

Abstract
We report the genome-wide mapping of ORC1 binding sites in mammals, by chromatin immunoprecipitation and parallel sequencing (ChIP-seq). ORC1 binding sites in HeLa cells were validated as active DNA replication origins (ORIs) using Repli-seq, a method that allows identification of ORI-containing regions by parallel sequencing of temporally ordered replicating DNA. ORC1 sites were universally associated with transcription start sites (TSSs) of coding or noncoding RNAs (ncRNAs). Transcription levels at the ORC1 sites directly correlated with replication timing, suggesting the existence of two classes of ORIs: those associated with moderate/high transcription levels (≥1 RNA copy/cell), firing in early S and mapping to the TSSs of coding RNAs; and those associated with low transcription levels (<1 RNA copy/cell), firing throughout the entire S and mapping to TSSs of ncRNAs. These findings are compatible with a scenario whereby TSS expression levels influence the efficiency of ORC1 recruitment at G1 and the probability of firing during S.
DNA replication is a highly orchestrated process that ensures fidelity of genomes during duplications, as well as their adaptation to variations in cell division, DNA damage, and, in metazoa, chromatin changes associated with development and differentiation. It initiates from multiple chromosomal loci, called replication origins (ORIs), which are selected in the G1 phase of the cell cycle by sequential recruitment of the origin recognition complex (ORC), CDC6, CDT1, and the MCM complex (the pre-replicative complex; pre-RC). Selected pre-RCs are then sequentially activated during the S phase, following a tight temporally ordered program (Mechali 2010).
In Saccharomyces cerevisiae, ORIs contain a 12-bp consensus for ORC binding (Bell and Stillman 1992). Genome-wide analyses of ORIs by chromatin immunoprecipitation and parallel sequencing (ChIP-seq) using anti-ORC or -MCM antibodies showed that this consensus is essential but not sufficient for origin activity and identified other features that influence selection and replication timing, including transcription and/or chromatin structure (Eaton et al. 2010). In metazoa, instead, pre-RC does not exhibit sequence specificity, and the number of potential ORIs is considerably larger, following a process of selection that differs according to cell type, functional status, or stress conditions (Mechali 2010). These further levels of complexity allow DNA replication to adapt to the unique expression patterns of individual cell types. Little is known, however, about the regulation of ORI selection and replication timing in metazoa.
Indirect evidence suggests a correlation between ORI selection and transcription, based on the observed enrichment of ORIs at gene promoters, usually in the proximity of transcription start sites (TSSs) (Mechali 2010). The extent of the reported ORI/promoter association (7%–44% in mammals, 64% in Drosophila) and the transcriptional status of the ORI-associated promoters, however, are controversial among different studies (Cadoret et al. 2008; Sequeira-Mendes et al. 2009; Karnani et al. 2010; MacAlpine et al. 2010; Cayrou et al. 2011; Martin et al. 2011). Mechanistically, transcription might facilitate ORC localization either indirectly, because of the enhanced chromatin accessibility associated with active promoters, or directly, through a subset of the transcription factors that allow RNA polymerase II (POLR2A) recruitment. At the same time, however, active gene transcription has been shown to prevent ORI activity inside that gene (Mechali 2010).
A correlation has been observed also between transcription and early replication, based on the findings that early-replicating genes can be either expressed or silent, while the majority of the late-replicating genes are silent (Maric and Prioleau 2010). It is not clear, however, whether replication timing correlates with transcription or transcription-associated chromatin modifications, or whether the influence of transcription on replication timing is exerted over large chromosomal domains (>100 kb), specific classes of genes, or ORI-associated promoters (Hiratani et al. 2008; Ryba et al. 2010; Eaton et al. 2011).
One major limitation to study metazoan ORIs is the lack of sensitive and stringent methods for genome-wide studies. In mammalian cells, available information is based on the isolation of DNA from short nascent-strand (SNS) or replication-bubble preparations, followed by hybridization to small fractions of the genome (∼1%–2%) (Mechali 2010; Cayrou et al. 2011; Mesner et al. 2011; Valenzuela et al. 2011), or parallel sequencing of SNSs (Martin et al. 2011). These studies confirmed ORI enrichment at promoters and gene bodies, although overlap between comparable ORI data sets is poor (∼10%–33%) (for review, see Gilbert 2010). We report here the first genome-wide ChIP-seq mapping of human ORIs, obtained by targeting ORC1, one of the critical players in the selection of active ORIs (DePamphilis et al. 2006), and using chromatin fractions enriched in pre-RC-bound DNA fragments. We validated the ORC1 sites as ORIs and analyzed their genomic position, replication timing, and association with transcription.
Results
ORIs segregate within high- and low-density fractions in an equilibrium density gradient
Ultracentrifugation of cross-linked chromatin in equilibrium density gradients allows separation of chromatin fragments based on their protein/DNA ratio and has been used to purify bulk chromatin (density: 1.42–1.39 g/cm3) from free DNA (≈1.69 g/cm3) or cross-linked proteins (≈1.25 g/cm3) (Solomon et al. 1988). Specific chromatin regions, such as enhancers, transcribed exons, or active promoters, show low density, due to higher protein/DNA ratios (Ip et al. 1988; Reneker and Brotherton 1991; Schwartz et al. 2005), suggesting that density centrifugation of sheared cross-linked chromatin can separate functional states of chromatin. Thus, we investigated whether ORI chromatin segregates from bulk chromatin in equilibrium density gradients. Chromatin from asynchronous HeLa cells was fractionated in a CsCl gradient (Fig. 1A,B) and analyzed for the distribution of pre-RC proteins and DNA sequences of known ORIs. ORC (1 and 2) and MCM (2, 5, and 7) proteins were all found in fractions with lower density than the bulk chromatin (fractions 6, 7), with MCMs showing weaker signals also in fractions 4 and 5 (Fig. 1C). The distribution of two known ORIs (PRKDC and LMNB2) was analyzed by Q-PCR, using probes specific for the ORIs or their flanking regions (±1–5 kb). While the latter were uniformly distributed along the gradient, the two ORIs were enriched in the low- and high-density fractions (6–7 and 13–16, respectively) (Fig. 1D). Together, these observations suggest that density centrifugation of cross-linked chromatin allows fractionation of ORIs in two states, as pre-RC-bound or protein-free ORI-DNA fragments, which segregate within low- and high-density fractions, respectively.
Figure 1.

ORI chromatin segregates from bulk chromatin in density gradients. (A,B) Cross-linked chromatin was fractionated by CsCl density-gradient centrifugation into 16 fractions and analyzed by gel electrophoresis. The densities (g/cm3) of fractions 1–16 are 1.23, 1.24, 1.26, 1.28, 1.30, 1.32, 1.34, 1.37, 1.39, 1.42, 1.45, 1.49, 1.52, 1.57, 1.62, and 1.67, respectively. (A) Protein distribution (Coomassie staining of 8% SDS PAGE). (B) DNA distribution (ethidium bromide staining of 1% agarose). (C) Western blot showing the distribution of ORC1 and 2, MCM2, 5, and 7 in the gradient fractions. (D) Q-PCR distribution of DNA fragments from the LMNB2 and PRKDC ORIs and flanking regions (LMNB2: −1, +3 kb; PRKDC: +1, +5 kb), expressed as “fold enrichment” over input DNA of each fraction. (E) Q-ChIP of ORC1 binding at LMNB2 and PRKDC ORIs using anti-ORC1 or anti-Flag (Mock) antibodies from total chromatin or low-density fractions (6, 7). (F) ChIP-seq profile of ORC1 at LMNB2 and PRKDC ORIs. The position of the Q-PCR probes used in E is shown below each ORC1 peak. (Scale bars) 1 kb. Illumina sequencing was performed with purified DNA from anti-ORC1 ChIP of fractions 6 and 7 (red empty box) or with total DNA of fractions 14–16 (shown in Supplemental Figs. S1, S2).
High-density chromatin fractions are enriched with nucleosome-free regions
High-density fractions have been reported to contain naked DNA, generally nucleosome free (Varshavsky et al. 1976; Schwartz et al. 2005). Since ORI DNA is nucleosome free (Eaton et al. 2010; Lubelsky et al. 2011), we investigated whether the high-density fractions can be used for the identification of human ORIs. Purified DNA from high-density fractions (fractions 14–16) and input DNA (prior to gradient centrifugation) were sequenced using the Illumina Genome Analyzer. Alignment of the obtained sequence tags to the human genome allowed identification of 53,274 enriched regions or peaks (P ≤ 0.05; height, h ≥ 10). Interestingly, these peaks included known ORIs (Supplemental Fig. S1), overlapped with HeLa DNase I hypersensitive (DH) sites (∼84%), and contained most of the HeLa transcriptionally active TSSs (∼87%) (Supplemental Fig. S2), suggesting that high-density fractions contain the most accessible (presumably nucleosome-free) regions of the genome, including active ORIs. Expectedly, functional analyses of six randomly selected peaks (by isolation and quantification of short DNA nascent strands; nascent strand abundance, or NSA, assay) (Giacca et al. 1997) revealed the presence of origin activity in only one (data not shown), thus indicating that high-density chromatin fractions are not enriched in ORI DNA.
Anti-ORC1 ChIP from low-density chromatin allows ORI identification
To investigate if the low-density fractions can be used in ChIP assays for ORI purification, we performed quantitative ChIP (Q-ChIP) of four known ORIs, using anti-ORC1 antibodies (Supplemental Fig. S3; Mendoza-Maldonado et al. 2010). As expected (Ladenburger et al. 2002), in the unfractionated chromatin, we detected highly significant ORC1 binding at the PRKDC ORI (Fig. 1E; Supplemental Fig. S4A) and no signal at the other three: LMNB2 (Fig. 1E), DBF4, and HPRT1 (data not shown). In the chromatin obtained from low-density fractions, instead, we detected a 10-fold increase of DNA recovery from the PRKDC ORI (Fig. 1E; Supplemental Fig. S4A) and significant ORC1 binding also to the other three—LMNB2 (Fig. 1E; Supplemental Fig. S4A), DBF4, and HPRT1 (data not shown)—demonstrating that low-density fractions contain (and are enriched with) pre-RC proteins bound to ORI DNA.
Thus, we sequenced purified DNA of anti-ORC1 ChIP from low-density fractions (anti-ORC1 ChIP-seq) and input DNA as control (i.e., total DNA of the low-density fractions, prior to ChIP). Alignment of the obtained sequence tags to the human genome (Supplemental Table S1) allowed identification of 13,604 ORC1 binding sites or peaks (P ≤ 0.05; h ≥ 4), including the four known ORIs tested (Fig. 1F; Supplemental Fig. S4B), with high reproducibility (as revealed by a second preparation from HeLa cells) (Supplemental Fig. S5). ChIP-seq data were validated by Q-ChIP (using independent chromatin preparations) of eight randomly selected peaks with different amplitude (A–H in Fig. 2A) and two control regions (C1, C2 in Fig. 2A). As expected, the amount of recovered DNA was consistent with peak amplitude or proximity of probes to the peak summit.
Figure 2.

High-resolution validation of newly identified ORIs. Q-ChIP of ORC1 (A) and MCM5 (B) binding to the PRKDC ORI, eight newly identified ORC1 peaks (A–H), and four control regions (C1–C4), using anti-ORC1, anti-MCM5, or anti-Flag (Mock) antibodies and chromatin of low-density fractions. (Error bars) SD; n = 2. The position of Q-PCR probes, relative to the summit of ORC1 peaks, is shown at the bottom of A. (Scale bar) 1 kb. (C) Distribution of DNA fragments from two ORC1 peaks (C, G) and their flanking regions (+5 and −1.5 kb, respectively). (D) NSA assay of the PRKDC ORI, A–K ORC1 sites, and their flanking regions (distance from the probe within the ORC1 peak, in kilobases, is shown below). The amplitude of the I–K peaks is shown in Supplemental Figure S3. The amounts of SNSs are relative to PRKDC ORI (=1).
Next, we investigated whether the newly identified ORC1 binding sites were associated with other pre-RC proteins and possessed physical properties of known ORIs. Anti-MCM5 ChIP using low-density chromatin revealed significant binding to all the ORC1 binding sites tested (8/8) and to the PRKDC ORI (Fig. 2B). Gradient distribution of two representative ORC1 sites showed enrichment in the low- and high-density fractions (Fig. 2C). Finally, we used the NSA assay to investigate whether the ORC1-binding regions were active ORIs. Results showed the presence of SNSs at 11/11 ORC1 sites (A–H and three additional peaks with smaller amplitude: I–K) (Fig. 2D; Supplemental Fig. S6), 10 of which revealed enrichment of early-replication intermediates at the ORC1 peak (A–F, H–K). Thus, the randomly selected ORC1 sites showed binding to pre-RC proteins and physical and functional properties of known ORIs, demonstrating that anti-ORC1 ChIP from low-density chromatin fractions allows ORI purification.
Genome-wide validation of the newly identified ORIs
Our finding that randomly selected ORC1 binding regions are active ORIs suggests that the ORC1 data set identifies ORIs on a genome-wide scale. Unfortunately, we could not test this hypothesis functionally since, to date, current methods for isolating early-replication intermediates do not seem to be robust enough to allow high-throughput mapping of human ORIs (Gilbert 2010; Hamlin et al. 2010).
A well-established genome-scale approach is instead available, which allows mapping of replication initiation regions within temporally ordered replicating DNA (Repli-seq) (Hansen et al. 2010). Briefly, replicating (BrdU-labeled) DNA is purified from six consecutive S-phase cell populations containing increasing DNA content (S1–S6), sequenced, and mapped to the genome, thus allowing visualization of replication progression. Replication-timing profiles are characterized by hundreds of symmetrical early-to-late transitions (named “inverted-Vs”) originating at replication initiation regions (the “inverted-V apexes”). Thus, any inverted-V apex should contain at least one ORC1 site. To test this hypothesis, we performed Repli-seq of HeLa cells (Fig. 3A–C) and found that 10 of the 11 validated ORIs (A–J in Fig. 2) mapped to the inverted-V apex of early-replicating regions (Supplemental Fig. S7). The remaining ORI mapped to a very-late (S6)–replicating region, where inverted-V apexes cannot be identified (Supplemental Fig. S7). These results demonstrate that inverted-V apexes contain ORIs, as predicted, and suggest that this genome-wide association can be used to functionally validate our data set of putative ORIs.
Figure 3.

Genomic validation of newly identified ORIs. Visualization of HeLa Repli-seq in the UCSC Genome Browser showing three genomic regions: two (A and B) from chromosome 8 (adjacent regions; same y-axis value ranges) and one (C) from chromosome 6, spanning a total of 18.2 Mb. ORC1 peaks (black vertical lines), S1–S6 sequence-tag densities, the algorithmically identified inverted-V apexes (red filled boxes), the s50 profile (see Methods), and RefSeq genes are shown. (A) One representative inverted-V is shown as two green arrows (the right one also corresponds to a temporal transition region). (Open red circles) ORC1 peaks mapping outside inverted-V apexes. (Scale bars) 5 Mb. (D) Q-ChIP of ORC1 binding to the PRKDC ORI, the newly identified ORC1 peaks mapping to TSS of known genes (A–G) and two control regions (C1 and C2, as in Fig. 2A), using anti-ORC1 or anti-Flag (Mock) antibodies and chromatin of low-density fractions from human activated CD4+ T cells (see text). (Error bars) SD; n = 2. The Q-PCR probes are as in Figure 2A.
To computationally identify all inverted-V apexes, we divided the human genome into nonoverlapping 50-kb windows and measured, for each of them, the “replication time estimator” (s50 ratio), defined as the fraction of the S phase (time) at which 50% of the sequence reads of each window are obtained (Chen et al. 2010). Analysis of the HeLa s50 genomic profile identified 2204 inverted-V apexes (Fig. 3A–C; Supplemental Table S2). As expected, they showed different replication timing, with a higher proportion detected in the early S phase (∼59% in S1 + S2) (Supplemental Table S2). Strikingly, ∼96% of the identified S1 inverted-V apexes (P < 2.2 × 10−16) and ∼78% of all inverted-V apexes (P < 2.2 × 10−16) contained at least one ORC1 site (Supplemental Table S2). Vice versa, ∼86% of the S1 ORC1 sites (P = 6.19 × 10−7) and ∼66% of all the ORC1 sites (P = 6.19 × 10−7) mapped within inverted-V apexes (Supplemental Table S2). The remaining ∼34% of sites were located within late-replicating regions, or within regions that separate early- from late-replicating chromosome domains (temporal transition regions) (Fig. 3A–C). The overlap between ChIP-seq and Repli-seq data sets during middle (S3 + S4) and late (S5 + S6) S was also highly significant, but smaller (Supplemental Table S2), probably due to the intrinsic difficulty of distinguishing late origin-containing regions from progressing forks originating from nearby early ORIs. However, we also found local enrichments of SNSs at ORC1 sites replicating in middle or late S, including late ORIs mapping outside the ORI-containing regions (Fig. 2D; Supplemental Fig. S7).
Finally, we investigated whether the newly identified ORIs were also selected in other cell types, in particular, in cells of non-cancer origin. To address this question, we performed anti-ORC1 Q-ChIP of low-density chromatin fractions from activated CD4+ T cells purified from the peripheral blood of a healthy donor. In these cells, we observed ORC1 binding in six out of seven ORIs identified in HeLa cells (Fig. 3D), thus suggesting that the same ORIs can be activated both in normal and cancer-derived (HeLa) cells.
ORC1 sites are expressed at variable levels and associate with TSSs of coding or noncoding RNAs
We then annotated the position of the identified ORC1 sites with respect to known genes (RefSeq and UCSC), HeLa RNA transcripts (RNA-seq) (see Supplemental Material, Section 3), and functional TSSs (TSS-seq). The TSS-seq data set contains position and expression levels of TSSs from 12 different human cell types obtained by parallel sequencing of oligo-capped full-length cDNAs (Yamashita et al. 2011).
Approximately 35% of the ORC1 sites (n = 4,794) mapped within proximal promoters of known genes (±2.5 kb from TSS of RefSeq and UCSC genes: “proximal-promoter sites”) (Fig. 4A–D; Supplemental Table S3), with the peak summit usually corresponding to the TSS (Supplemental Fig. S8). The remaining ∼65% were nearly equally distributed within gene-free regions (intergenic sites) or gene bodies (outside proximal promoters; intragenic sites) (Fig. 4E–J and 4K–O, respectively; Supplemental Table S3).
Figure 4.

Characterization of the ORC1-associated RNAs. ChIP-seq profiles of ORC1, K4me3, and POLR2A at newly identified ORIs, mapping within proximal promoters of known genes (A–D), intergenic (E–J), or intragenic (K–O) regions. Overlap with TSSs identified in the TSS-seq data set or with statistically significant K4me3 or POLR2A peaks is indicated below each panel. y-axis value ranges, where not indicated, are 1–12 (ORC1); 1–30 (POLR2A and K4me3). RefSeq genes and RNA-seq tags are as of March 2011. Dashed lines in the RNA-seq track indicate picture clipping. (Scale bar) 5 kb.
Approximately 72% of all sites (n = 9808) were associated with RNA-seq tags (RNA-seq+), with decreasing frequencies from proximal-promoter to intragenic and intergenic sites (∼95%, ∼75%, and ∼45%, respectively) (Fig. 5; Supplemental Table S3). Strikingly, ∼78% of these RNA-seq+ ORC1 sites were also identified in the TSS-seq data set (TSS-seq+; n = 7624; ∼56% of all sites) (Fig. 5; Supplemental Table S3), including sites mapping to annotated TSSs (∼96% of the proximal-promoter sites) and to nonannotated TSSs (i.e., only present in the TSS-seq data set; ∼62% of the intragenic plus intergenic sites). The remaining ∼22% RNA-seq+ sites were not found in the TSS-seq data set (TSS-seq−; n = 2184; ∼16% of all sites) (Fig. 5; Supplemental Table S3), although a small fraction (n = 183) mapped to TSSs of known genes. Indeed, the TSS-seq data set did not include HeLa cells and contained many cell-type-specific TSSs, most of which were associated with low transcription levels (<5 RNA copies/cell) (Yamashita et al. 2011). Notably, almost all the RNAseq+/TSSseq− HeLa sites (1973/2184; ∼90%) fell within the same range of expression (Supplemental Table S4). Visual inspection of 156 randomly selected RNA-seq+/TSS-seq− sites revealed the presence of RNA-seq transcripts always mapping below or within 2.5 kb from an ORC1 peak, suggesting that they represent HeLa-specific TSSs (Fig. 4H–J,O).
Figure 5.

Association of ORC1 sites with RNA-seq tags and functional TSSs. Pie diagram showing numbers and percentages of ORC1 peaks overlapping, or not, with HeLa RNA-seq tags (RNA-seq+ or RNA-Seq−), or with TSSs identified in the TSS-seq data set (TSS-seq+ or TSS-seq−) (Yamashita et al. 2011) or in the RefSeq/UCSC data set (TSS-RefSeq+ or TSS-RefSeq−). TSS-seq+ peaks can be associated, or not, with annotated TSS (TSS-RefSeq±): Among the 7624 RNA-seq+/TSS-RefSeq+, 4377 are TSS-RefSeq+ and 3247 TSS-RefSeq−; among the 1169 RNA-seq−/TSS-seq+, 138 are TSS-RefSeq+ and 1031 TSS-RefSeq−.
In conclusion, ∼72% (n = 9808) of the identified ORC1 sites mapped to transcriptionally active TSSs, ∼22% of which were associated with HeLa-specific TSSs (n = 2184). Notably, only 46.5% (n = 4560) of all the RNA-seq+ ORC1 sites were associated with proximal or internal promoters of annotated genes, while the majority (53.5%; n = 5248) mapped to nonannotated transcriptionally active TSSs (Supplemental Table S3).
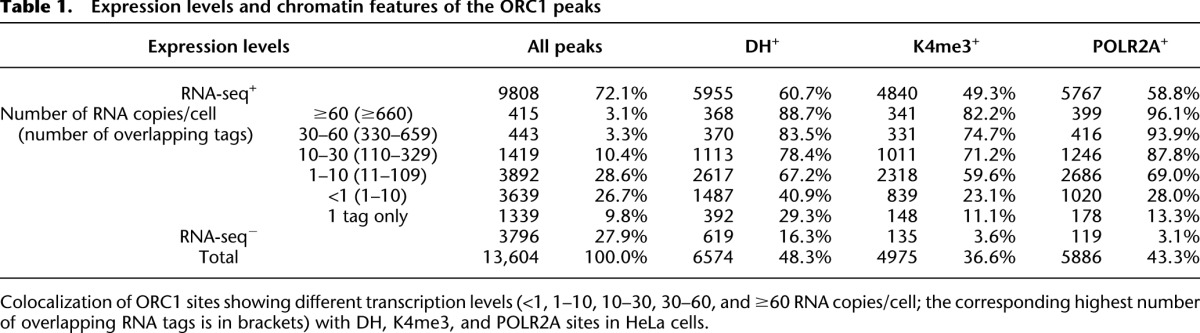
Since transcription is invariably associated with chromatin accessibility (Bell et al. 2011), we investigated the association of the RNA-seq+ ORC1 sites with DH sites, H3K4me3 deposition (K4), and POLR2A occupancy (Table 1). Surprisingly, only a fraction of the RNA-seq+ sites showed features of open chromatin: ∼61% overlapped with DH, ∼49% with K4, and ∼59% with POLR2A sites (P < 2.2 × 10−16 in all cases). Thus, we investigated whether chromatin accessibility correlates instead with levels of transcription at the ORC1 sites, measured as the highest number of overlapping RNA-seq tags within ±2.5 kb from the peak summit (Supplemental Fig. S9). Transcription levels at the ORC1 sites ranged from <1 up to a few thousand RNA copies/cell, as for the ribosomal protein genes (Table 1). Most of the ORC1 sites, however, were transcribed at relatively low levels: ∼55% showed <10 RNA copies/cell; in particular, ∼27% of all ORC1 sites showed <1 RNA copy/cell and ∼10% only 1 RNA-seq tag (Table 1). Among the most expressed ORC1 sites (>60 RNA copies/cell), ∼89%, ∼82%, and ∼96% colocalized with DH, K4, or POLR2A sites, respectively. These correlations, however, progressively weakened with decreasing transcription levels and were found in only a minority of the sites associated with 1 RNA-seq tag: ∼29% were DH+, ∼11% K4+, and ∼13% POLR2A+ (Table 1). Notably, visual inspection of ∼300 poorly transcribed sites showed that DH, K4, or POLR2A signals were frequently located immediately below our detection threshold or within ∼1–2 kb from the ORC1 peak with no overlap (Supplemental Fig. S10; data not shown). Together, these data demonstrate that levels of expression at ORC1 sites are highly variable, from <1 up to thousands of RNA copies/cell, and that the presence of detectable markers of open chromatin at ORIs correlate with expression levels.
Table 1.
Expression levels and chromatin features of the ORC1 peaks
The TSS-seq data set classifies >82% of the transcripts associated with intergenic TSSs as ncRNAs (Yamashita et al. 2011). Visual inspection (Fig. 4E–J) and sequence analysis (data not shown) of 300 RNA-seq+ randomly selected intergenic sites (including 198 TSS-seq+ and 102 TSS-seq− sites) confirmed the presence of short RNA transcripts (usually 200–300 nt) with the longest open reading frame (ORF) <100 amino acids, suggesting the presence of TSSs of ncRNAs. The same features were also observed in the transcripts of 207 of 300 randomly selected intragenic RNA-seq+ sites (Fig. 4M–O). Visual inspection of the other 93 intragenic sites revealed the presence of RNA-seq tags spliced to downstream coding exons, suggesting that they originated from internal unannotated TSSs of coding RNAs (Fig. 4K,L). In conclusion, almost all the intergenic and a large fraction of the intragenic ORC1 sites were associated with transcriptionally active TSSs of ncRNAs.
The remaining 3796 sites (∼28%) were not associated with RNA transcripts (RNA-seq−) (Fig. 5; Supplemental Table S3). However, one-third of them (n = 1265) mapped to functional TSSs (234 at proximal promoters, 475 within gene bodies, and 556 in intergenic regions) (Fig. 5; Supplemental Table S3), and a relatively small fraction colocalized with DH, K4, and POLR2A sites (∼16%, 4%, and 3%, respectively) (Table 1), suggesting that they might be expressed in HeLa at levels below RNA-seq sensitivity. Collectively, these data suggest that the association between ORC1 binding and transcriptional initiation is a universal feature of ORC1 sites, although levels of transcription differ significantly among them.
Transcription levels at ORC1 sites correlate with replication timing
We then investigated whether transcription levels at the ORC1 sites correlate with their replication timing. Replication timing was assigned to each site according to the s50 value of the corresponding 50-kb window. Of the 13,604 sites, ∼63% were early, ∼25% middle, and 12% late replicating (Supplemental Table S2). Comparison between replication timing and expression levels showed progressively lower transcription levels at ORC1 peaks firing later during S phase (Fig. 6A). However, analysis of replication timing within groups of sites with comparable expression showed that different levels of expression correspond to distinctive patterns of replication timing (Fig. 6B). The great majority of the highly (≥10 RNA copies/cell) and moderately (1–10 RNA copies/cell) transcribed sites replicated in early S (∼88% and ∼78%, respectively), with rare exceptions (∼1% and ∼3%, respectively, replicated in late S). Sites with low/undetectable expression (<1 RNA copy/cell), instead, replicated with similar frequencies during early or middle-late (S3–S6) S phase (∼47% vs. ∼53%, respectively) (Fig. 6B). Consistently, ORC1 sites with low/undetectable expression represented the vast majority (∼91%) of the ORIs firing in late S, while firing in early S was independent of expression levels (Fig. 6C). Thus, ORC1 sites with high/moderate expression (≥1 RNA copy/cell) fired earlier during the S phase, while late-firing ORIs invariably showed low/undetectable expression (<1 RNA copy/cell), suggesting that high/moderate expression at ORC1 sites correlates with early replication timing.
Figure 6.

Transcription levels at ORC1 peaks correlate with replication timing. (A) Boxplots of the transcription levels measured at ORC1 peaks replicating in the S1–S6 windows. (B) Frequency of early (S1 + S2), middle (S3 + S4), or late (S5 + S6) replicating ORC1 peaks within groups with homogenous expression (<1, 1–10, or ≥10 RNA copies/cell). (C) Frequency of ORC1 peaks with different transcription levels (<1, 1–10, or ≥10 RNA copies/cell) within groups of ORC1 sites homogeneous for replication timing. (D) Boxplots of the s50 values of ORC1 peaks with different transcription levels (<1, 1–10, or ≥10 RNA copies/cell) mapping within very-early-replicating (S1) inverted-V apexes. (E) Boxplots of the transcription levels measured at the ORC1 peaks mapping within proximal promoters of known genes, intragenic, or intergenic regions, as indicated. (F) Frequency of ORC1 peaks with different transcription levels (<1, 1–10, or ≥10 RNA copies/cell) for each genomic location, as indicated. (G) Frequency of early (S1 + S2), middle (S3 + S4), or late (S5 + S6) replicating ORC1 peaks for each genomic location, as indicated.
An apparent exception to this correlation is represented by the nearly half ORC1 sites with low/undetectable expression firing in early S (Fig. 6B). These sites, however, might represent ORIs that do not fire “autonomously” but that are “passively” activated by replicating forks that originate from sites with high/moderate transcription levels. This is consistent with the demonstration that progressing replication forks stimulate initiation in nearby unreplicated DNA (Lucas et al. 2000; Hyrien et al. 2003; Guilbaud et al. 2011). To test this hypothesis, we compared transcription levels and replication timing of the very early (S1) replicating ORC1 sites, by measuring the s50 values of ORC1 sites mapping within very early (S1) inverted-V apexes. Strikingly, while transcription levels increased, we observed a progressive decrease of the median s50 values of ORC1 peaks (i.e., a shift toward very early S) (Fig. 6D), with a highly significant difference between sites showing moderate (1–10 RNA copies/cell: median = 0.129) and high (≥10 RNA copies/cell: median = 0.123) transcription levels (P = 6.667 × 10−7). Thus, within early-replicating regions, the most expressed ORIs fire earlier than the least expressed, suggesting that (1) the latter are activated by the progressing replication forks; (2) ≥10 RNA copies/cell represents, in early S, the transcription level that distinguishes “autonomous” from “passively activated” ORIs. Taken together, these data support a model whereby ORC1 sites with high/moderate expression (≥1 RNA copy/cell) replicate in early S, while sites with low/undetectable expression (<1 RNA copy/cell) replicate in late S, unless activated by an incoming fork.
Finally, we investigated whether expression and replication timing correlate with the genomic position of the identified ORC1 sites. ORIs at the TSSs of proximal promoters were the most expressed (median expression: ∼5 RNA copies/cell) (Fig. 6E), with only ∼18% showing <1 RNA copy/cell (Fig. 6F). Intergenic sites, mostly mapping to TSSs of ncRNAs, were the least expressed, with ∼94% showing <1 RNA copy/cell (Fig. 6E,F). Intragenic sites, which mapped to TSSs of either coding RNAs or ncRNAs, showed intermediate expression levels (55% of sites with <1 RNA copy/cell) (Fig. 6E,F). Striking differences emerged when we compared the replication timing of the different classes: ∼97% of the promoter-associated sites replicated during early (∼80%) or middle (∼17%) S phase, while the intergenic sites fired almost uniformly throughout the entire S phase (∼46, ∼31, and ∼23% during early, middle, and late S phase, respectively) (Fig. 6G). The intragenic sites showed intermediate patterns of replication timing (Fig. 6G). In conclusion, these data demonstrate that the early-firing ORC1 sites are associated with highly expressed coding RNAs, and the late-firing sites with poorly expressed ncRNAs.
Discussion
This study reports the first genome-wide analysis in mammals of a component of the pre-RC. Our data set of ORC1 sites contains known ORIs, and, among the newly identified ones, several were validated for ORC1 and MCM5 binding and showed local enrichment of SNSs. Most notably, the majority of the identified ORC1 sites mapped within ORI-containing regions, as established by an independent genome-wide approach (the Repli-seq).
Previous attempts using antibodies against MCM or ORC and whole chromatin preparations were unsuccessful, probably owing to a lack of significant enrichment over background (Schepers and Papior 2010). This is also confirmed by our anti-ORC1 Q-ChIP analyses of four known ORIs in total versus fractionated HeLa chromatin, which showed significant ORC1 enrichment at the four ORIs only in the low-density chromatin fractions (Fig. 1F; Supplemental Fig. S4B). Most notably, anti-MCM5 ChIP-seq experiments allowed identification of genomic MCM5-binding sites, which largely overlap with the ORC1-binding sites (Supplemental Figs. S11, S12), further supporting our conclusion that the buoyant density of pre-RC-bound DNA is distinct from bulk chromatin and allows separation of ORI chromatin in equilibrium density centrifugation.
Previously reported genome-wide studies of human ORIs in HeLa were based on the mapping of early replication intermediates (Cadoret et al. 2008; Karnani et al. 2010) or restriction fragments containing replication bubbles (bubble-trap method) (Mesner et al. 2011) to the ENCODE genomic regions (∼1% of the human genome). These approaches, unfortunately, present several intrinsic difficulties, including purity and reproducibility of the nascent-strand DNA preparations (for review, see Hamlin et al. 2010) or low resolution of the bubble-trap method (for review, see Gilbert 2010). Accordingly, there was a modest overlap between the published data sets (ranging from ∼11% to 35%) (Supplemental Table S5), even when the same method was used in similar cells (<14%) (Gilbert 2010). The extent of overlap between the ORC1 peaks and each nascent-strand data set, or the bubble-trap data set, was also relatively modest: ∼11%–∼30% and ∼47%, respectively (Supplemental Table S5). Notably, however, when considering the replication timing of HeLa cells, the overlap with the nascent-strand or the bubble-trap data set in early S increased to 40% and ∼61%, respectively (Supplemental Table S6), suggesting that increased ORI efficiency and reduced heterogeneity with respect to ORI selection within cell populations (occurring in early S) allow easier identification of the same ORIs by different approaches.
Indirect evidence suggests that our data set of ORC1 sites does not contain all genomic ORIs. The number of ORC1 sites decreased progressively during the S phase (from 4097 in S1 to 721 in S6) (Supplemental Table S2). However, the interorigin spacing within the S1 inverted V-apexes is identical to that reported in HeLa cells (Supplemental Fig. S13A; Guilbaud et al. 2011), suggesting that the density of ORC1 sites in early S is consistent with the replication kinetics of HeLa cells. As the S phase progresses, instead, the interorigin spacing gradually increases (Supplemental Fig. S13A) due to the decreasing number of identified ORC1 sites, thus implying a parallel increase in DNA-polymerase processivity. Since interorigin spacing (∼30 kb) and fork velocity (∼0.7 kb/min) do not change in HeLa cells during S phase (Guilbaud et al. 2011), these data suggest that our anti-ORC1 ChIP-seq failed to identify a significant fraction of the late-S ORC1 sites. This observation might also apply to the Repli-seq, since the number of inverted V-apexes decreased progressively during S phase (from 568 in S1 to 229 in S5) (Supplemental Table S2).
Recent work emphasizes the highly variable percentage of cells in which a given ORI is, respectively, selected and then activated (so-called ORI efficiency: 5%–20% in metazoans) (Gilbert 2010; Tuduri et al. 2010). Consequently, success in identifying individual ORIs (by anti-ORC1 ChIP-seq) and ORI-containing regions (by Repli-seq) might largely depend on ORI efficiency. Since ORI efficiency negatively correlates with replication timing (Luo et al. 2010), anti-ORC1 ChIP-seq and Repli-seq analyses (or any other approach that uses populations of cells) might fail to detect the least-efficient and latest-firing ORIs. This is supported by three observations: (1) The amplitude (or height, h) of the ORC1 peaks, although highly heterogeneous (from 4 to 174), progressively decreased from the S1 to the S6 (Supplemental Fig. S13B). (2) The overlap between two anti-ORC1 ChIP-seq replicates in HeLa cells was ∼60%, yet peak amplitude was significantly higher in the common peaks (Supplemental Fig. S5B). (3) The overlap between ORC1 peaks and the published ORI data sets decreased during the S-phase progression (Supplemental Table S6).
We characterized the genomic positions of ORC1 sites and found a significant association with known genes and typical markers of transcription (POLR2A) or open chromatin (H3K4me3 and DH sites). However, this association held only for subsets of the ORC1 sites, as previously reported for known ORIs (Mechali 2010). Comparison with data sets of transcripts in HeLa cells (RNA-seq) and functional TSSs (TSS-seq), instead, suggests that association with active TSSs, within or outside annotated genes, represents a universal feature of ORC1 sites. We documented this association in ∼72% of the ORC1 sites, in which the associated transcripts were either found at previously identified TSSs or originated from within the summit of the ORC1 peaks. The same might also be true for the remaining 28%, where the absence of associated transcripts might be due to very low expression, below the sensitivity of RNA-seq. Indeed, ∼33% of the nonexpressed ORC1 sites mapped to known TSSs.
Although these observations do not clarify the functional significance of the association between transcription and replication, they suggest new scenarios. Up to now, promoters were thought to be associated with a relatively small subset of ORIs and to recruit the pre-RC through open chromatin or interaction with transcriptional factors. Since nearly half of the RNA-seq+ ORC1 sites were not associated with DH sites or K4 (Table 1), a stable open-chromatin structure might not necessarily be a prerequisite for ORC1 recruitment at expressed promoters. Transcription initiation, however, might favor recruitment of the pre-RC complex by generating transient states of accessibility to the transcription bubble itself. This mechanism might be critical for those genomic regions characterized by very infrequent transcription-initiation events and no detectable markers of open chromatin. Alternatively, transcription initiation (the transcriptional machinery itself or the nascent transcript) might be mechanistically linked to the initiation of DNA replication, as suggested (Hassan et al. 1994). Notably, inhibiting the recruitment of a multiprotein complex containing both transcription and replication factors to the human beta-globin locus control region prevents both its transcription and DNA replication (Karmakar et al. 2010). On the other hand, the nascent RNA itself might be involved in DNA replication (Mechali 2010): (1) The ORC is recruited to the ORI of the Epstein-Barr virus by an RNA-dependent interaction. (2) In Tetrahymena, rDNA amplification is regulated by ORC recruitment through a noncoding RNA (Mohammad et al. 2007). (3) In vertebrates, a specific class of RNAs (Y RNAs) has been implicated in replication initiation (Collart et al. 2011).
Regardless of the role of transcription initiation in the recruitment of the pre-RC and/or in the initiation of DNA replication, our data suggest a direct relationship between transcription levels at ORC1 sites and replication timing. A correlation between early replication and gene expression within large replicating domains has been previously reported (Maric and Prioleau 2010). However, the high number of exceptions argues against a direct relationship. For instance, in Drosophila and mouse, 10%–20% of expressed genes are late replicating, while ∼50% of nonexpressed genes are early replicating. On the contrary, here we showed that ∼88% of highly expressed (∼17% of all sites) and ∼78% of the moderately expressed (∼29% of all sites) ORC1 sites were early replicating, with only ∼1% and ∼3% being late replicating, respectively. Since the great majority (∼95%) of the highly/moderately expressed ORC1 sites were associated with annotated genes (proximal or intragenic TSSs), these data suggest that the reported incomplete association between gene expression and early replication is due to selection by ORC1 of a subset of highly/moderately expressed TSSs, and their constant activation during the early S phase. Expectedly, these ORC1 sites also showed the highest amplitude (Supplemental Fig. S13C), thus suggesting that they also correspond to the most efficient ORIs.
The remaining ORC1 sites (n = 7435, ∼55% of all sites) were all expressed at very low levels (<1 RNA copy/cell) and most frequently located at TSSs of ncRNAs, within genes or intergenic regions. Unlike the other ORC1 sites, they replicated throughout the entire S phase. Notably, these sites were only occasionally associated with open-chromatin markers (28% colocalized with DH sites, as opposed to 72% for the sites with ≥1 RNA copy/cell), and their amplitude was significantly lower (Supplemental Fig. S13C).
In summary, our data suggest the existence of two classes of ORIs: (1) Those that map to the TSSs of coding genes, are associated with moderate/high transcription levels (≥1 RNA copy/cell), and fire early during the S phase, likely representing the most-efficient ORIs; and (2) those that map to the TSSs of noncoding genes, are associated with very low transcription levels (<1 RNA copy/cell), and fire throughout the entire S, likely representing the least-efficient sites. These findings are compatible with a scenario whereby TSS expression levels influence the efficiency of pre-RC recruitment during G1 phase and the probability of firing during the subsequent S phase. Thus, the ORC1 sites associated with highly/moderately expressed coding RNAs would have a higher probability of firing (in a cell population) than those associated with poorly expressed ncRNAs. Yet, the least-expressed sites may be activated in early S by the incoming forks, while in late S, they are the only option available for replication initiation within gene-free or transcriptionally silenced regions. Notably, accumulating evidence suggests that transcription occurs in “factories” throughout interphase and that most of transcriptional initiation events are abortive (for review, see Mellor 2010). Thus, one can speculate that replication begins (at the G1/S border) at the TSSs of the most transcribed genes in these transcription factories, thus conferring early-firing properties to the whole locus, including the ORC1-bound TSSs of poorly expressed RNAs.
The molecular mechanisms that regulate selectivity of ORC1 recruitment to DNA remain unknown. Clearly, transcription per se is not sufficient for ORC1 recruitment, because ORC1 binds to only ∼39% of the annotated TSSs that are expressed in HeLa cells (Supplemental Fig. S14). Notably, the low-density chromatin fractions contain also the active promoters of HeLa, regardless of their binding to ORC1 (data not shown). Our preliminary analysis of the DNA sequences at ORC1 sites confirms the absence of obvious consensus sequences (data not shown). Thus, the selection of a subset of active TSSs might depend on their transcriptional activity during the time window of G1 phase at which ORC1 binds to chromatin, or might be conferred on ORC1 by other nuclear factors, which might also contribute to cell-type-specific activation of potential ORIs, as proposed (DePamphilis et al. 2006). The availability of robust assays, such as anti-ORC1 ChIP-seq, will allow analysis of ORIs in different cell types (both normal and cancer derived), significantly contributing to the resolution of this issue.
Methods
Cross-linking, sonication, density centrifugation, and analysis of fractions
HeLa S3 cells were grown to a density of 6 × 105 cells/mL in DMEM medium. Formaldehyde was added to the culture medium to a final concentration of 1% (4 min at room temperature). Cross-linking was stopped by addition of glycine to a final concentration of 125 mM. Cells were washed twice with PBS and lysed in SDS buffer: 100 mM NaCl, 50 mM Tris HCl (pH 8.1), 5 mM EDTA (pH 8), 0.5% SDS, and protease inhibitors. Chromatin lysate was then pelleted and washed once in IP buffer (100 mM NaCl, 100 mM Tris HCl at pH 8.1, 5 mM EDTA at pH 8, 0.3% SDS, 1.7% Triton X-100) and twice in sonication buffer (10 mM HEPES at pH 7.6, 1 mM EDTA, 0.5 mM EGTA, protease inhibitors). The cells were resuspended in sonication buffer to a concentration of 3 × 107 cells/mL. Sonication, sample preparation for equilibrium density centrifugation, dialysis of collected fractions, and analysis of DNA or protein content of each fraction were performed as previously described (Schwartz et al. 2005), with minor modifications. The sample volume was adjusted to 12 mL before centrifugation, transferred into a 14 × 89-mm Beckman Ultra-Clear centrifuge tube and spun for 120–144 h at +20°C (34,000 rpm in a Sorvall S52-ST rotor); 750-μL fractions were collected with a peristaltic pump from the top of the tube. The antibodies used for Western blot analyses of different Pre-RC proteins were the following: anti-ORC1 (Mendoza-Maldonado et al. 2010), anti-ORC2 (upstate 05-936), anti-MCM2 (ab4461), anti-MCM5 (ab17967), and anti-MCM7 (sc-9966).
Analysis of the distribution of DNA fragments along the gradient
The amount of a DNA fragment in each gradient fraction was determined by real-time PCR amplification using specific oligonucleotide primers and expressed as the percentage of the amount of the amplified DNA fragment from input DNA, prior to fractionation (“a” value). The amount of genomic DNA within each fraction was determined by measuring the DNA concentration of each fraction, and expressed as the percentage of the total DNA in the gradient (“b” value). The a:b ratio is indicated as “fold enrichment” in Figures 1D and 2C.
Isolation of human CD4+ T cells
Human CD4+ T cells were purified from peripheral blood mononuclear cells by negative selection with the CD4 T-Cell Isolation Kit (Miltenyi Biotec) and subsequently stimulated by immobilized anti-human CD3 mAb and soluble anti-human CD28 mAb. After 72 h, the cells were expanded in complete medium supplemented with recombinant human IL-2, and then processed like HeLa S3 cells.
ChIP assays
HeLa S3 cells were grown and processed for ORC1 and MCM5 Q-ChIP analyses as described above, using chromatin from dyalized low-density fractions: 80–100 μL/IP were adjusted to 1 mL with IP buffer prior to overnight incubation with anti-ORC1 or anti-MCM5 antibodies. For ORC1, MCM5, H3K4me3, and K79me2 Q-ChIP analyses of total chromatin, cells were lysed in SDS buffer and sonicated directly in IP buffer prior to overnight incubation with antibodies (anti-K4me3: Active Motif 39159; anti-K79me2: ab3594).
NSA assay
Genomic DNA was isolated as described (Rowntree and Lee 2006). Briefly, cells (1 × 106 cells/mL) were lysed with 0.4% NP-40 in RSB buffer. Purified nuclei (2.5 × 106 nuclei/mL) were digested overnight at 37°C in RSB with 0.25 mg/mL proteinase K, and total genomic DNA was phenol-extracted. The NSA assay was performed as described (Giacca et al. 1997). Nascent strands abundance was determined by real-time PCR and expressed as relative enrichment, normalized to the PRKDC ORI (Sibani et al. 2008).
Mapping of sequence reads and peak analysis
Data sets of RefSeq genes, DH sites, POLR2A, TSS-seq, and RNA-seq were obtained from the UCSC Genome Browser (links in Supplemental Material, Section 3). Illumina single-end sequencing reactions (36 nt) were performed with gel-excised DNA fragments (∼200 bp in length). Alignments were performed with bwa (version 0.5.7) (Li and Durbin 2010) to hg18 using default parameters. Reads for H3K4me3 were trimmed to 36 bp before alignment, using the FASTX-Toolkit (http://hannonlab.cshl.edu/fastx_toolkit/). Profiles for ChIP-seq data were generated using BEDtools v 2.7.1 (Quinlan and Hall 2010). Aligned data were analyzed with FindPeaks (FP4, versions 4.0.10–4.0.13) (Fejes et al. 2008). Parameters common to all analyses were the following: (1) “-dist_type 1 200” (triangular distribution model for fragment size, median fragment size = 200); (2) “-qualityfilter 1” (minimum mapping quality = 1); (3) “-duplicatefilter.” In addition, high-density fraction (14–16) data were analyzed using the “-subpeaks 0.2 option.”
ORC1 and high-density fraction (14–16) data were analyzed using the corresponding Input DNA as control. The H3K4me3 and POLR2A data sets were analyzed using FP4 with Montecarlo (MC) simulation with five iterations (“-iterations 5,” “-eff_frac 0.75”). The following filters were applied to the final files containing peaks: (1) P ≤ 0.01; (2) height as the value at which MC FDR = 0.
HeLa S3 Repli-seq analysis
The HeLa S3 Repli-seq procedure was performed as described (Hansen et al. 2010), with additional IPs using labeled unsorted cells as controls. To enhance the signal-to-noise ratio deriving from BrdU-positive regions, the data from all the anti-BrdU IPs (S1–S6 compartments and control) were processed with dspchip (v0.8.5, http://code.google.com/p/dspchip) (Fumagalli et al. 2012) as follows: Nonduplicated sequence-read tags with mapping quality higher than 15 were aligned to the human genome and used to compute raw profiles (options: “-nodup” and “-q 15”), which were then normalized using dspchip normalization facility (i.e., Power Normalization). The control signal was subtracted from the signal of each S-phase window (S1–S6), and the resulting six profiles were filtered using a Hanning Window low-pass filter (50 kb wide); negative values were discarded and set to 0 (option “-pl=NSFZ”).
Computation of s50 values and identification of inverted-V apexes
Each of the six (S1–S6) Repli-seq profiles (obtained with dspchip) was binned using 50-kb nonoverlapping intervals, and, for each bin, the mean value of the underlying signal was calculated. The signals deriving from each of the six compartments in which the S phase was divided (i.e., the signal in S1 indicates the amount of replicated DNA at 15% of S phase, the signal in S2 the amount of replicated DNA between 15% and 30% of S phase, etc.) were used to calculate the final (M: S1 + S2 + S3 + S4 + S5 + S6) and intermediate cumulative sums of bins (i.e., S1, S1 + S2, S1 + S2 + S3, etc.). The s50 value, i.e., the fraction of S phase at which 50% of the DNA of each bin (M/2 value) is replicated, was calculated by linear interpolation of the two cumulative sums closest to the M/2 value. The resulting s50 profile was smoothed using a Gaussian kernel (σ = 3.5). Local minima and flanking inflection points (defining the boundaries of the associated inverted-V apexes) were identified using a Sobel operator, included in scientific python (http://www.scipy.org). Adjacent inverted-V apexes within 50 kb were merged.
Statistical analyses are described in the Supplemental Material, Section 3.
Data access
Sequencing data have been submitted to the NCBI Gene Expression Omnibus (GEO) (http://www.ncbi.nlm.nih.gov/geo/) under accession no. GSE37583.
Acknowledgments
We thank L. Rotta and M. Riboni of the Sequencing Facility at the IFOM-IEO Campus; L. Mazzarella for help in the initial setup of Repli-seq; G. Penna and E. Mileti for help in the isolation of human T cells; H. Stunnenberg and E. Megens for parallel sequencing of anti-MCM5 ChIP; E. Colombo, M. Faretta, and L. Reavie for helpful discussions; and G. Natoli and P. Dalton for critical review of the manuscript. This study was supported by grants from AIRC, EC (EPITRON 518417, BLUEPRINT 282510), MIUR, and the Ministry of Health.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.142331.112.
References
- Bell SP, Stillman B 1992. ATP-dependent recognition of eukaryotic origins of DNA replication by a multiprotein complex. Nature 357: 128–134 [DOI] [PubMed] [Google Scholar]
- Bell O, Tiwari VK, Thoma NH, Schubeler D 2011. Determinants and dynamics of genome accessibility. Nat Rev Genet 12: 554–564 [DOI] [PubMed] [Google Scholar]
- Cadoret JC, Meisch F, Hassan-Zadeh V, Luyten I, Guillet C, Duret L, Quesneville H, Prioleau MN 2008. Genome-wide studies highlight indirect links between human replication origins and gene regulation. Proc Natl Acad Sci 105: 15837–15842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cayrou C, Coulombe P, Vigneron A, Stanojcic S, Ganier O, Peiffer I, Rivals E, Puy A, Laurent-Chabalier S, Desprat R, et al. 2011. Genome-scale analysis of metazoan replication origins reveals their organization in specific but flexible sites defined by conserved features. Genome Res 21: 1438–1449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CL, Rappailles A, Duquenne L, Huvet M, Guilbaud G, Farinelli L, Audit B, d'Aubenton-Carafa Y, Arneodo A, Hyrien O, et al. 2010. Impact of replication timing on non-CpG and CpG substitution rates in mammalian genomes. Genome Res 20: 447–457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collart C, Christov CP, Smith JC, Krude T 2011. The midblastula transition defines the onset of Y RNA-dependent DNA replication in Xenopus laevis. Mol Cell Biol 31: 3857–3870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePamphilis ML, Blow JJ, Ghosh S, Saha T, Noguchi K, Vassilev A 2006. Regulating the licensing of DNA replication origins in metazoa. Curr Opin Cell Biol 18: 231–239 [DOI] [PubMed] [Google Scholar]
- Eaton ML, Galani K, Kang S, Bell SP, MacAlpine DM 2010. Conserved nucleosome positioning defines replication origins. Genes Dev 24: 748–753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eaton ML, Prinz JA, MacAlpine HK, Tretyakov G, Kharchenko PV, MacAlpine DM 2011. Chromatin signatures of the Drosophila replication program. Genome Res 21: 164–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fejes AP, Robertson G, Bilenky M, Varhol R, Bainbridge M, Jones SJ 2008. FindPeaks 3.1: A tool for identifying areas of enrichment from massively parallel short-read sequencing technology. Bioinformatics 24: 1729–1730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fumagalli M, Rossiello F, Clerici M, Barozzi S, Cittaro D, Kaplunov JM, Bucci G, Dobreva M, Matti V, Beausejour CM, et al. 2012. Telomeric DNA damage is irreparable and causes persistent DNA-damage-response activation. Nat Cell Biol 14: 355–365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giacca M, Pelizon C, Falaschi A 1997. Mapping replication origins by quantifying relative abundance of nascent DNA strands using competitive polymerase chain reaction. Methods 13: 301–312 [DOI] [PubMed] [Google Scholar]
- Gilbert DM 2010. Evaluating genome-scale approaches to eukaryotic DNA replication. Nat Rev Genet 11: 673–684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilbaud G, Rappailles A, Baker A, Chen CL, Arneodo A, Goldar A, d'Aubenton-Carafa Y, Thermes C, Audit B, Hyrien O 2011. Evidence for sequential and increasing activation of replication origins along replication timing gradients in the human genome. PLoS Comput Biol 7: e1002322 doi: 10.1371/journal.pcbi.1002322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamlin JL, Mesner LD, Dijkwel PA 2010. A winding road to origin discovery. Chromosome Res 18: 45–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen RS, Thomas S, Sandstrom R, Canfield TK, Thurman RE, Weaver M, Dorschner MO, Gartler SM, Stamatoyannopoulos JA 2010. Sequencing newly replicated DNA reveals widespread plasticity in human replication timing. Proc Natl Acad Sci 107: 139–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassan AB, Errington RJ, White NS, Jackson DA, Cook PR 1994. Replication and transcription sites are colocalized in human cells. J Cell Sci 107: 425–434 [DOI] [PubMed] [Google Scholar]
- Hiratani I, Ryba T, Itoh M, Yokochi T, Schwaiger M, Chang CW, Lyou Y, Townes TM, Schubeler D, Gilbert DM 2008. Global reorganization of replication domains during embryonic stem cell differentiation. PLoS Biol 6: e245 doi: 10.1371/journal.pbio.0060245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyrien O, Marheineke K, Goldar A 2003. Paradoxes of eukaryotic DNA replication: MCM proteins and the random completion problem. Bioessays 25: 116–125 [DOI] [PubMed] [Google Scholar]
- Ip YT, Jackson V, Meier J, Chalkley R 1988. The separation of transcriptionally engaged genes. J Biol Chem 263: 14044–14052 [PubMed] [Google Scholar]
- Karmakar S, Mahajan MC, Schulz V, Boyapaty G, Weissman SM 2010. A multiprotein complex necessary for both transcription and DNA replication at the β-globin locus. EMBO J 29: 3260–3271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karnani N, Taylor CM, Malhotra A, Dutta A 2010. Genomic study of replication initiation in human chromosomes reveals the influence of transcription regulation and chromatin structure on origin selection. Mol Biol Cell 21: 393–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladenburger EM, Keller C, Knippers R 2002. Identification of a binding region for human origin recognition complex proteins 1 and 2 that coincides with an origin of DNA replication. Mol Cell Biol 22: 1036–1048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R 2010. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26: 589–595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubelsky Y, Sasaki T, Kuipers MA, Lucas I, Le Beau MM, Carignon S, Debatisse M, Prinz JA, Dennis JH, Gilbert DM 2011. Pre-replication complex proteins assemble at regions of low nucleosome occupancy within the Chinese hamster dihydrofolate reductase initiation zone. Nucleic Acids Res 39: 3141–3155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas I, Chevrier-Miller M, Sogo JM, Hyrien O 2000. Mechanisms ensuring rapid and complete DNA replication despite random initiation in Xenopus early embryos. J Mol Biol 296: 769–786 [DOI] [PubMed] [Google Scholar]
- Luo H, Li J, Eshaghi M, Liu J, Karuturi RK 2010. Genome-wide estimation of firing efficiencies of origins of DNA replication from time-course copy number variation data. BMC Bioinformatics 11: 247 doi: 10.1186/1471-2105-11-247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacAlpine HK, Gordan R, Powell SK, Hartemink AJ, MacAlpine DM 2010. Drosophila ORC localizes to open chromatin and marks sites of cohesin complex loading. Genome Res 20: 201–211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maric C, Prioleau MN 2010. Interplay between DNA replication and gene expression: A harmonious coexistence. Curr Opin Cell Biol 22: 277–283 [DOI] [PubMed] [Google Scholar]
- Martin MM, Ryan M, Kim R, Zakas AL, Fu H, Lin CM, Reinhold WC, Davis SR, Bilke S, Liu H, et al. 2011. Genome-wide depletion of replication initiation events in highly transcribed regions. Genome Res 21: 1822–1832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mechali M 2010. Eukaryotic DNA replication origins: Many choices for appropriate answers. Nat Rev Mol Cell Biol 11: 728–738 [DOI] [PubMed] [Google Scholar]
- Mellor J 2010. Transcription: From regulatory ncRNA to incongruent redundancy. Genes Dev 24: 1449–1455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendoza-Maldonado R, Paolinelli R, Galbiati L, Giadrossi S, Giacca M 2010. Interaction of the retinoblastoma protein with Orc1 and its recruitment to human origins of DNA replication. PLoS ONE 5: e13720 doi: 10.1371/journal.pone.0013720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesner LD, Valsakumar V, Karnani N, Dutta A, Hamlin JL, Bekiranov S 2011. Bubble-chip analysis of human origin distributions demonstrates on a genomic scale significant clustering into zones and significant association with transcription. Genome Res 21: 377–389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohammad MM, Donti TR, Sebastian Yakisich J, Smith AG, Kapler GM 2007. Tetrahymena ORC contains a ribosomal RNA fragment that participates in rDNA origin recognition. EMBO J 26: 5048–5060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM 2010. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 26: 841–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reneker JS, Brotherton TW 1991. Discrete regions of the avian β-globin gene cluster have tissue-specific hypersensitivity to cleavage by sonication in nuclei. Nucleic Acids Res 19: 4739–4745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowntree RK, Lee JT 2006. Mapping of DNA replication origins to noncoding genes of the X-inactivation center. Mol Cell Biol 26: 3707–3717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryba T, Hiratani I, Lu J, Itoh M, Kulik M, Zhang J, Schulz TC, Robins AJ, Dalton S, Gilbert DM 2010. Evolutionarily conserved replication timing profiles predict long-range chromatin interactions and distinguish closely related cell types. Genome Res 20: 761–770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schepers A, Papior P 2010. Why are we where we are? Understanding replication origins and initiation sites in eukaryotes using ChIP-approaches. Chromosome Res 18: 63–77 [DOI] [PubMed] [Google Scholar]
- Schwartz YB, Kahn TG, Pirrotta V 2005. Characteristic low density and shear sensitivity of cross-linked chromatin containing Polycomb complexes. Mol Cell Biol 25: 432–439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sequeira-Mendes J, Diaz-Uriarte R, Apedaile A, Huntley D, Brockdorff N, Gomez M 2009. Transcription initiation activity sets replication origin efficiency in mammalian cells. PLoS Genet 5: e1000446 doi: 10.1371/journal.pgen.1000446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sibani S, Rampakakis E, Di Paola D, Zannis-Hadjopoulos M 2008. Fine mapping and functional activity of the adenosine deaminase origin in murine embryonic fibroblasts. J Cell Biochem 104: 773–784 [DOI] [PubMed] [Google Scholar]
- Solomon MJ, Larsen PL, Varshavsky A 1988. Mapping protein–DNA interactions in vivo with formaldehyde: Evidence that histone H4 is retained on a highly transcribed gene. Cell 53: 937–947 [DOI] [PubMed] [Google Scholar]
- Tuduri S, Tourriere H, Pasero P 2010. Defining replication origin efficiency using DNA fiber assays. Chromosome Res 18: 91–102 [DOI] [PubMed] [Google Scholar]
- Valenzuela MS, Chen Y, Davis S, Yang F, Walker RL, Bilke S, Lueders J, Martin MM, Aladjem MI, Massion PP, et al. 2011. Preferential localization of human origins of DNA replication at the 5′-ends of expressed genes and at evolutionarily conserved DNA sequences. PLoS ONE 6: e17308 doi: 10.1371/journal.pone.0017308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varshavsky AJ, Bakayev VV, Ilyin YV, Bayev AA Jr, Georgiev GP 1976. Studies on chromatin. Free DNA in sheared chromatin. Eur J Biochem 66: 211–223 [DOI] [PubMed] [Google Scholar]
- Yamashita R, Sathira NP, Kanai A, Tanimoto K, Arauchi T, Tanaka Y, Hashimoto S, Sugano S, Nakai K, Suzuki Y 2011. Genome-wide characterization of transcriptional start sites in humans by integrative transcriptome analysis. Genome Res 21: 775–789 [DOI] [PMC free article] [PubMed] [Google Scholar]