

Abstract
The gastrointestinal microbiome undergoes shifts in species and strain abundances, yet dynamics involving closely related microorganisms remain largely unknown because most methods cannot resolve them. We developed new metagenomic methods and utilized them to track species and strain level variations in microbial communities in 11 fecal samples collected from a premature infant during the first month of life. Ninety six percent of the sequencing reads were assembled into scaffolds of >500 bp in length that could be assigned to organisms at the strain level. Six essentially complete (∼99%) and two near-complete genomes were assembled for bacteria that comprised as little as 1% of the community, as well as nine partial genomes of bacteria representing as little as 0.05%. In addition, three viral genomes were assembled and assigned to their hosts. The relative abundance of three Staphylococcus epidermidis strains, as well as three phages that infect them, changed dramatically over time. Genes possibly related to these shifts include those for resistance to antibiotics, heavy metals, and phage. At the species level, we observed the decline of an early-colonizing Propionibacterium acnes strain similar to SK137 and the proliferation of novel Propionibacterium and Peptoniphilus species late in colonization. The Propionibacterium species differed in their ability to metabolize carbon compounds such as inositol and sialic acid, indicating that shifts in species composition likely impact the metabolic potential of the community. These results highlight the benefit of reconstructing complete genomes from metagenomic data and demonstrate methods for achieving this goal.
The gut microbiome plays a critical role in determining human health. Gut microbes influence immune system development and function (Maslowski et al. 2009; Lathrop et al. 2011), process nutrients, and affect energy uptake by the host (Hooper and Gordon 2001; Ley et al. 2005). Inflammatory bowel disease (Xavier and Podolsky 2007) and necrotizing enterocolitis (Fell 2005) have been linked to abnormalities in the composition of the gut microbiome and inappropriate activation of the immune system responses directed against the gut microbiome. The vast majority of the human microbiota reside in the gastrointestinal (GI) tract (Savage 1977). The genomes of these microorganisms encode more than 3 million unique genes, more than two orders of magnitude larger than the number of genes in the human genome (Qin et al. 2010). More than 1000 species have been detected in the gut, with representatives from at least nine bacterial phyla and about 150 dominant species in the gut of each individual (Eckburg et al. 2005; Ley et al. 2006; Qin et al. 2010).
Various factors may cause rapid changes within the gut microbial community, including the availability of nutrients, drug consumption, phage blooms, and the presence of opportunistic pathogens (Reyes et al. 2010; Dethlefsen and Relman 2011; Fukuda et al. 2011; Gophna 2011). These changes may be particularly significant during the initial microbial colonization of the infant gut when the community is usually comparatively simple, sometimes consisting of only a few dominant species (Koenig et al. 2011; Morowitz et al. 2011). It has been previously suggested that biodiversity, which is particularly manifest at the species and strain level in the gut microbiome, can maintain or enhance ecosystem functioning in the face of environmental change (Backhed et al. 2005). Several studies published in recent years have demonstrated the significant functional differences associated with strain variation in medically relevant commensals. For example, only certain strains of Bifidobacterium longum were shown to provide protection against pathogens such as Escherichia coli due to the presence of genes encoding ATP-binding-cassette-type carbohydrate transporters (Fukuda et al. 2011). Virulence of Staphylococcus epidermidis varies among different strains (Gill et al. 2005) as does the ability to inhibit Staphylococcus aureus biofilm formation (Iwase et al. 2010).
As the significant role of species and strain level variation is increasingly recognized, it is clear that a better understanding of these variations in natural environments will enhance our understanding of the dynamics of microbial communities. Unfortunately, techniques used to date fall short of providing the strain-level resolution required for a comprehensive description of microbial communities. 16S rRNA gene surveys are widely used for characterizing microbial communities (Palmer et al. 2007; Mshvildadze et al. 2010; Jacquot et al. 2011; LaTuga et al. 2011; Mai et al. 2011) and may distinguish different species. Sequencing of microbial genomic DNA recovered from microbial community samples has the potential to provide information at the strain level. However, metagenomic analysis of unassembled or partially assembled data cannot provide information about the role of individual community members. In contrast, assembled and well-curated genomes reconstructed from such samples can provide metabolic insight at the species and strain levels.
In a recent paper (Morowitz et al. 2011), metagenomic data were used to provide a detailed description of a microbial community during gut colonization. Using 245 Mbp of pyrosequencing data, it was possible to reconstruct, de novo, the genomes of the dominant Serratia and Citrobacter species and a few plasmids and bacteriophages. The study revealed strain-level genotypic features that differentiated two Citrobacter strains and that may have explained strain abundance fluctuations over time. The study was limited in its ability to characterize less abundant organisms, yet, these organisms may have major significance for overall community function. The Illumina sequencing technology offers new opportunities for comprehensive study of microbial communities and the forces that govern their development. The orders-of-magnitude-greater amount of data from this technology make longitudinal time series studies with deep sampling of rare members possible, thus offering opportunities for the in-depth study of microbial community dynamics. Subject to sufficient sequencing coverage, the availability of accurate methods for the assembly of short read sequencing data, as well as methods for binning with fine resolution and high sensitivity, it should be possible to reconstruct complete genomes of all members in the community and to identify factors that explain their abundance.
In this paper, we present a time series study of relatively low complexity gut microbiome samples from a newborn premature infant. We developed novel approaches for sequence assembly and binning, and in so doing, were able to reconstruct complete and partial genome sequences of organisms with relative abundance as low as 0.05% of the community. This outcome indicates that metagenomic data have essentially the same potential to yield near-complete genomes as data from cultivated isolates. Our results provide insight into the metabolic differences associated with species and strain shifts and evidence for phage-based control of the strain composition of medically important species.
Results
Sample collection
We studied 11 fecal samples collected on post-natal days 15–24 from a female infant delivered by Caesarean section at 26 wk of gestation. Fecal samples were collected once or twice daily, as available, between days 15 and 24. This time frame was selected for study because it represents a critical colonization period (and one during which abnormalities can lead to neonatal necrotizing enterocolitis, although the latter did not develop in this infant) (Morowitz et al. 2010). See Methods for further details.
Microbial genome reconstruction
Raw data for this study consisted of ∼260 million 100-bp paired-end Illumina HiSeq reads from libraries with insert sizes of 400 (four samples) and 900 (seven samples) bp from the samples collected at the 11 time points (∼2.4 Gbp per sample) (see details in Supplemental Table S1). A novel pipeline was developed for analyzing the data in this study (Fig. 6, see below). The pipeline was designed to reconstruct high-quality complete genomes for those with coverage sufficient to ensure complete sampling and to assign all genome fragments longer than 500 bp to organisms accurately. A detailed description of the methods is provided in the Methods section and supporting online material.
We developed a new iterative approach in which different groups of genomes with similar levels of abundance are assembled simultaneously, separately from the rest of the data. This approach largely circumvents the complication that single genome assemblers (e.g., Velvet) (Zerbino and Birney 2008) encounter when mixtures of genomes with different levels of coverage are assembled using parameters suitable for just one coverage level. Data from all samples were coassembled together. We also developed new high-throughput processes for detecting assembly errors, removing gaps from the assembly, and a program for post-assembly manual genome curation that guides manual inspection to improve assemblies (e.g., where strain variation splits contigs). Typically, assembly errors in this study involved inappropriate linkage between fragments from the same genome, not chimeras involving genome segments from multiple organisms.
A critical component of the community metagenomic analysis is the assignment of genome fragments to clusters representing operational taxonomic units (OTUs), a process termed binning. Accurate binning is required for comparative genome and metabolic analysis. Here, we binned the data based on time series abundance profiles. Specifically, each scaffold is represented based on its relative abundances in the 11 samples and clustered with other scaffolds with similar abundance profiles. We used the Databionics implementation of ESOM (Ultsch and Moerchen 2005) to cluster all scaffolds longer than 500 bp, using abundance profile information (Fig. 1). This resulted in well-defined clusters of fragments with the same time series abundance patterns that represented either genomes or sets of sequences that belong to one of several strains (as in the case of S. epidermidis) (Fig. 1).
Figure 1.

Time series-based ESOM binning of the whole data set (left) and Staphylococcus scaffolds (right). (Left) An ESOM in which each point represents a 500- to 6000-bp segment. Data points are colored based on their scaffold's best hit from all published genomes in the NCBI nucleotide database (white data points have no significant hit). Note that the map is periodic. Clusters are: 1. Candida albicans; 2. Finegoldia magna; 3. Staphylococcus hominis; 4. Staphylococcus lugdunensis; 5. Leuconostoc citreum; 6. Peptoniphilus Carrol (novel species); 7. Staphylococcus aureus; 8. Propionibacterium Carrol (novel species); 8.1 Propionibacterium acnes; 9. Streptococcus; 10. Staphylococcus epidermidis, regions on the genome that are common to all strains; 10.1 strain 1 and some low abundance scaffolds that probably belong to rare S. epidermidis strains; 10.3. regions unique to S. epidermidis strain 3; 10.4. regions unique to S. epidermidis strain 4; 11. Enterococcus faecalis; and 12. Anaerococcus. (Right) An ESOM of the Staphylococcus genomes (numbers are the same as in the left panel), their plasmids (extension “P”) and infecting phage (A: phage 13; B: phage 14; and C: phage 46). For S. epidermidis strain 1, dark red represents segments unique to the strain, while light red represents regions common to both strain 1 and strain 3 (likewise dark/light green for strain 3).
Genome completeness
The fraction of the genome that was assembled was evaluated for all genomes based on the presence of a set of universal single copy genes (Raes et al. 2007) and other conserved genes (Supplemental Fig. S9), as well as scaffold connectivity and genome size comparison to closely related, published genomes (refer to the Methods section for more details).
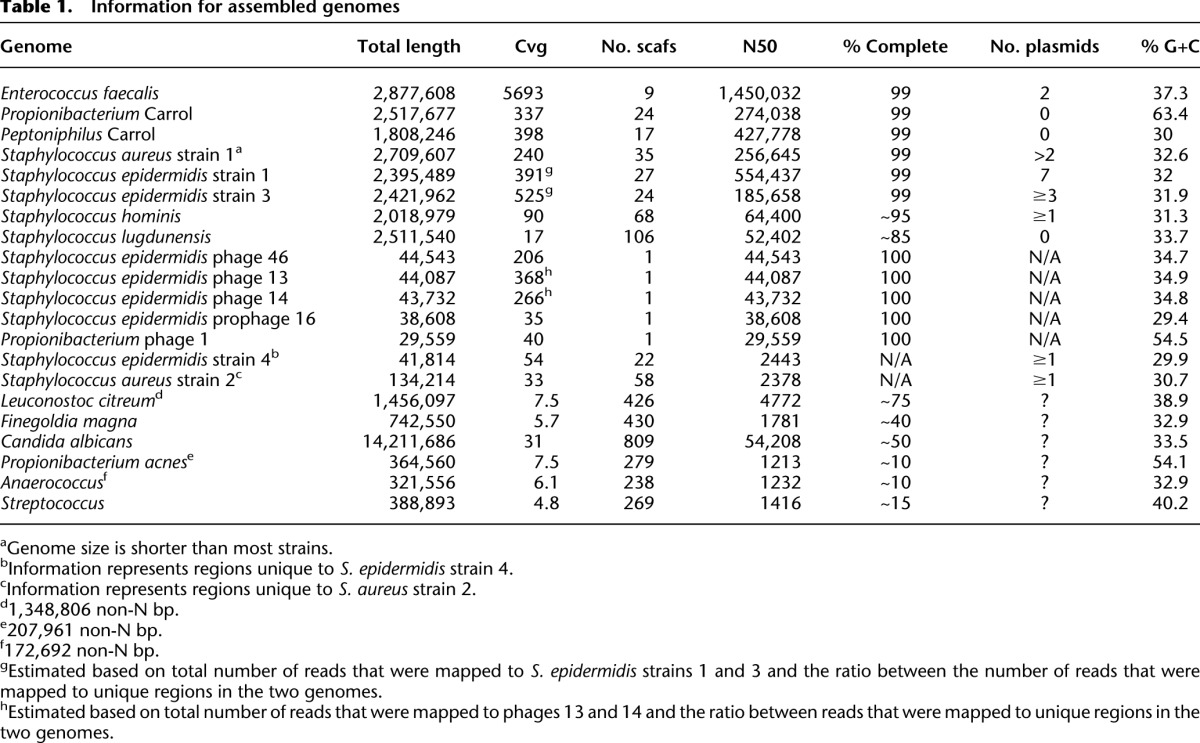
Supplemental Table S6 summarizes genome completeness. Complete genomes were reconstructed for four phages. One hundred percent connectivity was achieved for the genomes of Enterococcus faecalis, a Peptoniphilus species, a Propionibacterium species, and S. epidermidis strain 3. The genome of S. epidermidis strain 1 had 18 disconnected scaffold ends, all of which could be resolved with high confidence. These genomes are considered to be 99% complete (“essentially complete”). We refer to genomes estimated to be more than ∼80% complete as “near complete”. Other genomes are considered to be “partial.”
Species abundance changes and potential significance
Figure 2 describes time series abundance patterns for all genomes detected in our data set. Overall, the community was dominated by E. faecalis (phylum Firmicutes, family Enterococcaceae), a facultative anaerobic Gram-positive bacterium often detected in feces (Morowitz et al. 2011). Other dominant species included skin-associated microbes, consistent with previously observed microbial community structure in infants delivered via C-section (Dominguez-Bello et al. 2010). Four representatives of the Staphylococcus genus were identified (phylum Firmicutes, family Staphylococcaceae). Staphylococci are Gram-positive commensal bacteria that frequently colonize the skin and mucous membranes, such as the nasal cavities in humans and other mammals (Otto 2009). S. aureus appeared late, around day 22, and was represented by two strains. A low abundance phage was also recognized that clusters in the time series-based ESOM with the S. aureus strains and thus may infect them. S. epidermidis was relatively abundant throughout the time series, and strain abundances fluctuated significantly. Two other Staphylococcus species, S. hominis and S. lugdunensis, were present in low abundances. We also identified two species of Propionibacterium (phylum Actinobacteria, family Propionibacteriaceae), a genus whose membership includes Gram-positive aerotolerant anaerobes that are commonly found on the skin but have also been described in the gut (Mackie et al. 1999; Dworkin and Falkow 2006). Propionibacterium acnes is often associated with sebaceous follicles, where secreted lipids are utilized in a fermentation-based metabolism (Grice and Segre 2011) that produces the propionate that gives this genus its name (Dworkin and Falkow 2006). Growth status (Brzuszkiewicz et al. 2011) as well as strain type may influence whether P. acnes plays a role in skin homeostasis or causes disease, e.g., via immunostimulation (Nagy et al. 2005). A second species of Propionibacterium distinct from P. acnes was relatively abundant early and late in this colonization series. We refer to this species as Propionibacterium Carrol (or more simply, Propi. Carrol). In this notation, Carrol is a project designation, common to all genomes recovered in this study. A Peptoniphilus species Carrol (Pepto. Carrol) appears late in the colonization series. Peptinophilus species (phylum Firmicutes, family Clostridiales family XI. Incertae sedis) are also Gram-positive bacteria and are typically obligate anaerobes. Notably, this suggests a transition from facultative anaerobes toward a community dominated by obligate anaerobes. Several other genomes were detected at low abundances, in particular at the start and the end of the period, including Finegoldia magna, Anaerococcus (both are Firmicutes, Clostridiales family XI. Incertae sedis), Streptococcus (Firmicutes, Streptococcaceae), and P. acnes.
Figure 2.

Relative abundance in the community of abundant (left) and rare (right) species. Abundance was computed based on read mapping to unique regions on the assembled genomes.
Both Propi. Carrol and Pepto. Carrol represent species with no previously published genomes. A 16S rRNA-based search for related genomes revealed mostly hits from skin environments (Supplemental Tables S3, S4), which raises the possibility that both new genomes are from native skin bacteria. However, the late proliferation of both organisms suggests that they are well-adapted to the anaerobic conditions in the gut.
Community composition was also evaluated using 16S rRNA amplicon surveys (Supplemental Fig. S7 and supporting online material). These surveys generally reported similar bacterial species to those detected in the metagenomic analysis, with overall similar abundances.
Similarity of genomes to published genomes and mobile elements
Essentially-complete and near-complete genomes, except for those of Propi. Carrol and Pepto. Carrol, showed a remarkably high similarity to published genomes over most, but not all, of their length (Supplemental Table S5; Supplemental Fig. S7). All but these exceptions had orthologs for >89% of their genes, on average, in every published genome of their species at >96% amino acid (aa) level similarity. The similarity of S. epidermidis phage genomes to previously reported phage genomes varied greatly. However, several predicted proteins shared high sequence identity with predicted proteins in two published S. epidermidis phage genomes, even when no significant similarity could be detected at the DNA level.
The E. faecalis, Propi. Carrol, and Pepto. Carrol genomes contain very few mobile elements. E. faecalis has two plasmids, the other two species have no plasmids, and all have very few genes encoding transposases. S. aureus has at least two plasmids and several transposase genes, whereas the S. epidermidis strains have three to seven plasmids each and up to 15 transposase genes.
S. epidermidis
The disappearance of P. acnes and the late proliferation of Propi. Carrol, as well as changes in abundances of S. epidermidis, S. hominis, S. lugdunensis, and S. aureus, illustrate the importance of species-level shifts during the colonization period studied here. However, finer-scale changes in community composition could also be documented, illustrated best by consideration of the S. epidermidis strains.
We identified at least three strains of S. epidermidis (Fig. 1, left). This finding relied heavily on the time series-based binning approach, which allowed us to identify those regions that are common to all strains (cluster 10) (Fig. 1, left) and those that are unique to the different strains (clusters 10.1, 10.3, and 10.4) (Fig. 1, left). Cluster 10.1 contains sequences from at least two strains (predominantly strain 1) and a few phages. All strains have both unique genomic regions and plasmids (Fig. 1, right). The genomes of the two dominant strains, strain 1 and strain 3, were reassembled based on samples in which they were abundant (see Supplemental Material). The genomes were almost identical throughout ∼90% of their lengths, with only SNPs distinguishing them.
We evaluated evidence that might suggest that the opposing abundance patterns of the two dominant strains may be related to the phage infecting each of them (Fig. 3). Based on abundance patterns, we conclude that each of the three strains was infected by at least one phage, and that each phage infects a single host (see below). Strain 3, whose proliferation after day 16 is accompanied by the decline of strain 1, maintains a relatively high abundance after that day, whereas its infecting phage 46 declines. This suggests that the strain was able to rapidly develop phage resistance, unlike strain 1 whose abundance remained similar to its infecting phage throughout the whole time period. Both strains lack a CRISPR system that was reported in other S. epidermidis strains (Marraffini and Sontheimer 2008); however, comparative analysis revealed that two genes encoding the abortive infection (Abi) bacteriophage system proteins were present within the strain 3 genome but were missing from the strain 1 genome. The Abi bacteriophage system targets important steps in phage reproduction such as replication, transcription, or reproduction and also leads to cell death (Labrie et al. 2010). These genes have only been reported in two other strains of S. epidermidis (VCU144 and SK135).
Figure 3.

Abundance patterns of the Staphylococcus epidermidis strains (thick lines) and their infecting phage (thin dashed lines). All three phages exist primarily as prophages but also were identified as free-existing phages. Insertion positions are in regions that are shared by both abundant strains (and probably also by strain 4), but abundance patterns suggest that phage 13 and 14 infect strain 1, while phage 46 infects strain 3.
The two dominant strains also differ in a number of genes encoding drug resistance proteins. Strain 1 contains a complete mec region consisting of the mecA gene (which encodes the PBP2a protein associated with methicillin resistance) and its two regulating genes mecR1 and mecI (in this order), while strain 3 contains only mecA and part of mecR1 but lacks mecI and the last part of mecR1 including the PB site. Strains that carry the complete mec region may remain susceptible to methicillin when mecI fully represses the activation of mecA. The absence of mecI and the PB site of mecR1, on the other hand, results in constitutive expression of PBP2a leading to methicillin resistance (Petinaki et al. 2001). Strain 1 also contains a gene encoding a multidrug antimicrobial extrusion protein (MATE) that was lacking from strain 3, whereas the latter carries an aacA/aphD gentamicin and kanamycin resistance determinant (Ferretti et al. 1986) that is lacking from strain 1. Both strains, as well as the other staphylococci, carry several other drug resistance genes, which are, in some cases, also present in other genomes. The methicillin resistance gene femA (Hurlimann-Dalel et al. 1992) was found in all Staphylococcus genomes as well as the Propi. Carrol and E. faecalis genomes, but not the Peptoniphilus genome.
Strain 1 in our data set carries a bap gene, but strain 3 does not. The Bap protein has been directly linked to biofilm formation in various staphylococci, including S. epidermidis (Tormo et al. 2005). This gene is also present in the genome of the biofilm-positive strain RP62A but is absent from the biofilm-negative strain ATCC 12228. No toxin genes were found in either of the well-sampled S. epidermidis strains or in any other bacterium in the data set, except for S. aureus.
Our data provide evidence for lateral transfer of genes among closely related species, as several genes in S. epidermidis show unexpected high sequence similarity to genes of other genomes. For example, a complete arginine deiminase operon (one of two such operons in strain 1) shares 100% identity over its entire length with two published S. aureus genomes. In addition, this strain contains genes sometimes found on pathogenicity islands of S. aureus and other Staphylococcus species (see Supplemental Material). Each S. epidermidis strain has a few strain-specific transporters, including transporters for sulfate, nickel, and copper (strain 1), iron (transferring receptors), proline/betaine (strain 3), and a few transporters with uncertain function.
S. epidermidis phage
We assembled complete genomes of three phages and one prophage that infect at least three of the S. epidermidis strains in the community. All are primarily found as prophage, but three (phages 13, 14, and 46) are also found as free-existing phage (based on paired-end reads that connect both sides of the phage genomes). Integration sites for the phages are in regions that are common to both strains 1 and 3 (and possibly for the other strains as well). Based on matching time series abundance patterns for host and phage (and consistent with the integration site information), we infer that strain 1 is infected by phages 13 and 14, whereas strain 3 is infected by phage 46, and prophage 16 is integrated into the genome of strain 4 (Fig. 3). The abundance of phage 46 decreased dramatically after day 17, potentially due to abortive infection bacteriophage resistance genes in strain 3 (see above).
Phages 13 and 14 are closely related and share some sequence similarity to phage 46 (Supplemental Fig. S8). Phage 46 contains eight genes (all hypothetical) with no similarity to genes of any phage in any database. The genome of prophage 16 exhibited no similarity to any published genome throughout its entire length, except for a gene encoding a transposase. Interestingly, many of its predicted proteins showed high similarity, usually 90% identity or more, to published protein sequences.
We compared the genomes of the three S. epidermidis phages and the prophage to the genomes of two previously published S. epidermidis phages, PH15 and CNPH82 (Supplemental Fig. S10; Daniel et al. 2007). The genomes of the three phages exhibit similarity in terms of overall length and gene synteny (see Supplemental Material for a detailed discussion). Based on gene content, the three new genomes belong to the Siphoviridae family. Phage 46 differs from the other phage in a number of ways. In addition to differences in gene content, predicted tape measure protein (TMP) lengths (Katsura 1987) indicate that the tail of phage 46 is longer than all others in the comparison set.
A few short phage fragments are also clustered with the S. epidermidis strains, indicating that other low abundance phages were present in the data sets. All fragments exhibit some similarity to one of the sequenced phages.
Comparative analysis of two Propionibacterium species
We recovered essentially-complete and partial genomes of Propionibacterium species. Based on the partial genome, the rare P. acnes strain found only in the first two samples analyzed (Fig. 4, left) is most similar to P. acnes strain SK137 (Fig. 4, right). The dominant Propionibacterium species, Propi. Carrol, is related to both SK137 and Propionibacterium humerusii, which was isolated from a human patient undergoing revision of a shoulder arthroplasty (Butler-Wu et al. 2011). 16S rRNA gene identity relative to both P. acnes SK137 and P. humerusii P08 is 96.6%, (average aa identity for putative orthologous proteins 86.6%). The P. acnes SK137, P. humerusii, and Propi. Carrol genomes were aligned so that their gene content could be compared. The three genomes are essentially syntenous, so alignments highlight gene insertion/deletion events and assist in distinguishing orthologs from nonorthologous (nonsyntenous) genes that share sufficient sequence similarity to be brought into the analysis.
Figure 4.

Abundance patterns of Propionibacterium-related genomes and their alignment against P. acnes SK137. (Left) Abundance of Propi. Carrol (red) and P. acnes (blue) as well as one Propionibacterium phage (green). The phage could not be linked to any of the species, either through integration site or through its abundance pattern. (Right) Alignment of both Propi. Carrol and P. acnes species to the genome of the skin P. acnes SK137.
In all comparisons, many regulatory and transport genes are genotype-specific. In the case of transport genes, only Propi. Carrol has two ammonium transport genes and an adjacent gene for a nitrogen regulatory protein. This species is also the only one with transport and other genes required for arsenic resistance. In contrast, the P. acnes SK137 is enriched in genes coding for transporters for organic compounds such as glucitol/sorbitol (A,B,C components and glucitol operon activator protein) and cellobiose. Given this, it is interesting that SK137 encodes a larger number of glycosyl hydrolases (GH) than was found in the other two propionibacteria used in the comparison (families 1, 2, 3, 18, 20, 25, 31, 35, 85). Further, the representation of GH in the three genomes is markedly different. SK137 encodes a larger number of glycosyl hydrolases than was found in the other two propionibacteria and is enriched relative to Propi. Carrol in genes for compounds such as chitobiose, chitodextrins, and oligosaccharides (see Supplemental Materials).
Propi .Carrol and P. acnes SK137 differ in their sialic acid-related genes. P. acnes SK137 carries genes for sialic acid utilization, including a gene for a sialic acid transporter, three sialidases (neuraminidases), and for interconversion of N-acyl-D-glucosamine 6-phosphate and N-acyl-D-mannosamine 6-phosphate. The sialidase genes (EC:3.2.1.18) are absent from Propi. Carrol. Sialidase was also reported in other strains of P. acnes (Bruggemann et al. 2004) and is used for the degradation of host tissues (Nakatsuji et al. 2008). Propi. Carrol, on the other hand, has genes for sialic acid biosynthesis. Interestingly, cell surface sialic acid capsule production appears to be relatively rare in Gram-positive bacteria (Severi et al. 2007). The ability to produce this nine-carbon sugar acid may be important, as it is the predominant and terminal acid in complex glycans on mucins and glycoproteins associated with vertebrate cell membranes (where it may function in, for example, intercellular adhesion and cell signaling). It also may be produced in the gut in the course of inflammation (Severi et al. 2007).
Propi. Carrol is apparently unable to utilize myo-inositol. Inositol plays a role in eukaryotic cell messaging and can be derived from nutritional sources. In fact, breast milk (especially colostrum) is rich in inositol (Pereira et al. 1990). Infant formula contains less inositol, and solutions for intravenous nutrition generally lack it. Inositol diet supplementation for premature infants receiving parenteral nutrition has shown benefits for treatment of infants with respiratory difficulties (Hallman et al. 1992). It is intriguing that the dominant Propionibacterium species in the gut community studied here is apparently unable to utilize inositol. It is also apparently unable to use carnitine, a compound required for fatty acid utilization in eukaryotes. Carnitine is also a source of osmoprotectant (it dehydrates to form crotonobetaine), and can be utilized by microorganisms as a source of C and N and serve as an external electron acceptor in anaerobic respiration (Walt and Kahn 2002). Thus, comparative analyses suggest that utilization of carnitine by propionibacteria might only be important early in the gut colonization of the infant. Also intriguing is the lack of genes for molybdopterin biosynthesis and anaerobic dimethyl sulfoxide reductase and nitrate reductase complexes in the genome of Propi. Carrol. The absence of these genes may indicate a reduced capacity for anaerobic respiration in the Propi. Carrol genotype and is consistent with an overall transition in the community toward fermentation-based metabolism.
Interestingly, the Propi. Carrol genotype has genes to synthesize nicotinamide adenine dinucleotide (NAD), in contrast to SK137. Unlike SK137, it also has genes involved in glycerophospholipid metabolism, amino acid biosynthesis, terpenoid biosynthesis, maltose sugar transformation, Von Willebrand factor production (possibly involved in attachment), and diacylglycerol kinase synthesis that may convert diacylglycerol (DAG) to phosphatidic acid (PA), a compound possibly used for (lipid-based) signaling.
Notably, Propi. Carrol contains a CRISPR/Cas locus involved in phage resistance that is distinct from that in the P. humerusii genome and is present in some P. acnes strains but not in SK137. No spacer (crRNA) encoded by this locus matches genomically sampled Propionibacterium phage; thus, the locus probably does not confer resistance to this phage.
We also conducted comparative genomic analysis of Pepto. Carrol and the previously sequenced Peptoniphilus harei ACS-146-V-Sch2b. Details are provided in the Supplemental Material.
Discussion
We documented species and strain abundance variations, as well as associated genomic characteristics, during the early stages of microbial colonization of the human gut. At the strain level, we detected at least three strains of S. epidermidis, two of which were sufficiently abundant for their genomes to be completely assembled. It has been previously suggested that multiple strains provide the necessary genetic versatility that allows a species to withstand selective pressures such as phage blooms (Ley et al. 2006). The data presented in this paper, showing dramatic changes in S. epidermidis strain abundance, correlated with changes in host:phage abundance, may indicate that strain variation is, indeed, important for species stability as phage infectivity changes. Notably, the shift in the host:phage ratio for strain 3 from ∼1:1 to 10:1 around day 17 may suggest a change in either host resistance or phage infectivity, possibly associated with activation of the abortive infection bacteriophage system. The difference in genes encoding drug resistance, transporters, and regulatory proteins in strains 1 and 3 could also impact their relative abundances. Overall, these strains differ in ∼10% of their genes, some of which have similar homologs in other staphylococci, in particular S. aureus, but not in any published S. epidermidis genomes. This finding is consistent with previous reports (Chan et al. 2011), and suggests that other species of Staphylococcus can contribute to the gene pool that results in S. epidermidis strain variation.
The microbial community in our data set is mostly dominated by E. faecalis as well as skin-associated bacterial genera such as Propionibacterium (two species), Staphylococcus (four species and multiple strains), and Peptoniphilus. The early detection of a P. acnes strain is interesting and may suggest the derivation of this early colonist from the skin of a parent or caregiver. This is in line with previously observed microbial community structure in infants born by C-section (Dominguez-Bello et al. 2010). The proliferation of the novel Propionibacterium and Peptoniphilus species in later stages suggests that they are better adapted to the gut environment than the other early colonizers that did not proliferate. Comparative analysis reveals numerous differences between the Propi. Carrol and SK137 genomes, and we infer that these genotypic differences contribute to the apparently increased fitness of Propi. Carrol within the gut environment (Fig. 5). These differences include genes encoding different transporters, glycosyl hydrolases, and molecule utilization and resistance to metals and arsenic. One notable example of a strategy that differentiates Propionibacterium types involves genes related to sialic acid: P. acnes SK137 carries genes for sialic acid utilization, while Propi. Carrol carries genes for sialic acid biosynthesis. Production of cell surface sialic acid is known in pathogenic bacteria to allow the cells to evade the host's immune response (Severi et al. 2007). Thus, the transition from P. acnes to Propi. Carrol genotype in the infant studied here may be associated with a switch in the role of propionibacteria to production of sialic acid rather than consumption. This change could potentially alter the pathogenic significance of these organisms.
Figure 5.

Comparison of selected functionalities that differentiate Propi. Carrol and P. acnes SK137.
Until recently, sequencing capacities were only sufficient to enable the recovery of near-complete genomes from systems in which the dominant organisms were highly abundant (Tyson et al. 2004). Genome recovery has been accomplished in the gut environment but only for the most dominant genomes in low complexity communities (Morowitz et al. 2011). The introduction of high-throughput sequencing technologies provided the opportunity to scale these methods for analysis of more complex systems. Recently, the possibility of extracting one complete or near-complete genome from a high-throughput metagenomic data set was demonstrated (Iverson et al. 2012). The current study differs from these previous studies, first, in that eight essentially complete and near-complete microbial genomes were recovered and, second, in the comprehensiveness of the analysis: 96% of the reads were assembled into >500-bp scaffolds and binned. In fact, the vast majority of scaffolds were from genomes whose overall relative abundance was >0.05% in the data set, and these were accurately binned to the organism of origin. Time series-based binning proved to be both accurate and sensitive because sample compositions varied, and genomes had unique abundance patterns. This approach should be generally applicable for the binning of metagenomic data collected over a time course, even for more complex environments. Potential applications of this method may include samples collected along geochemical or other gradients, from different environments, or over longer periods of time, so long as the samples share the same genomes. However, the application will be limited if samples contain different strains of the same species.
Significant effort was invested in assuring the accuracy of the assembled genomes. This was achieved using new bioinformatic tools and manual curation guided by new programs. The resulting genomes are of very high quality, superior to draft genomes generated in most isolate genome sequencing projects. Overall, this study has demonstrated the potential for community genomic analyses to uncover, at high resolution, the patterns of organism abundance, phage abundance, and organism physiology that are important during establishment of the human microbiome.
Methods
Data analysis process overview
Figure 6 outlines the data analysis pipeline that was developed for this study. We provide here a concise description of the different steps. For a complete description, refer to the supporting online material.
Figure 6.

Analysis pipeline for metagenomic data employed in this study. Refer to the Methods section and the supporting online material for more details.
Data acquisition
Medical background of human subject
The subject of this study is a female infant who was delivered by Caesarean section at 26 wk of gestation at the Comer Children's Hospital at the University of Chicago. She was treated empirically with broad spectrum antibiotics (ampicillin/gentamicin) during the first 2 d of life but did not receive antibiotics during the remainder of the study period. She received fortified maternal breast milk as well as supplemental parenteral nutrition until caloric intake from enteral nutrition was adequate (post-natal day 20). The infant required mechanical ventilation during the first 6 wk of life due to respiratory distress syndrome and bronchopulmonary dysplasia but ultimately was weaned from mechanical ventilation and supplemental oxygen therapy. She was discharged to home at 3 mo of age.
Sample collection
Eleven fecal samples were collected between days 15 to 24 and sequenced on three lanes of an Illumina HiSeq2000 sequencer for 101 cycles from each end using TruSeq SBS sequencing kits version 2. Data were analyzed with pipeline 1.7 according to the manufacturer's instructions (Illumina). Overall, 254 million high-quality reads (24.5 Gbp) remained after trimming and removing human reads (refer to Supplemental Table S2 and Methods in the Supplemental Material).
Data preparation
Data were trimmed using an in-house script that removes all consecutive bases starting from the 3′ end of each read with quality values less than 7. Reads with <60 bp remaining after trimming were discarded. Average insert size and insert size standard deviation for each sample, which are required for the assembly, were determined based on mapping of reads from each sample to a preliminary, nonoptimized assembly.
Assembly
Data from all 11 samples were assembled together. We used a coverage-directed iterative approach that is based on separate assembly of different coverage bins, i.e., coverage ranges that include one or more genomes. This approach makes it possible to choose the optimal set of parameters (coverage-related parameters and k-mer size) for each coverage bin. For each iteration, assembly parameters that fit the coverage bin with the highest coverage remaining are used, and the data assembled using Velvet (Zerbino and Birney 2008) (ABySS [Simpson et al. 2009] was used for the last iteration). Scaffolds that fit only the coverage range that was defined for the current iteration are added to the overall resulting assembly, and the reads that map to these scaffolds are identified (using Bowtie [Langmead et al. 2009]) and removed from further analysis.
Assembly quality control
Misassemblies were detected based on the identification of positions that were not covered by any insert whose zero insert coverage was statistically significant. The significance of such positions was determined using a simple statistical framework that is based on Lander and Waterman (1988). Scaffolds containing such positions were split. Assembly gaps, represented by stretches of N's that were added as part of the scaffolding process in the assembly stage, were filled whenever possible based on the assembly of reads that were mapped to the edges of the gaps and their paired-end mates. Separate such assemblies were generated for both edges of each N-segment, and their consistency was confirmed.
Binning
We used ESOM with time series abundance profiles as follows. All scaffolds longer than 3 Kbp were broken into 3-Kbp (nonterminal) segments. Any remaining sequence was added to the adjacent 3-Kbp segment, forming sequences of up to 6 Kbp (terminal). These, together with the rest of the scaffolds that are longer than 500 bp, correspond to data points in the ESOM. Coverage was computed for each segment in each sample based on the number of reads from the sample that were mapped to it. These coverages were normalized twice: first, by the sum of reads for that sample and then by the sum of the segment's coverages across the 11 samples. The second normalization is required by ESOM in order to eliminate large differences in coverage that may skew the output. Parameters for ESOM were chosen based on Dick et al. (2009), clusters were extracted manually from the map shown in Figure 1, left. Data points were labeled and colored based on best blast hit of each data point's scaffold from the NCBI nucleotide database.
Assembly optimization
Manual curation
All complete and essentially complete genomes (Table 1) were manually curated in order to extend scaffolds, detect misassemblies, and verify assembly completeness. Examination of scaffold edges or suspect regions within the scaffolds was done based on local assemblies of reads that mapped to the region of interest and their paired-end mates.
Table 1.
Information for assembled genomes
Reassembly
For the genomes of S. epidermidis strains 1 and 3, we identified samples in which each of the strains was most abundant. These genomes were reassembled separately based on reads (and their mates) from these samples that were mapped to S. epidermidis scaffolds generated in the initial assembly.
Genome completeness
Completeness of all genomes that were not assembled into one piece was evaluated based on the following three indicators: (1) Presence of a set of 60 single-copy genes (Supplemental Fig. S9); (2) scaffold connectivity—genomes containing at least 95% of the single-copy genes went through connectivity inspection, in which scaffold ends were verified to be connected to other scaffolds in the same genome; and (3) a rough estimation for the completeness for the rest of the genomes was estimated based on size comparison to closely related genomes.
Gene prediction
Prodigal (Hyatt et al. 2010) was used for gene prediction, and an in-house pipeline was used for functional annotation. The pipeline includes BLAST-based similarity searches (against NR, KEGG, UniRef 90, COG) and HMM-based functional domain recognition searches (Quevillon et al. 2005).
Downstream analyses
ggKBase (see below) was used for metabolic analysis of the different genomes.
Data access
The entire data set is publicly available via an open knowledgebase (ggKBase: http://ggkbase.berkeley.edu/carrol/). Assemblies, genes, and predicted proteins are available at http://ggkbase.berkeley.edu/carrol/download/index.html. Read data sets were deposited in the NCBI Sequence Read Archive (SRA) (http://www.ncbi.nlm.nih.gov/sra) under accession number SRA052203.
Acknowledgments
This work was supported in part by NIH Grant 1R01AI092531-01, an EMBO long term fellowship (to I.S.), the Walter & Idun Berry Foundation (to E.K.C.), and the March of Dimes Foundation Research Grant 5-FY10-103 (to M.J.M.). D.A.R. is supported by a Distinguished Clinical Scientist Award from the Doris Duke Charitable Trust, by NIH Pioneer Award DP1OD000964, and by the Thomas C. and Joan M. Merigan Endowment at Stanford University.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.142315.112.
References
- Backhed F, Ley RE, Sonnenburg JL, Peterson DA, Gordon JI 2005. Host-bacterial mutualism in the human intestine. Science 307: 1915–1920 [DOI] [PubMed] [Google Scholar]
- Bruggemann H, Henne A, Hoster F, Liesegang H, Wiezer A, Strittmatter A, Hujer S, Durre P, Gottschalk G 2004. The complete genome sequence of Propionibacterium acnes, a commensal of human skin. Science 305: 671–673 [DOI] [PubMed] [Google Scholar]
- Brzuszkiewicz E, Weiner J, Wollherr A, Thurmer A, Hupeden J, Lomholt HB, Kilian M, Gottschalk G, Daniel R, Mollenkopf HJ, et al. 2011. Comparative genomics and transcriptomics of Propionibacterium acnes. PLoS ONE 6: e21581 doi: 10.1371/journal.pone.0021581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler-Wu SM, Sengupta DJ, Kittichotirat W, Matsen FA 3rd, Bumgarner RE 2011. Genome sequence of a novel species, Propionibacterium humerusii. J Bacteriol 193: 3678 doi: 10.1128/JB.05036-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan CX, Beiko RG, Ragan MA 2011. Lateral transfer of genes and gene fragments in Staphylococcus extends beyond mobile elements. J Bacteriol 193: 3964–3977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniel A, Bonnen PE, Fischetti VA 2007. First complete genome sequence of two Staphylococcus epidermidis bacteriophages. J Bacteriol 189: 2086–2100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dethlefsen L, Relman DA 2011. Incomplete recovery and individualized responses of the human distal gut microbiota to repeated antibiotic perturbation. Proc Natl Acad Sci (Suppl 1) 108: 4554–4561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dick GJ, Andersson AF, Baker BJ, Simmons SL, Thomas BC, Yelton AP, Banfield JF 2009. Community-wide analysis of microbial genome sequence signatures. Genome Biol 10: R85 doi: 10.1186/gb-2009-10-8-r85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez-Bello MG, Costello EK, Contreras M, Magris M, Hidalgo G, Fierer N, Knight R 2010. Delivery mode shapes the acquisition and structure of the initial microbiota across multiple body habitats in newborns. Proc Natl Acad Sci 107: 11971–11975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dworkin M, Falkow S 2006. The prokaryotes: A handbook on the biology of bacteria. Springer, New York [Google Scholar]
- Eckburg PB, Bik EM, Bernstein CN, Purdom E, Dethlefsen L, Sargent M, Gill SR, Nelson KE, Relman DA 2005. Diversity of the human intestinal microbial flora. Science 308: 1635–1638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fell JM 2005. Neonatal inflammatory intestinal diseases: Necrotising enterocolitis and allergic colitis. Early Hum Dev 81: 117–122 [DOI] [PubMed] [Google Scholar]
- Ferretti JJ, Gilmore KS, Courvalin P 1986. Nucleotide sequence analysis of the gene specifying the bifunctional 6′-aminoglycoside acetyltransferase 2″-aminoglycoside phosphotransferase enzyme in Streptococcus faecalis and identification and cloning of gene regions specifying the two activities. J Bacteriol 167: 631–638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda S, Toh H, Hase K, Oshima K, Nakanishi Y, Yoshimura K, Tobe T, Clarke JM, Topping DL, Suzuki T, et al. 2011. Bifidobacteria can protect from enteropathogenic infection through production of acetate. Nature 469: 543–547 [DOI] [PubMed] [Google Scholar]
- Gill SR, Fouts DE, Archer GL, Mongodin EF, Deboy RT, Ravel J, Paulsen IT, Kolonay JF, Brinkac L, Beanan M, et al. 2005. Insights on evolution of virulence and resistance from the complete genome analysis of an early methicillin-resistant Staphylococcus aureus strain and a biofilm-producing methicillin-resistant Staphylococcus epidermidis strain. J Bacteriol 187: 2426–2438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gophna U 2011. Microbiology. The guts of dietary habits. Science 334: 45–46 [DOI] [PubMed] [Google Scholar]
- Grice EA, Segre JA 2011. The skin microbiome. Nat Rev Microbiol 9: 244–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallman M, Bry K, Hoppu K, Lappi M, Pohjavuori M 1992. Inositol supplementation in premature infants with respiratory distress syndrome. N Engl J Med 326: 1233–1239 [DOI] [PubMed] [Google Scholar]
- Hooper LV, Gordon JI 2001. Commensal host-bacterial relationships in the gut. Science 292: 1115–1118 [DOI] [PubMed] [Google Scholar]
- Hurlimann-Dalel RL, Ryffel C, Kayser FH, Berger-Bachi B 1992. Survey of the methicillin resistance-associated genes mecA, mecR1-mecI, and femA-femB in clinical isolates of methicillin-resistant Staphylococcus aureus. Antimicrob Agents Chemother 36: 2617–2621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyatt D, Chen GL, Locascio PF, Land ML, Larimer FW, Hauser LJ 2010. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11: 119 doi: 10.1186/1471-2105-11-119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iverson V, Morris RM, Frazar CD, Berthiaume CT, Morales RL, Armbrust EV 2012. Untangling genomes from metagenomes: Revealing an uncultured class of marine Euryarchaeota. Science 335: 587–590 [DOI] [PubMed] [Google Scholar]
- Iwase T, Uehara Y, Shinji H, Tajima A, Seo H, Takada K, Agata T, Mizunoe Y 2010. Staphylococcus epidermidis Esp inhibits Staphylococcus aureus biofilm formation and nasal colonization. Nature 465: 346–349 [DOI] [PubMed] [Google Scholar]
- Jacquot A, Neveu D, Aujoulat F, Mercier G, Marchandin H, Jumas-Bilak E, Picaud JC 2011. Dynamics and clinical evolution of bacterial gut microflora in extremely premature patients. J Pediatr 158: 390–396 [DOI] [PubMed] [Google Scholar]
- Katsura I 1987. Determination of bacteriophage λ tail length by a protein ruler. Nature 327: 73–75 [DOI] [PubMed] [Google Scholar]
- Koenig JE, Spor A, Scalfone N, Fricker AD, Stombaugh J, Knight R, Angenent LT, Ley RE 2011. Succession of microbial consortia in the developing infant gut microbiome. Proc Natl Acad Sci (Suppl 1) 108: 4578–4585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labrie SJ, Samson JE, Moineau S 2010. Bacteriophage resistance mechanisms. Nat Rev Microbiol 8: 317–327 [DOI] [PubMed] [Google Scholar]
- Lander ES, Waterman MS 1988. Genomic mapping by fingerprinting random clones: A mathematical analysis. Genomics 2: 231–239 [DOI] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL 2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10: R25 doi: 10.1186/gb-2009-10-3-r25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lathrop SK, Bloom SM, Rao SM, Nutsch K, Lio CW, Santacruz N, Peterson DA, Stappenbeck TS, Hsieh CS 2011. Peripheral education of the immune system by colonic commensal microbiota. Nature 478: 250–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaTuga MS, Ellis JC, Cotton CM, Goldberg RN, Wynn JL, Jackson RB, Seed PC 2011. Beyond bacteria: A study of the enteric microbial consortium in extremely low birth weight infants. PLoS ONE 6: e27858 doi: 10.1371/journal.pone.0027858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley RE, Backhed F, Turnbaugh P, Lozupone CA, Knight RD, Gordon JI 2005. Obesity alters gut microbial ecology. Proc Natl Acad Sci 102: 11070–11075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley RE, Peterson DA, Gordon JI 2006. Ecological and evolutionary forces shaping microbial diversity in the human intestine. Cell 124: 837–848 [DOI] [PubMed] [Google Scholar]
- Mackie RI, Sghir A, Gaskins HR 1999. Developmental microbial ecology of the neonatal gastrointestinal tract. Am J Clin Nutr 69: 1035S–1045S [DOI] [PubMed] [Google Scholar]
- Mai V, Young CM, Ukhanova M, Wang X, Sun Y, Casella G, Theriaque D, Li N, Sharma R, Hudak M, et al. 2011. Fecal microbiota in premature infants prior to necrotizing enterocolitis. PLoS ONE 6: e20647 doi: 10.1371/journal.pone.0020647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marraffini LA, Sontheimer EJ 2008. CRISPR interference limits horizontal gene transfer in staphylococci by targeting DNA. Science 322: 1843–1845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maslowski KM, Vieira AT, Ng A, Kranich J, Sierro F, Yu D, Schilter HC, Rolph MS, Mackay F, Artis D, et al. 2009. Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature 461: 1282–1286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morowitz MJ, Poroyko V, Caplan M, Alverdy J, Liu DC 2010. Redefining the role of intestinal microbes in the pathogenesis of necrotizing enterocolitis. Pediatrics 125: 777–785 [DOI] [PubMed] [Google Scholar]
- Morowitz MJ, Denef VJ, Costello EK, Thomas BC, Poroyko V, Relman DA, Banfield JF 2011. Strain-resolved community genomic analysis of gut microbial colonization in a premature infant. Proc Natl Acad Sci 108: 1128–1133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mshvildadze M, Neu J, Shuster J, Theriaque D, Li N, Mai V 2010. Intestinal microbial ecology in premature infants assessed with non-culture-based techniques. J Pediatr 156: 20–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy A, Jedrychowski L, Gelencser E, Wroblewska B, Szymkiewicz A 2005. Induction of specific mucosal immune responses by viable or heat denatured probiotic bacteria of Lactobacillus strains. Acta Aliment 34: 33–39 [Google Scholar]
- Nakatsuji T, Liu YT, Huang CP, Zouboulis CC, Gallo RL, Huang CM 2008. Vaccination targeting a surface sialidase of P. acnes: Implication for new treatment of acne vulgaris. PloS one 3: e1551 doi: 10.1371/journal.pone.0001551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto M 2009. Staphylococcus epidermidis–the “accidental” pathogen. Nat Rev Microbiol 7: 555–567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer C, Bik EM, DiGiulio DB, Relman DA, Brown PO 2007. Development of the human infant intestinal microbiota. PLoS Biol 5: e177 doi: 10.1371/journal.pbio.0050177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira GR, Baker L, Egler J, Corcoran L, Chiavacci R 1990. Serum myoinositol concentrations in premature infants fed human milk, formula for infants, and parenteral nutrition. Am J Clin Nutr 51: 589–593 [DOI] [PubMed] [Google Scholar]
- Petinaki E, Arvaniti A, Dimitracopoulos G, Spiliopoulou I 2001. Detection of mecA, mecR1 and mecI genes among clinical isolates of methicillin-resistant staphylococci by combined polymerase chain reactions. J Antimicrob Chemother 47: 297–304 [DOI] [PubMed] [Google Scholar]
- Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, Nielsen T, Pons N, Levenez F, Yamada T, et al. 2010. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 464: 59–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quevillon E, Silventoinen V, Pillai S, Harte N, Mulder N, Apweiler R, Lopez R 2005. InterProScan: Protein domains identifier. Nucleic Acids Res 33: W116–W120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raes J, Korbel JO, Lercher MJ, von Mering C, Bork P 2007. Prediction of effective genome size in metagenomic samples. Genome Biol 8: R10 doi: 10.1186/gb-2007-8-1-r10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes A, Haynes M, Hanson N, Angly FE, Heath AC, Rohwer F, Gordon JI 2010. Viruses in the faecal microbiota of monozygotic twins and their mothers. Nature 466: 334–338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savage DC 1977. Microbial ecology of the gastrointestinal tract. Annu Rev Microbiol 31: 107–133 [DOI] [PubMed] [Google Scholar]
- Severi E, Hood DW, Thomas GH 2007. Sialic acid utilization by bacterial pathogens. Microbiology 153: 2817–2822 [DOI] [PubMed] [Google Scholar]
- Simpson JT, Wong K, Jackman SD, Schein JE, Jones SJ, Birol I 2009. ABySS: A parallel assembler for short read sequence data. Genome Res 19: 1117–1123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tormo MA, Knecht E, Gotz F, Lasa I, Penades JR 2005. Bap-dependent biofilm formation by pathogenic species of Staphylococcus: Evidence of horizontal gene transfer? Microbiology 151: 2465–2475 [DOI] [PubMed] [Google Scholar]
- Tyson GW, Chapman J, Hugenholtz P, Allen EE, Ram RJ, Richardson PM, Solovyev VV, Rubin EM, Rokhsar DS, Banfield JF 2004. Community structure and metabolism through reconstruction of microbial genomes from the environment. Nature 428: 37–43 [DOI] [PubMed] [Google Scholar]
- Ultsch A, Moerchen F 2005. ESOM-maps: Tools for clustering, visualization, and classification with emergent SOM. Technical Report No 46, Department of Mathematics and Computer Science, University of Marburg, Germany [Google Scholar]
- Walt A, Kahn ML 2002. The fixA and fixB genes are necessary for anaerobic carnitine reduction in Escherichia coli. J Bacteriol 184: 4044–4047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xavier RJ, Podolsky DK 2007. Unravelling the pathogenesis of inflammatory bowel disease. Nature 448: 427–434 [DOI] [PubMed] [Google Scholar]
- Zerbino DR, Birney E 2008. Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 18: 821–829 [DOI] [PMC free article] [PubMed] [Google Scholar]