation may lead to aberrant expression of genes required for neuronal homeostasis. Nevertheless, more studies need to be done to address the role of methylation, STAT5A, and C/EBPB in the regulation of neuronal genes.
Another sALS-related epigene, CTSZ warrants further investigation since its aberrant expression is associated with neurodegeneration by promoting neurotoxin elimination in the damaged cellular environment [62]. Expression of CTSZ as well as two other members of the cathepsin family, cathepsins B and D, increases in human and rodent ALS spinal cord and mutant SOD1 (G86R, G93A) mouse skeletal muscle suggesting they play an important role in ALS [63], [64].
Except for optineurin (OPTN) [65], which was identified as a hypo-methylated DMG without demonstrating changes in gene expression, loci known to be mutated in fALS were not present in our concordant epigenes [4]. This agrees with recent studies indicating that promoter regions of SOD1, VEGF, and metallothioneins I and II are not differentially methylated in sALS [66], [67]. When compared with the ALS Online genetics Database (ALSoD)-reported genes and other ALS-dependent methylation/gene expression of profiling studies [39], [41], [68]–[72], we observed a modest overlap of four concordant epigenes; Purkinje cell protein 4 (PCP4), catenin (CTNNAL1), fibroblast growth factor 18 (FGF18), and flavin containing monooxygenase 1 (FMO1). Furthermore, five of our concordant genes presented opposite direction of expression when compared to known ALS-dependent differentially expressed gene. Our data indicate that epigenetic mechanisms are potential regulators of these key genes in ALS.
Based on the large number of genes identified in the methylation (3,574 genes) and expression (1,182 genes) arrays, relatively few sALS-associated genes presented concordant direction between methylation and gene expression. The low occurrence of a small subset of genes potentially regulated by CpG modification in such a way that hyper-methylation promotes gene silencing and hypo-methylation promotes gene expression has been previously documented [73]. 5mC within promoter regions is associated with repression of gene expression by interfering with transcription factor binding or by providing a binding site for transcriptional repressors [10]. Interestingly, over half (55%) of the 251 common DMGs/DEGs presented same direction of 5mC and expression. In some cases, 5mC positively regulates gene transcription by promoting transcription factor binding at promoter regions [74] or, more commonly, by modifying intragenic CpG sites facilitating transcription efficiency, histone conformation, and regulating levels of sense and antisense mRNA [75]. Furthermore, 5 HmC, a highly enriched modification in brain, correlates with increased gene expression [10]. HM27K does not differentiate between 5mC and 5 HmC; therefore, some of the common epigenes presenting same direction of methylation and expression may be regulated by 5 HmC.
Although the high incidence of same direction sALS concordant epigenes parallels the high levels of global 5 HmC in spinal cord, loci specific 5 HmC modifications associated with sALS remain to be identified. Gene expression of non-common DEG (non-DMGs) could be determined, in part, by 5mC-dependent regulation of transcription factors. In addition to STAT5 and C/EBPB, we identified several transcription factors as concordant genes such as the transcription factor 7 (TCF7), RUNX3, IKAROS family zinc finger 1 (IKZF1), MSX2, and hypoxia inducible factor 3, alpha subunit (HIF3A). Furthermore, regulation of gene expression is a dynamic and complex mechanism and the interplay of several epigenetic pathways has been reported to modulate adult neurogenesis [76]. Therefore, alterations to epigenetic networks in conjunction with genetic predisposition may result in the development of sALS.
The prospect of identifying sALS epigenetic biomarkers in blood is exciting as it provides a minimally invasive alternative for sALS diagnostic and prognostic assessments. Although we did not detect significant global 5mC and 5 HmC differences in blood and inflammation-related epigene biomarkers may reflect systemic inflammatory changes rather than neuronal changes, further investigation of individual loci may provide potential epigenetic biomarkers for sALS.
There were several limitations to our study. First, a relatively small number of samples were analyzed and loci-specific 5 HmC analysis is still needed. Nevertheless, this is an initial step towards identifying epigenetic mechanisms altering key pathways leading to sALS, which will be validated in larger cohorts. Second, sALS postmortem tissue reflects the terminal disease stage rather than the pathogenic mechanisms leading to disease onset and progression. As sALS-affected motor neurons deteriorate at the terminal stage and heterogeneous tissue consisting of both gray and white matter was analyzed, our results may represent epigenetic regulation of the neuronal microenvironment, including microglia activation and the scarce neurons surviving the degenerative process [54], [72]. This may explain, in part the discrepancy in the direction of expression of common and concordant genes reported here with other sALS genome-wide expression profiles, as well as the heavily represented inflammation-related genes, in our concordant epigenes, which are not differentially expressed specifically in sALS motor neurons or ventral horns [68]. Finally, more studies are needed to concretely identify whether or not the genes identified in this study are involved in ALS pathogenesis.
Advances in identifying epigenetic regulators in disease states have led to new therapeutic approaches. Interestingly, demethylating agents have been extensively studied to reverse aberrant epigenetic changes associated with cancer [77] and more recently, histone deacetylase inhibitors have shown to have neuroprotective properties in animal models of neurodegenerative diseases [78]. These observations suggest reversible epigenetic modifications carry the potential for therapeutic treatment in sALS. We contend that environmental life exposures result in failure to maintain epigenetic homeostasis in the nervous system microenvironment leading to global and loci specific aberrant regulation of gene expression in sALS-affected tissue. Ascertaining the role of epigenetic regulation may provide a better understanding of the pathogenesis of sALS and new therapeutic targets.
Methods
Subjects and Tissue
Frozen human spinal cord samples from 12 Caucasian sALS subjects and 11 age and gender-matched neurologically-normal controls were obtained from the National Center for Child Health and Human Development (NICHD) Brain and Tissue Bank for Developmental Disorders at the University of Maryland, Baltimore, MD (Table 1). Whole blood was collected in EDTA tubes from a different cohort of 11 Caucasian sALS and 11 age- (5±years) and gender-matched neurologically-normal control subjects at the University of Michigan ALS Consortium (Table 4). Table S1 summarizes the samples used for each assay.
Ethics Statement
The participants donating blood reviewed and signed a written informed consent under a protocol reviewed and approved by the University of Michigan Medical School Institutional Review Board (HUM00028826).
Nucleotide Extraction and DNA Bisulfite Conversion
Genomic DNA was extracted from 50 mg of frozen postmortem spinal cord tissue (mostly grey matter including the ventral horn, but some white matter included) using the Promega Maxwell 16 Tissue DNA Purification kit and a Maxwell instrument (Promega Co, Madison, WI). Genomic DNA (1 µg) was bisulfite converted with an EZ DNA Methylation Kit (Zymo Research, Irvine, CA) according to the manufacturer’s instructions. Total RNA was extracted from the same tissue for methylation profiling (Table S1) using the RNeasy kit and treated with RNAse-free DNase1 according to the manufacturer’s instructions (Qiagen, Valencia, CA). Automated genomic DNA extraction from whole blood was performed at the Michigan Institute for Clinical & Health Research (MICHR) at the University of Michigan using Autogen FlexStar (Autogen, Holliston, MA) and Qiagen Flexigene reagents. Nucleotide concentration was assessed using a Nanodrop 2000 (Thermo Scientific) and RNA integrity was determined by microfluid electrophoresis with a 21000 Bioanalyzer (Agilent Technologies, Palo Alto, CA).
Global 5mC and 5 HmC
Differences in genomic DNA global methylation (Global 5mC) and hydroxymethylation (Global 5 HmC) from sALS and control spinal cord or whole blood were determined in duplicate using the colorimetric enzyme-linked immuno-sorbent assay (ELISA) MethylFlash (Methylated or Hydroxymethylated) DNA Quantification Kits according to the manufacturer’s directions (Epigentek Group Inc., Farmingdale, NY). The absorbance at 450 nm was captured in a Fluoroskan Ascent microplate reader (Labsystems, Vienna, Va). The percentage of Global 5mC and 5 HmC is expressed as mean ± standard error mean (SEM). The two-tailed t-test was used for statistical comparison. Graphs and statistical analysis were obtained with GraphPad Prism 5.
Methylation Profiling and Identification of DMGs
For high-throughput methylation profiling, 200 ng of bisulfite-converted DNA was whole-genome amplified (WGA), enzymatically fragmented, purified, and hybridized to the Infinium Human Methylation27 DNA BeadChip array (HM27K; Illumina, Inc., San Diego, CA) following the manufacturer’s instructions at the University of Michigan Sequencing Core. The HM27K quantitatively determines DNA methylation for 27,578 CpG sites spanning 14,495 genes. DMGs were identified using Illumina’s GenomeStudio software [79]. Single-base resolution corresponding to DNA methylation levels for each locus was reported and the methylation level is given by a beta (ß) value describing the percentage of the degree of methylation ranging from 0 (no methylation) to 1 (complete methylation). Any methylation value with a detection P-value >0.05 was excluded. Differential methylation of the selected CpG target regions of autosomal chromosomes between sALS and control groups were tested using Illumina Custom algorithm with multiple testing corrections applied. DiffScore, GenomeStudio’s statistical significance score for differential methylation, of >13 for hyper-methylation or <-13 for hypo-methylation, equivalent to False Discovery Rate (FDR) <5%, were used.
Genome-wide Expression Profiling
Microarray gene expression analysis was performed as previously described in our published protocols [80]. Briefly, RNA samples with an RNA integrity number (RIN) >6.4 were used for further microarray and real-time PCR analysis. Total RNA (75 ng) was amplified and biotin-labeled using the Ovation Biotin-RNA Amplification and Labeling System (NuGEN Technologies, Inc., San Carlos, CA) according to the manufacturer’s instructions. Amplification and hybridization was performed at the University of Michigan DNA Sequencing Core Affymetrix and Microarray Core Group (Ann Arbor, MI) using the Affymetrix GeneChip Human Genome U133 Plus 2.0 Array measuring over 47,000 transcripts representing over 20,000 human genes.
Affymetrix CEL files were analyzed using a local version of the GenePattern genomic analysis platform from the Broad Institute [81]. Samples were Robust Multi-array Average (RMA) normalized using the BrainArray Custom CDF HGU133Plus2_Hs_ENTREZG version 14 [82]. Microarray quality was assessed as previously published [80]. Briefly, probe-level modeling (PLM) and quality metrics provided by the BioConductor affy package were used to identify low-quality arrays [83]–[85]. Outlier arrays, skewed away from other arrays, identified by Principal Component Analysis (PCA) were excluded from further analyses. Intensity-Based Moderated T-statistic (IBMT) [86] was employed to identify DMGs with a 10% FDR cut-off between sALS and control samples.
Identification of Differentially Expressed Genes (DEGs)
Concordant epigenes are those exhibiting significant differential methylation (hyper- or hypo-methylation) and a parallel change of gene expression (under- or over-expression, respectively) between sALS and control. Differentially methylated (DMGs)/expressed (DEGs) genes were subjected to bioinformatics analyses.
Bioinformatics Analysis of Concordant Epigenes
Functional enrichment analysis
Database for Annotation, Visualization and Integrated Discovery (DAVID; http://david.abcc.ncifcrf.gov/) [87], [88] was used to identify enriched molecular biological functions and ALS-relevant pathways of concordant epigenes. A Benjamini-Hochberg corrected P-value of 0.05 was used as the cut-off for statistically significant over-representation.
Literature mining analysis
A literature mining approach was used to obtain a comprehensive list of potential ALS-associated targets (genes/proteins). SciMiner, a web-based literature mining tool [89], [90], retrieves, processes documents, and identifies potential ALS-associated targets from the ALS-related literature, defined by a PubMed-style query of “Amyotrophic Lateral Sclerosis”. The concordant epigenes were compared against the literature-derived ALS-associated targets that were observed in at least 2 or more papers and whose frequency (in terms of the number of papers) was significantly different from the background. Fisher’s exact test (p-value <0.05) was used to determine whether each gene’s frequency was significantly different from the complete collection of abstracts of over 20 million papers in PubMed. The concordant genes identified by the high-throughput arrays were compared with these literature-derived ALS-related genes to identify which known disease-relevant genes are most highly methylated/expressed and, consequently, likely involved in disease pathogenesis. The resulting genes were designated as literature-derived ALS-associated epigenes.
Transcriptional network analysis
To elucidate the functional relationships among the concordant epigenes, we generated transcriptional networks using Genomatix Pathway Systems (GePS; Genomatix Software GmbH, Munich, Germany) with a sentence-level co-citation filter. Two genes co-cited at the sentence level in the literature are linked, resulting in a co-citation network. Additionally, transcriptional regulatory information of predicted transcription factor binding sites (TFBS) in promoter regions of genes could be further incorporated. The network allows the visualization of concordant epigenes, their potential associations, and transcriptional regulation with each other. Therefore, it helps in the identification of key genes that are highly connected to genes, and which play potentially important roles in the pathogenesis of sALS. Potential TFBSs of two highly connected genes in the network, STAT5A and C/EBPB, were searched among the promoters of the concordant epigenes using MatInspector (Genomatix) [91].
Pyrosequencing
To validate the HM27K arrays, we assessed gene-specific methylation of three selected cytokine genes based on the fact that immune response is associated with the pathogenesis of sALS [92] and two transcription factors. Amplicons of the promoter regions of the genes coding for the CKLF-like MARVEL transmembrane domain-containing proteins 2 and 3 (CMTM2 and CMTM3), the chemokine (C-X-C motif) ligand 12 (CXCL12), signal transducer and activator of transcription 5A (STAT5A), and CCAAT/enhancer binding protein beta (C/EBPB) were generated in 30 ml reactions using the PyroMark kit (Qiagen, Valencia, CA) with 4.8 pmol of the forward non-biotinylated primer, 2.4 pmol of the reverse biotinylated primer (Table S3), and 25 ng of bisulfite converted genomic DNA as previously described [93]. PCR conditions: 95°C for 15 min, 50 cycles [95°C for 30 s, 40–50°C for 30 s, 72°C for 20 s], 72°C for 10 min. Ten ml of the amplicon was Streptavidin Sepharose (Amersham Bioscience, Uppsala, Sweden) were purified, denatured with 0.2 M NaOH, and pyrosequenced using 0.5 mM of sequencing primer in a PSQ96 HS System (Qiagen) following the manufacturer’s protocol. Percent methylation of the region analyzed containing the identified Illumina methylation site or individual sites are presented as mean ± SEM with a two-sample equal variance t-test using GraphPad Prism 5.
RT-PCR
cDNA was generated by reverse transcription from total RNA isolated for microarray analysis using an iScript cDNA Synthesis Kit (Bio-Rad, Hercules, CA). RT-PCR was performed in triplicate using sequence-specific primers (Table S4) with SYBR Green PCR reagents (Bio-Rad, Hercules, CA). The PCR amplification profile was as follows: 95°C for 5 min, [denaturation at 95°C for 30 s, annealing at 55–60°C for 60 s, and extension at 72°C for 30 s] x40 cycles, and a final phase of 72°C for 5 min. The fluorescence threshold CT value, representing mRNA expressed in sALS samples, was calculated by the iCycler iQ system software. mRNA levels were normalized to an endogenous reference (ΔCT) and then relative to the control group (ΔΔCT). Levels of PCR products are demonstrated as mean ± SEM and a two-sample equal variance t-test was performed using GraphPad Prism 5 to confirm that mRNA levels were significantly different between sALS and control.
Supporting Information
Validation of HM27K arrays using pyrosequencing. Amplicons to the promoter regions identified by HM27K of cytokines CXCL12, CMTM3, CMTM2, C/EBPB, and STAT5A were generated using bisulfite-converted genomic DNA from human postmortem spinal cord and used as templates for pyrosequencing (ALS n = 11; Ctrl n = 11) Results are presented as mean of percent methylation of all CpG sites within the area tested on each gene (A) or as percent methylation of individual sites for STAT5A; site 2 was identified with the HM27K (B). Results are presented as mean ± SEM and a two-sample equal variance t-test was used. *p<0.05, **p<0.01 compared to control group (Ctrl).
(EPS)
RT-PCR confirmation of concordant epigenes in spinal cord. Total RNA was extracted from postmortem human spinal cord tissue used for the methylation analysis from sALS subjects (n = 8–11) and controls (n = 8–11) and subjected to RT-PCR. Results were normalized to housekeeping genes [TATA-box Binding Protein (TBP) for CTSZ, FCER1G, TREM2, NRN1 and NNAT; ribosomal 18S subunit for CHI3L2, H19, PEG10, and LUM], and are presented as fold-changes calculated by the 2−ΔΔC T method. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001.
(EPS)
Samples used for methylation and expression analyses.
(DOC)
Detailed comparison of concordant and common epigenes to other high-throughput data sets.
(DOC)
Human oligonucleotide sequences of primers used for pyrosequencing.
(DOC)
Human oligonucleotide sequences of primers used for real-time RT-PCR.
(DOC)
Acknowledgments
We are grateful to the participants in this study and their families, as well as to Stacey Sakowski, PhD, Lisa McLean, J. Simon Lunn, PhD, and Yu Hong for critical revisions, suggestions, and assistance in the submission of the manuscript. We thank Crystal Pacut for technical assistance and Aaron C. Goldstrohm, PhD for allowing us to use his automated Maxwell 16 System Instrument.
Funding Statement
This work was supported by the University of Michigan A. Alfred Taubman Medical Research Institute (http://www.taubmaninstitute.org); the Program for Neurology Research and Discovery (to E.L.F.); the National Institutes of Health (NIH) (R01-ES012933 to D.M.R.; AG020628, AG028268, and ES017885 to R.Y.; the Michigan Institute for Clinical & Health Research (UL1RR024986)); the American Diabetes Association Junior Faculty Award to B.C.C.; and the Juvenile Diabetes Research Foundation Postdoctoral Fellowship to J.H. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. de Carvalho M, Swash M (2011) Amyotrophic lateral sclerosis: an update. Curr Opin Neurol 24: 497–503. [DOI] [PubMed] [Google Scholar]
- 2. Ferraiuolo L, Kirby J, Grierson AJ, Sendtner M, Shaw PJ (2011) Molecular pathways of motor neuron injury in amyotrophic lateral sclerosis. Nat Rev Neurol 7: 616–630. [DOI] [PubMed] [Google Scholar]
- 3. Beers DR, Henkel JS, Xiao Q, Zhao W, Wang J, et al. (2006) Wild-type microglia extend survival in PU.1 knockout mice with familial amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A 103: 16021–16026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.ALSoD: ALS Online Genetic Database. Available: http://alsod.iop.kcl.ac.uk/. Accessed1 August 2012.
- 5. Siddique T, Ajroud-Driss S (2011) Familial amyotrophic lateral sclerosis, a historical perspective. Acta Myol 30: 117–120. [PMC free article] [PubMed] [Google Scholar]
- 6.van Blitterswijk M, van Es MA, Koppers M, van Rheenen W, Medic J, et al. (2012) VAPB and C9orf72 mutations in 1 familial amyotrophic lateral sclerosis patient. Neurobiol Aging 33: 2950 e2951–2954. [DOI] [PubMed]
- 7. Ahmed A, Wicklund MP (2011) Amyotrophic lateral sclerosis: what role does environment play? Neurol Clin 29: 689–711. [DOI] [PubMed] [Google Scholar]
- 8. Qureshi IA, Mehler MF (2011) Advances in epigenetics and epigenomics for neurodegenerative diseases. Curr Neurol Neurosci Rep 11: 464–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cortessis VK, Thomas DC, Levine AJ, Breton CV, Mack TM, et al. (2012) Environmental epigenetics: prospects for studying epigenetic mediation of exposure-response relationships. Hum Genet 131: 1565–1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dawson MA, Kouzarides T (2012) Cancer epigenetics: from mechanism to therapy. Cell 150: 12–27. [DOI] [PubMed] [Google Scholar]
- 11. Irier HA, Jin P (2012) Dynamics of DNA methylation in aging and Alzheimer’s disease. DNA Cell Biol 31 Suppl 1S42–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ito S, Shen L, Dai Q, Wu SC, Collins LB, et al. (2011) Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science 333: 1300–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, et al. (2009) Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 324: 930–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chen ZX, Riggs AD (2011) DNA methylation and demethylation in mammals. J Biol Chem 286: 18347–18353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chestnut BA, Chang Q, Price A, Lesuisse C, Wong M, et al. (2011) Epigenetic regulation of motor neuron cell death through DNA methylation. J Neurosci 31: 16619–16636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Calvo A, Moglia C, Balma M, Chio A (2010) Involvement of immune response in the pathogenesis of amyotrophic lateral sclerosis: a therapeutic opportunity? CNS Neurol Disord Drug Targets 9: 325–330. [DOI] [PubMed] [Google Scholar]
- 17. Hashioka S, Klegeris A, Schwab C, McGeer PL (2009) Interferon-gamma-dependent cytotoxic activation of human astrocytes and astrocytoma cells. Neurobiol Aging 30: 1924–1935. [DOI] [PubMed] [Google Scholar]
- 18. Makrantonaki E, Zouboulis CC (2007) The skin as a mirror of the aging process in the human organism–state of the art and results of the aging research in the German National Genome Research Network 2 (NGFN-2). Exp Gerontol 42: 879–886. [DOI] [PubMed] [Google Scholar]
- 19. Bae SI, Cheriyath V, Jacobs BS, Reu FJ, Borden EC (2008) Reversal of methylation silencing of Apo2L/TRAIL receptor 1 (DR4) expression overcomes resistance of SK-MEL-3 and SK-MEL-28 melanoma cells to interferons (IFNs) or Apo2L/TRAIL. Oncogene 27: 490–498. [DOI] [PubMed] [Google Scholar]
- 20. Crispi S, Sanzari E, Monfregola J, De Felice N, Fimiani G, et al. (2004) Characterization of the human STAT5A and STAT5B promoters: evidence of a positive and negative mechanism of transcriptional regulation. FEBS Lett 562: 27–34. [DOI] [PubMed] [Google Scholar]
- 21. DeBaun MR, Niemitz EL, Feinberg AP (2003) Association of in vitro fertilization with Beckwith-Wiedemann syndrome and epigenetic alterations of LIT1 and H19. Am J Hum Genet 72: 156–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kim JH, Kang HS, Kim TW, Kim SJ (2011) Differential methylation hybridization profiling identifies involvement of STAT1-mediated pathways in breast cancer. Int J Oncol 39: 955–963. [DOI] [PubMed] [Google Scholar]
- 23. Lee YM (2011) Control of RUNX3 by histone methyltransferases. J Cell Biochem 112: 394–400. [DOI] [PubMed] [Google Scholar]
- 24. Inoue K, Shiga T, Ito Y (2008) Runx transcription factors in neuronal development. Neural Dev 3: 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Marron TU, Guerini V, Rusmini P, Sau D, Brevini TA, et al. (2005) Androgen-induced neurite outgrowth is mediated by neuritin in motor neurones. J Neurochem 92: 10–20. [DOI] [PubMed] [Google Scholar]
- 26. Medina M, Marinescu RC, Overhauser J, Kosik KS (2000) Hemizygosity of delta-catenin (CTNND2) is associated with severe mental retardation in cri-du-chat syndrome. Genomics 63: 157–164. [DOI] [PubMed] [Google Scholar]
- 27. Ramos C, Robert B (2005) msh/Msx gene family in neural development. Trends Genet 21: 624–632. [DOI] [PubMed] [Google Scholar]
- 28. Teijido O, Casaroli-Marano R, Kharkovets T, Aguado F, Zorzano A, et al. (2007) Expression patterns of MLC1 protein in the central and peripheral nervous systems. Neurobiol Dis 26: 532–545. [DOI] [PubMed] [Google Scholar]
- 29. Wang Q, Lu QJ, Xiao B, Zheng Y, Wang XM (2011) Expressions of Axl and Tyro-3 receptors are under regulation of nerve growth factor and are involved in differentiation of PC12 cells. Neurosci Bull 27: 15–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zheng S, Chou AH, Jimenez AL, Khodadadi O, Son S, et al. (2002) The fetal and neonatal brain protein neuronatin protects PC12 cells against certain types of toxic insult. Brain Res Dev Brain Res 136: 101–110. [DOI] [PubMed] [Google Scholar]
- 31. Chen H, Dzitoyeva S, Manev H (2012) Effect of aging on 5-hydroxymethylcytosine in the mouse hippocampus. Restor Neurol Neurosci 30: 237–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Frijters R, Heupers B, van Beek P, Bouwhuis M, van Schaik R, et al. (2008) CoPub: a literature-based keyword enrichment tool for microarray data analysis. Nucleic Acids Res 36: W406–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nestor CE, Ottaviano R, Reddington J, Sproul D, Reinhardt D, et al. (2012) Tissue type is a major modifier of the 5-hydroxymethylcytosine content of human genes. Genome Res 22: 467–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. van den Hove DL, Chouliaras L, Rutten BP (2012) The role of 5-hydroxymethylcytosine in aging and Alzheimer’s disease: current status and prospects for future studies. Curr Alzheimer Res 9: 545–549. [DOI] [PubMed] [Google Scholar]
- 35. Yang HH, Hu N, Wang C, Ding T, Dunn BK, et al. (2010) Influence of genetic background and tissue types on global DNA methylation patterns. PLoS One 5: e9355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bollati V, Galimberti D, Pergoli L, Dalla Valle E, Barretta F, et al. (2011) DNA methylation in repetitive elements and Alzheimer disease. Brain Behav Immun 25: 1078–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Coppede F (2011) An overview of DNA repair in amyotrophic lateral sclerosis. ScientificWorldJournal 11: 1679–1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kisby GE, Milne J, Sweatt C (1997) Evidence of reduced DNA repair in amyotrophic lateral sclerosis brain tissue. Neuroreport 8: 1337–1340. [DOI] [PubMed] [Google Scholar]
- 39. Dangond F, Hwang D, Camelo S, Pasinelli P, Frosch MP, et al. (2004) Molecular signature of late-stage human ALS revealed by expression profiling of postmortem spinal cord gray matter. Physiol Genomics 16: 229–239. [DOI] [PubMed] [Google Scholar]
- 40. Ishigaki S, Niwa J, Ando Y, Yoshihara T, Sawada K, et al. (2002) Differentially expressed genes in sporadic amyotrophic lateral sclerosis spinal cords–screening by molecular indexing and subsequent cDNA microarray analysis. FEBS Lett 531: 354–358. [DOI] [PubMed] [Google Scholar]
- 41. Malaspina A, Kaushik N, de Belleroche J (2001) Differential expression of 14 genes in amyotrophic lateral sclerosis spinal cord detected using gridded cDNA arrays. J Neurochem 77: 132–145. [DOI] [PubMed] [Google Scholar]
- 42. Eklow C, Makrygiannakis D, Backdahl L, Padyukov L, Ulfgren AK, et al. (2008) Cellular distribution of the C-type II lectin dendritic cell immunoreceptor (DCIR) and its expression in the rheumatic joint: identification of a subpopulation of DCIR+ T cells. Ann Rheum Dis 67: 1742–1749. [DOI] [PubMed] [Google Scholar]
- 43. Miah SM, Hughes TL, Campbell KS (2008) KIR2DL4 differentially signals downstream functions in human NK cells through distinct structural modules. J Immunol 180: 2922–2932. [DOI] [PubMed] [Google Scholar]
- 44. Piccio L, Buonsanti C, Mariani M, Cella M, Gilfillan S, et al. (2007) Blockade of TREM-2 exacerbates experimental autoimmune encephalomyelitis. Eur J Immunol 37: 1290–1301. [DOI] [PubMed] [Google Scholar]
- 45. Butovsky O, Siddiqui S, Gabriely G, Lanser AJ, Dake B, et al. (2012) Modulating inflammatory monocytes with a unique microRNA gene signature ameliorates murine ALS. J Clin Invest 122: 3063–3087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zhang R, Gascon R, Miller RG, Gelinas DF, Mass J, et al. (2005) Evidence for systemic immune system alterations in sporadic amyotrophic lateral sclerosis (sALS). J Neuroimmunol 159: 215–224. [DOI] [PubMed] [Google Scholar]
- 47. Smith SA, Miller RG, Murphy JR, Ringel SP (1994) Treatment of ALS with high dose pulse cyclophosphamide. J Neurol Sci 124 Suppl: 84–87 [DOI] [PubMed] [Google Scholar]
- 48. Tanaka K, Kanno T, Yanagisawa Y, Yasutake K, Hadano S, et al. (2011) Bromocriptine methylate suppresses glial inflammation and moderates disease progression in a mouse model of amyotrophic lateral sclerosis. Exp Neurol 232: 41–52. [DOI] [PubMed] [Google Scholar]
- 49. Van Den Bosch L, Tilkin P, Lemmens G, Robberecht W (2002) Minocycline delays disease onset and mortality in a transgenic model of ALS. Neuroreport 13: 1067–1070. [DOI] [PubMed] [Google Scholar]
- 50. Werdelin L, Boysen G, Jensen TS, Mogensen P (1990) Immunosuppressive treatment of patients with amyotrophic lateral sclerosis. Acta Neurol Scand 82: 132–134. [DOI] [PubMed] [Google Scholar]
- 51. Shaw PJ (2005) Molecular and cellular pathways of neurodegeneration in motor neurone disease. J Neurol Neurosurg Psychiatry 76: 1046–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Valentine JS, Doucette PA, Zittin Potter S (2005) Copper-zinc superoxide dismutase and amyotrophic lateral sclerosis. Annu Rev Biochem 74: 563–593. [DOI] [PubMed] [Google Scholar]
- 53. Ramji DP, Vitelli A, Tronche F, Cortese R, Ciliberto G (1993) The two C/EBP isoforms, IL-6DBP/NF-IL6 and C/EBP delta/NF-IL6 beta, are induced by IL-6 to promote acute phase gene transcription via different mechanisms. Nucleic Acids Res 21: 289–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Valente T, Mancera P, Tusell JM, Serratosa J, Saura J (2012) C/EBPbeta expression in activated microglia in amyotrophic lateral sclerosis. Neurobiol Aging 33: 2186–2199. [DOI] [PubMed] [Google Scholar]
- 55. Deforges S, Branchu J, Biondi O, Grondard C, Pariset C, et al. (2009) Motoneuron survival is promoted by specific exercise in a mouse model of amyotrophic lateral sclerosis. J Physiol 587: 3561–3572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ting CH, Lin CW, Wen SL, Hsieh-Li HM, Li H (2007) Stat5 constitutive activation rescues defects in spinal muscular atrophy. Hum Mol Genet 16: 499–514. [DOI] [PubMed] [Google Scholar]
- 57. Xu J, Chen S, Chen H, Xiao Q, Hsu CY, et al. (2009) STAT5 mediates antiapoptotic effects of methylprednisolone on oligodendrocytes. J Neurosci 29: 2022–2026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Bacon C, Endris V, Rappold G (2009) Dynamic expression of the Slit-Robo GTPase activating protein genes during development of the murine nervous system. J Comp Neurol 513: 224–236. [DOI] [PubMed] [Google Scholar]
- 59. Pellikka M, Tanentzapf G, Pinto M, Smith C, McGlade CJ, et al. (2002) Crumbs, the Drosophila homologue of human CRB1/RP12, is essential for photoreceptor morphogenesis. Nature 416: 143–149. [DOI] [PubMed] [Google Scholar]
- 60. Jun G, Moncaster JA, Koutras C, Seshadri S, Buros J, et al. (2012) delta-Catenin is genetically and biologically associated with cortical cataract and future Alzheimer-related structural and functional brain changes. PLoS One 7: e43728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kudo LC, Parfenova L, Vi N, Lau K, Pomakian J, et al. (2010) Integrative gene-tissue microarray-based approach for identification of human disease biomarkers: application to amyotrophic lateral sclerosis. Hum Mol Genet 19: 3233–3253. [DOI] [PubMed] [Google Scholar]
- 62. Ratovitski T, Chighladze E, Waldron E, Hirschhorn RR, Ross CA (2011) Cysteine proteases bleomycin hydrolase and cathepsin Z mediate N-terminal proteolysis and toxicity of mutant huntingtin. J Biol Chem 286: 12578–12589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Gonzalez de Aguilar JL, Niederhauser-Wiederkehr C, Halter B, De Tapia M, Di Scala F, et al. (2008) Gene profiling of skeletal muscle in an amyotrophic lateral sclerosis mouse model. Physiol Genomics 32: 207–218. [DOI] [PubMed] [Google Scholar]
- 64. Offen D, Barhum Y, Melamed E, Embacher N, Schindler C, et al. (2009) Spinal cord mRNA profile in patients with ALS: comparison with transgenic mice expressing the human SOD-1 mutant. J Mol Neurosci 38: 85–93. [DOI] [PubMed] [Google Scholar]
- 65. Maruyama H, Morino H, Ito H, Izumi Y, Kato H, et al. (2010) Mutations of optineurin in amyotrophic lateral sclerosis. Nature 465: 223–226. [DOI] [PubMed] [Google Scholar]
- 66. Morahan JM, Yu B, Trent RJ, Pamphlett R (2007) Are metallothionein genes silenced in ALS? Toxicol Lett 168: 83–87. [DOI] [PubMed] [Google Scholar]
- 67. Oates N, Pamphlett R (2007) An epigenetic analysis of SOD1 and VEGF in ALS. Amyotroph Lateral Scler 8: 83–86. [DOI] [PubMed] [Google Scholar]
- 68. Jiang YM, Yamamoto M, Kobayashi Y, Yoshihara T, Liang Y, et al. (2005) Gene expression profile of spinal motor neurons in sporadic amyotrophic lateral sclerosis. Ann Neurol 57: 236–251. [DOI] [PubMed] [Google Scholar]
- 69. Wang XS, Simmons Z, Liu W, Boyer PJ, Connor JR (2006) Differential expression of genes in amyotrophic lateral sclerosis revealed by profiling the post mortem cortex. Amyotroph Lateral Scler 7: 201–210. [DOI] [PubMed] [Google Scholar]
- 70. Lederer CW, Torrisi A, Pantelidou M, Santama N, Cavallaro S (2007) Pathways and genes differentially expressed in the motor cortex of patients with sporadic amyotrophic lateral sclerosis. BMC Genomics 8: 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Morahan JM, Yu B, Trent RJ, Pamphlett R (2009) A genome-wide analysis of brain DNA methylation identifies new candidate genes for sporadic amyotrophic lateral sclerosis. Amyotroph Lateral Scler 10: 418–429. [DOI] [PubMed] [Google Scholar]
- 72. Kirby J, Ning K, Ferraiuolo L, Heath PR, Ismail A, et al. (2011) Phosphatase and tensin homologue/protein kinase B pathway linked to motor neuron survival in human superoxide dismutase 1-related amyotrophic lateral sclerosis. Brain 134: 506–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Lokk K, Vooder T, Kolde R, Valk K, Vosa U, et al. (2012) Methylation markers of early-stage non-small cell lung cancer. PLoS One 7: e39813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Rishi V, Bhattacharya P, Chatterjee R, Rozenberg J, Zhao J, et al. (2010) CpG methylation of half-CRE sequences creates C/EBPalpha binding sites that activate some tissue-specific genes. Proc Natl Acad Sci U S A 107: 20311–20316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Shenker N, Flanagan JM (2012) Intragenic DNA methylation: implications of this epigenetic mechanism for cancer research. Br J Cancer 106: 248–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Szulwach KE, Li X, Smrt RD, Li Y, Luo Y, et al. (2010) Cross talk between microRNA and epigenetic regulation in adult neurogenesis. J Cell Biol 189: 127–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Connolly R, Stearns V (2012) Epigenetics as a Therapeutic Target in Breast Cancer. J Mammary Gland Biol Neoplasia. [DOI] [PMC free article] [PubMed]
- 78. Gray SG, Dangond F (2006) Rationale for the use of histone deacetylase inhibitors as a dual therapeutic modality in multiple sclerosis. Epigenetics 1: 67–75. [DOI] [PubMed] [Google Scholar]
- 79.Illumina I (2008) GenomeStudio Gene Expression Module v1.0 User Guide. Illumina. 97–107.
- 80. Hur J, Sullivan KA, Pande M, Hong Y, Sima AA, et al. (2011) The identification of gene expression profiles associated with progression of human diabetic neuropathy. Brain 134: 3222–3235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Reich M, Liefeld T, Gould J, Lerner J, Tamayo P, et al. (2006) GenePattern 2.0. Nat Genet 38: 500–501. [DOI] [PubMed] [Google Scholar]
- 82. Dai M, Wang P, Boyd AD, Kostov G, Athey B, et al. (2005) Evolving gene/transcript definitions significantly alter the interpretation of GeneChip data. Nucleic Acids Res 33: e175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Gautier L, Cope L, Bolstad BM, Irizarry RA (2004) affy–analysis of Affymetrix GeneChip data at the probe level. Bioinformatics 20: 307–315. [DOI] [PubMed] [Google Scholar]
- 84. Bolstad BM, Irizarry RA, Astrand M, Speed TP (2003) A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics 19: 185–193. [DOI] [PubMed] [Google Scholar]
- 85. Irizarry RA, Bolstad BM, Collin F, Cope LM, Hobbs B, et al. (2003) Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res 31: e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Sartor MA, Tomlinson CR, Wesselkamper SC, Sivaganesan S, Leikauf GD, et al. (2006) Intensity-based hierarchical Bayes method improves testing for differentially expressed genes in microarray experiments. BMC Bioinformatics 7: 538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Huang da W, Sherman BT, Lempicki RA (2009) Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 37: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Huang da W, Sherman BT, Lempicki RA (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4: 44–57. [DOI] [PubMed] [Google Scholar]
- 89. Hur J, Schuyler AD, States DJ, Feldman EL (2009) SciMiner: web-based literature mining tool for target identification and functional enrichment analysis. Bioinformatics 25: 838–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Hur J, Sullivan KA, Schuyler AD, Hong Y, Pande M, et al. (2010) Literature-based discovery of diabetes- and ROS-related targets. BMC Med Genomics 3: 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Cartharius K, Frech K, Grote K, Klocke B, Haltmeier M, et al. (2005) MatInspector and beyond: promoter analysis based on transcription factor binding sites. Bioinformatics 21: 2933–2942. [DOI] [PubMed] [Google Scholar]
- 92. McGeer PL, McGeer EG (2002) Inflammatory processes in amyotrophic lateral sclerosis. Muscle Nerve 26: 459–470. [DOI] [PubMed] [Google Scholar]
- 93. Estecio MR, Gharibyan V, Shen L, Ibrahim AE, Doshi K, et al. (2007) LINE-1 hypomethylation in cancer is highly variable and inversely correlated with microsatellite instability. PLoS One 2: e399. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Validation of HM27K arrays using pyrosequencing. Amplicons to the promoter regions identified by HM27K of cytokines CXCL12, CMTM3, CMTM2, C/EBPB, and STAT5A were generated using bisulfite-converted genomic DNA from human postmortem spinal cord and used as templates for pyrosequencing (ALS n = 11; Ctrl n = 11) Results are presented as mean of percent methylation of all CpG sites within the area tested on each gene (A) or as percent methylation of individual sites for STAT5A; site 2 was identified with the HM27K (B). Results are presented as mean ± SEM and a two-sample equal variance t-test was used. *p<0.05, **p<0.01 compared to control group (Ctrl).
(EPS)
RT-PCR confirmation of concordant epigenes in spinal cord. Total RNA was extracted from postmortem human spinal cord tissue used for the methylation analysis from sALS subjects (n = 8–11) and controls (n = 8–11) and subjected to RT-PCR. Results were normalized to housekeeping genes [TATA-box Binding Protein (TBP) for CTSZ, FCER1G, TREM2, NRN1 and NNAT; ribosomal 18S subunit for CHI3L2, H19, PEG10, and LUM], and are presented as fold-changes calculated by the 2−ΔΔC T method. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001.
(EPS)
Samples used for methylation and expression analyses.
(DOC)
Detailed comparison of concordant and common epigenes to other high-throughput data sets.
(DOC)
Human oligonucleotide sequences of primers used for pyrosequencing.
(DOC)
Human oligonucleotide sequences of primers used for real-time RT-PCR.
(DOC)