

The role of Y14 in regulating mRNA decay and P-body formation is described. Y14 interacts with Dcp2 and inhibits mRNA decapping. Moreover, Y14 prevents mRNA degradation and is essential for P-body formation. Y14 may function independent of the EJC to counteract mRNA decay in mRNA metabolism.
Abstract
The exon-junction complex (EJC) deposited on a newly spliced mRNA plays an important role in subsequent mRNA metabolic events. Here we show that an EJC core heterodimer, Y14/Magoh, specifically associates with mRNA-degradation factors, including the mRNA-decapping complex and exoribonucleases, whereas another core factor, eIF4AIII/MLN51, does not. We also demonstrate that Y14 interacts directly with the decapping factor Dcp2 and the 5′ cap structure of mRNAs via different but overlapping domains and that Y14 inhibits the mRNA-decapping activity of Dcp2 in vitro. Accordingly, overexpression of Y14 prolongs the half-life of a reporter mRNA. Therefore Y14 may function independently of the EJC in preventing mRNA decapping and decay. Furthermore, we observe that depletion of Y14 disrupts the formation of processing bodies, whereas overexpression of a phosphomimetic Y14 considerably increases the number of processing bodies, perhaps by sequestering the mRNA-degradation factors. In conclusion, this report provides unprecedented evidence for a role of Y14 in regulating mRNA degradation and processing body formation and reinforces the influence of phosphorylation of Y14 on its activity in postsplicing mRNA metabolism.
INTRODUCTION
During splicing of precursor mRNAs (pre-mRNAs), a multicomponent exon-junction complex (EJC) is deposited ∼20 nucleotides upstream of every ligated exon junction of mRNAs (Le Hir et al., 2000). The EJC influences several downstream mRNA metabolic events, including mRNA export, surveillance/quality control, and translation (Le Hir et al., 2001; Gatfield and Izaurralde, 2002; Kashima et al., 2006; Singh et al., 2007; Lee et al., 2009). In eukaryotic cells, a specialized cytoplasmic mRNA surveillance mechanism termed nonsense-mediated mRNA decay (NMD) eliminates mRNAs that contain nonsense mutations or acquire premature termination codons owing to aberrant splicing. NMD thus prevents the production of truncated or potentially harmful proteins (Le Hir et al., 2001; Kashima et al., 2006; Singh et al., 2007). The EJC facilitates recruitment of factors involved in NMD and therefore plays a critical role in mRNA quality control.
The RNA-binding protein Y14 is a phylogenetically conserved protein. It contains an RNA-recognition motif (RRM) in the central region and two consecutive arginine–serine (RS) dipeptides in the C-terminal domain; those two RS repeats serve as the primary phosphorylation sites of Y14 in cells (Hsu et al., 2005). Y14 forms a stable heterodimer with the Mago nashi (Mago) protein (Fribourg et al., 2003; Tange et al., 2005). In Drosophila, the Y14/Mago complex is involved in localization and translational control of specific maternal mRNAs at the posterior pole of the oocyte. In mammals, the Y14/Mago homologue (Magoh) heterodimer constitutes a part of the EJC core and interacts with several key factors involved in NMD (Tange et al., 2005; Kashima et al., 2006; Singh et al., 2007). Y14 participates in NMD via its interaction with the NMD factor Upf3 (Gehring et al., 2003). Depletion of Y14 abolishes NMD, indicating that Y14 is essential for this process (Gehring et al., 2003). The C-terminal arginine/glycine/serine–rich domain of Y14 can be posttranslationally modified; the two consecutive arginine–serine dipeptides in this domain are the primary phosphorylation sites (Hsu et al., 2005). We previously reported that hypophosphorylated Y14 has higher affinity for the EJC/NMD factors, but it is intriguing that a nonphosphorylatable Y14 mutant was not considerably different from wild-type Y14 in its ability to activate NMD (Hsu et al., 2005). After the mRNA surveillance step, Y14 remains associated with the mRNA and then is removed during productive rounds of translation (Dostie and Dreyfuss, 2002). In addition, Y14 may also promote translation (Lee et al., 2009). Nevertheless, the details of how Y14 is involved in postsplicing mRNA metabolism remain to be investigated.
In this study, we detected a direct interaction between Y14 and the mRNA-decapping factors, which implicates Y14 in mRNA degradation. Regulation of mRNA turnover is an important step in gene expression. Degradation of eukaryotic mRNAs is generally initiated by 3′-end deadenylation, followed by enzymatic removal of the 5′ 7-methyl guanosine (m7GpppG) cap that then allows mRNA degradation by 5′-to-3′ exoribonucleases (Larimer et al., 1992; Hsu and Stevens, 1993). The decapping enzyme Dcp2 cleaves between the α and β phosphates of the cap and releases N7-methyl GDP and a 5′ monophosphorylated mRNA body (van Dijk et al., 2002; Wang et al., 2002; Piccirillo et al., 2003). Dcp2 forms a stable complex with Dcp1 (Lykke-Andersen, 2002; She et al., 2008). Dcp1 not only accelerates the catalytic step of decapping, but it also links other decapping activators to Dcp2 (She et al., 2004, 2008). These activators, including Dhh1/RCK/DDX6, Edc3, Edc4/Hedls/Ge-1, and Pat1, are generally evolutionarily conserved from yeast to mammals (Fischer and Weis, 2002; Kshirsagar and Parker, 2004; Coller and Parker, 2005; Fenger-Gron et al., 2005; Tritschler et al., 2007, 2008; Tritschler et al., 2009; Harigaya et al., 2010), yet how these factors individually facilitate decapping remains unclear.
The mRNA-decapping factors are located within dynamic cytoplasmic foci termed processing bodies (P-bodies; Sheth and Parker, 2003; Cougot et al., 2004; Teixeira and Parker, 2007). Translationally repressed mRNAs are targeted to P-bodies for degradation or may return to translation (Cougot et al., 2004; Franks and Lykke-Andersen, 2007). P-bodies also contain the deadenylation factors, 5′-to-3′ exoribonucleases, and factors involved in regulating the various mRNA decay or translational control pathways (Sheth and Parker, 2003, 2006; Kshirsagar and Parker, 2004; Yu et al., 2005). Several P-body factors interact directly with the decapping complex and thereby modulate mRNA biogenesis. For example, the tristetraprolin protein that acts as a destabilizing factor of AU-rich element–mediated mRNA decay recruits the decapping factors to promote decapping and subsequent degradation of target mRNAs (Fenger-Gron et al., 2005; Franks and Lykke-Andersen, 2007). The NMD effector Upf1 also recruits the decapping complex to P-bodies for translation repression and degradation of NMD substrates (Sheth and Parker, 2006; Franks et al., 2010). Moreover, the pool of untranslated mRNAs can regulate the size and number of P-bodies (Teixeira et al., 2005). Blocking mRNA decay and translation at different stages differentially affects P-body formation (Cougot et al., 2004; Brengues et al., 2005; Teixeira et al., 2005; Franks and Lykke-Andersen, 2007). Although the components of P-bodies have been largely unveiled by recent biochemical and cell biology studies, their assembly mechanism and exact function remain to be investigated.
In this study, we found that, among the EJC components, Y14/Magoh could particularly interact with the decapping and mRNA degradation factors. Y14 antagonized mRNA decay by inhibiting the decapping activity of Dcp2 and also modulated P-body formation. We deduced a function of Y14 in preventing mRNA degradation during mRNA biogenesis.
RESULTS
Y14 specifically associates with the mRNA-degradation complexes
To investigate whether the EJC core factors function individually in mRNA metabolism, we compared their interaction with several postsplicing factors. Using anti-FLAG, we examined whether FLAG-tagged EJC core proteins (Y14, Magoh, eIF4AIII, and MLN51) could coimmunoprecipitate epitope-tagged postsplicing factor (Upf3b, Dcp1a, or mRNA export receptor TAP) or endogenous Dcp2, Edc4, or the TAP-interacting RNA helicase DDX3 in HEK293 cells. Immunoprecipitation was performed in the presence of RNase A. Immunoblotting using antibodies against each epitope tag or specific antibodies revealed that all EJC factors coprecipitated hemagglutinin (HA)-tagged Upf3b and TAP, but only Y14 and Magoh could coprecipitate myc-Dcp1a and endogenous Dcp2 and Edc4 (Figure 1A). However, none of the EJC core proteins interacted with DDX3 (Figure 1A). Additional immunoprecipitation of FLAG-Y14 was performed, and the result confirmed its specific interaction with Dcp1a, DDX6, and exoribonuclease Xrn1 (Supplemental Figure S1). Because the expression level of FLAG-Y14 was similar to that of endogenous Y14 (see later discussion of Figure 5B), we could exclude the possibility that the interactions of FLAG-Y14 with the tested factors were simply due to its surplus expression. In conclusion, our finding indicated that the EJC subcore complex Y14/Magoh is possibly distinct from eIF4AIII/MLN51 with respect to its association with factors involved in postsplicing mRNA biogenesis, implying a unique role for Y14/Magoh in mRNA degradation.
FIGURE 1:
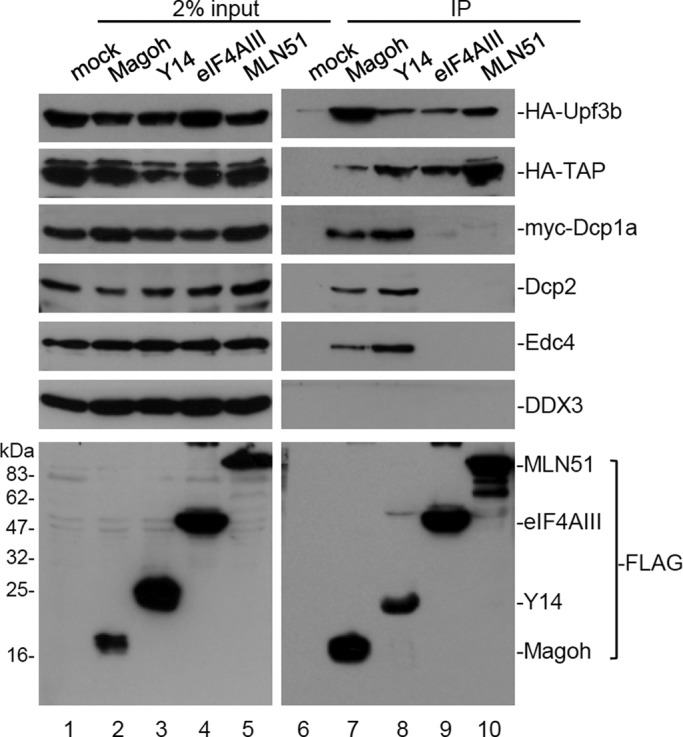
Y14/Magoh specifically associates with mRNA-decapping factors. HEK293 cells were transfected with the expression vector encoding a FLAG-tagged EJC core alone or together with a vector for expressing HA-Upf3b, HA-TAP, or myc-Dcp1a. Immunoprecipitation was performed in the presence of RNase A. Cell lysates were subjected to immunoprecipitation (IP) using anti-FLAG. Immunoblotting was subsequently performed using antibodies against HA or c-myc epitope, or Dcp2, Edc4, or DDX3. Bottom, one set of representative data for anti-FLAG immunoblotting.
FIGURE 5:

Y14 modulates the formation of processing bodies. (A) HeLa cells were transfected with siRNA targeting to Y14 or eIF4AIII. Indirect immunofluorescence was performed using antibodies against Y14, eIF4AIII, Dcp1a, and Edc4. Arrowhead and arrow, respectively, indicate Y14- and eIF4AIII-depleted cells. Scale bar, 8 μm. Cell lysates were subjected to immunoblotting with antibodies against Y14, eIF4AIII, or α-tubulin. Immunoblotting shows the level of Y14 and eIF4AIII in mock-transfected and siRNA-transfected cells; α-tubulin was used as control. The bar graph shows the average number of P-bodies in mock, Y14-depleted (siY14), and eIF4AIII-depleted (sieIF4AIII) cells (n > 200); asterisks indicate p < 0.01. (B) HeLa cells were transiently transfected with an expression vector encoding FLAG-tagged Y14 (WT), Y14SA, or Y14SE. Immunofluorescence was performed using anti-FLAG and anti-Edc4; arrowhead indicates Y14-overexpressed cells. Immunoblotting was performed using anti-Y14 to detect both FLAG-tagged Y14 (wild-type and mutants) and endogenous Y14 proteins. The bar graph shows the average number of P-bodies in mock and different Y14-overexpressing cells (n > 200).
Y14 interacts directly with Dcp2 and inhibits its decapping activity
The foregoing immunoprecipitation showed that Y14 associated with the decapping factors (Figure 1). Using a pull-down assay, we observed that the GST-Y14 fusion protein could interact with both in vitro–translated Dcp1a and Dcp2 (unpublished data). Because Dcp2 is the catalytic subunit of the decapping complex (van Dijk et al., 2002; Wang et al., 2002; Piccirillo et al., 2003), we then focused on the interaction of Y14 with Dcp2 and investigated whether Y14 could even modulate mRNA decapping via such an interaction. Indeed, endogenous Y14 could be immunoprecipitated by anti-Dcp2, but not by anti-DDX3, in the presence of RNase (Supplemental Figure S2). Moreover, the in vitro pull-down experiment confirmed the interaction between recombinant histidine (His)-tagged Dcp2 and glutathione S-transferase (GST)–Y14, which was also RNase resistant (Figure 2A, lane 3). We then mapped the domain(s) of Y14 involved in the interaction with Dcp2. Using terminally truncated Y14 proteins fused to GST as bait, we observed that the C-terminal 23–amino acid, but not the N-terminal 55–amino acid, deletion of Y14 abolished its interaction with Dcp2 (Figure 2A, lanes 5 and 6). Therefore the RRM and C-terminal domain of Y14 might be engaged in the interaction with Dcp2. Moreover, we observed that both phosphorylation of Y14 at the RS dipeptides (Y14p) and heterodimerization of Y14 with Magoh also impeded the Y14–Dcp2 interaction (Figure 2A, lanes 4 and 7). The intriguing observation that the Y14/Magoh heterodimer failed to bind Dcp2 perhaps suggested a mutually exclusive interaction between Y14 and Magoh, but structural details of their interaction need further investigation.
FIGURE 2:

Y14 interacts directly with Dcp2 and inhibits its decapping activity. (A) Diagram shows the domains of full-length Y14 and truncated forms of Y14. Y14 can be phosphorylated at the serine residues of RSRS near the C-terminus. Recombinant GST or one of the various GST-Y14 proteins (including the full-length nonphosphorylated and phosphorylated Y14, Y14∆N, or Y14∆C, and GST-Y14/His-Magoh) was incubated with His-Dcp2, followed by affinity chromatography using glutathione–Sepharose. Sepharose-bound His-Dcp2 was detected by immunoblotting using anti-Dcp2. The bait proteins were detected by Coomassie blue staining. (B) The decapping reaction was performed by incubating His-Dcp2 and GST (lane 2) or increasing amounts of GST-Y14 (lanes 3–8) with a 176-nucleotide RNA containing a 32P-labeled 5′ cap and then analyzed by TLC. Lane 1 shows RNA only; ori indicates the origin of the TLC plate. Relative decapping efficiency is indicated below the gel. (C) The Dcp2 decapping assay was performed as in B; the reactions did not contain any additional recombinant protein (lane 2) or contained GST (lane 3), GST fused to full-length or truncated Y14 (lanes 4–7; as in A), or GST-Y14/His-Magoh (lane 8). Asterisk indicates an unidentified nucleotide; its level varied among different batches of recombinant Y14 proteins. The bar graph shows relative inhibition of decapping for the different GST-Y14 forms; the mean and SD were obtained from three independent experiments, and asterisks indicate p < 0.01.
Next we examined whether Y14 could modulate the decapping activity of Dcp2. Dcp2 hydrolyzes m7GpppG-capped mRNAs to release N7-methyl GDP (van Dijk et al., 2002; Wang et al., 2002; Piccirillo et al., 2003). The decapping assay was performed by incubating His-Dcp2 with the 164-nt PIP85∆i RNA fragment containing a 32P-labeled m7G*pppG (the asterisk denotes the 32P label). In general, ∼50% of the RNA substrate was decapped by Dcp2. GST-Y14 inhibited the decapping activity of Dcp2 in a dose-dependent manner, whereas GST did not (Figure 2B). We also assessed the ability of various structural forms of Y14 to inhibit Dcp2-mediated decapping. Figure 3C shows that, in addition to wild-type Y14, only Y14∆N inhibited decapping reproducibly, with slightly higher activity than the wild type (Figure 2C, lane 6). However, phosphorylated Y14, Y14∆C, and Y14/Magoh had no or only minimal activity on decapping inhibition (Figure 2C, lanes 5, 7, and 8). Another recombinant RNA-binding protein, His-tagged RBM4, did not show any decapping-inhibition activity (Supplemental Figure S3), excluding the possibility that the decapping activity of Dcp2 was nonspecifically competed off by any RRM-containing proteins. Our data apparently indicate that the ability of Y14 to inhibit decapping correlated strongly with its capacity to interact with Dcp2.
FIGURE 3:

Y14 binds directly to the mRNA cap structure. (A) For UV cross-linking, 5 μg of recombinant GST or GST-Y14 was incubated with 32P-labeled capped PIP85a∆i RNA (m7G*pppG-; lanes 3 and 4) or 32P-labeled free cap (m7G*pppGp; lanes 5 and 6). After UV radiation and RNase digestion, proteins were resolved by SDS–PAGE and detected by autogradiography (lanes 3–6) and Coomassie blue staining (lanes 1 and 2). (B) UV cross-linking of GST or GST-Y14 (full-length, ∆C, or ∆N) proteins was performed as in A using 32P-labeled m7G*pppG-RNA (lanes 5–8) or noncapped RNA (*pG-; lanes 9–12) as substrate. Lanes 1–4, Coomassie blue staining of the recombinant Y14 proteins used. (C) UV cross-linking of GST, GST-Y14, or GST-Y14∆C was performed using the m7G*pppG-capped RNA as substrate in the absence (–) or presence (+) of m7GpppG. (D) UV cross-linking was performed with the m7G*pppG-capped RNA and 5 μg of His-tagged Y14 (lane 1) or His-Magoh alone (lane 5) or 5 μg of His-Y14 together with 1.25, 2.5, or 5 μg of His-Magoh (lanes 2–4, respectively). (E) FLAG-tagged Y14 and CBP80 were each transiently expressed in HEK293 cells and immunopurified from the lysates using anti-FLAG Agarose. Purified proteins (lanes 1–3) were then subjected to affinity chromatography using m7GTP-Sepharose in the absence (lanes 4–6) or presence (lanes 7–9) of m7GpppG. Chromatography was performed using GTP-agarose (lanes 10–12). FLAG-tagged protein input (lanes 1–3), and proteins bound to the m7GTP (lanes 4–9) or GTP (lanes 10–12) were analyzed by immunoblotting with anti-FLAG.
Y14 binds directly to the mRNA cap structure
Next we used ultraviolet (UV) cross-linking to assess the ability of Y14 to bind to the cap structure. Purified GST-Y14 was incubated with the 32P-labeled RNA substrate used in the decapping assay (m7G*pppG-; see earlier description), followed by UV cross-linking and RNase digestion. Figure 3A shows that GST-Y14, but not GST, was cross-linked to the 5′-mRNA cap structure (Figure 3A, lanes 3 and 4). The cross-linking was also detected with His-tagged Y14 (Figure 3D, lane 1). Cross-linking was then performed using a 32P-labeled free cap (m7G*pppGp; see Materials and Methods) as substrate. The result showed that that Y14, like Dcp2 (Piccirillo et al., 2003), was unable to bind a free cap (Figure 3A, lane 6). Perhaps, through binding to the RNA body, Y14 might more efficiently interact with the cap. Next we examined two truncated GST-Y14 proteins in their cap-binding ability and whether Y14 could discriminate the 5′-end structure of the mRNA. The result showed that full-length Y14 and Y14∆C, but not ∆N, could cross-link to the m7GpppG cap (Figure 3B, lanes 6–8), indicating that the N-terminal domain of Y14 is critical for cap binding. However, using the noncapped RNA substrate, we observed that full-length Y14 still cross-linked to the 5′ phosphate of the RNA, although Y14∆C could not (Figure 3B, lanes 11 and 12). Therefore we assumed that recombinant Y14 might access the 5′ end of RNA, irrespective of the cap structure, due to the presence of its unmodified C-terminal domain, which could nonspecifically bind RNA (Fribourg et al., 2003). To evaluate this possibility, we performed a competition assay. The result showed that an excess amount of free m7GpppG cap could completely disrupt the cross-linking of Y14∆C to the m7GpppG-capped RNA, although a residual amount of full-length Y14 was still 32P labeled (Figure 3C). From these results, we concluded that Y14 might have considerable specificity to bind the 5′ cap structure of mRNA.
Next we examined whether Magoh could bind the cap or modulate the cap-binding activity of Y14. Of interest, in the absence of Y14, His-Magoh was not cross-linked to the cap (Figure 3D, lane 5), but when the reactions were performed with His-Y14 and increasing amounts of His-Magoh, both proteins could be cross-linked to the cap (Figure 3D, lanes 2–4). Therefore the binding of His-Magoh to the cap was dependent on the presence of Y14, suggesting that Y14 might bring Magoh close to the cap.
We also evaluated whether Y14 in HEK293 cell extracts can bind to the mRNA cap. FLAG-tagged Y14 was transiently expressed in HEK293 cells and then immunopurified from the extracts. Using affinity chromatography, we observed that FLAG-Y14, as the positive control FLAG-CBP80, bound to N7-methyl GTP (m7GTP)-linked Sepharose (Figure 3E, lanes 5 and 6), but FLAG-REF could not (Supplemental Figure S4). Moreover, an excess of m7GpppG competed off the binding of Y14 and CBP80 to m7GTP-Sepharose (Figure 3E, lanes 8 and 9), and neither of them could bind to GTP-agarose (Figure 3E, lanes 11 and 12), indicating the specificity of Y14 in binding to the mRNA cap structure.
In conclusion, our results demonstrate the potential and direct binding of Y14 to the cap structure of mRNAs. It appears that the domains of Y14 involved in cap binding and Dcp2 interaction are somewhat different but not completely mutually exclusive. Taken together, our findings provide a hint that Y14 may be located closely to the mRNA 5′ end and function to prevent decapping during mRNA metabolism.
Overexpression of Y14 inhibits decay of a reporter mRNA
The foregoing results prompted us to examine whether Y14 is capable of preventing mRNA decay. To test this possibility, we used a β-globin (βG) reporter in which the 3′ untranslated region (UTR) was derived from tumor necrosis factor α (TNFα; Chang and Tarn, 2009; Figure 4A, diagram). This reporter was transcriptionally controlled by a tetracycline-responsive promoter and produced mRNA with a short half-life due to its AU-rich elements (AREs) in the 3′ UTR. To evaluate the effect of various EJC factors on βG reporter mRNA degradation, we cotransfected the vector expressing a FLAG-EJC factor and the reporter in HeLa cells. In mock-transfected cells, we observed a gradual decrease of the βG mRNA level after the addition of the tetracycline analogue doxycycline to turn off reporter expression (Figure 4A, lanes 1–4). This decline was slowed by overexpression of FLAG-Y14 (Figure 4A, lanes 5–8) but not by Magoh (Figure 4B, lanes 4–6), eIF4AIII, or an EJC-associated and Y14-interacting protein, PYM (Figure 4A, lanes 9–16). The effect of Y14 overexpression in slowing mRNA degradation was confirmed by Northern blotting analysis (Supplemental Figure S5). The prolonged half-life of the reporter mRNA upon Y14 overexpression (Figure 4) suggested a role of Y14 in preventing mRNA degradation. Because the C-terminally truncated Y14 (Y14∆C) was unable to inhibit decapping, we examined whether it also loses the activity in preventing reporter mRNA decay. As compared with the wild-type Y14, Y14∆C showed a minor effect on protecting the reporter mRNA from degradation (Figure 4B, lanes 7–9, and Figure 4C).
FIGURE 4:

Overexpression of Y14 prevents the decay of a reporter mRNA. (A) HeLa tet-off cells were cotransfected with vectors expressing the TNFα-3′ UTR–containing βG reporter (diagram) and the reference GFP and the mock or FLAG-protein expression vector for 24 h. After doxycycline addition for 0, 1, 2, or 4 h, total RNA was prepared from each transfectant and then subjected to an RNase protection assay using 32P-labeled probes (rightmost lane) specific to the βG or GFP mRNA. Top, a representative autoradiogram of the RNase protection assay. (B) The mRNA degradation assay was performed as in A with the effectors Magoh (lanes 4–6) and Y14 (∆C and full-length, lanes 7–12). Immunoblotting in both A and B was performed using anti-FLAG. M, DNA markers. (C) The semilogarithmic graph shows the rate of βG mRNA decay in each transfectant. Each percentage was calculated by dividing the βG level (normalized to the GFP level) of each time point to that of time zero. The t1/2 was calculated using the function ln(0.5)/b, where b is the slope of the trend line obtained from the equation y = ne−bx. The results were obtained from three independent experiments; however, SD was invisible in the logarithmic scale.
Next we evaluated whether Y14 could still prevent mRNA decay when artificially tethered to a reporter mRNA. We engineered a β-globin reporter in which the 5′ UTR contained six copies of the bacteriophage MS2 coat protein (MCP)–binding site (Supplemental Figure S6A, diagram). Both RNase protection and Northern blotting analyses showed that overexpression of the MCP-Y14 fusion protein in HeLa cells could slow the degradation of the MS2-containing reporter RNA (Supplemental Figures S6A and 5B). Because inhibition of mRNA decapping may cause accumulation of deadenylated degradation intermediates (Fenger-Gron et al., 2005), we examined whether Y14-stablized mRNAs may lack the poly(A) tail. Using oligo(dT)-bound RNAs as template, we performed reverse transcription-PCR to detect the MS2-containing mRNA. The result showed that ∼50% of the reporter mRNA from MCP-Y14–expressed cells was lost after selection by oligo(dT) cellulose chromatography (Supplemental Figure S6C). This observation was reproducible and suggested that at least a part of Y14-stablized mRNAs was deadenylated.
Y14 depletion impairs the formation of P-bodies
Our foregoing results implicated a role of Y14 in mRNA decapping and decay. Therefore we attempted to examine whether Y14 may modulate P-body formation. However, we initially could not detect endogenous Y14 in P-bodies (Figure 5A; see Y14-positive cells). We reasoned that this might result from transient localization of the EJC/NMD factors in P-bodies (Durand et al., 2007; Franks et al., 2010). Nevertheless, we still used an Y14-specific small interfering RNA (siRNA) to knock down the expression of Y14 and examined P-bodies by using indirect immunofluorescence with anti-Dcp1a or anti-Edc4 in HeLa cells. Immunoblotting indicated that the overall level of Y14 was reduced to ∼20% of the mock-treated cells (Figure 5A, immunoblotting). However, immunostaining with anti-Y14 could clearly reveal Y14-depleted cells (as indicated by arrowhead). We observed that very fewer P-bodies were microscopically detectable in Y14-negative cells as compared with Y14-containing cells (Figure 5A). This observation coincided with the foregoing results indicating a role of Y14 in mRNA degradation. Nevertheless, siRNA-mediated depletion of eIF4AIII did not affect the number of P-bodies (arrowed cells). Moreover, when we examined stress granules in Y14-depleted and arsenite-treated cells, we did not detect any significant effect of Y14 depletion in stress granule formation (Supplemental Figure S7). This observation reinforced that Y14 might have a specific and essential role in P-body formation.
Next we examined the effect of overexpression of Y14 in P-body formation. When FLAG-tagged wild-type Y14 was overexpressed in HeLa cells, the number of P-bodies was only minimally increased (Figure 5B). Surprisingly, when we overexpressed a phosphomimetic Y14 mutant (Y14SE), within which the two serine residues (S166 and S168) in the duplicated RS repeats were changed to glutamates (Hsu et al., 2005), we observed a marked accumulation of P-bodies in Y14SE-overexpressed cells (Figure 5B). However, overexpression of the nonphosphorylatable Y14 mutant (Y14SA; Hsu et al., 2005) did not much significantly increase the P-body number, similar to that observed with wild-type Y14 (Figure 5B). This observation reinforced a role for Y14 in P-body formation and furthermore suggested that the phosphorylation status of Y14 could modulate its activity in P-body induction.
Phosphomimetic Y14 sequesters mRNA and the mRNA degradation machinery
Our foregoing data showed that phosphorylated Y14 was unable to interact with Dcp2 and could not inhibit decapping in vitro (Figure 2). Therefore the observation that P-body formation was induced by phosphomimetic Y14 was somewhat unexpected. To unveil its possible mechanism, we initially examined whether Y14SE can function in inhibition of mRNA decay in cells. Using the ARE-containing reporter, we observed that overexpression of FLAG-Y14SE could slow reporter mRNA degradation, as did the wild-type Y14 (Figure 6A). Using anti-FLAG for immunoprecipitation, we reproducibly observed that slightly higher amounts of the mRNA-degradation factors, including Dcp1a, Dcp2, DDX6, Edc4, Xrn1, and Rrp6, were coprecipitated with FLAG-Y14SE than with the wild-type Y14 (Figure 6B) or Y14SA (Supplemental Figure S8) in HEK293 cell lysates. We thus suspected that Y14SE has a slower turnover rate from its associated mRNA-degradation complex. Finally, we evaluated whether Y14SE also binds to spliced mRNAs with a higher affinity. We prepared nuclear extracts from HEK293 cells that contained overexpressed FLAG-tagged Y14 or Y14SE for in vitro splicing of a precursor mRNA. Immunoprecipitation of the reaction mixtures showed that FLAG-Y14 preferentially bound the spliced mRNA rather than the precursor or intermediate RNAs, as expected (Figure 6C, lane 5). FLAG-Y14SE bound to the spliced mRNA even with a higher affinity than the wild-type (Figure 6C, lanes 5, 6, 8, and 9). Therefore the tighter association of Y14SE with target mRNAs and cofactors might be consistent with Y14SE-induced P-body formation.
FIGURE 6:

Phosphomimetic Y14 inhibits mRNA decay and binds to the mRNA degradation complex and spliced mRNA with high affinity. (A) The in vivo mRNA degradation assay and following RNase protection assay were performed as in Figure 4. HeLa tet-off cells were mock transfected (lanes 1–3) or transfected with the wild-type Y14 (lanes 4–6) or Y14SE (lanes 7–9) expression vector. Immunoblotting was performed using anti-FLAG. (B) HEK293 cells were transiently transfected with the mock vector or the expression vector encoding FLAG-Y14 or FLAG-Y14SE. Cell lysates were subjected to immunoprecipitation using anti-FLAG, followed by immunoblotting using antibodies against each indicated protein. (C) In vitro splicing of the PIP85a pre-mRNA was performed in nuclear extracts prepared from mock, FLAG-Y14-expressing, or FLAG-Y14SE-expressing HEK293 cells (lanes 1–3). The reactions were subjected to immunoprecipitation using anti-FLAG; the resins were then washed with buffer containing 150 mM NaCl (lanes 4–6) or 300 mM NaCl (lanes 7–9). RNA was then extracted from the resins and fractioned on a denaturing gel, followed by autoradiography. Immunoblotting was performed using anti-FLAG. M, DNA markers. From top to bottom the bands represent the following RNA species: unspliced pre-mRNA, lariat intron-exon 2, lariat intron, and ligated exons.
DISCUSSION
In this study, we uncovered a novel function of Y14 in inhibiting the activity of Dcp2 and regulating P-body formation and inferred that Y14 might prevent mRNA decay in a step of mRNA metabolism.
In general, mRNA decapping can be positively regulated by mRNA-degradation factors and negatively regulated by translation factors (Schwartz and Parker, 1999, 2000; Vilela et al., 2000; She et al., 2004, 2008). To prevent mRNA decay, the eukaryotic translation initiation factor 4E (eIF4E) competes with Dcp1 for binding to the cap structure and may also inhibit the decapping activity of Dcp2 (Vilela et al., 2000). The poly(A)-binding protein PABP associates with the 5′ cap via its interaction with eIF4G and also inhibits Dcp2 decapping activity (Khanna and Kiledjian, 2004). In addition to the above-mentioned translation factors, the testis-specific variable-charge X-linked protein VCX-A directly binds Dcp2 and inhibits its activity, thereby stabilizing mRNAs (Jiao et al., 2006). Y14 is thus similar to VCX-A in binding and inactivating Dcp2 (Figure 2). Our results indicated that Y14 uses different but overlapping domains to interact with Dcp2 and the mRNA cap (Figures 2, 3, and 7). Nevertheless, the interaction of Y14 with Dcp2 strongly correlated well with its activity in inhibiting decapping (Figure 2). Moreover, the observation that Y14, but not Y14/Magoh, could bind and inhibit Dcp2 implies that dissociation of Magoh from Y14 may allow Y14 to interact with Dcp2 (Figure 2). Finally, we found that the N-terminal domain of Y14, which contributes to cap binding, shares minimal sequence similarity with the cap-binding domain of eIF4E (unpublished data); whether such conservation has any structural and/or functional significance with respect to the binding of Y14 to the cap structure needs to be studied by future experiments.
FIGURE 7:

Model of Y14 function in inhibition of mRNA degradation. (A) Schematic representation of the domains of Y14 and their functional roles in the inhibition of mRNA decapping. (B) A simplified model shows Y14 in postsplicing complexes and its possible role in the inhibition of mRNA degradation. Y14 associates with the EJC complex after pre-mRNA splicing and is exported with mature mRNAs to the cytoplasm and functions in NMD. Phosphorylation of Y14 may trigger its dissociation from the EJC/NMD complexes and allow the recruitment of the mRNA degradation factors. Subsequent dephosphorylation of Y14 activates its function in mRNA decapping. While dephosphorylation of Y14 is blocked, the mRNA degradation complex may accumulate in P-bodies.
We show that overexpression of Y14 could stabilize a reporter mRNA (Figure 4). However, this apparent function of Y14 appears to be opposite to the role of the EJC in NMD, which promotes the decay of mRNAs bearing premature stop codons (Gehring et al., 2003; Kashima et al., 2006). To initiate NMD, the EJC sequentially recruits the Upf2/3 proteins and the NMD key effector Upf1 (Lykke-Andersen, 2002; Kashima et al., 2006; Cheng et al., 2007; Singh et al., 2007; Chamieh et al., 2008; Isken et al., 2008; Ivanov et al., 2008). Upf1 inhibits translation initiation and recruits the mRNA-degradation machineries to destroy NMD substrates (Kashima et al., 2006; Isken et al., 2008). In this study, we found that the two EJC core dimers, that is, Y14/Magoh and eIF4AIII/MLN51, are distinct in their interaction affinities toward the mRNA-degradation factors (Figure 1). In particular, Y14 by itself directly interacted with the decapping factor Dcp2 and inhibited decapping (Figure 2). Because Y14 may remain associated with the mRNA after surveillance (Dostie and Dreyfuss, 2002), we infer that Y14-mediated decapping inhibition occurs during translation to prevent mRNA decay and hence, as reported (Dostie and Dreyfuss, 2002; Lee et al., 2009), to promote mRNA translation. In addition, the possibility that Y14 functions as an antagonistic factor of Dcp2 to control the stability of Dcp2-specific target transcripts (Li et al., 2008) remains to be tested.
We previously reported that Y14 is a phosphoprotein in cells, and its two tandem RS dipeptides can be phosphorylated by the SR protein-specific kinase SRPK1 in vitro (Hsu et al., 2005). However, unphosphorylated Y14 has higher affinity for the EJC/NMD factors (Hsu et al., 2005). Therefore we hypothesize that the newly assembled EJC complex contains hypophosphorylated Y14, which is then exported with messenger ribonucleoproteins (mRNPs) to the cytoplasm and recruits NMD factors. On the basis of our present data, we deduced that phosphorylation of Y14 and conformational rearrangements of the EJC/NMD complexes occur during or immediately after the step of mRNA surveillance and allow Y14 (or Y14/Magoh) to recruit the mRNA-degradation factors (Figure 1), but the eventual dephosphorylation of Y14 is necessary to induce its direct interaction with Dcp2 (Figure 2). We previously reported that the nonphosphorylatable Y14 mutant associates with the ribosomes with a higher affinity than the wild-type Y14 (Hsu et al., 2005). It is plausible to deduce that, after dephosphorylation, Y14-associated mRNPs are subjected to translation, in which Y14 prevents mRNA degradation (Figure 4) and enhances translation (Lee et al., 2009). However, we noted that Y14SE, which cannot be dephosphorylated, still slowed mRNA decay (Figure 6), and suspected that this was due to the capacity of Y14-SE in sequestration of the degradation factors; this intriguing observation requires future examination. In conclusion, Y14 may undergo sequential phosphorylation and dephosphorylation during the postsplicing mRNA biogenesis pathway, which in turn dictates the function of Y14 in mRNA metabolism, perhaps similar to that observed with the NMD key factor Upf1 (Kashima et al., 2006; Isken et al., 2008). Nevertheless, future investigations are required to unveil the details of putative iterative phosphorylation–dephosphorylation of Y14 during the mRNA biogenesis pathway.
Although most of the NMD factors cycle through P-bodies, their association with P-bodies is transient (Durand et al., 2007; Franks et al., 2010). Y14 is primarily located in the nucleus; its localization in the cytoplasmic P-bodies was minimal but could be marginally enhanced by blockade of NMD complex disassembly (Franks et al., 2010). Although we did not detect stable Y14 foci in the cytoplasm, our data indicate that Y14 may play a role in P-body biogenesis, and its phosphorylation status substantially affects the number of P-bodies (Figure 5). We thus infer that depletion of Y14 impedes P-body formation or accumulation, probably by preventing the targeting of NMD substrates or mRNA degradation factors to P-bodies. In contrast, overexpression of the phosphomimetic Y14 mutant resulted in P-body accumulation. This may be rationalized by the observation that this mutant remained sequestered with the mRNA-degradation factors on the mRNAs (Figure 6). Without subsequent (and ultimate) dephosphorylation of Y14, all those factors are accumulated in P-bodies. Overall our results indicate that phosphorylation of Y14 plays an important role in remodeling Y14-containing postsplicing mRNPs and their fate during mRNA biogenesis.
MATERIALS AND METHODS
Plasmid construction
The bacterial expression vectors encoding GST-Y14 and GST-Y14∆C, and the His-tagged proteins Y14, Magoh, and SRPK1, were described previously (Hsu et al., 2005; Chuang et al., 2011). The GST-Y14∆N expression vector was made analogously to the GST-Y14 vector (Hsu et al., 2005) and produced a GST fusion to the truncated Y14 lacking the N-terminal 55 residues. The bacterial expression vector encoding His-tagged Dcp2 was constructed by inserting the human Dcp2 cDNA in frame with the 6×His epitope in the pET29b vector (Novagen, Gibbstown, NJ). The mammalian vectors for expression of the FLAG-tagged proteins Y14, Y14SA, Magoh, MLN51, and eIF4AIII, and the HA-tagged proteins Upf3b and TAP, were described previously (Hsu et al., 2005; Chuang et al., 2011). The expression vectors encoding FLAG-CBP80 or PYM and 6×c-myc–tagged Dcp1a were constructed by subcloning the respective cDNA into pcDNA3-derived plasmids (Invitrogen, Carlsbad, CA). The vectors for expressing FLAG-tagged Y14SE (S166E/S168E) or Y14SA (S166A/S168A) were generated by site-directed mutagenesis to generate mutants from their respective parental vectors. Constructs and probes used for RNase protection assays were as described (Chang and Tarn, 2009).
Purification of recombinant proteins
GST-fusion proteins were expressed and purified using glutathione–Sepharose as described (Hsu et al., 2005; Chuang et al., 2011). The procedure for purification of the GST-Y14/His-Magoh heterodimer has also been described (Chuang et al., 2011). Phosphorylated GST-Y14 was obtained by coexpression of His-tagged SRPK1 and GST-Y14 in bacteria as described (Hsu et al., 2005); ∼90% of purified GST-Y14 was phosphorylated. Recombinant His-Dcp2 was overexpressed in Escherichia coli strain BL21 (Novagen) by induction with 0.8 mM isopropyl-1-thio-β-d-galactopyranoside (Protech) and purified using His•Bind Resin (Novagen) under denaturing conditions (4 M urea) as described (Piccirillo et al., 2003). Purified proteins were renatured by dialyzing overnight against a buffer containing 50 mM Tris-HCl, 5 mM EDTA, 500 mM l-arginine, 1 mM oxidized glutathione, 5 mM reduced glutathione, and 0.4% polyethylene glycol 4000 and then in a storage buffer containing 10 mM Tris, 2 mM magnesium acetate, 100 mM NaCl, 1 mM DTT, and 10% glycerol at 4°C overnight.
In vitro pull-down assay
For each recombinant GST-fusion protein, 3 μg was incubated with 2 μg of His-Dcp2 in a 180-μl mixture at 30°C for 30 min. The mixture was then incubated with glutathione–Sepharose (GE Healthcare, Piscataway, NJ) in 50 mM Tris-HCl (pH 7.4) containing 150 mM NaCl and 0.1% NP-40 at 4°C for 2 h. Bound proteins were resolved by SDS–PAGE and subsequently stained with Coomassie blue or analyzed by immunoblotting using monoclonal anti-Dcp2 (0.4 μg/ml; Abnova, Taipei City, Taiwan).
Cell culture, transfection, immunoprecipitation, and cap-affinity chromatography
Culture and transient transfection of HEK293 cells were carried out essentially as described (Chuang et al., 2011). Immunoprecipitation was performed essentially as described (Chuang et al., 2011) in the presence of RNase A (Sigma-Aldrich). For cap-affinity chromatography, FLAG-tagged Y14 and CBP80 were overexpressed in HEK293 cells and immunoprecipitated from cell lysates using anti-FLAG Agarose (Sigma-Aldrich) and eluted from the resin by competition with the FLAG peptide (Sigma-Aldrich). Cap-affinity chromatography was performed essentially as described (Kiriakidou et al., 2007) with minor modifications. Immunopurified FLAG-proteins were then incubated with m7GTP-Sepharose (GE Healthcare) or GTP-agarose (Sigma-Aldrich) in 20 mM Tris-HCl (pH 7.4) containing 100 mM NaCl, 2.5 mM MgCl2, 1 mM dithiothreitol, and 1 mg/ml heparin at 4°C for 2 h. The cap analogue m7GpppG (New England BioLabs, Ipswich, MA) was used as a competitive inhibitor at a concentration of 75 μM.
Antibodies
The monoclonal antibody used was anti–α-tubulin (0.3 μg/ml) from NeoMarkers (Fremont, CA). The polyclonal antibodies used were against DDX3 (Lai et al., 2008), Dcp1a (0.5 μg/ml), Xrn1 (0.5 μg/ml), and Y14 (0.4 μg/ml) from Bethyl (Montgomery, TX), eIF4AIII (0.5 μg/ml), DDX6 (0.1 μg/ml), and Edc4 (0.2 μg/ml) from Abcam (Cambridge, MA), hRrp6 (1 μg/ml), Dcp2 (1 μg/ml), and FLAG epitope (0.4 μg/ml) from Sigma-Aldrich, c-myc epitope (0.5 μg/ml) from Upstate (Millipore, Billerica, CA), and HA epitope (0.5 μg/ml) from Covance (Berkeley, CA).
RNA decapping assay and UV cross-linking
The intron-less PIP85a (Li et al., 2003) RNA substrate (PIP85a∆i) was produced by in vitro transcription using T7 RNA polymerase (Promega, Madison, WI), extracted with phenol/chloroform, and recovered by ethanol precipitation. PIP85a∆i was cap labeled by vaccinia virus capping enzyme (Epicentre Biotechnologies, Madison, WI) at 37°C for 1 h in the presence of [α-32P]GTP (3000 Ci/mmol). The methylated cap was produced in the same reaction containing 0.1 mM S-adenosyl methionine. To remove the RNA body, the 32P-labeled and methyl-capped PIP85a∆i was digested with RNase T1 (Roche, Indianapolis, IN) at 37°C for 30 min; the resulting m7G*pppGp was confirmed by polyethyleneimine-cellulose TLC (Sigma-Aldrich) analysis (Supplemental Figure S9). To prepare end-labeled, uncapped RNA, 5′ triphosphates of in vitro–transcribed PIP85a∆i were removed by alkaline phosphatase (Roche), followed by end labeling with [γ-32P]ATP (3000 Ci/mmol) using T4 polynucleotide kinase (New England BioLabs). All labeled RNAs were gel purified. The decapping assay was carried out essentially as described (Zhang et al., 1999). In brief, 1 μg of GST-fusion protein (∼25 pmol) was first incubated with 0.5 × 104 cpm (∼7.5 fmol) of cap-labeled PIP85a∆i in a reaction containing 50 mM Tris-HCl (pH 7.9), 30 mM (NH4)2SO4, and 1 mM MgCl2 at 4°C for 10 min. Subsequently, 0.5 μg (∼10 pmol) of His-Dcp2 was added into the reaction and incubation continued at 37°C for 40 min. The reaction was terminated by adding EDTA to a final concentration of 50 mM, and an aliquot of each sample was analyzed by polyethyleneimine-cellulose TLC developed in 0.45 M (NH4)2SO4. For UV cross-linking, 15 fmol of PIP85a∆i RNA was incubated with 5 μg of one of the various GST-Y14, His-Y14, or His-Magoh at 4°C for 15 min, followed by UV irradiation (Stratagene, Santa Clara, CA). The reaction was then treated with 1 mg/ml RNase A at 37°C for 30 min. The cross-linked products were resolved by 12% SDS–PAGE, followed by autoradiography.
RNA protection assay
The RNase protection assay was performed essentially as described (Chang and Tarn, 2009). HeLa tet-off cells were transfected with the pTRE-TNFα-UTR reporter vector, the green fluorescent protein (GFP) control vector, and FLAG-Y14, Y14∆C, Magoh, PYM, or eIF4AIII. After 24 h, 10 μg/ml doxycycline (Sigma-Aldrich) was added to stop transcription of the reporter, and cells were harvested at various times. For each reaction, 5 μg RNA and 2 × 105 cpm of the probe were used. Supplemental Figure S10 demonstrates that an excess of the probes and sufficient amount of RNase T1 were used. The resulting products were resolved on 6% denaturing polyacrylamide–urea gels, followed by autoradiography.
In vitro splicing and immunoprecipitation
HEK293 cells were transiently transfected with the FLAG-Y14 or FLAG-Y14SE expression vector using calcium phosphate, followed by nuclear extract preparation as described (Lee et al., 2010). The PIP85a pre-mRNA was in vitro transcribed and α-32P labeled as described (Li et al., 2003). In vitro splicing using the intron-containing PIP85a pre-mRNA as the substrate was performed in mock or Y14-overexpressing HEK293 nuclear extracts. For immunoprecipitation of FLAG-Y14-containing mRNPs, the splicing reaction was performed at 30°C for 2 h, and the reaction mixtures were subjected to immunoprecipitation using anti-FLAG Agarose (Lee et al., 2010). Coprecipitated RNAs were recovered from the resins and analyzed by electrophoresis on 6% denaturing polyacrylamide gels.
Indirect immunofluorescence
HeLa cells were transiently transfected with the vector encoding FLAG-tagged wild-type, SA, or SE mutant of Y14, Y14-targeting small interfering RNA (5′-ggguauacucuaguugaaa-3′, Invitrogen), or eIF4AIII-targeting small interfering RNA (5′-uugaguuucacgaaccugaauaucc-3′, Invitrogen) using Lipofectamine 2000 (Invitrogen). The primary antibodies used for immunofluorescence included monoclonal anti-Y14 (Abcam), anti-Dcp1a (Sigma-Aldrich), and anti-FLAG (Sigma-Aldrich) and polyclonal anti-eIF4AIII, anti-Dcp1a, and anti-Edc4. Cells were incubated with antibodies at an appropriate concentration and subsequently washed with phosphate-buffered saline as described (Chang and Tarn, 2009). Cells were then incubated with fluorescein isothiocyanate (FITC)–conjugated anti–rabbit immunoglobulin G (IgG; Cappel Laboratories, Downingtown, PA), FITC-conjugated anti–mouse IgG (Cappel), rhodamine-conjugated anti–rabbit IgG (Cappel Laboratories), or rhodamine-conjugated anti–mouse IgG (Cappel Laboratories). Nuclei were counterstained with Hoechst 33258 (Sigma-Aldrich). The specimens were observed using a laser confocal microscope (LSM 510 META; Carl Zeiss, Jena, Germany) coupled with an image analysis system. The P-bodies in Y14-deficient or Y14-overexpressing cells were counted in 25 randomly selected areas, which in total contained >200 cells in individual transfections. Three independent experiments were performed.
Supplementary Material
Acknowledgments
We greatly appreciate the work of Yu-Shiou Lu and Pey-Jey Peng in the initial stage of this study. Funding was provided by Grant NSC 98-2628-B-001-007-MY3 of the National Science Council of Taiwan.
Abbreviations used:
- ARE
AU-rich element
- βG
β-globin
- EJC
exon-junction complex
- eIF4E
eukaryotic translation initiation factor 4E
- FITC
fluorescein isothiocyanate
- GFP
green fluorescent protein
- GST
glutathione S-transferase
- HA
hemagglutinin
- IP
immunoprecipitation
- m7GpppG
7-methyl guanosine
- mRNP
messenger ribonucleoprotein
- NMD
nonsense-mediated mRNA decay
- P-bodies
processing bodies
- pre-mRNA
precursor mRNA
- RRM
RNA-recognition motif
- siRNA
small interfering RNA
- TNFα
tumor necrosis factor α
- UTR
untranslated region
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E12-03-0217) on October 31, 2012.
REFERENCES
- Brengues M, Teixeira D, Parker R. Movement of eukaryotic mRNAs between polysomes and cytoplasmic processing bodies. Science. 2005;310:486–489. doi: 10.1126/science.1115791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamieh H, Ballut L, Bonneau F, Le Hir H. NMD factors UPF2 and UPF3 bridge UPF1 to the exon junction complex and stimulate its RNA helicase activity. Nat Struct Mol Biol. 2008;15:85–93. doi: 10.1038/nsmb1330. [DOI] [PubMed] [Google Scholar]
- Chang WL, Tarn WY. A role for transportin in deposition of TTP to cytoplasmic RNA granules and mRNA decay. Nucleic Acids Res. 2009;37:6600–6612. doi: 10.1093/nar/gkp717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Z, Muhlrad D, Lim MK, Parker R, Song H. Structural and functional insights into the human Upf1 helicase core. EMBO J. 2007;26:253–264. doi: 10.1038/sj.emboj.7601464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang TW, Peng PJ, Tarn WY. The exon junction complex component Y14 modulates the activity of the methylosome in biogenesis of spliceosomal small nuclear ribonucleoproteins. J Biol Chem. 2011;286:8722–8728. doi: 10.1074/jbc.M110.190587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coller J, Parker R. General translational repression by activators of mRNA decapping. Cell. 2005;122:875–886. doi: 10.1016/j.cell.2005.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cougot N, Babajko S, Seraphin B. Cytoplasmic foci are sites of mRNA decay in human cells. J Cell Biol. 2004;165:31–40. doi: 10.1083/jcb.200309008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dostie J, Dreyfuss G. Translation is required to remove Y14 from mRNAs in the cytoplasm. Curr Biol. 2002;12:1060–1067. doi: 10.1016/s0960-9822(02)00902-8. [DOI] [PubMed] [Google Scholar]
- Durand S, Cougot N, Mahuteau-Betzer F, Nguyen CH, Grierson DS, Bertrand E, Tazi J, Lejeune F. Inhibition of nonsense-mediated mRNA decay (NMD) by a new chemical molecule reveals the dynamic of NMD factors in P-bodies. J Cell Biol. 2007;178:1145–1160. doi: 10.1083/jcb.200611086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenger-Gron M, Fillman C, Norrild B, Lykke-Andersen J. Multiple processing body factors and the ARE binding protein TTP activate mRNA decapping. Mol Cell. 2005;20:905–915. doi: 10.1016/j.molcel.2005.10.031. [DOI] [PubMed] [Google Scholar]
- Fischer N, Weis K. The DEAD box protein Dhh1 stimulates the decapping enzyme Dcp1. EMBO J. 2002;21:2788–2797. doi: 10.1093/emboj/21.11.2788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franks TM, Lykke-Andersen J. TTP and BRF proteins nucleate processing body formation to silence mRNAs with AU-rich elements. Genes Dev. 2007;21:719–735. doi: 10.1101/gad.1494707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franks TM, Singh G, Lykke-Andersen J. Upf1 ATPase-dependent mRNP disassembly is required for completion of nonsense-mediated mRNA decay. Cell. 2010;143:938–950. doi: 10.1016/j.cell.2010.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fribourg S, Gatfield D, Izaurralde E, Conti E. A novel mode of RBD-protein recognition in the Y14-Mago complex. Nat Struct Biol. 2003;10:433–439. doi: 10.1038/nsb926. [DOI] [PubMed] [Google Scholar]
- Gatfield D, Izaurralde E. REF1/Aly and the additional exon junction complex proteins are dispensable for nuclear mRNA export. J Cell Biol. 2002;159:579–588. doi: 10.1083/jcb.200207128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehring NH, Neu-Yilik G, Schell T, Hentze MW, Kulozik AE. Y14 and hUpf3b form an NMD-activating complex. Mol Cell. 2003;11:939–949. doi: 10.1016/s1097-2765(03)00142-4. [DOI] [PubMed] [Google Scholar]
- Harigaya Y, Jones BN, Muhlrad D, Gross JD, Parker R. Identification and analysis of the interaction between Edc3 and Dcp2 in Saccharomyces cerevisiae. Mol Cell Biol. 2010;30:1446–1456. doi: 10.1128/MCB.01305-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu CL, Stevens A. Yeast cells lacking 5′→3′ exoribonuclease 1 contain mRNA species that are poly(A) deficient and partially lack the 5′ cap structure. Mol Cell Biol. 1993;13:4826–4835. doi: 10.1128/mcb.13.8.4826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu IW, Hsu M, Li C, Chuang TW, Lin RI, Tarn WY. Phosphorylation of Y14 modulates its interaction with proteins involved in mRNA metabolism and influences its methylation. J Biol Chem. 2005;280:34507–34512. doi: 10.1074/jbc.M507658200. [DOI] [PubMed] [Google Scholar]
- Isken O, Kim YK, Hosoda N, Mayeur GL, Hershey JW, Maquat LE. Upf1 phosphorylation triggers translational repression during nonsense-mediated mRNA decay. Cell. 2008;133:314–327. doi: 10.1016/j.cell.2008.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov PV, Gehring NH, Kunz JB, Hentze MW, Kulozik AE. Interactions between UPF1, eRFs, PABP and the exon junction complex suggest an integrated model for mammalian NMD pathways. EMBO J. 2008;27:736–747. doi: 10.1038/emboj.2008.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao X, Wang Z, Kiledjian M. Identification of an mRNA-decapping regulator implicated in X-linked mental retardation. Mol Cell. 2006;24:713–722. doi: 10.1016/j.molcel.2006.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashima I, Yamashita A, Izumi N, Kataoka N, Morishita R, Hoshino S, Ohno M, Dreyfuss G, Ohno S. Binding of a novel SMG-1-Upf1-eRF1-eRF3 complex (SURF) to the exon junction complex triggers Upf1 phosphorylation and nonsense-mediated mRNA decay. Genes Dev. 2006;20:355–367. doi: 10.1101/gad.1389006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khanna R, Kiledjian M. Poly(A)-binding-protein-mediated regulation of hDcp2 decapping in vitro. EMBO J. 2004;23:1968–1976. doi: 10.1038/sj.emboj.7600213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiriakidou M, Tan GS, Lamprinaki S, De Planell-Saguer M, Nelson PT, Mourelatos Z. An mRNA m7G cap binding-like motif within human Ago2 represses translation. Cell. 2007;129:1141–1151. doi: 10.1016/j.cell.2007.05.016. [DOI] [PubMed] [Google Scholar]
- Kshirsagar M, Parker R. Identification of Edc3p as an enhancer of mRNA decapping in Saccharomyces cerevisiae. Genetics. 2004;166:729–739. doi: 10.1534/genetics.166.2.729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai MC, Lee YH, Tarn WY. The DEAD-box RNA helicase DDX3 associates with export messenger ribonucleoproteins as well as tip-associated protein and participates in translational control. Mol Biol Cell. 2008;19:3847–3858. doi: 10.1091/mbc.E07-12-1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larimer FW, Hsu CL, Maupin MK, Stevens A. Characterization of the XRN1 gene encoding a 5′ → 3′ exoribonuclease: sequence data and analysis of disparate protein and mRNA levels of gene-disrupted yeast cells. Gene. 1992;120:51–57. doi: 10.1016/0378-1119(92)90008-d. [DOI] [PubMed] [Google Scholar]
- Le Hir H, Gatfield D, Izaurralde E, Moore MJ. The exon-exon junction complex provides a binding platform for factors involved in mRNA export and nonsense-mediated mRNA decay. EMBO J. 2001;20:4987–4997. doi: 10.1093/emboj/20.17.4987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Hir H, Izaurralde E, Maquat LE, Moore MJ. The spliceosome deposits multiple proteins 20-24 nucleotides upstream of mRNA exon-exon junctions. EMBO J. 2000;19:6860–6869. doi: 10.1093/emboj/19.24.6860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HC, Choe J, Chi SG, Kim YK. Exon junction complex enhances translation of spliced mRNAs at multiple steps. Biochem Biophys Res Commun. 2009;384:334–340. doi: 10.1016/j.bbrc.2009.04.123. [DOI] [PubMed] [Google Scholar]
- Lee KM, Hsu W, Tarn WY. TRAP150 activates pre-mRNA splicing and promotes nuclear mRNA degradation. Nucleic Acids Res. 2010;38:3340–3350. doi: 10.1093/nar/gkq017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Lin RI, Lai MC, Ouyang P, Tarn WY. Nuclear Pnn/DRS protein binds to spliced mRNPs and participates in mRNA processing and export via interaction with RNPS1. Mol Cell Biol. 2003;23:7363–7376. doi: 10.1128/MCB.23.20.7363-7376.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Song MG, Kiledjian M. Transcript-specific decapping and regulated stability by the human Dcp2 decapping protein. Mol Cell Biol. 2008;28:939–948. doi: 10.1128/MCB.01727-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lykke-Andersen J. Identification of a human decapping complex associated with hUpf proteins in nonsense-mediated decay. Mol Cell Biol. 2002;22:8114–8121. doi: 10.1128/MCB.22.23.8114-8121.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piccirillo C, Khanna R, Kiledjian M. Functional characterization of the mammalian mRNA decapping enzyme hDcp2. RNA. 2003;9:1138–1147. doi: 10.1261/rna.5690503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz DC, Parker R. Mutations in translation initiation factors lead to increased rates of deadenylation and decapping of mRNAs in Saccharomyces cerevisiae. Mol Cell Biol. 1999;19:5247–5256. doi: 10.1128/mcb.19.8.5247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz DC, Parker R. mRNA decapping in yeast requires dissociation of the cap binding protein, eukaryotic translation initiation factor 4E. Mol Cell Biol. 2000;20:7933–7942. doi: 10.1128/mcb.20.21.7933-7942.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- She M, Decker CJ, Sundramurthy K, Liu Y, Chen N, Parker R, Song H. Crystal structure of Dcp1p and its functional implications in mRNA decapping. Nat Struct Mol Biol. 2004;11:249–256. doi: 10.1038/nsmb730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- She M, Decker CJ, Svergun DI, Round A, Chen N, Muhlrad D, Parker R, Song H. Structural basis of dcp2 recognition and activation by dcp1. Mol Cell. 2008;29:337–349. doi: 10.1016/j.molcel.2008.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheth U, Parker R. Decapping and decay of messenger RNA occur in cytoplasmic processing bodies. Science. 2003;300:805–808. doi: 10.1126/science.1082320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheth U, Parker R. Targeting of aberrant mRNAs to cytoplasmic processing bodies. Cell. 2006;125:1095–1109. doi: 10.1016/j.cell.2006.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh G, Jakob S, Kleedehn MG, Lykke-Andersen J. Communication with the exon-junction complex and activation of nonsense-mediated decay by human Upf proteins occur in the cytoplasm. Mol Cell. 2007;27:780–792. doi: 10.1016/j.molcel.2007.06.030. [DOI] [PubMed] [Google Scholar]
- Tange TO, Shibuya T, Jurica MS, Moore MJ. Biochemical analysis of the EJC reveals two new factors and a stable tetrameric protein core. RNA. 2005;11:1869–1883. doi: 10.1261/rna.2155905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teixeira D, Parker R. Analysis of P-body assembly in Saccharomyces cerevisiae. Mol Biol Cell. 2007;18:2274–2287. doi: 10.1091/mbc.E07-03-0199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teixeira D, Sheth U, Valencia-Sanchez MA, Brengues M, Parker R. Processing bodies require RNA for assembly and contain nontranslating mRNAs. RNA. 2005;11:371–382. doi: 10.1261/rna.7258505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tritschler F, Braun JE, Eulalio A, Truffault V, Izaurralde E, Weichenrieder O. Structural basis for the mutually exclusive anchoring of P body components EDC3 and Tral to the DEAD box protein DDX6/Me31B. Mol Cell. 2009;33:661–668. doi: 10.1016/j.molcel.2009.02.014. [DOI] [PubMed] [Google Scholar]
- Tritschler F, Eulalio A, Helms S, Schmidt S, Coles M, Weichenrieder O, Izaurralde E, Truffault V. Similar modes of interaction enable Trailer Hitch and EDC3 to associate with DCP1 and Me31B in distinct protein complexes. Mol Cell Biol. 2008;28:6695–6708. doi: 10.1128/MCB.00759-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tritschler F, Eulalio A, Truffault V, Hartmann MD, Helms S, Schmidt S, Coles M, Izaurralde E, Weichenrieder O. A divergent Sm fold in EDC3 proteins mediates DCP1 binding and P-body targeting. Mol Cell Biol. 2007;27:8600–8611. doi: 10.1128/MCB.01506-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dijk E, Cougot N, Meyer S, Babajko S, Wahle E, Seraphin B. Human Dcp2: a catalytically active mRNA decapping enzyme located in specific cytoplasmic structures. EMBO J. 2002;21:6915–6924. doi: 10.1093/emboj/cdf678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilela C, Velasco C, Ptushkina M, McCarthy JE. The eukaryotic mRNA decapping protein Dcp1 interacts physically and functionally with the eIF4F translation initiation complex. EMBO J. 2000;19:4372–4382. doi: 10.1093/emboj/19.16.4372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Jiao X, Carr-Schmid A, Kiledjian M. The hDcp2 protein is a mammalian mRNA decapping enzyme. Proc Natl Acad Sci USA. 2002;99:12663–12668. doi: 10.1073/pnas.192445599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu JH, Yang WH, Gulick T, Bloch KD, Bloch DB. Ge-1 is a central component of the mammalian cytoplasmic mRNA processing body. RNA. 2005;11:1795–1802. doi: 10.1261/rna.2142405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Williams CJ, Wormington M, Stevens A, Peltz SW. Monitoring mRNA decapping activity. Methods. 1999;17:46–51. doi: 10.1006/meth.1998.0706. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.