

Abstract
Establishing if species contractions were the result of natural phenomena or human induced landscape changes is essential for managing natural populations. Fishers (Martes pennanti) in California occur in two geographically and genetically isolated populations in the northwestern mountains and southern Sierra Nevada. Their isolation is hypothesized to have resulted from a decline in abundance and distribution associated with European settlement in the 1800s. However, there is little evidence to establish that fisher occupied the area between the two extant populations at that time. We analyzed 10 microsatellite loci from 275 contemporary and 21 historical fisher samples (1880–1920) to evaluate the demographic history of fisher in California. We did not find any evidence of a recent (post-European) bottleneck in the northwestern population. In the southern Sierra Nevada, genetic subdivision within the population strongly influenced bottleneck tests. After accounting for genetic subdivision, we found a bottleneck signal only in the northern and central portions of the southern Sierra Nevada, indicating that the southernmost tip of these mountains may have acted as a refugium for fisher during the anthropogenic changes of the late 19th and early 20th centuries. Using a coalescent-based Bayesian analysis, we detected a 90% decline in effective population size and dated the time of decline to over a thousand years ago. We hypothesize that fisher distribution in California contracted to the two current population areas pre-European settlement, and that portions of the southern Sierra Nevada subsequently experienced another more recent bottleneck post-European settlement.
Introduction
Over the past 100 years there has been a marked reduction in many species geographic ranges. For rare or hard to observe species, it is often unclear if their absence is a response to a changing landscape, or if they have been absent from an area for an extended period of time. If they were considered present early during the last epoch, but are now unable to be detected, this is seen as a natural range contraction [1]. On the other hand, if they were considered present until the last century, but are now unable to be detected, this is often viewed as caused by human induced disturbances. Establishing if contractions of species were the result of natural causes or human-induced landscape changes is essential for managing natural populations. Mistakes associated with misidentifying the geographic range of a species and misattributing declines in geographic range can have large effects on the allocation of scarce conservation resources [2].
Traditionally, the historical distribution of a species has been based on accounts of explorers, naturalists, and indigenous peoples that are verified by specimens preserved in museum collections. Recently, technological and laboratory advances in molecular genetics have created the ability to extract DNA from historical specimens and examine the population genetic signals obtained, providing a new tool by which we can test ideas proposed by these early naturalists [3], [4]. Historical and contemporary genetic information can provide insight into the nature of population expansions or declines [5], [6], the loss of genetic diversity [7], [8], temporal changes in population connectivity [9], or the historical range of a species [10], [11].
Prior to European settlement, fishers (Martes pennanti) were distributed widely in both Canada and the northern U.S. forests [12]. In the late 1800's and early 1900's, fisher populations dramatically declined due to a combination of fur trapping, logging, and predator control and by the early 1900's were extirpated from large portions of their historic range [13]. Reintroductions and expansions from refugia populations have been successful in reestablishing fisher populations in the eastern and Rocky Mountain states [14]–[18]. However, West Coast populations have not experienced the same degree of recovery. There are 5 geographically disjunct fisher populations present on the West Coast: two native populations in California [19], [20], a reintroduced population established in the 1950's in Oregon, and two recently reintroduced populations (one on the Olympic Peninsula in Washington State and one in California [21], [22]).
The two native fisher populations in California are geographically and genetically isolated [19], [23], [24]. Conservation concerns are particularly acute for fisher in the southern Sierra Nevada Mountains because its population size is estimated at less than 300 adults [25]. The majority of information about the history of fisher in California comes from the work of the naturalist Joseph Grinnell. Grinnell et al. [26] used information from extensive surveys, collecting expeditions, trapping records, and local knowledge from approximately 1910–1930 to create distribution maps for 21 species of carnivores. Grinnell's range maps show the historical fisher range as continuous from the northwestern Klamath and Siskiyou Mountains to the southern tip of the Sierra Nevada (Fig. 1).
Figure 1. Historical range map for fisher in California.

Fisher locations used by Grinnell et al. [26] to document the distribution of fisher in California. Locations are based primarily on reports of trappers and collecting expeditions from 1919–1924. Grinnell wrote that “spots [black dots] indicate, almost all of them with certainty, the locality of capture; probably some indicate the residence of post office or trapper”. The outlined area is the Grinnell et al. [26] assessment of the range of fisher in California from ∼1850–1925.
Fisher populations in California are thought to have declined precipitously in both abundance and distribution over the last 150 years due to habitat alteration and fur trapping associated with the European settlement of California beginning with the gold rush in 1848 [27]. Currently, the two areas that maintain native populations of fisher in California are separated by a 420 km gap, which is more than four times the maximum dispersal distance of fisher [19], [27]. The reason for this gap is not well understood. The majority of habitat in this area is contiguously forested and appears, at least superficially, to be suitable for fisher occupancy. Grinnell's range map shows only a few records of fisher in the central Sierra Nevada and none in the northern Sierra Nevada (Fig. 1), but despite these facts this gap is considered part of the historical range of the species [26].
The accepted hypothesis for the lack of records in the gap area is that the northern and central Sierra Nevada had experienced a greater degree of anthropogenic change at the time of the Grinnell surveys than the southern Sierra Nevada and that the species was already extirpated from the gap by the early 1900's [27]. The central and northern Sierra was the main area of human development as a result of the gold rush. Yet, in a study of the history of forest conditions in the Sierra Nevada, McKelvey and Johnston [28] found that due to transportation limitations, logging at the turn of the century was relatively limited in the central and northern Sierra. At this time even the most heavily affected National Forest in this area still had 50% virgin forest and therefore, likely retained areas of large trees that are associated with fisher habitat in California [29]–[31]. Based on such information, it is unclear why fisher would have been completely extirpated from the gap prior to Grinnell's surveys.
An alternative hypothesis is that this distributional gap may not be the result of recent human influences but rather is a historical discontinuity in fisher distribution that existed prior to the European settlement of California. Fishers are thought to have colonized the West Coast of the United States in a relatively recent range expansion from British Columbia southward in a series of stepwise founder events during the mid to late Holocene [12], [23], [32]. Evidence of an early peninsular expansion is found in the gradient of genetic diversity decreasing from north to south down the West Coast [23], and the existence of a shared haplotype between British Columbia and a historical sample from northwestern California [32]. However, evidence indicates there has been little gene flow between the two regions in the time since colonization with high genetic divergence in nuclear DNA (F ST = 0.48–0.60) and the absence of a shared mtDNA haplotype between northwestern California and the southern Sierra Nevada [23], [24].
There are important conservation concerns regarding the southern Sierra Nevada fisher population's risk of extinction stemming from its small population size, isolation, and low genetic diversity. Determining whether the isolation of fisher in the southern Sierra Nevada has occurred recently (within the last 150 years), or if the population has been persisting in long-term isolation, are important alternative hypotheses that need to be distinguished to inform future conservation decisions. Discussions of how to manage this population to support long-term persistence have included the potential need for translocations to augment populations or reintroductions into the current gap region to re-establish connectivity [33], [34]. If population decline and isolation occurred recently then potential risk from inbreeding depression due to small population size may be an important consideration for the southern Sierra Nevada and aggressive measures to restore genetic connectivity may in fact be prudent. Conversely, detection of a more ancient timeline for isolation would indicate the potential for significant local adaptations within the population and that creating genetic connectivity with northwestern California fishers could actually trigger a reduction in fitness due to outbreeding depression [35], [36].
Recent research has attempted to address the historical continuity of fisher populations in California using mtDNA. Knaus et al. [24] sequenced the entire mtDNA genome for 40 fisher samples and found the southern Sierra Nevada to be fixed for a single haplotype that is different from the closest haplotype in northwestern California by 9 base-pair substitutions. The absence of a shared mtDNA haplotype between northwestern California and the southern Sierra Nevada and the amount of genetic differentiation between haplotypes indicates long term isolation. Using a molecular clock approach, they estimated the divergence between these two populations occurred thousands of years ago [24].
While the results of Knaus et al. [24] are striking, mtDNA is maternally transmitted and consequently only provides insight into female mediated gene flow. This may be especially problematic for species such as fisher that exhibit female philopatry where most of the large movements are made by males [37]. This would result in primarily male mediated gene flow across long distances. As nuclear DNA is bipaternally inherited, it may show different genetic signals from mtDNA that reflect the influence of males on connectivity. Numerous studies have shown discord between estimates of divergence from mtDNA versus nuclear DNA and emphasized the importance of analyzing both mtDNA and nuclear DNA prior to making conservation decisions [38]–[40].
Our objective is to use nuclear DNA to distinguish between the alternate hypotheses that the geographic isolation of the two California fisher populations occurred before or after the European settlement of California. We also wish to more precisely date this divergence. The hypothesis that fisher decline and isolation in California occurred prior to 1850 would be supported by lack of evidence of a recent bottleneck and contraction in population size greater than 160 years ago. Conversely, if the hypothesis that isolation occurred after 1850 is correct, we would expect to see evidence of a recent population bottleneck and a contraction in population size within the last ∼160 years. Evidence of post-European isolation would be at odds with mtDNA analyses [24] and indicate male mediated gene flow between California fisher populations. In a broader sense, this research is also aimed at showing the importance of understanding historical biogeographic patterns to better understand and manage contemporary patterns of species on the landscape.
Materials and Methods
Ethics Statement
All necessary permits were obtained for the described field studies. These included a Scientific Research and Collecting Permit from the U.S. Department of the Interior, National Park Service (SEKI-2008-SCI-0014).
Samples
We obtained both historical (H) and contemporary (C) genetic samples from the extant range of fisher in California which includes one area in northwestern California (NW) and a second area in the southern Sierra Nevada (SSN) (Fig. 2). The NW and SSN populations were defined a priori based on previous research that indicated that these populations are geographically isolated due to an unoccupied 420 km gap between them [19], [27], as well as genetically isolated [23], [24]. We genotyped 127 individuals from hair samples collected in the SSNC through the U.S. Forest Service Sierra Nevada Carnivore Monitoring Program [41]. In the NWC we obtained genotypes from 148 individuals based on hair, scat, and tissue samples collected in collaboration with a number of existing research projects in the region. Genetic samples from both regions were collected from 2006–2009. Historical samples were located by searching databases of museum collections. We found 41 fisher specimens from 1884–1920 in the collections of the Smithsonian National Museum of Natural History and the Museum of Vertebrate Zoology at the University of California, Berkeley (Table S1). We collected maxilloturbinal bones from inside the nasal cavity to maximize the probability of obtaining high quality DNA while minimizing damage to specimens [42], [43]. We also collected tissue from pelts, bone fragments, or muscle when available. In total, 17 historical specimens were obtained from the NWH and 24 from the SSNH. We did not find any historical fisher specimens from the current gap in fisher distribution.
Figure 2. Sample locations.

Locations of the historical (H) and contemporary (C) genetic samples from the northwestern mountains (NW) and southern Sierra Nevada (SSN) of California. Sample size is as follows: NWH n = 5, SSNH n = 16, NWC n = 148, SSNC n = 127. Grinnell's assumed historical range as adapted by Davis et al. [115] is shown in gray.
Laboratory Analysis
We extracted DNA from museum specimens in a separate laboratory used exclusively for the extraction and processing of genetic material from museum specimens following recommended ancient DNA protocols [42], [44]. We analyzed the samples at 10 microsatellite loci. MP0059, MP0144, MP0175, MP0197, MP0200, and MP0247 were developed from tissue samples from the SSN [45]. Loci MA1 [46], GGU101, GGU216 [47], and LUT733 [48], were developed in other mustelid species [marten (Martes americana), wolverine (Gulo gulo), and otter (Lutra lutra), respectively].
The quality and quantity of DNA obtained from historical and non-invasive samples can vary considerably because of age and different methods of preservation and storage. The potential for degraded or low quantity DNA increases the likelihood of genotyping errors such as allelic dropout or false alleles [49]. To address this potential for error, we ran samples a minimum of three times per locus and accepted genetic data only if the samples produced consistent genotype scores [50], [51]. If the genotype differed in one or more of these amplifications, we conducted an additional round of 3 amplifications. If multiple inconsistencies were found in the genotype at a locus we removed that sample from the analysis. We also checked for genotyping errors using the software DROPOUT [52].
Statistical analyses
We tested microsatellite genotypes for departures from Hardy-Weinberg proportions at each locus and gametic disequilibrium for each pair of loci using Fisher's exact test in Genepop 4.0 [53], [54]. We also used Genepop 4.0 to calculate expected heterozygosity (H E), proportional excess of homozygotes (F IS), F ST [55], R ST [56], and conduct tests for genetic differentiation between sample groups. The amount of genetic diversity present in the sample groups was compared using paired t-tests of arcsine-transformed H E, and A R [57]. We used sequential Bonferroni corrections to correct for multiple comparisons when assessing statistical significance [58].
Detecting bottlenecks
We used three methods to determine whether fisher in California had experienced a recent reduction in population size. We first tested for heterozygosity excess which is characteristic of bottlenecked populations using BOTTLENECK 1.2.02 [59]. This heterozygosity excess exists because rare alleles are lost more rapidly during a bottleneck but have little impact on heterozygosity [60]. Heterozygosity excess is transient and will only persist for 0.2 – 4Ne generations after the bottleneck. The average expected heterozygosity at mutation-drift equilibrium was calculated using 5000 replications assuming a two-phase mutational model. We conducted analyses with both 5% and 20% of mutations set as multistep mutations in the two-phase model with a variance of 12 to encompass the range of multistep mutations observed in natural populations [61]. The observed heterozygosity was then tested against the equilibrium expected heterozygosity using the Wilcoxon signed-rank test. We also conducted the test excluding all loci that were out of Hardy-Weinberg, as such loci can create bias, but doing so did not significantly change the results.
Second, we also used BOTTLENECK to test for a shift in the mode of the distribution of allele frequencies. This mode shift distortion is transient and can only be detected for a few dozen generations. Luikart et al. [62] found using simulations that the graphical mode shift method is likely (P>.80) to detect a bottleneck of up to 20 breeding individuals using 8–10 microsatellite loci. The mode shift test could not be applied to the historical samples because at least 30 individuals are needed to avoid high type 1 error rates.
The third method used detects reductions in effective population size (N e) using the M-Ratio which is defined as M = k/r where k is the total number of alleles and r is the range in allele size [63]. Because a bottleneck causes a greater reduction in the number of alleles than in the range of allele sizes, M is smaller in reduced populations. Garza and Williamson [63] found that a reduction in population size can be detected using M for 125 generations if the population rebounded quickly in size or 500 generations if the population remained reduced. We used the software M_P_Val to calculate M and the software M_Critical to determine the cutoff value for statistical significance [63]. We set model parameters at 90% single-step mutations and 10% multi-step mutations (ps) and the average size of multistep mutation (Δg) of 3.5 with the mutation rate μ held constant at 5×10−4. In this model θ = 4N eμ so if μ is held constant different values of θ are representative of different starting (pre-decline) N e. As the equilibrium N e for fisher in California is not known, we calculated M and M-Critical values for four different values of θ (1, 2, 5, and 10) which represent a wide range of pre-decline N e (500, 1000, 2500, and 5000 respectively).
The presence of unaccounted for genetic subdivision has the potential to bias bottleneck tests [64]. While genetic subdivision has not been previously detected in the NWC population, past research has shown significant subdivision in the SSNC [23]. To assess the influence of this subdivision, we divided the SSNC into three genetic groups and assessed the influence of this on the bottleneck tests. The subdivisions between demes in the SSNC roughly correspond to the areas north of the Kings River (North), between the Kings River and Middle Fork of the North Fork of the Tule River (Central), south of the Middle Fork of the North Fork Tule River (South) (Fig. 3). Previous research on fisher populations in southern Ontario has found rivers to be a major barrier to genetic connectivity [18], [65]. These subdivision boundaries are also supported by data from a recent population genetic analysis of the SSNC showing moderate subdivision (F ST 0.05–0.13) between these areas (J.M. Tucker unpublished data).
Figure 3. Population subdivision in the southern Sierra Nevada.
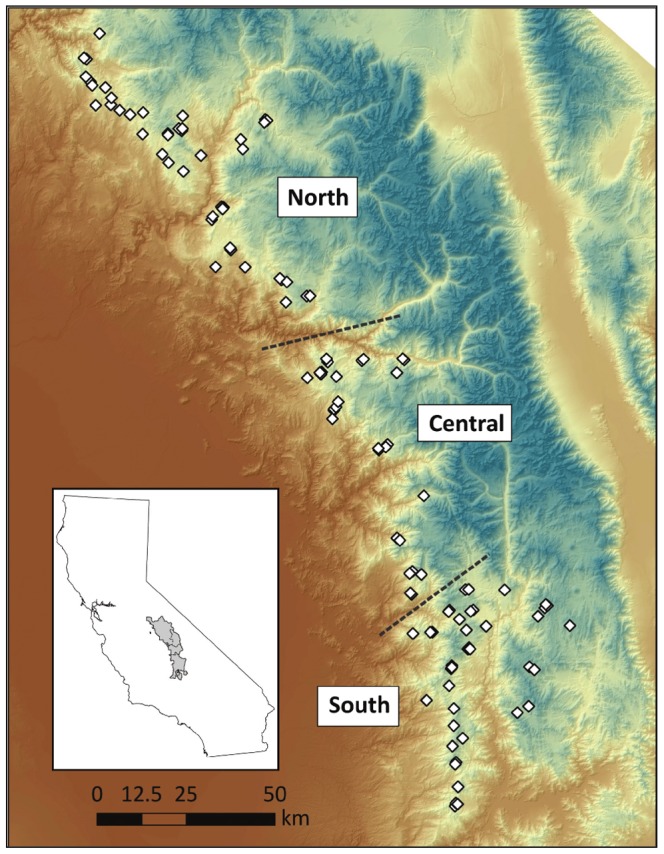
Approximate location of population subdivisions used in bottleneck analyses within the contemporary southern Sierra Nevada.
Demographic history models
We employed a coalescent-based Bayesian analysis to assess the most recent major change in N e and to estimate the date of the change. This model assumes that an ancestral N e (N 1) changed to the current N e (N 0), at a time T generations ago [66], [67]. This model uses a stepwise mutation model and assumes a mutation rate scaled in terms of the current populations size such that that θ = 2N 0μ, where μ is the per locus mutation rate. While this method does employ a strict stepwise mutation model, it has been found to be robust to moderate departures created by the presence of multistep mutations [68]. The method then estimates the posterior distributions of N 1, N 0, T, and θ that describe the genealogical and demographic history of the sample, assuming either linear or exponential size change. Prior distributions for N 1, N 0, T, and θ are assumed to be log normal with their means and standard deviations drawn from hyperprior distributions truncated at zero. We conducted the analysis using MSVAR 1.3 [67] which uses Markov Chain Monte Carlo (MCMC) simulations to estimate the posterior distribution of each parameter.
We conducted 6 independent simulations of the model varying the prior and hyperprior distributions with a range of biologically realistic distribution values to examine their effect on the posterior distributions. These variations of the priors had little effect on the posterior distribution of the models so prior distributions for all other analyses were set to the parameters of simulation 1 (Table S2). To check for the convergence of model we conducted five replications of the simulations for each data set. Each simulation was performed for 2×109 iterations with parameter values recorded every 1×105 iterations resulting in 20,000 records.
We removed the first 10% of data from each chain as burn-in and assessed chain convergence using the Brooks, Gelman, and Rubin Convergence Diagnostic test [69], [70]. We conducted convergence diagnostics in R version 2.11.1 [71] using the package BOA version 1.1.7 [72]. The test statistic is a multivariate potential scale reduction factor (MPSRF) that assesses the convergence of a set of parameters simultaneously. The MPSRF value for all parameters was ∼1.0 indicating acceptable chain convergence. We then combined the last 50% of the data from each chain (10,000 records/chain, 50,000 total records) and calculated the mode and 90% highest posterior densities (HPD) of the posterior distributions of each parameter using the R-package Locfit 1.5–6 [73]. We evaluated the strength of evidence for population expansion versus decline by calculating the Bayes factor for each of the models [74], [75] as described by Storz and Beaumont [67]. The Bayes factor indicates the following levels of support for the model; BF<0.33 = false detection of contraction/expansion, 0.33–3 = no support, 3–10 = substantial support, and ≥10 = strong support [74].
While the generation time (average age of reproduction) for fisher has not been well studied, the average age of first reproduction is estimated at 2–3 years, with high reproductive rates documented in 5–7 year old females [76], and successful reproduction found in females as old as 10 years (C. Thompson personal communication). We used a generation interval of five years. Parameter estimates of T can easily be adjusted for different generation times by multiplying accordingly. We ran the simulations for all data sets using both the exponential and linear models.
Results
We successfully obtained genotypes at a minimum of seven loci for 127 individuals in the SSNC, 148 individuals in the NWC, 16 individuals from the SSNH, and five individuals from the NWH (Table 1). The dates of the historical samples that successfully yielded microsatellite genotypes ranged from 1884–1920, which represents the overall timeframe of available historical samples (Table S1). Nine of the 10 microsatellite loci were polymorphic in all samples. The exception was the MA1 locus which was monomorphic in the NWC. Tests for Hardy-Weinberg proportions showed deviation from expected values at MP200 and MP59 in the SSNC. However, these deviations are non-significant after accounting for genetic population structure. We also found MP200 deviated in the SSNH to have a homozygote excess compared to expected Hardy-Weinberg proportions. To assess the influence of this locus, we conducted SSNH analyses both with and without this locus but did not find any notable difference in results.
Table 1. Estimates of genetic diversity for the northwest (NW) and southern Sierra Nevada (SSN) at 10 microsatellite loci: sample size (n), expected heterozygosity (H E), proportional excess of homozygotes (F IS), mean number of alleles (A), and allelic richness (A R).
| n | H E | F IS | A | A R | |
| NW-Historical | 5 | 0.635 | 0.028 | 3.60 | 3.34 |
| NW-Contemporary | 148 | 0.431 | 0.028 | 3.75 | 2.17 |
| SSN-Historical | 16 | 0.590 | 0.046** | 3.60 | 2.81 |
| SSN-Contemporary | 127 | 0.565 | 0.101*** | 3.50 | 2.51 |
Allelic richness is based on a minimum size of 4 individuals which represents the number of individuals with genotypes at all 10 loci in the historical NW sample.
P<0.05;
P<0.001.
While we did not find any evidence for departure from Hardy-Weinberg proportions at individual loci, we did find some important patterns over all loci within each sample group. F IS values were small and statistically insignificant in both the NWH and NWC samples, but had significant p-values in both the SSNH and SSNC. Most notably, the SSNC showed a large deficit of heterozygotes (F IS = 0.101, p<0.001) (Table 1). This is indicative of the potential presence of the Wahlund effect [77] in the SSN, in which unaccounted for population subdivision in a sample generates a deficit of heterozygotes relative to expected Hardy-Weinberg proportions.
Tests for gametic disequilibrium did not find any strong associations between loci. After correcting for multiple comparisons statistically significant gametic disequilibrium was found between two pairs of loci in the SSNC (MP197/MP200, and MA1/MP144), one pair in the NWC (MP175/LUT733), and none in either historical sample group. No pairs of loci were consistently significant across sample groups indicating that the loci used were assorting independently.
We did not find any difference in the amount of genetic diversity within sample groups with paired t-tests showing no significant differences in H E, or A R. However, all metrics of genetic diversity were lowest in the NWC (Table 1). H E was markedly lower in the NWC (0.431) compared to all other samples (0.57–0.64). Allelic richness (A R) was higher in both historical samples (NWH = 3.34, SSNH = 2.81) than in either contemporary sample (NWC = 2.17, SSNC = 2.51). Samples in the NWC were monomorphic at locus MA1, and have extremely low diversity at the MP200 locus (2 of 3 alleles at 1% frequency). When these two loci were removed from calculations the NWC H E increases to 0.54 which is similar to the value for the other sample groups at 8 loci (NWH = 0.55, SSNH = 0.60, SSNC = 0.55) and A R in the two contemporary populations becomes equal (SSNC(8loci) = NWC(8loci) = 2.46).
We found each group to be significantly genetically different. Tests for genic differentiation between sample groups were significant at P<0.001. F ST and R ST values were moderate between historical sample groups (NWH/SSNH: F ST = 0.10, R ST = 0.20) but increased over time with contemporary samples showing increased divergence (SSNC/NWC: F ST = 0.37, R ST = 0.58). We also found temporal divergence over time with moderate F ST values between temporally spaced samples in the same geographic location (SSNH/SSNC = 0.17, NWH/NWC = 0.20) (Table 2). R ST values were considerably higher than F ST values indicating that when variation in allele length is accounted for genetic divergence between samples groups is even greater.
Table 2. Pairwise comparisons of genetic differentiation between samples with R ST above the diagonal and F ST below the diagonal.
| NWH | NWC | SSNH | SSNC | |
| NWH | – | 0.321 | 0.195 | 0.500 |
| NWC | 0.198 | – | 0.363 | 0.581 |
| SSNH | 0.098 | 0.291 | – | 0.265 |
| SSNC | 0.208 | 0.374 | 0.170 | – |
H denotes historical samples and C denotes contemporary samples. All pairwise comparisons shown in the table are significant at P<0.01.
Population bottlenecks
We did not find any signal of a recent population bottleneck for either the historical or contemporary NW samples. Both NW samples had non-significant results for the Wilcoxon heterozygosity excess test and the NWC was also negative for the shifted mode test. Bottleneck tests for the SSN were mixed. For the SSNC the heterozygosity excess test was statistically significant regardless of the proportion of multistep mutations in the two-phase model (5%: p = 0.04, 20%: p<0.001), but showed no evidence of a shifted mode. The SSNH was significant at α = 0.05 for heterozygosity excess but only when using 20% multistep mutations (5%: p = 0.10, 20%: p = 0.05). We found no evidence of a population bottleneck for any sample group using the M-Ratio method (Table 3).
Table 3. Results of BOTTLENECK tests including the p values for the Wilcoxon heterozygosity (H E) excess test with two different proportions of multistep mutations in the two phase model (TPM), shifted mode test, and M-Ratio value and M-Critical values.
| n | H E Excess TPM 20% | H E Excess TPM 5% | Shifted Mode | M-Ratio | M-Critical θ = 1 | M-Critical θ = 10 | |
| NWH | 5 | 0.19 | 0.22 | - | 0.92 | 0.71 | 0.55 |
| NWC | 148 | 0.08 | 0.22 | No | 0.91 | 0.77 | 0.71 |
| SSNH | 16 | 0.05 | 0.10 | - | 0.87 | 0.78 | 0.64 |
| SSNC | 127 | 0.00 | 0.04 | No | 0.89 | 0.78 | 0.72 |
| SSNC – North | 44 | <0.001 | <0.001 | Yes | 0.83 | 0.78 | 0.69 |
| SSNC – Central | 32 | <0.001 | <0.001 | Yes | 0.82 | 0.78 | 0.68 |
| SSNC – South | 51 | 0.08 | 0.19 | No | 0.85 | 0.78 | 0.70 |
θ = 1 represents an initial (pre-decline) N e of 500 and θ = 10 an N e of 5000. M-Ratio values that fall below the M-Critical value are considered statistically significant at α = 0.05. Results incorporating population structure in the SSNC are shown on the last 3 lines where H denotes historical samples and C denotes contemporary samples.
The mixed results in the SSNC were clarified after accounting for genetic population subdivision. Both the North and Central SSNC samples showed strong evidence of a recent bottleneck with significant heterozygosity excess tests (p<0.001) and shifted modes. The South SSNC sample showed no evidence of a recent bottleneck in either the heterozygosity excess or shifted mode tests. After accounting for populations subdivision there was still no evidence of a bottleneck in the SSNC using the M-Ratio method (Table 3).
Demographic history
We were unable to obtain consistent results for the demographic change analysis in the NWH due to small sample size (n = 5) and therefore, did not include these results in our analyses. However, results from the other three sample groups consistently indicate that there was a large population decline with current N e estimates over 90% lower than the estimates of the ancestral N e. These results were consistent across a variety of prior distributions and both demographic models (exponential and linear). Bayes factor values were >10 for all models indicating strong evidence for a population decline (Table 4).
Table 4. The mode and 90% highest posterior density (in parentheses) of the posterior distributions for the Storz and Beaumont [67] models.
| Sample | BF | Scale | N 0 | N 1 | Time (T) |
| Historical-SSN | 10.9 | Exp | 154 (1–2160) | 1862 (454–7952) | 442 (96–25249) |
| Historical-SSN | 13.5 | Linear | 102 (1–1993) | 1922 (457–7838) | 1054 (109–61941) |
| Contemporary-SSN | 36.1 | Exp | 167 (23–838) | 1613 (383–7102) | 1693 (60–23307) |
| Contemporary-SSN | 65.2 | Linear | 139 (17–692) | 1405 (358–8143) | 3134 (160–73610) |
| Contemporary-NW | 41.1 | Exp | 129 (23–513) | 1698 (288–12302) | 2884 (162–37153) |
| Contemporary-NW | 45.5 | Linear | 128 (27–547) | 1640 (246–19639) | 8549 (373–353012) |
The Bayes factor (BF) indicates the strength of evidence for a population decline with values greater than 10 representing very strong support. N 0 and N 1 are the current and ancestral N e respectively. Time (T) represents the date of the change in population size from N 0 to N 1.
The ratio of the posterior distributions of current and ancestral population sizes (r = N 0/N 1) indicates the direction of demographic change where r = 1 signifies population stability, r<1 population decline, and r>1 population expansion. Combining all simulations for all data sets for the exponential model we found the 90% highest posterior density (HPD) of the ratio r to be 0.011–0.095 with a mode of 0.081, and for the linear model an HPD of 0.010–0.066 with a mode of 0.062. These r values indicate that the current N e is estimated to be less than 10% of the ancestral N e and show an unambiguous signal of population decline for fisher in California (Fig. 4A).
Figure 4. MSVAR results: population size change.

A) ratio of current and ancestral population sizes (r = N 0/N 1) where r = 1 signifies population stability, r<1 decline, and r>1 expansion. 4B–D) Posterior distributions of the current (N 0) and ancestral (N 1) effective population size using both the exponential (thick lines) and linear (thin lines) models: B) northwestern-historical, C) southern Sierra Nevada-contemporary, and D) southern Sierra Nevada-historical. The dotted line shows the prior distribution for N 0 and N 1.
The modes of the 90% HPD of the posterior distributions for ancestral effective population size (N1) for the exponential model were SSNH = 1862, SSNC = 1613, and NWC = 1698 compared to modal values for current effective population sizes (N 0) of 154, 167, and 129 respectively (Table 4, Fig. 4 B–D). Estimates for N 0 and N 1 were similar but slightly lower for the linear model. Estimates of the time of population contraction varied between populations, but all showed support for population decline occurring well prior to the European settlement of California (T- SSNC = 1693 years before present [YBP], T-NWC = 2884 YBP, T-SSNH = 442 YBP). We adjusted the time estimates for the SSNH data to reflect the increased age of samples by adding the average age of the sample (95 years) to the estimate. Estimates for the timing of the decline were longer for the linear model than for the exponential model for all sample groups (Table 4, Fig. 5). We put more emphasis on the results of the exponential model because it is likely more realistic when modeling population dynamics [66].
Figure 5. MSVAR results: time of population decline.

Posterior distribution of time of decline (T) for the linear (thin line) and exponential (thick line) models. A) Time (in years before present) for the contemporary SSN (solid lines) and NW (dashed lines). B) Time for the historical SSN. The vertical dotted line shows the approximate time of the European settlement of California (∼1850) relative to the age of each of the samples.
Population subdivision can also bias demographic history models by creating a spurious signal of population decline. The potential bias is greatest for highly subdivided populations (high F ST), highly variable markers, and species with large N e [78]. The recommended ad hoc method to counteract any potential bias created by population subdivision is to sample equally across demes [78]. We followed this ad hoc approach by conducting the MSVAR analysis in the SSNC with numerous samples from all three of the identified demes such that each of the North, Central, and South groups were well represented in our sample. Considering the characteristics of the data used in this analysis (moderate F ST values, low variability markers, and small population size) reduce the potential for biased results, combined with our use of the ad hoc method of sampling across demes, we feel our results are robust to the potential bias created by population subdivision.
Discussion
Population contraction and isolation
Our analyses supports the hypothesis that the NW and SSN fisher populations became isolated far before the European settlement of California and that the absence of fisher in the northern Sierra Nevada is likely a long standing gap in this species' historical range. We found a genetic signal for a more than 90% reduction in N e of fisher and estimated that this decline occurred over a thousand years ago. A decline of this magnitude is consistent with a major range contraction. There is a positive correlation between changes in abundance and distribution, where species' abundance decreases its range also decreases [79]–[82]; species with the strongest declines exhibit the largest range contractions [79]. While the positive correlation between abundance and range size is not universal [79], [83], the extreme decline in N e detected in our analyses makes the idea of concurrent stability in range size unlikely. While the 90% highest posterior density of 3 of the 6 models did not definitively exclude a post-settlement decline (Table 4), the vast majority of the mass of the distribution of the time parameter (T) support pre-European settlement, with an average of 90% of the contemporary and 81% of historical MCMC chains indicating a time of contraction prior to 1850.
In addition to an ancient population contraction that isolated the SSN from the NW, our analyses indicate the SSN has also undergone a more recent population bottleneck likely associated with the impact of human development in the late 19th and early 20th century. The presence of a bottleneck signal only in the north and central portions of the SSNC and not in the south reflects differences in the extent of anthropogenic influence across the Sierra Nevada. The majority of human settlement, and its associated impacts, occurred in the central and northern Sierra Nevada. Settlement in the southern Sierra was minimal in comparison due to the absence of gold deposits and steeper topography that restricted access to forest lands. Our results indicate that the area at southern tip of the Sierra Nevada may have acted as a refuge for fisher during the era of extensive logging and development that began with the gold rush and continued into the first half of the twentieth century [84]. This area appears to have maintained a stable population size while fisher in the rest of SSN was in decline.
The window of time that the heterozygosity excess and shifted mode tests can detect a bottleneck is shorter than the timeframe for the M-Ratio test. The magnitude of the reduction in the M-Ratio from equilibrium values is also highly dependent on the pre-bottleneck population size. Accordingly, simulation studies have shown the M-Ratio test performs well if the pre-bottleneck population size was large, the bottleneck was of long duration, or the population had time to recover [85]. The length of time that the M-Ratio is informative can vary considerably (125–500 generations) depending on the bottleneck characteristics in terms of severity, duration, and post-bottleneck recovery. Assuming a generation interval for fisher of 5 years, significantly reduced M-Ratios would be indicative of decline that occurred anywhere from 625–2500 years ago. However, in permanently reduced populations the M-Ratio will recover over time, whereas allelic diversity does not [63]. Consequently, a population with low allelic diversity but a high M-Ratio, such as was observed in this study, is indicative of a population that has been small for a very long time. This conclusion is further supported by the fact that we found all sample groups to have low genetic diversity, and did not find any significant difference in diversity between contemporary and historical samples (collected between 1880 and 1920). This suggests that a population reduction, and its concurrent reduction in genetic diversity, occurred prior to the dates of the historical samples.
Our data suggests continual isolation of the NW and SSN populations during the last century. The increase in F ST from 0.10 in the early 1900s to 0.37 in 2006–2009 shows the genetic isolation of the populations during the intervening years. However, the F ST estimates between historical NW and SSN samples are likely biased considering the number of samples available from each population was small and from a relatively limited geographic subset of each area. Genotypic differentiation was strong across all spatial and temporal samples, and the amount of within population genetic differentiation over time period was similar in both areas (F ST: SSNH-SSNC = 0.17, NWH-NWC = 0.20) which can be attributed to the effects of genetic drift in small populations over time.
Considerations for bottleneck tests
Recent studies have found that bottleneck detection methods sometimes perform poorly at detecting very recent or weak population declines [68], [86], [87]. This creates a concern that a post-settlement decline would not be detected even if it had occurred. Girod et al. [68] used simulations to evaluate the ability of MSVAR to detect expansion/declines assessing performance using Bayes factors. Their analyses of populations with recent and/or weak declines resulted in very low Bayes factors (≤3) indicating no support for the detection of a decline. Accordingly, if the decline in the California fisher population was very recent we would expect MSVAR to produce a model with little support (low Bayes factors) reflecting the supposed poor ability of the method to detect recent declines. However, our MSVAR analyses produced high Bayes factors (≥10) for all models showing strongly supported signals of decline. Such high Bayes factors are in agreement with the results of the Girod et al. [68] for more ancient times of contraction (≥50 generations). The poor performance of the heterozygosity excess and M-Ratio tests detected in the Girod et al. [68] study is likely due to their simulation being conducted under a strict stepwise mutational model which has been identified as an unrealistically conservative model for microsatellite loci that may not have much power to detect bottlenecks that have actually occurred [88]. Other studies have shown these two methods to have a much higher power to detect bottlenecks [85], [88].
An important consideration in the interpretation of bottleneck tests is the potential influence of isolation by distance (IBD) within populations. While the SSNC has been found to exhibit a significant isolation by distance pattern across the entire population, tests for IBD were non-significant within each of the North, Central, and South subpopulations (J.M. Tucker unpublished data). The clustered distribution of samples in the NWC and SSNH and the small sample size of the NWH prevented us from testing for IBD in these populations. However, IBD has been found to have little effect on the heterozygosity excess method implemented in BOTTLENECK [89]. IBD does influence the M-Ratio such that both equilibrium and post bottleneck values of M are depressed compared with a non-IBD population. Thus, IBD can result in M values in non-bottlenecked populations that are lower than the Garza and Williamson's [63] recommended M-Critical cutoff value of 0.68 providing a false signal of a bottleneck. However, given the consistently high M values detected in this study (M = 0.82–0.92, Table 3) we do not feel that IBD biased our M-Ratio analyses.
Effective population size estimates
The similarity between the estimates of N e in the NWC and SSNC populations is surprising given that the NWC is thought to have a larger total population size (N) than the SSNC. There are no published estimates of N in the NWC, but unofficial estimates place it at between 1000–2000 individuals (C. Carroll personal communication cited in [90]) compared to estimates of 160–360 for the SSNC [91]. The ratio of N e/N is not well understood and can vary considerably between populations or species due to factors such as fluctuating population size, variance in reproductive success, unequal sex ratio, or population density [92]–[95]. Predicted values of the N e/N ratio in the literature vary widely; Nunney [96] estimated that theoretically the N e/N ratio should be 0.5, Nunney and Elam [97] found the average ratio across empirical data from 13 species to be 0.73, and Frankham [92] found the mean ratio across 102 species to be 0.11. Consequently, it is difficult to interpret what the estimated values of N e mean in terms of N in relation to each population. Extrapolating the modal values of the exponential model for N 0 across a wide range of possible N e/N ratio values of 0.05–0.5, the total population size for the NW could range from 258–2850 and SSN from 334–3380. Both of these population size ranges encompass the current possible estimates of N for both areas.
Biogeographical influences
The population contraction detected in this study and in the ancient mitochondrial divergence date reported by Knaus et al. [24] may reflect a shift in habitat distribution or community composition associated with one of a number of potentially significant climate shifts during the end of the Pleistocene or Holocene epochs. There are many well-known hypotheses about the cause of the mass extinctions and major shifts in species distribution that occurred at the end of the Pleistocene including temperature increases, changes in precipitation, or shifts in the ecological balance due to the arrival of human hunters in North America [98]. In more recent climactic history there are two well documented “mega-droughts” that occurred in California that have not been matched in severity or duration since. These droughts were first described by Stine [99] and were estimated to have lasted over 200 and 140 years each from 832–1074 and 1122–1299 AD respectively [100]. These droughts fall into a period of warmer temperatures referred to as the Medieval Warm Period [101] or Medieval Climate Anomaly [99]. While the divergence dates reported by Knaus et al. [24] would support a late Pleistocene climate shift as a possible cause of the divergence of California fisher populations, the results of this study found dates that support a more recent event, such as the aforementioned mega-droughts as a potential cause of the population contraction. Neither method allows for precise dating of the demographic shift. Nevertheless, both studies show that the contraction of the fisher populations in California pre-dated the gold rush and was not a direct result of the European settlement of California.
The reason fisher would be absent from the central and northern Sierra is perplexing, considering that there is no obvious geographic feature that marks a significant break in the topography or vegetative composition of the Sierra Nevada. However, a number of other species such as the great gray owl (Strix nebulosa), wolverine (Gulo gulo), and foxtail pine (Pinus balfouriana) have also been found to have long term genetic and geographic isolation in the southern Sierra Nevada [11], [102], [103] indicating that there are perhaps unique vegetative, climactic, or topographic elements in this region that are absent from the northern Sierra Nevada. A recent climate assessment has shown the southern Sierra Nevada to be somewhat resistant to climate changes observed elsewhere in California due to the extreme elevation of the mountains in this region [104].
The Sierra Nevada is characterized by a gradual change in its maximum elevation and average slope, such that the elevation of the Sierran crest and average slope is highest in the south. The area of the Sierra Nevada occupied by fisher is at the southernmost extent of its range where the weather is hotter and drier than in other areas. To mitigate the effects of high heat and low humidity, fisher may use cool and damp microhabitats characterized by dense canopies, large diameter trees, steep slopes, and close proximity to water [30]. One possible explanation for fisher presence in the southern Sierra is that the steep topography in this portion of the mountain range facilitates the creation and persistence of these essential microhabitat areas.
Relatively high amounts of subdivision have been reported in other parts of the fisher's range. Kyle et al. [105] found the amount of genetic subdivision observed between fisher populations (global F ST = 0.137) was much higher than for other closely related carnivore species of American marten (F ST = 0.0198) or wolverine (F ST = 0.0427) [106], [107]. A linear regression of genetic versus geographic distance found that fisher have twice the subdivision per unit distance than martens and 5 times more per unit distance than wolverine [108]. The high amounts of subdivision observed in fisher may result from being habitat specialists which makes them especially vulnerable to habitat fragmentation [109], [110]. Strikingly, this study found the structure per unit distance between the SSNC and NWC to be to be ∼10 times greater (0.961/1000 km) and between then SSNH and NWH to be ∼4 times greater (0.348/1000 km) than Kyle et al. [105] found for fisher populations across North America (0.092/1000 km). However, high subdivision is not universal among fisher populations. Populations in southern Ontario, Canada have been found to have weak subdivision and high genetic connectivity attributed to high amounts of gene flow along expansion fronts in a growing population [16], [111].
Conservation Implications
Our results provide a historical perspective for contemporary conservation and management decisions for fisher in California. There are ongoing debates as to whether efforts should be made to restore connectivity between the NW and SSN and thereby increase genetic diversity in the isolated SSN. The results of this study show that both populations have persisted in isolation far prior to the European settlement of California. Therefore, attempting to restore connectivity between them would be inconsistent with the historical record and run the risk of losing local adaptations that evolved in each population [36]. Given their long term isolation, the NW and SSN fisher populations should be considered independently for management and conservation decisions.
In 2004, the west coast population of fisher (southern Oregon, northwestern California, and southern Sierra Nevada of California) was found warranted but precluded for listing as a single distinct population segment (DPS) under the federal Endangered Species Act [112]. Among the criteria for considering a population as a DPS it must be markedly separated from other populations of the same taxon (discrete) and differ from other populations in its ecological setting or genetic characteristics (significant) [113]. As both of these criteria can be met by quantitative measures of genetic discontinuity or genetic uniqueness [113], the detection of long term genetic isolation of the southern Sierra Nevada fisher population has important implications for its legal status. The observed genetic differentiation coupled with observed differences in diet, home range size, and habitat associations between the SSN and NW [29], [114], [115] speaks to the potential of the SSN population itself as a DPS.
Supporting Information
Location and collection date of historical fisher genetic samples. Samples were collected from the Smithsonian National Museum of Natural History (SNM) and the Museum of Vertebrate Zoology at the University of California, Berkeley (MVZ). Samples that successfully genotyped at a minimum of 7 of 10 microsatellite loci are shown in bold.
(DOCX)
Prior and hyperprior parameters for runs of the Storz and Beaumont (2002) analysis implemented in MSVAR. Columns 3–6 show the starting values for the mean and variance of the prior distributions. Columns 7–10 show the means and variances (and their means and variances) of the hyperprior distributions. Parameters listed are generation interval (g), current N e (N 0), ancestral N e (N 1), mutation rate scaled in terms of current population size (θ), and time (T). All values are in a log10 scale.
(DOCX)
Acknowledgments
We extend our appreciation to the collaborators that provided or assisted in obtaining fisher genetic samples including S.Yaeger, L.Findley, S.Farber, S.Matthews, M.Higley, R.Callas, T. Engstrom, R.Powell, L.Diller, B.Zielinski, K.Slauson, J.Bolis, and the numerous field technicians who collected these samples. We thank C.Conroy and the Museum of Vertebrate Zoology, and R.Fisher and the Smithsonian National Museum of Natural History for access to their collections. We thank K.McKelvey and K.Aubry who collected the historical samples from the Smithsonian, C.Engkjer and P.Minton-Edison for laboratory assistance, and D.Patterson and M.Ellis for their assistance in statistical analyses in R. We would also like to thank the anonymous reviewers as well as M.Hebblewhite, M.Mitchell, B.Zielinski, and S.Hegg for their comments on this manuscript.
Funding Statement
This work was funded by the U.S. Forest Service Pacific Southwest Region, Rocky Mountain Research Station and the University of Montana. Additional funding was provided by the National Fish and Wildlife Foundation. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Lyons SK (2003) A quantitative assessment of the range shifts of Pleistocene mammals. J Mammal 84: 385–402. [Google Scholar]
- 2. McKelvey KS, Aubry KB, Schwartz MK (2008) Using anecdotal occurrence data for rare or elusive species: the illusion of reality and a call for evidentiary standards. Bioscience 58: 549–555. [Google Scholar]
- 3. Schwartz MK (2007) Ancient DNA confirms native Rocky Mountain fisher (Martes pennanti) avoided early 20th Century extinction. J Mammal 88: 921–925. [Google Scholar]
- 4. Wandeler P, Hoeck PEA, Keller LF (2007) Back to the future: museum specimens in population genetics. Trends Ecol Evol 22: 634–642. [DOI] [PubMed] [Google Scholar]
- 5. Goossens B, Chikhi L, Ancrenaz M, Lackman-Ancrenaz I, Andau P, et al. (2006) Genetic signature of anthropogenic population collapse in orangutans. PLoS Biology 4: 285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Okello J, Wittemyer G, Rasmussen H, Arctander P, Nyakaana S, et al. (2008) Effective population size dynamics reveal impacts of historic climatic events and recent anthropogenic pressure in African elephants. Mol Ecol 17: 3788–3799. [DOI] [PubMed] [Google Scholar]
- 7. Smulders MJM, Snoek LB, Booy G, Vosman B (2003) Complete loss of MHC genetic diversity in the common hamster (Cricetus cricetus) population in The Netherlands. Consequences for conservation strategies. Conserv Genet 4: 441–451. [Google Scholar]
- 8. Johnson JA, Tingay RE, Culver M, Hailer F, Clarke ML, et al. (2009) Long-term survival despite low genetic diversity in the critically endangered Madagascar fish-eagle. Mol Ecol 18: 54–63. [DOI] [PubMed] [Google Scholar]
- 9. Martinez-Cruz B, Godoy J, Negro J (2007) Population fragmentation leads to spatial and temporal genetic structure in the endangered Spanish imperial eagle. Mol Ecol 16: 477–486. [DOI] [PubMed] [Google Scholar]
- 10. Ross JD, Arndt AD, Smith RFC, Johnson JA, Bouzat JL (2006) Re-examination of the historical range of the greater prairie chicken using provenance data and DNA analysis of museum collections. Conserv Genet 7: 735–751. [Google Scholar]
- 11. Schwartz MK, Aubry KB, McKelvey KS, Pilgrim KL, Copeland JP, et al. (2007) Inferring geographic isolation of wolverines in California using historical DNA. J Wildl Manage 71: 2170–2179. [Google Scholar]
- 12.Graham RW, Graham MA (1994) Late quaternary distrubution of Martes in North America. In: Buskirk SW, Harestad AS, Raphael MG, and Powell RA, editors. Martens, Sables, and Fishers: Biology and Conservation. Ithica: Cornell University Press. pp. 26–58.
- 13.Powell RA (1993) The fisher: Life history, ecology, and behavior.Minneapolis : University of Minnesota Press. 237 p.
- 14. Williams RN, Rhodes OE, Serfass TL (2000) Assessment of genetic variance among source and reintroduced fisher populations. J Mammal 81: 895–907. [Google Scholar]
- 15. Vinkey RS, Schwartz MK, McKelvey KS, Foresman KR, Pilgrim KL, et al. (2006) When reintroductions are augmentations: the genetic legacy of fishers (Martes pennanti) in Montana. J Mammal 87: 265–271. [Google Scholar]
- 16. Carr D, Bowman J, Kyle CJ, Tully SM, Koen EL, et al. (2007) Rapid homogenization of multiple sources: genetic structure of a recolonizing population of fishers. J Wildl Manage 71: 1853–1861. [Google Scholar]
- 17. Carr D, Bowman J, J Wilson P (2007) Density-dependent dispersal suggests a genetic measure of habitat suitability. Oikos 116: 629–635. [Google Scholar]
- 18. Hapeman P, Latch EK, Fike JA, Rhodes OE, Kilpatrick CW (2011) Landscape genetics of fishers (Martes pennanti) in the Northeast: Dispersal barriers and historical influences. J Hered 102: 251. [DOI] [PubMed] [Google Scholar]
- 19. Zielinski WJ, Kucera TE, Barrett RH (1995) Current distribution of the fisher, Martes pennanti, in California. Calif Fish Game 81: 104–112. [Google Scholar]
- 20. Aubry KB, Lewis JC (2003) Extirpation and reintroduction of fishers (Martes pennanti) in Oregon: implications for their conservation in the Pacific states. Biol Conserv 114: 79–90. [Google Scholar]
- 21.Lewis JC, Hayes GE (2004) Feasibility assessment for reintroducing fishers to Washington. Olympia: Washington Department of Fish and Wildlife. 70 p.
- 22. Callas RL, Figura P (2008) Translocation plan for the reintroduction of fishers (Martes pennanti) to lands owned by Sierra Pacific Industries in the northern Sierra Nevada of California. California Department of Fish and Game 80. [Google Scholar]
- 23. Wisely SM, Buskirk SW, Russell GA, Aubry KB, Zielinski WJ (2004) Genetic diversity and structure of the fisher (Martes pennanti) in a peninsular and peripheral metapopulation. J Mammal 85: 640–648. [Google Scholar]
- 24. Knaus BJ, Cronn R, Pilgrim K, Schwartz MK (2011) Mitochondrial genome sequences illuminate maternal lineages of conservation concern in a rare carnivore. BMC Ecol 11: 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Spencer W, Rustigian-Romsos H, Strittholt J, Scheller R, Zielinski W, et al. (2011) Using occupancy and population models to assess habitat conservation opportunities for an isolated carnivore population. Biol Conserv 144: 788–803. [Google Scholar]
- 26.Grinnell J, Dixon JS, Linsdale JM (1937) Fur-bearing mammals of California: their natural history, systematic status, and relations to man. Univ.California Press, Berkeley. 375 p.
- 27. Zielinski WJ, Truex RL, Schlexer FV, Campbell LA, Carroll C (2005) Historical and contemporary distributions of carnivores in forests of the Sierra Nevada, California, USA. J Biogeogr 32: 1385–1407. [Google Scholar]
- 28.McKelvey KS, Johnston JD (1992) Historical perspectives on forests of the Sierra Nevada and the transverse ranges of Southern California: Forest conditions at the turn of the century. J. Verner, KS McKelvey, BR Noon, RJ Gutiérrez, GI Gould Jr, and TW Beck, technical coordinators. The California Spotted Owl: A technical assessment of its current status. USDA Forest Service, Pacific Southwest Research Station, General Technical Report: pp 225–246.
- 29. Zielinski WJ, Truex RL, Schmidt GA, Schlexer FV, Schmidt KN, et al. (2004) Home range characteristics of fisher in California. J Mammal 85: 649–657. [Google Scholar]
- 30. Zielinski WJ, Truex RL, Schmidt GA, Schlexer FV, Schmidt KN, et al. (2004) Resting habitat selection by fishers in California. J Wildl Manage 68: 475–492. [Google Scholar]
- 31. Purcell KL, Mazzoni AK, Mori SR, Boroski BB (2009) Resting structures and resting habitat of fishers in the southern Sierra Nevada, California. For Ecol Manage 258: 2696–2706. [Google Scholar]
- 32. Drew RE, Hallett JG, Aubry KB, Cullings KW, Koepf SM, et al. (2003) Conservation genetics of the fisher (Martes pennanti) based on mitochondrial DNA sequencing. Mol Ecol 12: 51–62. [DOI] [PubMed] [Google Scholar]
- 33.Powell RA, Zielinski WJ (2005) Evaluating the demographic factors that affect the success of reintroducing fishers (Martes pennanti), and the effects of removals on a source population. Final Report to the U S Fish and Wildlife Service. 20 p.
- 34.Sierra Pacific Industries, United States Fish and Wildlife Service (2007) Candidate Conservation Agreement with Assurances for fisher for the Stirling Management Area. 41 p.
- 35. Edmands S (2007) Between a rock and a hard place: evaluating the relative risks of inbreeding and outbreeding for conservation and management. Mol Ecol 16: 463–475. [DOI] [PubMed] [Google Scholar]
- 36. Frankham R, Ballou JD, Eldridge MDB, Lacy RC, Ralls K, et al. (2011) Predicting the probability of outbreeding depression. Conserv Biol 25: 465–475. [DOI] [PubMed] [Google Scholar]
- 37.Aubry KB, Wisely SM, Raley C, Buskirk SW (2004) Zoogeography, spacing patterns, and dispersal in fishers: insights gained from combining field and genetic data. In: Harrison D, J, Fuller AK, Proulx G, editors. Martens and fishers (Martes) in human-altered environments: an international perspective. New York: Springer Science. pp. 201–220.
- 38. Waits L, Taberlet P, Swenson JE, Sandegren F, Franzén R (2000) Nuclear DNA microsatellite analysis of genetic diversity and gene flow in the Scandinavian brown bear (Ursus arctos). Mol Ecol 9: 421–431. [DOI] [PubMed] [Google Scholar]
- 39. Pardini AT, Jones CS, Noble LR, Kreiser B, Malcolm H, et al. (2001) Sex-biased dispersal of great white sharks. Nature 412: 139–140. [DOI] [PubMed] [Google Scholar]
- 40. Yang D-, Kenagy G (2009) Nuclear and mitochondrial DNA reveal contrasting evolutionary processes in populations of deer mice (Peromyscus maniculatus). Mol Ecol 18: 5115–5125. [DOI] [PubMed] [Google Scholar]
- 41. Zielinski WJ, Baldwin JA, Truex RL, Tucker JM, Flebbe PA (2012) Estimating trend in occupancy for the southern Sierra fisher (Martes pennanti) population. J Fish Wildl Manage In Press. [Google Scholar]
- 42. Fleischer RC, Olson SL, James HF, Cooper AC (2000) Identification of the extinct Hawaiian eagle (Haliaeetus) by mtDNA sequence analysis. Auk 117: 1051–1056. [Google Scholar]
- 43. Wisely S, Maldonado JE, Fleische R (2004) A technique for sampling ancient DNA that minimizes damage to museum specimens. Conserv Genet 5: 105–107. [Google Scholar]
- 44. Gilbert MTP, Bandelt HJ, Hofreiter M, Barnes I (2005) Assessing ancient DNA studies. Trend Ecol Evol 20: 541–544. [DOI] [PubMed] [Google Scholar]
- 45. Jordan MJ, Higley JM, Mattews SM, Rhodes OE, Schwartz MK, et al. (2007) Development of 22 new microsatellite loci for fishers (Martes pennanti) with variability results from across their range. Mol Ecol Notes 7: 797–801. [Google Scholar]
- 46. Davis CS, Strobeck C (1998) Isolation, variability, and cross-species amplification of polymorphic microsatellite loci in the family Mustelidae. Mol Ecol 7: 1776–1778. [DOI] [PubMed] [Google Scholar]
- 47. Duffy AJ, Landa A, O'Connell M, Stratton C, Wright JM (1998) Four polymorphic microsatellite in wolverine, Gulo gulo . Anim Genet 29: 63–72. [PubMed] [Google Scholar]
- 48. Dallas J, Piertney S (1998) Microsatellite primers for the Eurasian otter. Mol Ecol 7: 1248–1251. [PubMed] [Google Scholar]
- 49. Taberlet P, Luikart G (1999) Non-invasive genetic sampling and individual identification. Biol J Linn Soc 68: 41–55. [Google Scholar]
- 50. Eggert LS, Eggert JA, Woodruff DS (2003) Estimating population sizes for elusive animals: the forest elephants of Kakum National Park, Ghana. Mol Ecol 12: 1389–1402. [DOI] [PubMed] [Google Scholar]
- 51. McKelvey KS, Schwartz M (2004) Genetic errors associated with population estimation using non-invasive molecular tagging: problems and new solutions. J Wildl Manage 68: 438–448. [Google Scholar]
- 52. McKelvey KS, Schwartz MK (2005) Dropout: a program to identify problem loci and samples for noninvasive genetic samples in a capture-mark-recapture framework. Mol Ecol Notes 5: 716–718. [Google Scholar]
- 53. Raymond M, Rousset F (1995) GENEPOP (version 1.2): population genetics software for exact tests and ecumenicism. J Hered 86: 248–249. [Google Scholar]
- 54. Rousset F (2008) GENEPOP'007: a complete re-implementation of the GENEPOP software for Windows and Linux. Mol Ecol Resour 8: 103–106. [DOI] [PubMed] [Google Scholar]
- 55. Weir BS, Cockerham CC (1984) Estimating F-statistics for the analysis of population structure. Evolution 38: 1358–1370. [DOI] [PubMed] [Google Scholar]
- 56. Slatkin M (1995) A measure of population subdivision based on microsatellite allele frequencies. Genetics 139: 457–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Archie JW (1985) Statistical analysis of heterozygosity data: independent sample comparisons. Evolution 623–637. [DOI] [PubMed] [Google Scholar]
- 58. Rice WR (1989) Analyzing tables of statistical tests. Evolution 43: 223–225. [DOI] [PubMed] [Google Scholar]
- 59. Piry S, Luikart G, Cornuet J (1999) Computer note. BOTTLENECK: a computer program for detecting recent reductions in the effective size using allele frequency data. J Hered 90: 502–503. [Google Scholar]
- 60. Cornuet JM, Luikart G (1996) Description and power analysis of two tests for detecting recent population bottlenecks from allele frequency data. Genetics 144: 2002–2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Di Rienzo A, Peterson AC, Garza JC, Valdes AM, Slatkin M, et al. (1994) Mutational processes of simple-sequence repeat loci in human populations. Proc Natl Acad Sci USA 91: 3166–3170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Luikart G, Allendorf F, Cornuet J, Sherwin W (1998) Distortion of allele frequency distributions provides a test for recent population bottlenecks. J Hered 89: 238. [DOI] [PubMed] [Google Scholar]
- 63. Garza J, Williamson E (2001) Detection of reduction in population size using data from microsatellite loci. Mol Ecol 10: 305–318. [DOI] [PubMed] [Google Scholar]
- 64. Broquet T, Angelone S, Jaquiery J, Joly P, Lena JP, et al. (2010) Genetic bottlenecks driven by population disconnection. Conserv Biol 24: 1596–1605. [DOI] [PubMed] [Google Scholar]
- 65. Garroway CJ, Bowman J, Wilson PJ (2011) Using a genetic network to parameterize a landscape resistance surface for fishers, Martes pennanti . Mol Ecol 20: 3978–3988. [DOI] [PubMed] [Google Scholar]
- 66. Beaumont MA (1999) Detecting population expansion and decline using microsatellites. Genetics 153: 2013–2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Storz JF, Beaumont MA (2002) Testing for genetic evidence of population expansion and contraction: an empirical analysis of microsatellite DNA variation using a hierarchical Bayesian model. Evolution 56: 154–166. [DOI] [PubMed] [Google Scholar]
- 68. Girod C, Vitalis R, Leblois R, Fréville H (2011) Inferring population decline and expansion from microsatellite data: a simulation-based evaluation of the Msvar method. Genetics 188: 165–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Gelman A, Rubin DB (1992) Inference from iterative simulation using multiple sequences. Statistical Science 7: 457–472. [Google Scholar]
- 70. Brooks SP, Gelman A (1998) General methods for monitoring convergence of iterative simulations. J Comput Graph Stat 7: 434–455. [Google Scholar]
- 71.R Development Core Team (2010) R: A language and environment for statistical computing. R Foundation for Statistical Computing. Vienna, Austria.
- 72. Smith BJ (2007) boa: an R package for MCMC output convergence assessment and posterior inference. J Stat Softw 21: 1–37. [Google Scholar]
- 73. Loader C (2007) Locfit: Local regression, likelihood and density estimation. R Package Version 1.5–4. [Google Scholar]
- 74.Jeffreys H (1961) Theory of probability. Oxford, UK: Oxford University Press. 447 p.
- 75. Kass RE, Raftery AE (1995) Bayes factors. J Am Stat Assoc 90: 773–795. [Google Scholar]
- 76.Lofroth EC, Raley CM, Higley JM, Truex RL, Yaeger JS, et al.. (2010) Conservation of fishers in south-central British Columbia, western Washington, western Oregon, and California- Volume I: conservation assessment. Denver: USDI Bureau of Land Management. 117 p.
- 77. Wahlund S (1928) Zusammensetzung von Populationen und Korrelationserscheinungen vom Standpunkt der Vererbungslehre aus betrachtet. Hereditas 11: 65–106. [Google Scholar]
- 78. Chikhi L, Sousa VC, Luisi P, Goossens B, Beaumont MA (2010) The confounding effects of population structure, genetic diversity and the sampling scheme on the detection and quantification of population size changes. Genetics 186: 983–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Fuller R, Gregory R, Gibbons D, Marchant J, Wilson J, et al. (1995) Population declines and range contractions among lowland farmland birds in Britain. Conserv Biol 9: 1425–1441. [Google Scholar]
- 80. Gaston KJ, Blackburn TM, Lawton JH (1997) Interspecific abundance-range size relationships: an appraisal of mechanisms. J Anim Ecol 579–601. [DOI] [PubMed] [Google Scholar]
- 81. Newton I (1997) Links between the abundance and distribution of birds. Ecography 20: 137–145. [Google Scholar]
- 82. Rodríguez JP (2002) Range contraction in declining North American bird populations. Ecol Appl 12: 238–248. [Google Scholar]
- 83. Gaston KJ, Curnutt J (1998) The dynamics of abundance-range size relationships. Oikos 81: 38–44. [Google Scholar]
- 84.Beesley D (1996) Reconstructing the landscape: an environmental history, 1820–1960. In: Sierra Nevada Ecosystem Project Final Report to Congress, volume II, assessments and scientific basis for management options. Davis: University of California Centers for Water and Wildlife Resources. pp 3–24.
- 85. Williamson-Natesan EG (2005) Comparison of methods for detecting bottlenecks from microsatellite loci. Conserv Genet 6: 551–562. [Google Scholar]
- 86. Mock KE, Latch E, Rhodes O (2004) Assessing losses of genetic diversity due to translocation: long-term case histories in Merriam's turkey (Meleagris gallopavo merriami). Conserv Genet 5: 631–645. [Google Scholar]
- 87. Busch JD, Waser PM, DeWoody JA (2007) Recent demographic bottlenecks are not accompanied by a genetic signature in banner-tailed kangaroo rats (Dipodomys spectabilis). Mol Ecol 16: 2450–2462. [DOI] [PubMed] [Google Scholar]
- 88. Luikart G, Cornuet JM (1998) Empirical evaluation of a test for identifying recently bottlenecked populations from allele frequency data. Conserv Biol 12: 228–237. [Google Scholar]
- 89. Leblois R, Estoup A, Streiff R (2006) Genetics of recent habitat contraction and reduction in population size: does isolation by distance matter? Mol Ecol 15: 3601–3615. [DOI] [PubMed] [Google Scholar]
- 90.Greenwald DN, Carlton J, Schneider B (2000) Petition to list the fisher (martes pennanti) as an endangered species in its west coast range. Tucson: Petition submitted to the U.S. Fish and Wildlife Service by the Center for Biological Diversity and Sierra Nevada Forest Protection Campaign. 91 p.
- 91.Spencer W, Rustigian H, Scheller R, Syphard A, Strittholt J, et al.. (2008) Baseline evaluation of fisher habitat and population status, and effects of fires and fuels management on fishers in the southern Sierra Nevada. Final report for the USDA Forest Service, Pacific Southwest Region. 107 p.
- 92. Frankham R (1995) Effective population size/adult population size ratios in wildlife: a review. Genet Res 66: 95–107. [DOI] [PubMed] [Google Scholar]
- 93. Wade MJ (1980) Effective population size: the effects of sex, genotype, and density on the mean and variance of offspring numbers in the flour beetle, Tribolium castaneum . Genet Res 36: 1–10. [DOI] [PubMed] [Google Scholar]
- 94. Ardren WR, Kapuscinski AR (2003) Demographic and genetic estimates of effective population size (Ne) reveals genetic compensation in steelhead trout. Mol Ecol 12: 35–49. [DOI] [PubMed] [Google Scholar]
- 95. Hare MP, Nunney L, Schwartz MK, Ruzzante DE, Burford M, et al. (2011) Understanding and estimating effective population size for practical application in marine species management. Conserv Biol 25: 438–449. [DOI] [PubMed] [Google Scholar]
- 96. Nunney L (1993) The influence of mating system and overlapping generations on effective population size. Evolution 1329–1341. [DOI] [PubMed] [Google Scholar]
- 97. Nunney L, Elam DR (1994) Estimating the effective population size of conserved populations. Conserv Biol 8: 175–184. [Google Scholar]
- 98. Barnosky AD, Koch PL, Feranec RS, Wing SL, Shabel AB (2004) Assessing the causes of Late Pleistocene extinctions on the continents. Science 306: 70–75. [DOI] [PubMed] [Google Scholar]
- 99. Stine S (1994) Extreme and persistent drought in California and Patagonia during mediaeval time. Nature 369: 546–549. [Google Scholar]
- 100. Cook ER, Seager R, Heim RR Jr, Vose RS, Herweijer C, et al. (2010) Megadroughts in North America: placing IPCC projections of hydroclimatic change in a long-term palaeoclimate context. Journal of Quaternary Science 25: 48–61. [Google Scholar]
- 101. Lamb HH (1965) The early medieval warm epoch and its sequel. Palaeogeogr, Palaeoclimatol, Palaeoecol 1: 13–37. [Google Scholar]
- 102. Hull JM, Keane JJ, Savage WK, Godwin SA, Shafer JA, et al. (2010) Range-wide genetic differentiation among North American great gray owls (Strix nebulosa) reveals a distinct lineage restricted to the Sierra Nevada, California. Mol Phylogenet Evol [DOI] [PubMed] [Google Scholar]
- 103. Eckert AJ, Tearse BR, Hall BD (2008) A phylogeographical analysis of the range disjunction for foxtail pine (Pinus balfouriana, Pinaceae): the role of Pleistocene glaciation. Mol Ecol 17: 1983–1997. [DOI] [PubMed] [Google Scholar]
- 104.Moser S, Franco G, Pittiglio S, Chou W, Cayan D (2009) The future is now: An update on climate change science impacts and response options for California. California Climate Center Report CEC-500-2008-071. Sacramento: California Energy Commission. 114 p.
- 105. Kyle CJ, Robitaille JF, Strobeck C (2001) Genetic variation and structure of fisher (Martes pennanti) populations across North America. Mol Ecol 10: 2341–2347. [DOI] [PubMed] [Google Scholar]
- 106. Kyle C, Davis C, Strobeck C (2000) Microsatellite analysis of North American pine marten (Martes americana) populations from the Yukon and Northwest Territories. Can J Zool 78: 1150–1157. [Google Scholar]
- 107. Kyle CJ, Strobeck C, Bradley RD (2002) Connectivity of peripheral and core populations of North American wolverines. J Mammal 83: 1141–1150. [Google Scholar]
- 108. Kyle C, Strobeck C (2001) Genetic structure of North American wolverine (Gulo gulo) populations. Mol Ecol 10: 337–347. [DOI] [PubMed] [Google Scholar]
- 109.Buskirk SW, Powell RA (1994) Habitat ecology of fishers and america martens. In: Buskirk SW, Harestad AS, Raphael MG, Powell RA, editors. Martens, sables, and fishers: biology and conservation. Ithaca: Cornell University Press. pp. 283–296.
- 110. Weir RD, Corbould FB (2010) Factors affecting landscape occupancy by fishers in north-central British Columbia. J Wildl Manage 74: 405–410. [Google Scholar]
- 111. Garroway CJ, Bowman J, Carr D, Wilson PJ (2008) Applications of graph theory to landscape genetics. Evol Appl 1: 620–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. U.S. Department of the Interior, Fish and Wildlife Service (2004) 12-month Finding for a Petition To List the West Coast Distinct Population Segment of the Fisher (Martes pennanti); Proposed Rule. Fed Regist 69: 18770–18792. [Google Scholar]
- 113. United States Fish and Wildlife Service, National Marine Fisheries Service (1996) Policy regarding the recognition of distinct vertebrate population segments under the Endangered Species Act. Fed Regist 61: 4722–4. [Google Scholar]
- 114. Zielinski WJ, Duncan NP, Farmer EC, Truex RL, Clevenger AP, et al. (1999) Diet of fishers (Martes pennanti) at the southernmost extent of their range. J Mammal 80: 961–971. [Google Scholar]
- 115. Davis FW, Seo C, Zielinski WJ (2007) Regional variation in home-range-scale habitat models for fisher (Martes pennanti) in California. Ecol Appl 17: 2195–2213. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Location and collection date of historical fisher genetic samples. Samples were collected from the Smithsonian National Museum of Natural History (SNM) and the Museum of Vertebrate Zoology at the University of California, Berkeley (MVZ). Samples that successfully genotyped at a minimum of 7 of 10 microsatellite loci are shown in bold.
(DOCX)
Prior and hyperprior parameters for runs of the Storz and Beaumont (2002) analysis implemented in MSVAR. Columns 3–6 show the starting values for the mean and variance of the prior distributions. Columns 7–10 show the means and variances (and their means and variances) of the hyperprior distributions. Parameters listed are generation interval (g), current N e (N 0), ancestral N e (N 1), mutation rate scaled in terms of current population size (θ), and time (T). All values are in a log10 scale.
(DOCX)