

Endoplasmic reticulum (ER) stress induces an mRNA decay pathway termed regulated Ire-dependent decay (RIDD). This study shows that in Drosophila cells, ER association is sufficient for targeting mRNAs to RIDD and most membrane-associated mRNAs are degraded more rapidly during ER stress. It is also reported that a small number of mRNAs are specifically protected from this default pathway.
Abstract
Ire1 is an endoplasmic reticulum (ER) transmembrane protein that senses disturbances in protein folding homeostasis and contributes to a multifaceted response to stress. The nuclease activity of Ire1, in addition to splicing the mRNA encoding the transcription factor Xbp1, mediates mRNA degradation in response to ER stress through a pathway termed regulated Ire1-dependent decay (RIDD). We previously showed that ER targeting of substrates is necessary for RIDD; in this paper, we show that ER localization is also sufficient to induce decay in a normally unaffected mRNA. Using microarrays, we also measured relative mRNA degradation in the presence and absence of ER stress in Drosophila S2 cells, and determined mRNA membrane association using detergent fractionation. The vast majority of mRNAs that were strongly associated with the ER were degraded faster during ER stress in an Ire1-dependent manner, suggesting that RIDD is the default pathway for ER-localized mRNAs during stress. We also show that the mRNA encoding plexin A remains highly polysome associated during stress and escapes degradation by RIDD, and that its 5′ untranslated region can protect a strong RIDD target from degradation. These results suggest that while translation is generally attenuated during ER stress, continued translation of certain messages can protect them from degradation by RIDD.
INTRODUCTION
As the entry point for the secretory pathway, the endoplasmic reticulum (ER) must accommodate changes in the flux of proteins that need to be folded and processed. Large increases in the ER protein-folding burden or disruptions to ER function, often referred to as ER stress, are associated with many pathologies, while both sustained and temporary increases in folding burden occur regularly in secretory cell types. The unfolded protein response (UPR) is a collection of pathways that work to restore and enhance ER function, both during stress and during the development and normal functioning of secretory cells (Walter and Ron, 2011; Moore and Hollien, 2012). Three signaling proteins, Ire1, Atf6, and Perk, form the core of the metazoan UPR and work to increase the folding capacity of the ER and to decrease the burden of incoming proteins. Ire1, the most broadly conserved of these signaling proteins, is an ER transmembrane protein with a cytosolic endoribonuclease activity activated by ER stress (Cox et al., 1993; Mori et al., 1993). Ire1 cleaves the mRNA encoding Xbp1, leading to removal of a regulatory intron and formation of the in-frame template for this transcription factor (Shen et al., 2001; Yoshida et al., 2001; Calfon et al., 2002). Atf6 is proteolytically cleaved during stress, and the released cytoplasmic domain also functions as a transcription factor (Haze et al., 1999; Wang et al., 2000). Perk decreases the load on the ER by phosphorylating eIF2α, thus attenuating general translation (Shi et al., 1998; Harding et al., 1999). This phosphorylation event also leads to increased translation of Atf4, a third UPR transcription factor, which is regulated by upstream open reading frames (uORFs) in its 5′ untranslated region (UTR; Harding et al., 2000).
We previously found that in addition to cleaving and thereby activating Xbp1, Ire1 mediates the degradation of a subset of mRNAs in Drosophila S2 and mammalian cells during ER stress via a pathway termed regulated Ire1-dependent decay (RIDD; Hollien and Weissman, 2006; Hollien et al., 2009). The targets of RIDD are highly enriched for mRNAs encoding proteins with signal sequences and/or transmembrane domains, and mutation or removal of the signal sequences in reporter mRNAs prevents their decay (Hollien and Weissman, 2006). Localization to the ER therefore appears to be necessary for degradation by RIDD. However, additional sequence or structural elements important for substrate selection have been elusive. We therefore hypothesized that localization to the ER is also sufficient for RIDD, predicting that most ER-localized mRNAs are RIDD targets. Although several genome-wide experiments have previously examined the effects of ER stress (Travers et al., 2000; Okada et al., 2002; Harding et al., 2003; Shen et al., 2005), the vast transcriptional remodeling of the ER under stress has precluded a complete view of the effects of mRNA turnover. In this study, we used reporter mRNAs and microarray analysis of mRNA decay in Drosophila S2 cells to provide support for the hypothesis that RIDD is the default pathway for ER-localized mRNAs during stress.
RESULTS
Targeting the mRNA encoding green fluorescent protein (GFP) to the ER membrane results in degradation by RIDD
To test whether signal sequence–mediated localization to the ER membrane is sufficient for RIDD, we compared the decay of mRNA reporters encoding either GFP alone or GFP with an N-terminal signal sequence in the presence and absence of ER stress. While the two reporters were degraded with similar kinetics in the absence of stress, addition of dithiothreitol (DTT), a reducing agent that strongly induces ER stress, increased the decay rate of the signal sequence–containing RNA, but not that of the cytosolic RNA (Figure 1A). This increase in degradation was Ire1-dependent, as RNA interference (RNAi) of Ire1 abolished the effect (Figure 1B). Thus the mRNA encoding signal sequence GFP is a target of RIDD.
FIGURE 1:
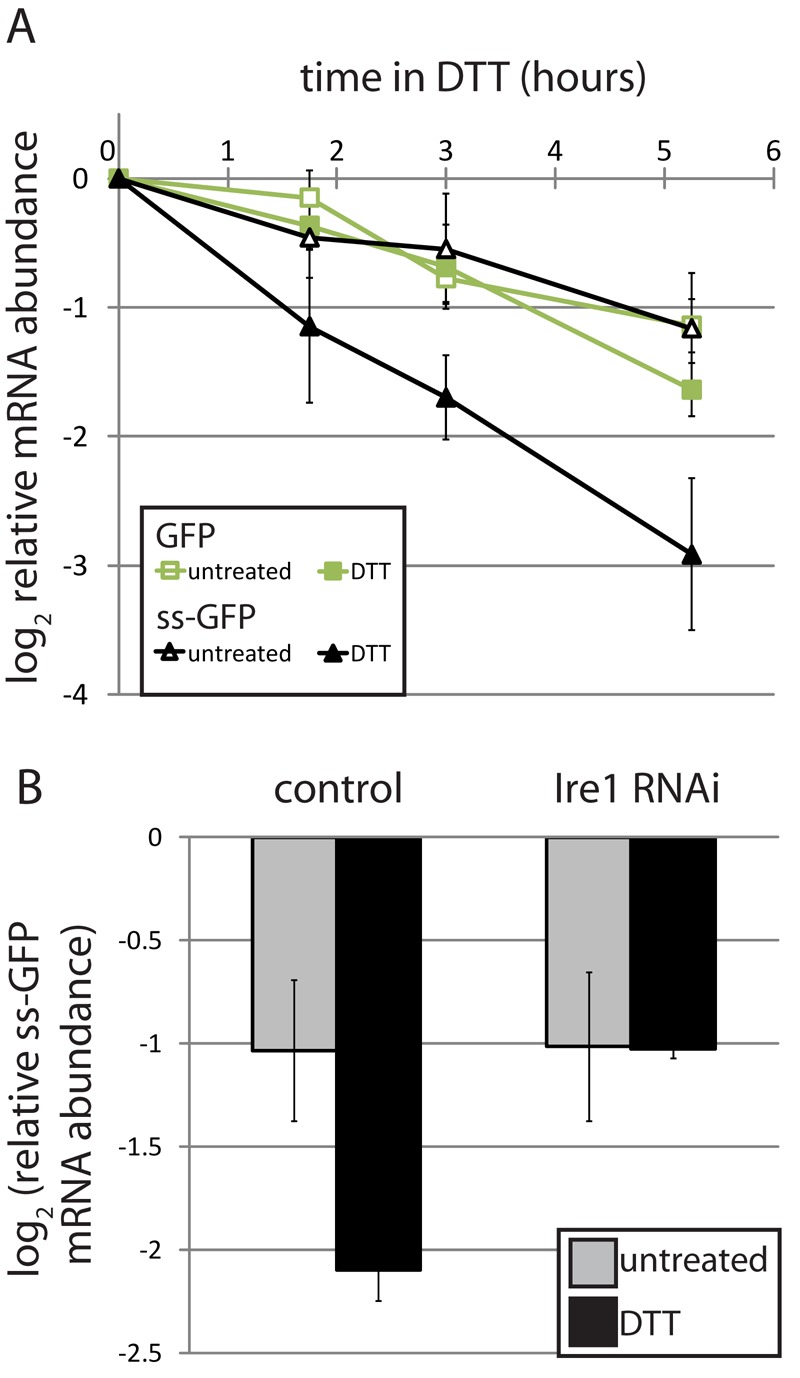
Targeting of GFP mRNA to the ER membrane results in degradation by RIDD. (A) GFP and an ER-targeted GFP containing the signal sequence of Drosophila Hsp70-3 (ss-GFP) were placed under the control of the copper-inducible metallothionein promoter and stably transfected into Drosophila S2 cells. We induced expression for 3 h, removed copper, and monitored mRNA levels over time in the absence and presence of the ER stressor DTT (2 mM), by real-time qPCR. GFP mRNA levels were normalized to that of Rpl19. Shown are the average and SD of four independent experiments. The copper wash-out was effective, as no additional decay was observed when we also added a copper chelator or general transcription inhibitor (see Figure S1). (B) We measured the relative mRNA degradation of the ss-GFP reporter from (A) following depletion of Ire1 by RNAi. Shown are the relative mRNA levels 5 h after removal of copper, in the absence and presence of DTT (2 mM), for two independent experiments (average and SD).
RIDD broadly degrades membrane-bound mRNAs
The hypothesis that localization to the ER membrane is necessary and sufficient for targeting an mRNA to the RIDD pathway predicts that the turnover rates of essentially all membrane-localized mRNAs should increase when Ire1 is activated during ER stress. To test this, we measured mRNA decay in the presence and absence of ER stress and of Ire1 using spotted DNA microarrays. We treated S2 cells with actinomycin D to inhibit transcription; this was followed by DTT to induce ER stress. We then used microarrays to compare RNA levels in untreated cells with those treated with actinomycin D alone or actinomycin D and DTT. Overall mRNA levels decreased after 4.5 h in actinomycin D, as expected, although many RNAs were stable over this time. To take into account the overall signal decrease in actinomycin D samples, we used the levels of mRNAs encoding ribosomal proteins to normalize signals for individual mRNAs in each array (see Materials and Methods). This normalization procedure resulted in the expected levels of decay for previously characterized RIDD targets. In parallel, we carried out this procedure for cells depleted of Ire1 by RNAi and for control cells to which no double-stranded RNA (dsRNA) was added.
To distinguish between membrane-associated and cytosolic mRNAs, we permeabilized S2 cells using digitonin to extract cytosolic mRNAs, then with Triton X-100 to extract membrane-bound RNAs, using a method developed by Nicchitta and colleagues (Stephens et al., 2008). After measuring RNA levels by microarray, we calculated the fraction membrane association (FM = mRNA in Triton supernatant/sum of mRNA in digitonin and Triton supernatants) for each measurable mRNA. To test the hypothesis that all membrane-associated mRNAs are degraded by RIDD, we first examined the set of mRNAs that had high FM values (>0.75) and were predicted to contain signal sequence coding regions. Of these mRNAs, 99% clustered together as RIDD targets, displaying ER stress–dependent and Ire1-dependent increases in degradation (Figure 2A).
FIGURE 2:
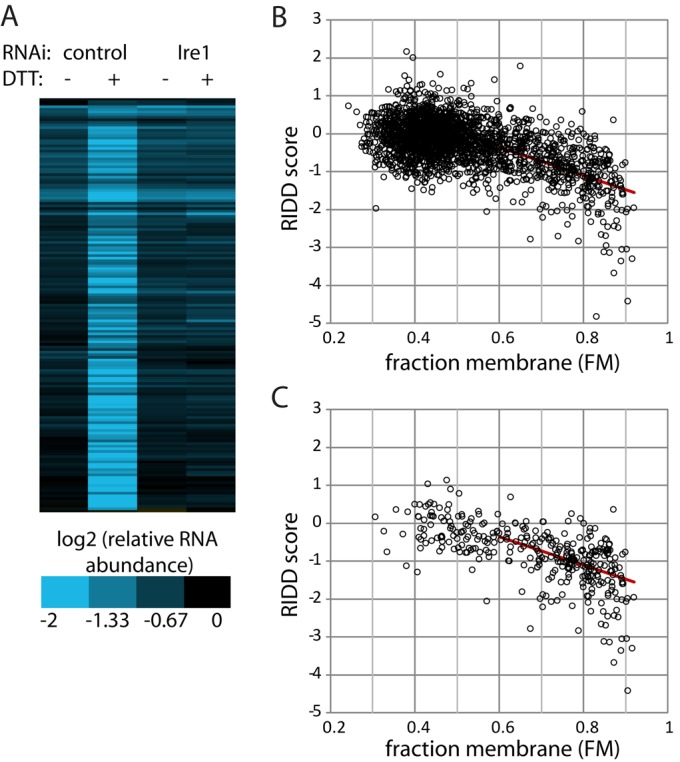
RIDD broadly degrades membrane-associated mRNAs. (A) After either mock-treating or depleting Drosophila S2 cells of Ire1 using RNAi, we blocked transcription with actinomycin D (1 μg/ml) and measured mRNA decay in the absence and presence of ER stress (2 mM DTT, 4.5 h). RNA levels relative to untreated controls were measured using spotted DNA microarrays and normalized to mRNAs encoding ribosomal proteins (see Materials and Methods). In parallel, we measured membrane association using detergent extraction and calculated the FM for each mRNA. Shown is a cluster analysis of mRNAs with a high experimental FM (>0.75) and containing predicted signal sequence-coding regions. (B) RIDD score (see Results; lower values indicate degradation via RIDD) vs. FM for mRNAs that were stable (degraded less than threefold 4.5 h following actinomycin D treatment) in the absence of ER stress. (C) RIDD score vs. FM as in (B), but for mRNAs containing predicted signal sequence-coding regions. (B and C) The red line indicates a linear fit of the entire data set with FM > 0.54 (R2 = 0.29).
The extent of RIDD targeting appeared to depend on the degree of membrane association. For each measurable mRNA, we calculated a RIDD score, equal to the log2 value of DTT-dependent degradation in control cells (no RNAi) minus the log2 value of DTT-dependent degradation in cells depleted of Ire1 by RNAi. As seen in Figure 2B, mRNAs with high FM values had more negative RIDD scores, indicating that mRNAs more strongly associated with the ER membrane displayed stronger degradation in an ER stress–dependent and Ire1-dependent manner. The effect of RIDD became statistically significant at FM ∼ 0.54, as determined by paired t tests comparing actinomycin-treated samples in the presence versus absence of DTT and comparing DTT-treated samples with versus without depletion of Ire1 (Supplemental Table S1). RIDD scores were described well by a linear fit in this region (FM > 0.54), although this did not fully account for the range of RIDD scores, as described in the next section and in Tables S2 and S3. In particular, RIDD scores at very high FM tended to be lower than predicted from the linear fit; this may be the result of membrane association that is stronger than we are able to measure using our fractionation technique.
The correlation between RIDD targeting and membrane association was independent of traditional ER-targeting sequences within the RNA. Although mRNAs with signal sequences were more likely to have high FM values, there were many with low FM values, potentially because the proteins they encode are posttranslationally inserted into the ER; these were not RIDD targets (Figure 2C). Conversely, mRNAs lacking signal sequences and transmembrane domains but displaying high FM values followed the same trend as those containing identifiable targeting sequences (Supplemental Figure S2A). Thus it appears that the localization of the mRNA, and not the encoded protein, is a major factor in RIDD targeting.
Ire1 is known to cleave Xbp1 at specific stem-loop structures, and the seven-member loop has several conserved nucleotides. In addition, RIDD targets in mammalian cells often display similar stem-loops (Han et al., 2009; Hur et al., 2012). To test whether the presence of an Xbp1-like stem-loop influences the ability of an mRNA to be degraded by RIDD, we analyzed all transcripts associated with the mRNAs measured in our array data that were stable (degraded less than threefold after 4.5 h) in the absence of stress (i.e., those shown in Figure 2B). Of these 2368 mRNAs, 240 contained a predicted Ire1 cleavage site, defined as a stem of at least five base pairs, allowing for GC, AU, and GU pairs, and a loop with the sequence CnGCnGn. The presence of an Ire1 site, however, did not appear to influence RIDD targeting, as the distribution of mRNAs containing these sites was similar to that for the data set as a whole (Figure S2B). In contrast, using the same criteria, we identified potential cleavage sites in nine of the 11 confirmed RIDD targets in mammals (Table S4), representing a very strong enrichment.
Plexin A is protected from RIDD via its 5′ UTR
Although membrane association is important for RIDD targeting, this factor alone does not explain the full range of RIDD scores: some RNAs displayed especially strong or especially weak RIDD. We were particularly interested in those RNAs that displayed weaker RIDD targeting than expected based on their membrane association (Table S2), as these may reveal specific protection mechanisms. We chose several of these potentially protected mRNAs and used quantitative PCR (qPCR) to measure their decay rates in the presence and absence of ER stress. Unlike the typical membrane-associated mRNA, these mRNAs did not decay significantly faster in the presence of ER stress, even when we inhibited transcription using higher concentrations of actinomycin D or with 5,6-dichloro-1-β-d-ribofuranosylbenzimidazole, which inhibits RNA polymerase II more specifically (Figure S3).
Two features of the protected RNAs were immediately apparent. First, the weakest RIDD targets are more likely to lack traditional ER signal sequence–coding regions (5/10) when compared with the strongest targets (2/19; Tables S2 and S3), although as described in the preceding section this effect does not appear to be a major factor when the full data set is considered (Figure S2A). Second, the weakest RIDD targets were enriched for mRNAs encoding proteins with neuronal functions, such as axon guidance (Table S2).
One mechanism that could underlie this protection is the potential dissociation of the non-RIDD targets from the ER during stress. However, when we repeated our fractionation procedure with and without DTT (2 mM, 20 min), we found that most RNAs, including very strong RIDD targets, were more extractable with digitonin during stress (Figure 3A). A notable exception to this shift was the RIDD-protected mRNA encoding plexin A (plexA), a semaphorin receptor involved in axon guidance.
FIGURE 3:

PlexA remains ER- and polysome-associated during ER stress. (A) Shown are the FM values for the indicated mRNAs in the presence and absence of DTT (2 mM, 20 min), measured using detergent extraction (see Materials and Methods). Actin is shown as a cytosolic control, sparc and Hydr2 are RIDD targets, and the remaining mRNAs are protected from RIDD. (B–F) We either treated S2 cells with DTT (2 mM, 20 min, black bars) or left them untreated (gray bars), and then compared polysome association of the designated mRNAs using sucrose gradient fractionation. We collected RNA samples from the monosome peak (mono), the first four polysome peaks (low-density polysome, LP), and the denser polysome peaks (high-density polysome, HP), and analyzed mRNA content by qPCR. Shown is the average and SD of two to three independent experiments. Note the different scale for the y-axis for plexA. (G) FM values as in (A), but after longer DTT treatment.
We hypothesized that plexA escapes translational repression, thus allowing this RNA to remain strongly membrane-associated during stress and perhaps also leading to its protection from degradation. To test this, we measured the association of plexA and other messages with polysomes in the presence and absence of stress using sucrose gradient fractionation (Figure 3, B–F). As expected, the overall effect during ER stress was a shift of rRNA from polysome to monosome fractions, and BM-40-SPARC (referred to as sparc), a strong RIDD target, and ManF, a weak RIDD target, followed this trend (Figures 3, B and C, and S4). Atf4 and Gadd34 shifted from monosomes to polysomes in the presence of stress (Figure 3, D and E), as expected based on their previously described translational up-regulation (Harding et al., 2000; Lee et al., 2009). PlexA, while not displaying a significant shift during stress, was highly enriched in dense polysome fractions (Figure 3F). The continued ribosome and membrane association of the plexA mRNA may be a consequence of regulation of translation initiation or elongation, or of the potentially long time it would take to finish translating plexA after the initiation of ER stress. Although plexA is a large protein (1945 amino acids; Winberg et al., 1998), longer DTT treatments did not affect the continued membrane association of its mRNA (Figure 3G).
Hypothesizing that continued translation initiation is a key factor in protecting plexA from RIDD, we analyzed the effect of the 5′ UTR of plexA. We constructed reporters to test whether this region of plexA is sufficient to protect a strong RIDD target from degradation (Figure 4). We previously showed that the coding sequence of sparc is sufficient to direct it to the RIDD pathway during ER stress (Hollien and Weissman, 2006). When we replaced the 5′ UTR of this sparc reporter with the plexA 5′ UTR, degradation during ER stress was dramatically reduced. The 5′ UTR of Gadd34 was also protective, as predicted based on its ability to up-regulate translation initiation during stress (Lee et al., 2009). Continued translation during stress is often mediated by regulatory uORFs, and the 5′ UTR of plexA contains several putative uORFs. We therefore mutated the most upstream of these sequences, replacing the double start codon ATGATG with GTAGAT in the plexA sparc reporter. This mRNA was degraded more rapidly during stress, suggesting that the protective effects of the plexA 5′ UTR are mediated by uORFs (Figure 4).
FIGURE 4:
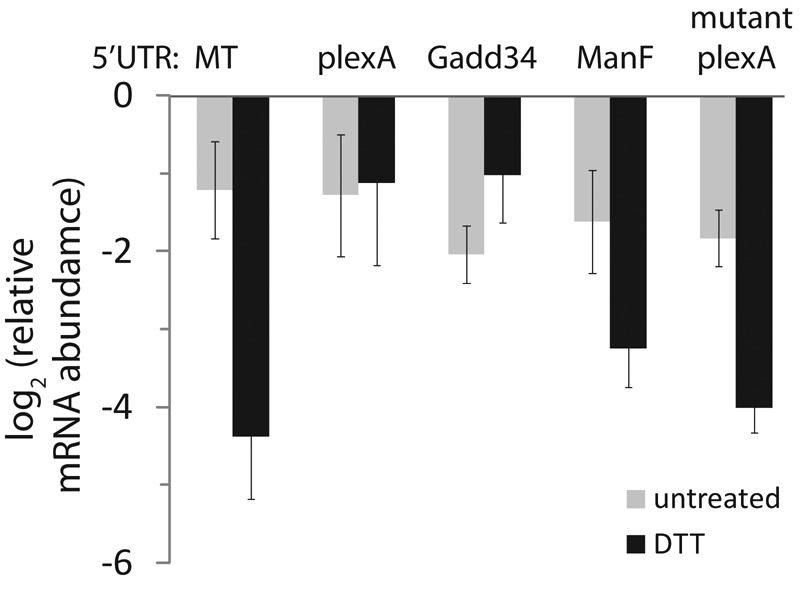
The 5′ UTRs of PlexA and Gadd34 confer protection from RIDD. We constructed reporter plasmids containing the metallothionein promoter and coding sequence of sparc with various 5′ UTRs: metallothionein (MT), plexA, Gadd34, ManF, or plexA with the first putative uORF mutated. We transiently transfected these plasmids into Drosophila S2 cells, induced expression overnight, removed copper, and monitored mRNA levels in the absence and presence of the DTT (2 mM, 4.5 h) by qPCR. Shown are the average and SD of at least three independent transfections.
Our data support a model in which plexA is protected from RIDD via continued translation during stress, as mediated by uORFs in its 5′ UTR. As other mRNAs that are protected from RIDD appeared to be extractable with digitonin and translationally attenuated during ER stress (Figure 3, A and F), we predicted that the 5′ UTRs of these mRNAs would not confer protection to sparc. Indeed, the 5′ UTR of ManF was not protective (Figure 4), suggesting that other mechanisms exist for protecting certain mRNAs from degradation during ER stress.
DISCUSSION
On the basis of the results described here, we propose that in Drosophila cells, RIDD is the default pathway for ER-bound mRNAs during stress, and decay occurs with minimal sequence specificity. The increase in decay for this large class of mRNAs applies even to mRNAs that are robustly up-regulated transcriptionally by the UPR, as shown previously for BiP in mammalian cells (Han et al., 2009). The advantages of initiating two seemingly opposed gene expression programs may include the ability to time the two responses. In S2 cells, both RIDD and Xbp1 splicing appear to be initiated immediately upon treatment of cells with DTT, but the increase in mRNA levels for Xbp1 targets is delayed by 1–1.5 h (Hollien and Weissman, 2006). This could reflect the time required for translation and activation of the Xbp1 protein, or the immediate increase in mRNA turnover may contribute to this lag phase. Either way, Ire1 may represent a means to time the two general outputs, such that the initial step relieves acute stress, whereas the second step restores homeostasis. A second advantage may be that the increased turnover of ER mRNAs would ensure sensitivity to changes in the transcriptional program. Increasing decay rates on a broad scale would allow mRNAs not actively transcribed during ER stress to be replaced with those that are, thus enhancing the effects of the transcriptional up-regulation of mRNAs encoding ER chaperones and other proteins with important ER functions.
This broad targeting of mRNAs to the RIDD pathway did not appear to be affected by the presence of a traditional Ire1 recognition site in the substrate mRNA. This was somewhat unexpected, as the RIDD targets identified in mammals appear to contain Xbp1-like sites (Han et al., 2009; Hur et al., 2012). As we have not carried out a genome-wide measurement of stress-dependent decay rates in mammalian cells, the exact scope of mammalian substrates remains to be determined. However, based on the available data, the RIDD pathway in mammals may be more restricted to particular transcripts, and thus may be more important in restricting expression of particular proteins, rather than in relieving general stress or enhancing the overall sensitivity to transcriptional regulation.
Our results also indicate that a small group of mRNAs are protected from RIDD. These include a subset of mRNAs that lack traditional ER-targeting motifs, despite physical association with membranes. As many of these mRNAs encode proteins that function at the ER or the plasma membrane, it is likely that they are indeed localized to the ER, and perhaps the specifics of their interactions with the ER and the ribosome affect their ability to be degraded. Previous reports have demonstrated that certain mRNAs encoding cytosolic proteins localize to the ER membrane (Pyhtila et al., 2008; Chen et al., 2011), and p180 was recently shown to be involved in the translation-independent localization of certain ER-associated mRNAs (Cui et al., 2012), indicating there are multiple mechanisms by which mRNAs can be localized to the ER. Despite this enrichment in the mRNAs protected from RIDD, however, the mechanism of ER localization does not appear to be a key factor in RIDD in general, as transcripts with and without traditional signal sequences appear to follow the same trends (Figure S2). Transcripts protected from RIDD are also enriched for those encoding proteins functioning in axon guidance. It is not clear to us why neuronal mRNAs would be protected from RIDD, although the number of genes is small and they may have alternate functions in nonneuronal cells, such as S2 cells.
We have shown for one mRNA, plexA, that protection from RIDD can be mediated by uORFs in the 5′ UTR. Most mRNAs are translationally repressed during ER stress, and we speculate that this release from extensive polysome association may make the typical mRNA more accessible to nucleases such as Ire1, while highly translated mRNAs such as plexA would be insensitive. This proposed relationship between translation and RIDD predicts that inhibition of Perk-mediated translational attenuation (Harding et al., 1999) would inhibit RIDD. Although we previously reported that knockdown of Perk in S2 cells does not affect RIDD (Hollien and Weissman, 2006), this result may be explained by minimal residual eIF2α phosphorylation, or by the fact that in S2 cells, translational attenuation can still occur when Perk is inhibited, through a largely uncharacterized mechanism (Garrey et al., 2010). Thus the relationship between RIDD and Perk in Drosophila cells remains somewhat unclear. Notably, however, other mechanisms for protection from RIDD must exist, as protection of ManF does not appear to be mediated by its 5′ UTR.
Overall, we have found that RIDD reflects the interdependence of localization, translation, and mRNA decay. We propose that these features of RIDD allow the overburdened ER to focus on folding proteins that are important for stress recovery, by removing mRNAs lacking specific mechanisms for up-regulation at either the transcriptional or translational level and thus allowing the ER to rapidly reestablish homeostasis in the presence of stress.
MATERIALS AND METHODS
Plasmids
For GFP reporters, we subcloned EGFP into a Drosophila expression vector containing the metallothionein promoter, as described previously (Hollien and Weissman, 2006). We targeted the GFP reporter mRNA to the ER by including the coding sequence for the first 23 amino acids of Drosophila BiP (CG4147), using fusion PCR. For reporters in Figure 4, we amplified the 5′ UTRs of target genes plexA (CG11081), ManF (CG7013), and Gadd34 (CG3825), using S2 cDNA as a template. The PCR products, containing MscI and KpnI restriction sites, were then cloned into a reporter containing the Drosophila metallothionein gene promoter and sparc (CG6378) coding sequence.
Cell culture, transfections, RNAi, actinomycin D, and DTT treatments
We cultured Drosophila S2 cells (Invitrogen, Carlsbad, CA) at room temperature in Schneider's media (Invitrogen) supplemented with 10% fetal bovine serum and antibiotics. We transiently and stably transfected S2 cells with 2 μg plasmid DNA using Cellfectin II (Invitrogen). We generated polyclonal cell lines by cotransfecting our expression plasmids with a hygromycin resistance plasmid (pCoHygro; Invitrogen) and selecting for resistant cells. To monitor decay of mRNAs expressed from reporter constructs, we treated cells with CuSO4 (100 μM, 3 h for GFP reporters in Figure 1; 300 μM, overnight for the sparc reporters in Figure 4) to induce expression, washed cells to remove the CuSO4, left cells untreated or added DTT (2 mM), and collected RNA samples over time.
To test the Ire1 dependence of mRNA decay rates, we used RNAi to deplete S2 cells of Ire1 (CG4583). We amplified a 643-nucleotide region of the protein-coding sequence from S2 cDNA using primers that contained T7 RNA polymerase sites on the 5′ ends. We then used this product as a template for dsRNA synthesis, using the Megascript T7 kit (Ambion, Austin, TX). We incubated S2 cells with the dsRNA in serum-free media for 45 min, replaced the serum, and allowed cells to recover for 3 d. This procedure was repeated once for the array samples and twice for the analysis of GFP reporter mRNAs. We subjected cells to ER stress 1 d following the final dsRNA treatment.
To measure the extent of decay of endogenous mRNAs in the absence and presence of ER stress, we either left cells untreated or treated them with actinomycin D (1 μg/ml) with or without 2 mM DTT for the indicated times. For all RNA analyses, we isolated total RNA using Trizol reagent (Invitrogen), and used these samples as templates for cDNA synthesis.
Digitonin fractionation
We used a modified version of the protocol developed by Stephens and Nicchitta for separation of membrane and cytosolic mRNAs (Stephens et al., 2008). We incubated S2 cells with or without DTT (2 mM, 20 min or 4.5 h) and then added cycloheximide (35 μM) for 10 min. After collecting cells by centrifugation, we resuspended cells in cytosol buffer (150 mM KOAc, 20 mM HEPES, pH 7.5, 2.5 mM Mg(OAc)2, 200 U/ml RNaseOUT, 35 μM cycloheximide) and immediately permeabilized them with 1 mg/ml digitonin (15 min on ice). We then centrifuged the cell lysates (800 × g, 5 min at 4°C) and collected the supernatant as the cytosolic fraction. We resuspended the pellet in cytosol buffer with 1% Triton X-100 (15 min on ice), centrifuged as above, and collected the supernatant as the membrane-bound fraction.
Real-time qPCR
To digest any contaminating plasmid DNA from transient transfections, we pretreated RNA samples with DNaseI (NEB, Ipswich, MA). We then synthesized cDNA from 2 μg total RNA using Superscript II (Invitrogen) or M-MuLV reverse transcriptase (NEB). We measured relative mRNA abundance by real-time qPCR using the Mastercycler ep realplex (Eppendorf, Hamburg, Germany) with SYBR Green as the fluorescent dye. We measured each sample in triplicate and normalized to the Rpl19 mRNA. To control for contaminating plasmid or genomic DNA, we also measured samples to which no reverse transcriptase was added. The primers used for qPCR are given in Table S5.
Microarray samples, hybridization, and analysis
We carried out labeling and hybridization of cDNA samples for microarray analysis as described previously, using the same array design and platform (Hollien and Weissman, 2006). We removed irregular spots (by visual inspection), spots that had low signal intensity (signal < twofold background), and spots with poor color consistency (R2 < 0.6 for linear relationship between Cy5 vs. Cy3 intensities across all pixels). We then used the ratio of medians for Cy5/Cy3 for further analysis. For samples derived from actinomycin-treated cells, standard normalization procedures were not appropriate, given the lack of transcription and expected decay of a large percentage of mRNAs. To normalize among different samples, we therefore divided the ratio of medians for each spot on a given array by the average of the 51 spots corresponding to mRNAs encoding ribosomal proteins, which displayed little to no decay relative to bulk rRNA levels in the timescale of this experiment. To ensure that only quality data were included, we removed all spots that were missing data for more than 3 of the 24 samples. After normalization, we divided the ratio for each spot in the actinomycin D (−/+ DTT) samples by the corresponding spot in the untreated sample from that experiment. We then averaged the data between independent experiments (five for the control experiment, three for the Ire1 RNAi experiment).
For the digitonin fraction samples, we hybridized each sample on two to three arrays and used the average for analysis. We removed poor-quality spots as described above and used a standard normalization by setting the average ratio of medians to 1.0 for each array. We carried out two independent fractionations for analysis by microarray. Taken together, our quality control measures resulted in a final 2556 spots representing 2419 different genes that were included in further analyses. We used the Signal P 4.0 (Petersen et al., 2011) and TMHMM (Krogh et al., 2001) servers to predict signal sequences and transmembrane domains, respectively.
Sucrose gradient fractionation
We incubated S2 cells with or without DTT (2 mM, 20 min) and then added cycloheximide (35 μM) for 10 min to lock ribosomes in place. We lysed cells in a low-salt buffer (20 mM TrisBase, 10 mM NaCl, 3 mM MgCl, 200 mM sucrose, 0.3% Triton X-100, pH 7.4) and added lysate to the top of a 15–50% sucrose gradient. After centrifuging the gradients with a SW55Ti rotor (47,000 rpm, 60 min, 4°C) we collected samples using a gradient fractionator equipped with an absorbance detector (ISCO, Lincoln, NE) and used Trizol to purify the RNA. We synthesized cDNA from 1.8 μg of each sample and used qPCR to analyze samples.
Supplementary Material
Acknowledgments
We thank Stewart Barlow for conducting pilot experiments measuring mRNA decay by microarray and Neal Tolley, Andrew Weyrich, and Steve Blair for technical advice and use of equipment. We also thank Kristin Moore, Joshua Plant, and Kael Fischer for critical reading of the manuscript. This work was supported by the University of Utah and a National Institutes of Health R00 grant (GM081255) to J.H.
Abbreviations used:
- dsRNA
double-stranded RNA
- DTT
dithiothreitol
- ER
endoplasmic reticulum
- FM
fraction membrane
- GFP
green fluorescent protein
- plexA
plexin A
- qPCR
quantitative PCR
- RIDD
regulated Ire1-dependent decay
- RNAi
RNA interference
- sparc
BM-40-SPARC
- uORFs
upstream open reading frames
- UPR
unfolded protein response
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E12-06-0491) on November 7, 2012.
REFERENCES
- Calfon M, Zeng H, Urano F, Till JH, Hubbard SR, Harding HP, Clark SG, Ron D. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature. 2002;415:92–96. doi: 10.1038/415092a. [DOI] [PubMed] [Google Scholar]
- Chen Q, Jagannathan S, Reid DW, Zheng T, Nicchitta CV. Hierarchical regulation of mRNA partitioning between the cytoplasm and the endoplasmic reticulum of mammalian cells. Mol Biol Cell. 2011;22:2646–2658. doi: 10.1091/mbc.E11-03-0239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox JS, Shamu CE, Walter P. Transcriptional induction of genes encoding endoplasmic reticulum resident proteins requires a transmembrane protein kinase. Cell. 1993;73:1197–1206. doi: 10.1016/0092-8674(93)90648-a. [DOI] [PubMed] [Google Scholar]
- Cui XA, Zhang H, Palazzo AF. p180 promotes the ribosome-independent localization of a subset of mRNA to the endoplasmic reticulum. PLoS Biol. 2012;10:e1001336. doi: 10.1371/journal.pbio.1001336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrey JL, Lee YY, Au HH, Bushell M, Jan E. Host and viral translational mechanisms during cricket paralysis virus infection. J Virol. 2010;84:1124–1138. doi: 10.1128/JVI.02006-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han D, Lerner AG, Vande Walle L, Upton JP, Xu W, Hagen A, Backes BJ, Oakes SA, Papa FR. IRE1α kinase activation modes control alternate endoribonuclease outputs to determine divergent cell fates. Cell. 2009;138:562–575. doi: 10.1016/j.cell.2009.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding HP, Novoa I, Zhang Y, Zeng H, Wek R, Schapira M, Ron D. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol Cell. 2000;6:1099–1108. doi: 10.1016/s1097-2765(00)00108-8. [DOI] [PubMed] [Google Scholar]
- Harding HP, Zhang Y, Ron D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature. 1999;397:271–274. doi: 10.1038/16729. [DOI] [PubMed] [Google Scholar]
- Harding HP, et al. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol Cell. 2003;11:619–633. doi: 10.1016/s1097-2765(03)00105-9. [DOI] [PubMed] [Google Scholar]
- Haze K, Yoshida H, Yanagi H, Yura T, Mori K. Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol Biol Cell. 1999;10:3787–3799. doi: 10.1091/mbc.10.11.3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollien J, Lin JH, Li H, Stevens N, Walter P, Weissman JS. Regulated Ire1-dependent decay of messenger RNAs in mammalian cells. J Cell Biol. 2009;186:323–331. doi: 10.1083/jcb.200903014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollien J, Weissman JS. Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response. Science. 2006;313:104–107. doi: 10.1126/science.1129631. [DOI] [PubMed] [Google Scholar]
- Hur KY, So JS, Ruda V, Frank-Kamenetsky M, Fitzgerald K, Koteliansky V, Iwawaki T, Glimcher LH, Lee AH. IRE1α activation protects mice against acetaminophen-induced hepatotoxicity. J Exp Med. 2012;209:307–318. doi: 10.1084/jem.20111298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogh A, Larsson B, von Heijne G, Sonnhammer EL. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol. 2001;305:567–580. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- Lee YY, Cevallos RC, Jan E. An upstream open reading frame regulates translation of GADD34 during cellular stresses that induce eIF2α phosphorylation. J Biol Chem. 2009;284:6661–6673. doi: 10.1074/jbc.M806735200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore KA, Hollien J. The unfolded protein response in secretory cell function. Annu Rev Genet. 2012;46:165–183. doi: 10.1146/annurev-genet-110711-155644. [DOI] [PubMed] [Google Scholar]
- Mori K, Ma W, Gething MJ, Sambrook J. A transmembrane protein with a cdc2+/CDC28-related kinase activity is required for signaling from the ER to the nucleus. Cell. 1993;74:743–756. doi: 10.1016/0092-8674(93)90521-q. [DOI] [PubMed] [Google Scholar]
- Okada T, Yoshida H, Akazawa R, Negishi M, Mori K. Distinct roles of activating transcription factor 6 (ATF6) and double-stranded RNA-activated protein kinase-like endoplasmic reticulum kinase (PERK) in transcription during the mammalian unfolded protein response. Biochem J. 2002;366:585–594. doi: 10.1042/BJ20020391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen TN, Brunak S, von Heijne G, Nielsen H. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods. 2011;8:785–786. doi: 10.1038/nmeth.1701. [DOI] [PubMed] [Google Scholar]
- Pyhtila B, Zheng T, Lager PJ, Keene JD, Reedy MC, Nicchitta CV. Signal sequence- and translation-independent mRNA localization to the endoplasmic reticulum. RNA. 2008;14:445–453. doi: 10.1261/rna.721108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen X, et al. Complementary signaling pathways regulate the unfolded protein response and are required for C. elegans development. Cell. 2001;107:893–903. doi: 10.1016/s0092-8674(01)00612-2. [DOI] [PubMed] [Google Scholar]
- Shen X, Ellis RE, Sakaki K, Kaufman RJ. Genetic interactions due to constitutive and inducible gene regulation mediated by the unfolded protein response in C. elegans. PLoS Genet. 2005;1:e37. doi: 10.1371/journal.pgen.0010037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Vattem KM, Sood R, An J, Liang J, Stramm L, Wek RC. Identification and characterization of pancreatic eukaryotic initiation factor 2 α-subunit kinase, PEK, involved in translational control. Mol Cell Biol. 1998;18:7499–7509. doi: 10.1128/mcb.18.12.7499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens SB, Dodd RD, Lerner RS, Pyhtila BM, Nicchitta CV. Analysis of mRNA partitioning between the cytosol and endoplasmic reticulum compartments of mammalian cells. Methods Mol Biol. 2008;419:197–214. doi: 10.1007/978-1-59745-033-1_14. [DOI] [PubMed] [Google Scholar]
- Travers KJ, Patil CK, Wodicka L, Lockhart DJ, Weissman JS, Walter P. Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER-associated degradation. Cell. 2000;101:249–258. doi: 10.1016/s0092-8674(00)80835-1. [DOI] [PubMed] [Google Scholar]
- Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334:1081–1086. doi: 10.1126/science.1209038. [DOI] [PubMed] [Google Scholar]
- Wang Y, Shen J, Arenzana N, Tirasophon W, Kaufman RJ, Prywes R. Activation of ATF6 and an ATF6 DNA binding site by the endoplasmic reticulum stress response. J Biol Chem. 2000;275:27013–27020. doi: 10.1074/jbc.M003322200. [DOI] [PubMed] [Google Scholar]
- Winberg ML, Noordermeer JN, Tamagnone L, Comoglio PM, Spriggs MK, Tessier-Lavigne M, Goodman CS. Plexin A is a neuronal semaphorin receptor that controls axon guidance. Cell. 1998;95:903–916. doi: 10.1016/s0092-8674(00)81715-8. [DOI] [PubMed] [Google Scholar]
- Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell. 2001;107:881–891. doi: 10.1016/s0092-8674(01)00611-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.