

Interaction of vimentin filaments (vIFs) and force-bearing adhesions is essential for endoplasm spreading. For adhesions to be connected to a contractile network involved in endoplasm spreading, vIFs are needed. Thus endoplasm spreading and microtubule stabilization in the periphery require a multicomponent actin network anchored at adhesions.
Abstract
For cells to develop long-range forces and carry materials to the periphery, the microtubule and organelle-rich region at the center of the cell—the endoplasm—needs to extend to near the cell edge. Depletion of the actin cross-linking protein filamin A (FlnA) causes a collapse of the endoplasm into a sphere around the nucleus of fibroblasts and disruption of matrix adhesions, indicating that FlnA is involved in endoplasmic spreading and adhesion growth. Here, we report that treatment with the calpain inhibitor N-[N-(N-acetyl-l-leucyl)-l-leucyl]-l-norleucine (ALLN) restores endoplasmic spreading as well as focal adhesion (FA) growth on fibronectin-coated surfaces in a Fln-depleted background. Addback of calpain-uncleavable talin, not full-length talin, achieves a similar effect in Fln-depleted cells and indicates a crucial role for talin in endoplasmic spreading. Because FA maturation involves the vimentin intermediate filament (vIF) network, we also examined the role of vIFs in endoplasmic spreading. Wild-type cells expressing a vimentin variant incapable of polymerization exhibit deficient endoplasmic spreading as well as defects in FA growth. ALLN treatment restores FA growth despite the lack of vIFs but does not restore endoplasmic spreading, implying that vIFs are essential for endoplasm spreading. Consistent with that hypothesis, vIFs are always displaced from adhesions when the endoplasm does not spread. In Fln-depleted cells, vIFs extend beyond adhesions, nearly to the cell edge. Finally, inhibiting myosin II–mediated contraction blocks endoplasmic spreading and adhesion growth. Thus we propose a model in which myosin II–mediated forces and coalescence of vIFs at mature FAs are required for endoplasmic spreading.
INTRODUCTION
Cell motility and polarization rely on physical connections between cellular compartments to move the cell and its organelles in a given direction. These connections are the basis of the coherent cytoskeleton and allow forces to be transmitted throughout the cytoplasm (Cai and Sheetz, 2009; Cai et al., 2010). Observing the initial stages of cell spreading on a substrate reveals a well-known but poorly understood example of cellular coherence in which the endoplasm—the central, organelle-rich compartment of the cell—moves roughly in concert with the cell edge despite several microns separating the two regions. Although the displacement and structure of the cell edge are frequently studied topics, the mechanism underlying the coordinated movement of the endoplasm with the cell edge remains elusive.
Our previous work elucidated a role for filamins (Flns) in cell spreading, namely that depletion of the main Fln isoforms in fibroblasts, FlnA and FlnB, blocked endoplasmic spreading and destabilized force-bearing adhesions (Lynch et al., 2011). Force-bearing adhesions, for the purposes of this article, refer to adhesions that can support high levels of force, grow, and mature, supporting associated protrusions without collapse, as seen previously (Lynch et al., 2011). In accordance with Fln's actin cross-linking and bundling capabilities (Hartwig and Shevlin, 1986; Cunningham et al., 1992; Gorlin et al., 1993; Flanagan, 2001), we also observed dramatic disruption of the actin cytoskeleton, including a reduction in dorsal stress fibers and transverse arcs surrounding the endoplasm. Thus the physical linkage connecting structural elements of the endoplasm with adhesive structures was lost and the endoplasm did not spread. However, the endoplasmic spreading defect was also accompanied by changes in microtubule (MT) extension and guidance, as well as in deficient adhesion maturation, indicating a more complex mechanism involving multiple cytoskeletal components.
Given the numerous phenotypes associated with Fln depletion, determining a single, causative trait that leads to defective endoplasmic spreading is a difficult undertaking. To address this issue, we sought to isolate a single phenotype associated with Fln depletion, that of deficient adhesion growth (Xu et al., 2010). Adhesion growth and maturation occurs in a hierarchical manner (Zaidel-Bar et al., 2003, 2004), with many additional proteins being recruited to an adhesive structure over the course of its lifetime (Zaidel-Bar et al., 2007). Conversely, adhesion turnover involves proteolysis of adhesion proteins, such as talin (Franco et al., 2004), Fln (Gorlin et al., 1990), and paxillin (Cortesio et al., 2011), by the calcium-regulated protease calpain. Adhesion maturation and turnover therefore directly affect the size of an adhesive structure.
Another factor influencing the size of an adhesion is its association with cytoskeletal polymers. Actin stress fibers connect to maturing adhesions (Hotulainen and Lappalainen, 2006; Oakes et al., 2012), whereas MT colocalization with an adhesion tends to promote adhesion disassembly (Kaverina et al., 1999; Krylyshkina et al., 2002, 2003). A third cytoskeletal component, the vimentin intermediate filament (vIF) network, is associated with adhesion maturation and growth (Tsuruta, 2003). Thus cytoskeletal polymers differentially affect adhesion maturation and the size of an adhesive structure.
In this study, we explored the role of adhesion growth and maturation in endoplasmic spreading with both chemical and genetic means. Our findings implicate adhesion growth and vIFs in the endoplasmic spreading mechanism, with the coalescence of vIFs at adhesive structures being critical for the coherent spreading of the endoplasm.
RESULTS
N-[N-(N-Acetyl-l-leucyl)-l-leucyl]-l-norleucine restores focal adhesion growth and endoplasmic spreading in Fln-depleted mouse embryonic fibroblasts
Recent studies indicated a role for FlnA in stabilizing focal adhesions, since FlnA depletion led to increased rates of disassembly (Xu et al., 2010; Lynch et al., 2011). The turnover appeared to be protease dependent, since the calpain inhibitor N-[N-(N-acetyl-l-leucyl)-l-leucyl]-l-norleucine (ALLN) reversed the increased rates of adhesion turnover in Fln-depleted cells and subsequently restored adhesion maturation (Xu et al., 2010). Therefore we tested whether ALLN would restore adhesions in Fln-depleted mouse embryonic fibroblasts (MEFs). In this system, addition of ALLN restored normal peripheral FAs (Figure 1C) in a manner similar to the expression of full-length (FL) FlnA (Figure 1A) in Fln-depleted MEFs that had smaller adhesions (Figure 1B). Quantification showed that peripheral adhesion area in Fln-depleted MEFs was increased threefold by ALLN treatment and twofold by expressing FL FlnA (Figure 1D). A similar effect was observed with quantification of total adhesion area (Supplemental Figure S1). Adhesion growth occurred primarily during the contractile phase, P2, of cell spreading (Döbereiner et al., 2004; Figure 1E), similar to untreated wild-type (WT) MEFs.
FIGURE 1:

Calpain inhibition rescues endoplasmic spreading in Fln-depleted MEFs. (A) Fln-depleted MEFs were transfected with FL FlnA, (B) treated with DMSO, or (C) treated with 50 μM ALLN for 22 h, plated on 10 μg/ml FN-coated glass for 30 min, fixed, stained for paxillin, and imaged with confocal microscopy. GFP signal was associated with successful FlnA shRNA transfection and expression (scale, 20 μm). (D) Areas of peripheral adhesion plaques were measured manually from a single confocal slice in each condition (Fln-depleted with FL FlnA, 7 cells, 128 adhesions; Fln-depleted with DMSO, 10 cells, 305 adhesions; Fln-depleted with ALLN, 12 cells, 323 adhesions; *p < 0.001). (E) Peripheral adhesion areas were measured at various time points using the same method as in D (with DMSO, 10 min: 16 cells, 139 adhesions; with DMSO, 20 min: 15 cells, 174 adhesions; with DMSO, 30 min: 12 cells, 198 adhesions; with ALLN, 10 min: 16 cells, 295 adhesions; with ALLN, 20 min: 9 cells, 168 adhesions; with ALLN, 30 min: 10 cells, 213 adhesions). (F) Fln-depleted MEFs were transfected with FL FlnA, (G) treated with DMSO, or (H) treated with 50 μM ALLN for 22 h, plated on FN-coated glass for 30 min, and fixed (scale, 20 μm). (I) Endoplasm/whole-cell area was determined by measuring RFP-ER area and whole-cell area in fluorescence and DIC channels, respectively (with DMSO, 40 cells; with ALLN, 41 cells; three separate experiments; *p < 0.001).
If increased turnover of adhesions was linked to the Fln-depleted endoplasmic spreading defect, then treatment of Fln-depleted MEFs with ALLN should restore endoplasmic spreading. Differential interference contrast (DIC) micrographs of Fln-depleted MEFs showed a dense endoplasm clustered near the center of the cell (Figure 1G) compared with cells expressing FL FlnA (Figure 1F). On treatment with ALLN, the endoplasm spread in a manner similar to control cells (Figure 1H). Quantification of the endoplasm/whole-cell area ratio, measured as described previously (Lynch et al., 2011), showed that the ratio in Fln-depleted MEFs was increased to control levels by treatment with ALLN (Figure 1I; absolute values of endoplasm and whole-cell areas can be found in Supplemental Figure S2, A and B). Taken together, these results indicate that the defect in focal adhesion growth was correlated with the endoplasmic spreading deficiency previously reported in Fln-depleted MEFs, and Fln was not necessary for endoplasm spreading. This raised the possibility that another adhesion protein was turning over more rapidly as a result of Fln depletion.
Expression of a calpain-resistant talin variant rescues endoplasmic spreading in Fln-depleted MEFs
Because calpain-mediated proteolysis of talin was an important step in focal adhesion turnover, preventing talin cleavage may have promoted adhesion growth and endoplasmic spreading. To test this possibility, we transfected Fln-depleted MEFs with the green fluorescent protein (GFP)-calpain-uncleavable talin (NC talin), which can stabilize adhesions for further maturation (Franco et al., 2004). In Fln-depleted MEFs, expression of NC talin restored endoplasm spreading (Figure 2B), whereas expression of FL talin appeared to have no effect (Figure 2A). Endoplasm/whole-cell area ratios measured in Fln-depleted MEFs expressing NC talin were indistinguishable from those in the Fln-depleted MEFs expressing FL FlnA (Figure 2C; absolute values of endoplasm and whole-cell area can be found in Supplemental Figure 2, A and B). Like Fln-depleted MEFs treated with ALLN, expression of NC talin produced larger, more elongated adhesions (Figure 2E) than did expression of FL talin (Figure 2D). Likewise, peripheral adhesion area was significantly greater in Fln-depleted MEFs transfected with NC talin compared with Fln-depleted MEFs with or without expressed FL talin (Figure 2F). Adhesion growth and total adhesion area showed a similar trend (Figure 2G and Supplemental Figure S1). Thus, inducing adhesion growth using two distinct approaches—ALLN treatment and NC talin expression—rescued Fln-depleted endoplasmic spreading, indicating that the increase in the cleavage of talin upon Fln depletion (Xu et al., 2010) was responsible for the loss of endoplasmic spreading.
FIGURE 2:
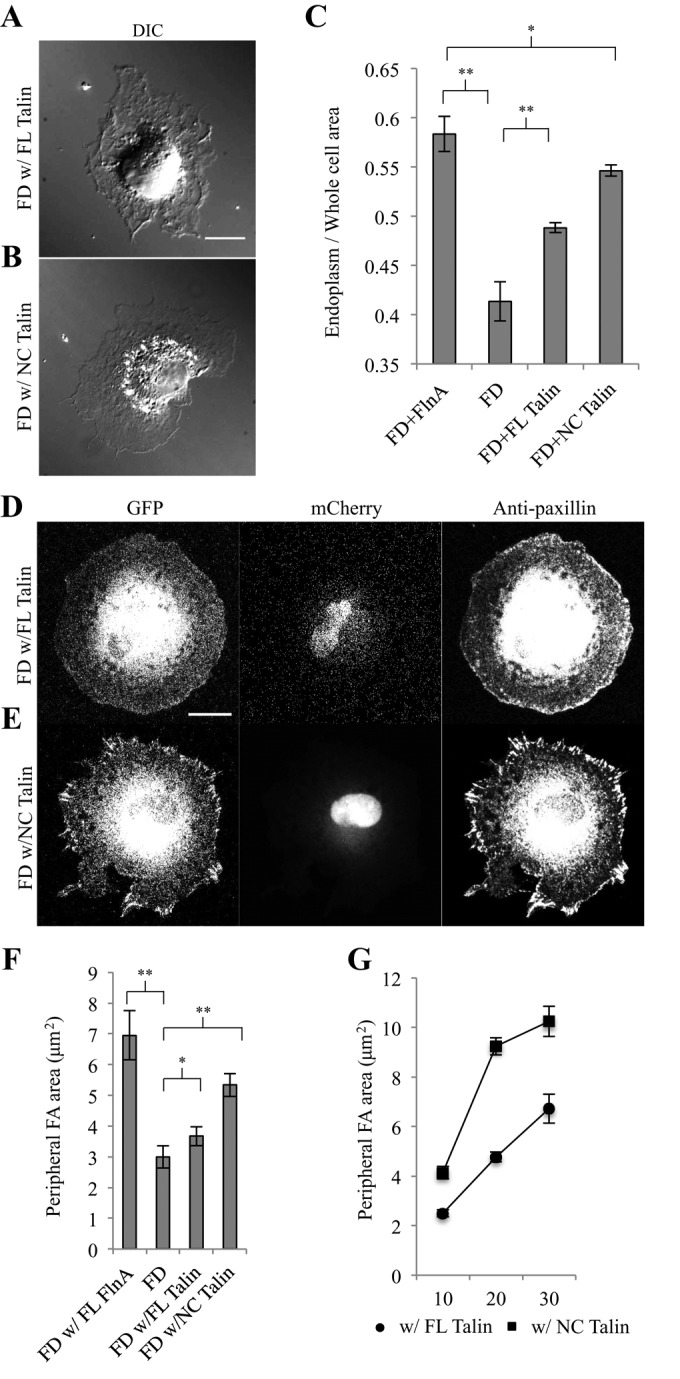
Calpain-uncleavable talin expression rescues endoplasmic spreading in Fln-depleted MEFs. (A) Fln-depleted MEFs were cotransfected with GFP-tagged FL talin or (B) calpain-uncleavable talin (NC talin), spread for 30 min, and fixed (scale, 20 μm). (C) Endoplasm/whole-cell area ratios were measured using DIC images for Fln-depleted MEFs cotransfected with GFP-tagged wild-type FlnA, FL talin, or NC talin (at least 33 cells analyzed/experimental condition; *p > 0.1, **p < 0.001). (D) Fln-depleted MEFs, expressing mCherry as an shRNA marker, were cotransfected with GFP-tagged wild-type or (E) calpain-uncleavable talin, spread on FN for 30 min, fixed, and stained for paxillin (scale, 20 μm). (F) Areas of peripheral adhesion plaques were measured manually from a single confocal slice in each condition (Fln-depleted with FL FlnA, 7 cells, 128 adhesions; Fln-depleted, 10 cells, 305 adhesions; Fln-depleted with FL talin, 11 cells, 166 adhesions; Fln-depleted with NC talin, 5 cells, 82 adhesions; *p > 0.1, **p < 0.01). (G) Peripheral adhesion areas were measured at various time points using the same method as in F (75 adhesions measured across 5 cells per time point for each cell type).
Endoplasmic spreading requires vimentin intermediate filaments
Because adhesions associated with vimentin intermediate filaments (vIFs) tend to be larger and more stable (Tsuruta, 2003; Burgstaller et al., 2010), we investigated the role of vIFs in endoplasmic spreading. We transfected wild-type control MEFs with either a full-length vimentin-GFP construct (GFP-FL Vim) or a GFP-tagged dominant-negative vimentin variant expressing only amino acids 1–138 (GFP–Vim 1-138; Chang et al., 2009). These amino acids code only for the vimentin globular head domain and a small portion of the α-helical rod domain, so their incorporation specifically into growing vIFs halts elongation. Spread cells expressing GFP–Vim 1-138 lacked polymerized vIFs (Figure 3B) compared with controls expressing GFP-FL Vim (Figure 3A). Similar to the Fln-depleted system, cells expressing GFP–Vim 1-138 were unable to spread the endoplasm (Figure 3B) and had endoplasm/whole-cell area ratios that were significantly lower than those of controls (Figure 3H; absolute values of endoplasm and whole-cell area can be found in Supplemental Figure 2, C and D). In addition, cells transfected with GFP–Vim 1-138 did not round up during the spreading process (Supplemental Figure S3). FAs were also smaller and more punctate, as in Fln-depleted MEFs (Figure 3, D and E), resulting in peripheral adhesion areas and total adhesion areas that were significantly less than for controls (Figure 3I and Supplemental Figure S1). Taken together, these results demonstrated marked phenotypic similarities between the Fln-depleted system and cells expressing GFP–Vim 1-138 and indicated that vIFs were required for endoplasmic spreading.
FIGURE 3:

Vimentin intermediate filaments are required for endoplasmic spreading. (A) RPTPα+/+ (control) MEFs were transfected with GFP–FL Vim, (B) transfected with GFP Vim 1-138, or (C) transfected with GFP–Vim 1-138 and treated with ALLN, spread for 30 min on FN-coated glass, and fixed (scale, 20 μm). (D–G) Control MEFs were transfected and spread as in A–C, with subsequent immunostaining for paxillin (scale, 20 μm). (H) Endoplasm/whole-cell ratios were measured as in Figure 2C (control, 30 cells; with FL Vim, 39 cells; with Vim1-138, 30 cells; with Vim 1-138 + ALLN, 45 cells; *p < 0.001, **p > 0.1). (I) Peripheral adhesion areas were measured manually from a single confocal slice in each condition (with FL Vim, 6 cells, 120 adhesions; with Vim 1-138, 6 cells, 120 adhesions; with Vim 1-138 + ALLN, 9 cells, 222 adhesions; *p < 0.001). (J) Peripheral adhesion plaque areas were measured at various time points using the same method as in I (with FL Vim, 10 min: 4 cells, 42 adhesions; 20 min: 6 cells, 114 adhesions; 30 min: 170 adhesions; with Vim1-138, 10, 20, 30 min: 5 cells each, 100 adhesions each).
Both vimentin intermediate filaments and mature focal adhesions are required for endoplasmic spreading
Because expression of GFP–Vim 1-138 inhibited FA growth as well as vIF assembly, there was a question of whether both or only one of these factors was sufficient for endoplasmic spreading. However, Fln-depleted MEFs have vIFs with a contracted endoplasm, indicating that vIFs are not sufficient for endoplasmic spreading. To determine whether mature FAs are sufficient for endoplasmic spreading in the absence of vIFs, we transfected wild-type MEFs with GFP–Vim 1-138 and treated them with ALLN overnight. After spreading on fibronectin (FN)-coated glass, vIFs did not form (Figure 3C), but adhesions grew and matured (Figure 3G) similar to controls (Figure 3F). Peripheral and total adhesion areas increased via a similar trend (Figure 3J and Supplemental Figure S1). Of interest, the endoplasm did not spread with ALLN addition (Figure 3C), and endoplasm/whole-cell area ratios were unchanged from the GFP–Vim 1-138–expressing cells without ALLN (Figure 3H; absolute values of endoplasm and whole-cell area can be found in Supplemental Figure S2, C and D). Thus adhesion growth was not sufficient for endoplasmic spreading. Both vIFs and large, mature FAs were needed for endoplasmic spreading to occur.
Vimentin intermediate filaments must coalesce at mature focal adhesions for efficient endoplasmic spreading
If endoplasmic spreading required both vIFs and adhesion growth, then it followed that vIFs and adhesions might associate during the endoplasmic spreading process. In control cells transfected with paxillin-GFP and stained for vimentin, vIFs coalesced at FA sites in a directed manner (Figure 4F), and vIFs contacted 75–80% of total FAs (Figure 4E). In Fln-depleted cells, vIFs extended into the periphery without coalescing at FAs (Figure 4G), and only ∼35% of FAs in Fln-depleted MEFs were in contact with vIFs (Figure 4E). To further test the role of vIFs, we treated control MEFs with withaferin A (WFA), a highly specific, vimentin-binding inhibitor of vimentin cytoskeleton spreading that allowed vIFs to form while blocking their extension to the periphery (Thaiparambil et al., 2011). In WFA-treated cells, vIFs formed but were restricted to the perinuclear region (Figure 4, B and I) compared with controls (Figure 4, A and H). As in Fln-depleted MEFs, vIFs in WFA-treated cells contacted only ∼30% of total FAs (Figure 4E). it is intriguing that WFA-treated cells also showed a defect in adhesion growth (Figure 4I) and an unspread endoplasm (Figure 4C), with endoplasm/whole-cell area ratios that were ∼30% lower than for controls (Figure 4D; absolute values of endoplasm and whole-cell area can be found in Supplemental Figure S2, C and D). Similar to transfection with GFP–Vim 1-138, treatment with WFA did not cause cells to round up during the spreading process (Supplemental Figure S3) and both vIFs and FAs were observed at early time points (Supplemental Figure S4). Thus, under multiple conditions, endoplasmic spreading was correlated with coalescence of vIFs at large, mature FAs. Cells deficient in either component were unable to spread the endoplasm.
FIGURE 4:

Endoplasmic spreading requires coalescence of vimentin intermediate filaments at mature focal adhesions. (A) RPTPα+/+ (control) MEFs were treated with DMSO or (B) 500 nM WFA overnight, spread on FN-coated glass for 30 min, fixed, stained for vimentin, and subsequently subjected to fluorescence confocal imaging (green, anti-vimentin; red, phalloidin). (C) Cells were treated with WFA as in A, fixed, and imaged in DIC. (D) Endoplasm/whole-cell area ratios were significantly lower for cells treated with WFA than controls (30 cells measured for each condition; *p < 0.001). (E) Each cell type was plated on FN-coated glass for 30 min, fixed, and stained for vimentin and paxillin. FA plaques colocalizing with vIF tips were quantified as a percentage of total FA plaques (control, 13 cells; Fln-depleted, 13 cells; control with DMSO, 13 cells; control with WFA, 13 cells; *p < 0.001). (F–I) Sample images used to generate E. Cells were prepared as described in E and subjected to confocal imaging.
Microtubules and RhoA/Rho-associated, coiled-coil–containing protein kinase–dependent cytoskeletal tension are necessary but not independently sufficient for vIFs extending past FAs in Fln-depleted MEFs
Because there appeared to be a need for association of vIFs with FAs, we carefully examined vIF distribution in Fln-depleted MEFs and their controls. Fln-depleted MEFs had a more extensive vIF network, covering a larger fraction of the cell area (Figure 5B), compared with controls (Figure 5A). The average distance between the cell edge and areas containing vIFs was nearly half that of controls in Fln-depleted MEFs (Figure 5G). Much of the vIF network extended to regions without FAs in the absence of Fln. Given that the extent of vIF spread area is dependent on the extent of MT spread area (Colakoglu and Brown, 2009; Burgstaller et al., 2010), we observed MTs in FlnA−/− MEFs from the start of spreading (Figure 5H). MTs extended to near the cell edge at early time points but were retracted from the edge after ∼10 min of spreading (Figure 5I), which is in agreement with our previous results (Lynch et al., 2011).
FIGURE 5:

VIFs extend closer to the cell edge in Fln-depleted MEFs in a microtubule-dependent manner. (A–F) In each condition, MEFs were plated on FN-coated glass for 30 min, fixed, and stained for vimentin and phalloidin, with GFP signal corresponding with shRNA expression. Cells treated with nocodazole and/or Y-27632 were exposed 30 min before spreading (scale, 20 μm). (G) Distances of vIFs to the cell edge were determined by averaging the distance from the vIF boundary to the cell edge at 10 points in the cell, using at least six cells per cell type (*p < 0.001, **p < 0.05). (H) FlnA−/− MEFs were transfected with p3xGFP-EMTB and visualized in TIRF. Shown is the initial stage of spreading (∼2 min from initiation) (scale bar, 20 μm). (I) Kymograph generated across white line from F. Yellow dotted line indicates MT boundary. Note that MTs stay confined even as the cell continues to spread after ∼10 min; scale, 20 μm, 5 min.
Nocodazole reversed the overextension of vIFs in Fln-depleted cells (Figure 5D) compared with controls (Figure 5C), causing confinement of vIFs to the cell center and increasing the distance between vIFs and the cell edge (Figure 5G). Because nocodazole is both an inhibitor of MT polymerization and an activator of RhoA/Rho-associated, coiled-coil–containing protein kinase (ROCK; Chang et al., 2008), we treated Fln-depleted MEFs with nocodazole and Y-27632, a ROCK inhibitor, simultaneously and observed a phenotype similar to that seen in Fln-depleted MEFs treated with nocodazole alone (Figure 5, E and G). Thus MTs are necessary for the overextension of vIFs in Fln-depleted MEFs.
Of interest, treating Fln-depleted MEFs with Y-27632 alone also reversed the overextension of vIFs in Fln-depleted MEFs (Figure 5, F and G). Therefore ROCK-dependent activation of myosin and the resulting cytoskeletal tension is also necessary for overextension of vIFs in Fln-depleted MEFs.
Endoplasmic spreading requires myosin II contractile activity
Because cytoskeletal tension was affecting coalescence of vIFs at FAs, we sought to determine whether contraction played a role in endoplasmic spreading. We allowed control MEFs to spread in the presence of blebbistatin, a myosin II inhibitor (Straight et al., 2003), and found that the endoplasm was not spread after 30 min (Figure 6A). Endoplasm/whole-cell area ratios in blebbistatin-treated cells were lower than for controls (Figure 6B; absolute values of endoplasm and whole cell area can be found in Supplemental Figure S2, C and D). In accordance with the endoplasmic spreading deficiency observed in Fln-depleted MEFs, cells expressing GFP–Vim 1-138, and WFA-treated cells, this deficiency was coupled with a defect in adhesion growth (Figure 6D) compared with controls (Figure 6C). Thus, inhibiting myosin II blocked endoplasmic spreading, indicating that force generation, in addition to coalescence of vIFs at FAs, was necessary for endoplasmic spreading.
FIGURE 6:
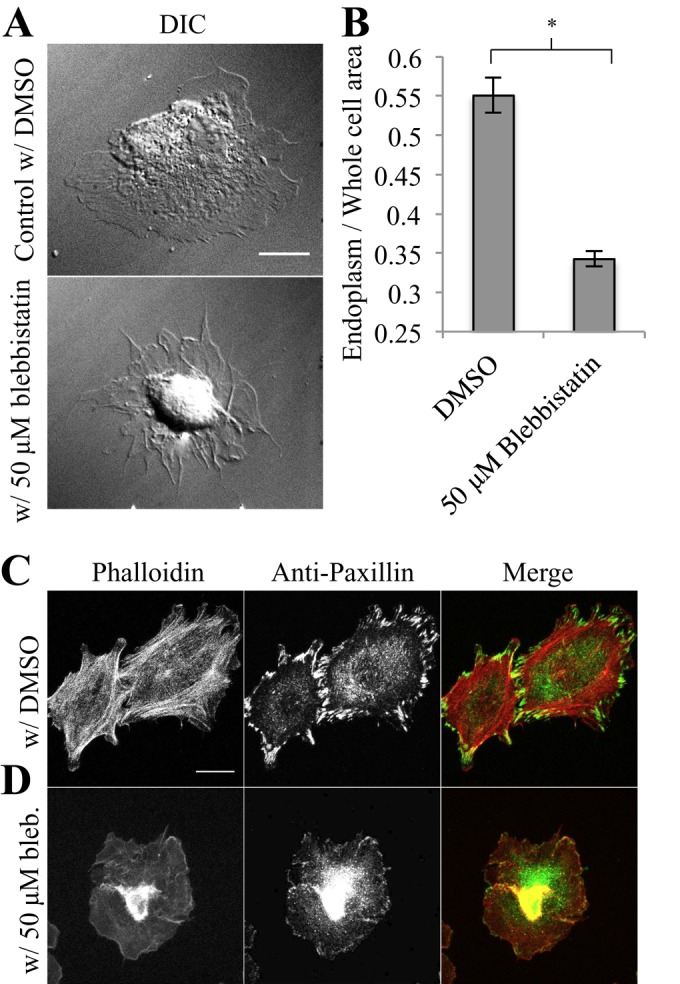
Endoplasmic spreading requires myosin II–mediated contraction. (A) Control MEFs were treated with 50 μM blebbistatin or an equal volume of DMSO and plated on 10 μg/μl FN for 30 min (scale, 20 μm). (B) Endoplasm/whole-cell ratios show a reduction in endoplasmic spreading upon blebbistatin treatment (50 cells/cell type, n = 2 experiments, *p < 0.01). (C) Cells were treated and spread as in A and then fixed and stained for phalloidin and paxillin (scale, 20 μm).
DISCUSSION
These findings indicate that the effect of Fln depletion upon endoplasm spreading that was reported earlier (Lynch et al., 2011) may be indirect. Indeed, either ALLN or NC talin rescues endoplasmic spreading in a Fln-depleted background (Figures 1I and 2C). Thus Fln levels are not necessary for endoplasmic spreading, but large, mature adhesions are critical, and those require contractile force to form (Figure 6D). In addition, endoplasm spreading appears to depend upon the coalescence of vIFs at these force-bearing adhesions. We show that vIFs are associated with FAs when the endoplasm spreads, and disruption of vIFs or their associations with FAs leaves the endoplasm unspread. Thus we suggest that both force-bearing adhesions and the coalescence of vIFs to the adhesions are needed for endoplasmic spreading.
In a model suggested recently (Xu et al., 2010), mature adhesions can form only when there is competition between talin and Fln for calpain-mediated proteolysis. When Fln levels are depleted, calpain quickly turns over talin at the adhesion, precluding further maturation and growth. Adding FL FlnA back to the Fln-depleted system restores talin–Fln competition for calpain proteolysis, thereby dividing calpain cleavage between talin and Fln and allowing further adhesion growth. Inhibiting calpain cleavage in Fln-depleted cells allows further growth because talin now has a longer half-life at the adhesion. Overexpressing NC talin achieves the same result but bypasses the calpain cleavage step. Of interest, FL talin expression does not rescue the Fln-depleted endoplasmic spreading defect (Figure 2, A and C). This could be explained by suggesting that only the talin in adhesions is relevant and/or the cleavage of talin leads to irreversible steps that prevent adhesion growth. It seems unlikely that ALLN treatment of Fln-depleted cells or overexpression of NC talin would make the ∼15% remaining cellular FlnA (Lynch et al., 2011) sufficient to allow adequate adhesion maturation to occur. Thus adhesion growth and some of the many proteins involved in that process are needed for endoplasm spreading, perhaps through the anchorage of vIFs.
The effects of vIF disruption have been reported in a number of systems, including a loss of polarization and a raised central cellular region (Ivanova et al., 1976; Eckert, 1986; Goldman et al., 1996). Our results with GFP–Vim 1-138 quantify the latter effect and correlate it with the inhibition of endoplasmic spreading (Figure 3H), and reduced endoplasmic spreading necessarily results in a thicker central cellular region. In addition, vIFs promote adhesion maturation and stabilization (Tsuruta, 2003), and we find that GFP–Vim 1-138–transfected cells have decreased adhesions, similar to Fln-depleted cells and Fln-depleted cells transfected with FL talin (Figure 3, H and I). This raises the question of whether vIF disassembly is acting through its inhibition of adhesion growth or another aspect such as the mechanical role that vIFs can play. It seems that adhesion growth is not the critical element, since ALLN treatment caused adhesions to grow in the absence of vIFs (Figure 3G), and endoplasmic spreading was not rescued (Figure 3H). Therefore neither the presence of vIFs nor adhesion growth is sufficient for endoplasmic spreading on its own. This raises the point that an integration of the vIFs and the adhesions may be critical.
The necessity for both vIFs and mature FAs in the endoplasmic spreading mechanism leads to the logical conclusion that they most likely associate in some way during the process. This interaction has been shown in other systems (Bershadsky et al., 1987; Kreis et al., 2005) but has never been approached in the context of endoplasmic spreading. The GFP–Vim 1-138 system was unsuitable to probe this issue, as vIFs do not polymerize in the presence of this variant. WFA treatment provided an alternative, since WFA inhibited endoplasmic spreading (Figure 4C) and adhesion growth (Figure 4I) and yet still allowed vIF formation (Figure 4B). Even so, vIFs did not interact with adhesions in WFA-treated cells (Figure 4I). Further, the vIFs do not coalesce at adhesions in Fln-depleted cells (Figure 4G) but extend beyond them to the cell periphery. Thus we suggest that the coalescence of vIFs at FAs is required for endoplasmic spreading.
The association of vIFs with adhesions could be through several different proteins, including plectins, zyxin, or Fln. Further, because vimentin filament fragments are transported along MTs and aggregate at adhesions before they form vIFs (Burgstaller et al., 2010), disruption of the MT-adhesion interaction could disrupt proper vIF–adhesion association. Plectins are members of the plakin family of proteins, which bind to all cytoskeletal filaments (MTs, IFs, and actin filaments), as well as to integrins (Jefferson et al., 2004), making them ideal candidates for linking the vIFs to the adhesions. However, we did not observe plectin isoform P1f in any of our wild-type or experimental systems (unpublished data). Alternatively, there are other proteins that associate with mature adhesions and bind vimentin that could link vIFs to adhesions. Zyxin is one such candidate (Lee et al., 2004); however, we detected no differences in zyxin localization between Fln-depleted cells and their controls (unpublished data). Another protein that associates with both adhesions and vimentin is Fln (Kim et al., 2010a, b). Because endoplasm spreading was rescued in Fln-depleted cells by ALLN treatment and NC talin expression, it appears that Fln is dispensable for endoplasmic spreading and adhesion growth. However, Fln-depleted cells still express ∼15% of cellular FlnA levels (as well as relatively small quantities of FlnC; Baldassarre et al., 2009), and, though unlikely, the treatments could potentially inhibit the turnover of the remaining Fln to enable proper adhesion development. This is an area that needs further study and could be aided by superresolution analyses of the adhesion structure.
Because endoplasm spreading appears to involve significant force generation (Lynch et al., 2011), as does adhesion maturation, we tested whether myosin II contraction is necessary for spreading of the endoplasm. Indeed, blebbistatin treatment blocks endoplasmic spreading and adhesion growth (Figure 6). The essential association of vIFs with large, mature adhesions strengthens the connection between contractile elements surrounding the endoplasm and adhesions, thus allowing force generation and subsequent spreading of the endoplasm. As presented previously (Burgstaller et al., 2010; Lynch et al., 2011), a cage-like structure of actin interspersed with IFs surrounds the endoplasm. MTs typically extend toward the periphery as MT motors transport vimentin fragments outward. As adhesions begin to grow, a vimentin-binding protein associated with mature adhesions can anchor the aggregation of vIFs at adhesions and subsequent elongation toward the central cage along with actin filaments. Once vIFs interact with the central cage, any contraction of myosin attached to the cage on actin filaments from adhesions or in connected stress fibers will result in endoplasmic spreading through such strong, stable connections between the cage and the substrate. Thus we suggest that actin filaments anchored at force-bearing adhesions are needed for endoplasmic spreading, and the vIFs could be involved in developing the proper anchorage (Figure 7).
FIGURE 7:

Endoplasmic spreading is a continuous process dependent on coalescence of vimentin filaments at force-bearing adhesions. A cage-like structure surrounds the endoplasm from the beginning of spreading. As microtubules migrate outward, vimentin fragments are transported out to early, maturing adhesions. Vimentin fragments collect at growing adhesions, subsequently forming vimentin filaments that can interact with the central endoplasmic cage. Myosin II contraction in the endoplasmic cage and associated actomyosin structures causes the endoplasm to pull forward on the vIF-strengthened connection to the growing adhesion, resulting in further growth. Vimentin fragments continue to be transported to the periphery on microtubule tracks toward the next layer of growing adhesions. Vimentin filaments collect at the next round of adhesions, connecting with the spreading endoplasmic cage. The process repeats, allowing the endoplasm to spread in any direction that the cell spreads.
Although some flattening of the endoplasm occurs during the initial spreading of cells before major contractions (Lynch et al., 2011), the major increase in endoplasm area is driven by myosin contraction. Several steps are required to build the contractile network to spread the endoplasm, including the initial spreading of MTs to transport vIF subunits to the adhesions (Figure 5) and adhesion growth with vIFs and links to the contractile network. At later times, the continuous extension of MTs into the periphery promotes a new round of vimentin aggregation at more peripheral, “younger” adhesions. These adhesions would then connect with the now more expansive central cage, supporting contractile elements that exert force on the substrate and spreading the endoplasm further (Figure 7). Of interest, this model might also provide an explanation for the plateauing of endoplasm spreading at ∼60–70% of cell area because the endoplasm would only spread into regions exhibiting mature adhesions and vIFs.
In summary, these findings provide a deeper understanding of the steps involved in endoplasmic spreading. Numerous studies documented aspects of adhesion maturation and vIF assembly that are shown here to be important for endoplasmic spreading. Many questions remain, however, including how the contractile network interacts with the vIFs in the endoplasm cage and with growing adhesions at early stages of spreading as well as at later times. We feel that the lack of endoplasm spreading and physical separation of the adhesions and endoplasm can cause many indirect effects on cell functions such as compromising signaling processes that occur through peripheral adhesions and the trapping of secretory vesicles in the endoplasm.
MATERIALS AND METHODS
Antibodies and reagents
Mouse monoclonal anti-paxillin, mouse monoclonal anti-vimentin, rhodamine–phalloidin, and anti-zyxin were purchased from BD Biosciences (San Diego, CA), Abcam (Cambridge, MA), Molecular Probes (Eugene, OR), and Abcam, respectively. Anti-plectin P1f was obtained from Gerhard Wiche (University of Vienna, Vienna, Austria; Burgstaller et al., 2010). ALLN, WFA, blebbistatin, and nocodazole were all purchased from Sigma-Aldrich (St. Louis, MO).
Constructs and transfection
pSilencer H1-3.1 puro expression vector (Ambion, Austin, TX) was used as described previously (Cai et al., 2006) but targeting Mus musculus flnA with the sequence 5′-CCATACTTACTGTATCCGA-3′. Insert design was conducted using the pSilencer insert design tool on the Ambion website. For transfection detection, this vector was engineered to express mCherry in place of GFP. pRFP-ER was obtained from Clontech (Mountain View, CA). FL and NC paxillin-GFP, as well as FL/NC talin-GFP, were obtained from Anna Huttenlocher (University of Wisconsin-Madison, Madison, WI; Franco et al., 2004; Cortesio et al., 2011). FL/NC FlnA-GFP was obtained from Donald Ingber (Children's Hospital and Harvard Medical School, Boston, MA; Mammoto et al., 2007). Full-length GFP-Vim and GFP–Vim 1-138 were obtained from Robert Goldman (Northwestern University, Evanston, IL; Chang et al., 2009). pEGFP-3xEMTB was obtained from J. Chloe Bulinski (Columbia University, New York, NY). Transfections were accomplished using the Nucleofector system (Lonza, Walkersville, MD). FlnB−/− MEFs were transfected according to the manufacturer's protocol, incubated for 1 d, exposed to puromycin at 1.75 μg/ml (Sigma-Aldrich) in DMEM for 3 d, and used for experimentation. Transfected cells were selected for analysis based on GFP or mCherry expression, depending on the short hairpin RNA (shRNA) vector used. Control cells were allowed at least 24 h of recovery time before trypsinization and spreading. Transfected cells were selected for analysis based on expression of fluorescent protein if applicable or randomly if used as an untransfected control.
Cell culture
FlnA−/−, FlnB−/−, and RPTPα+/+ (wild-type control) MEFs were maintained in high-glucose DMEM (Life Technologies, Carlsbad, CA) plus 10% fetal bovine serum (Life Technologies) at 37°C.
Chemical treatment
Fln-depleted and negative control MEFs, as well as RPTPα+/+ MEFs (control) transfected with GFP-Vim and GFP–Vim 1-138 were incubated in 50 μM ALLN in dimethyl sulfoxide (DMSO) in serum media (fetal bovine serum plus DMEM) for 22 h before experimentation. Fln-depleted MEFs were incubated with 10 μM nocodazole (an equivalent volume of DMSO for control cells) in serum-free media for 30 min. Control MEFs were incubated with 50 μM blebbistatin (an equivalent volume of DMSO for control cells) in serum-free media for 30 min. Control MEFs were incubated with 500 nM WFA (an equivalent volume of DMSO for control cells) in serum media for 22 h. Cell spreading assays were then conducted with either live-cell imaging or fixation and immunostaining.
Coverglass treatment and spreading assay preparation
On the day of the experiment, silanized coverglasses were treated with a droplet of FN (Roche) at 10 μg/ml and were incubated at 37°C for 1 h. Meanwhile, cells were trypsinized and resuspended in serum-free DMEM (without phenol red; Life Technologies) for 30 min before experimentation. DIC and epifluorescence microscopy were performed on an Olympus IX70 microscope (Olympus, Center Valley, PA) mounted with a PlanApo 60×/1.40 numerical aperture (NA) objective (Olympus) and a Roper Scientific Cool-Snap FX cooled charge-coupled device camera (Photometrics, Tucson, AZ). Imaging software used was SimplePCI (Hamamatsu Corporation, Sewickley, PA).
Immunostaining and microscopy
Cells were fixed with 3.7% formaldehyde in phosphate-buffered saline (PBS), quenched with 50 mM ammonium chloride in PBS, and permeabilized with 0.1% Triton X-100 in PBS. Primary and secondary antibody stainings were performed at 37°C for 1.5 h each. Confocal imaging was completed on either an Olympus IX81 microscope with a UPlanApo 60×/1.40 NA oil immersion objective and FluoView imaging software (Olympus) or a Zeiss LSM 700 microscope with a Zeiss Plan-Apochromat 63 × 1.4 NA oil DIC objective and Zeiss Zen Imaging Suite imaging software (Carl Zeiss, Jena, Germany). For time-course assays, immunostaining was begun after 10, 20, and 30 min. For live-cell total internal reflection fluorescence (TIRF)imaging, an Olympus IX81 microscope with a Plan Apo 60×/1.45 NA oil immersion TIRF objective mounted with a Cascade II camera (Photometrics) was used.
Image analysis and statistics
Endoplasm/whole-cell area ratios were measured by the method previously reported (Lynch et al., 2011). In brief, endoplasm area was defined as the central cellular region seen in DIC that contains the cell's organelles and vesicles. Whole-cell area was determined by tracing the cell edge in ImageJ (National Institutes of Health, Bethesda, MD). Both areas were measured using the Area function in ImageJ. Focal adhesion areas were analyzed by hand using the Area measurement tool in ImageJ. Paxillin signals <0.5 μm2 were not considered. Between 10 and 15 representative peripheral FA areas per cell were quantified after thresholding. Measurements of peripheral FA area refer to the average area of individual peripheral adhesions, and measurements of total FA area refer to the total adhesive area of the cell after thresholding and subtraction of background signal from the perinuclear region. Determination of vIF connection with FAs was quantified by summing the total adhesions per cell and assessing how many were contacted by vIFs. The ratio of those FAs that colocalized with a vimentin IF to the total number of FAs was then calculated. Average distance between vIFs and the cell edge was assessed by measuring 10 regions around each cell for the distance from the tip of a vIF to the cell edge (orthogonal to the cell edge). All experiments were carried out at least three times unless otherwise noted. All p values were generated using the two-tailed Student's t test, and all graphs were generated in Excel (Microsoft, Redmond, WA).
Supplementary Material
Acknowledgments
We thank Yuanyi Feng and Christopher Walsh for FlnA−/− MEFs, Xianghua Zhou and Levent Akyürek for FlnB−/− MEFs, Akiko Mammoto and Donald Ingber for the pEGFP-FL FlnA plasmid, Anna Huttenlocher for FL/NC talin-GFP and FL paxillin plasmids, Robert Goldman for GFP–FL Vim and GFP–Vim 1-138 plasmids, J. Chloe Bulinski for the pEGFP-3xEMTB plasmid, Michael Partridge for the pLNCX-paxillin-RFP plasmid, and Gerhard Wiche for anti-P1f antibody. We also thank all past and present members of the Sheetz laboratory who assisted with our study.
Abbreviations used:
- ALLN
N-[N-(N-acetyl-l-leucyl)-l-leucyl]-l-norleucine
- DIC
differential interference contrast
- DMSO
dimethyl sulfoxide
- EMTB
ensconsin microtubule binding
- ER
endoplasmic reticulum
- FA
focal adhesion
- FL
full length
- Fln
filamin
- FN
fibronectin
- GFP
green fluorescent protein
- IF
intermediate filament
- MEF
mouse embryonic fibroblast
- MT
microtubule
- NC
calpain-noncleavable
- RFP
red fluorescent protein
- ROCK
RhoA/Rho-associated, coiled coil–containing protein kinase
- RPTP
receptor protein tyrosine phosphatase
- shRNA
short hairpin RNA
- Vim
vimentin
- vIF
vimentin intermediate filament
- WFA
withaferin A
- WT
wild type
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E12-05-0377) on October 31, 2012.
*These authors contributed equally to this work.
REFERENCES
- Baldassarre M, Razinia Z, Burande CF, Lamsoul I, Lutz PG, Calderwood DA. Filamins regulate cell spreading and initiation of cell migration. PLoS One. 2009;4:e7830. doi: 10.1371/journal.pone.0007830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bershadsky AD, Tint IS, Svitkina TM. Association of intermediate filaments with vinculin-containing adhesion plaques of fibroblasts. Cell Motil Cytoskeleton. 1987;8:274–283. doi: 10.1002/cm.970080308. [DOI] [PubMed] [Google Scholar]
- Burgstaller G, Gregor M, Winter L, Wiche G. Keeping the vimentin network under control: cell-matrix adhesion-associated plectin 1f affects cell shape and polarity of fibroblasts. Mol Biol Cell. 2010;21:3362–3375. doi: 10.1091/mbc.E10-02-0094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Y, et al. Nonmuscle myosin IIA-dependent force inhibits cell spreading and drives F-actin flow. Biophys J. 2006;91:3907–3920. doi: 10.1529/biophysj.106.084806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Y, Rossier O, Gauthier NC, Biais N, Fardin M-A, Zhang X, Miller LW, Ladoux B, Cornish VW, Sheetz MP. Cytoskeletal coherence requires myosin-IIA contractility. J Cell Sci. 2010;123:413–423. doi: 10.1242/jcs.058297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Y, Sheetz MP. Force propagation across cells: mechanical coherence of dynamic cytoskeletons. Curr Opin Cell Biol. 2009;21:47–50. doi: 10.1016/j.ceb.2009.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang L, Barlan K, Chou Y-H, Grin B, Lakonishok M, Serpinskaya AS, Shumaker DK, Herrmann H, Gelfand VI, Goldman RD. The dynamic properties of intermediate filaments during organelle transport. J Cell Sci. 2009;122:2914–2923. doi: 10.1242/jcs.046789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang Y-C, Nalbant P, Birkenfeld J, Chang Z-F, Bokoch GM. GEF-H1 couples nocodazole-induced microtubule disassembly to cell contractility via RhoA. Mol Biol Cell. 2008;19:2147–2153. doi: 10.1091/mbc.E07-12-1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colakoglu G, Brown A. Intermediate filaments exchange subunits along their length and elongate by end-to-end annealing. J Cell Biol. 2009;185:769–777. doi: 10.1083/jcb.200809166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortesio CL, Boateng LR, Piazza TM, Bennin DA, Huttenlocher A. Calpain-mediated proteolysis of paxillin negatively regulates focal adhesion dynamics and cell migration. J Biol Chem. 2011;286:9998–10006. doi: 10.1074/jbc.M110.187294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham CC, Gorlin JB, Kwiatkowski DJ, Hartwig JH, Janmey PA, Byers HR, Stossel TP. Actin-binding protein requirement for cortical stability and efficient locomotion. Science. 1992;255:325–327. doi: 10.1126/science.1549777. [DOI] [PubMed] [Google Scholar]
- Döbereiner H-G, Dubin-Thaler B, Giannone G, Xenias HS, Sheetz MP. Dynamic phase transitions in cell spreading. Phys Rev Lett. 2004;93:108105. doi: 10.1103/PhysRevLett.93.108105. [DOI] [PubMed] [Google Scholar]
- Eckert B. Alteration of the distribution of intermediate filaments in PtK1 cells by acrylamide II: effect on the organization of cytoplasmic organelles. Cell Motil Cytoskeleton. 1986;6:15–24. doi: 10.1002/cm.970060104. [DOI] [PubMed] [Google Scholar]
- Flanagan LA. Filamin A, the Arp2/3 complex, and the morphology and function of cortical actin filaments in human melanoma cells. J Cell Biol. 2001;155:511–518. doi: 10.1083/jcb.200105148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco SJ, Rodgers MA, Perrin BJ, Han J, Bennin DA, Critchley DR, Huttenlocher A. Calpain-mediated proteolysis of talin regulates adhesion dynamics. Nat Cell Biol. 2004;6:977–983. doi: 10.1038/ncb1175. [DOI] [PubMed] [Google Scholar]
- Goldman RD, Khuon S, Chou YH, Opal P, Steinert PM. The function of intermediate filaments in cell shape and cytoskeletal integrity. J Cell Biol. 1996;134:971–983. doi: 10.1083/jcb.134.4.971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorlin JB, Henske E, Warren ST, Kunst CB, D'Urso M, Palmieri G, Hartwig JH, Bruns G, Kwiatkowski DJ. Actin-binding protein (ABP-280) filamin gene (FLN) maps telomeric to the color vision locus (R/GCP) and centromeric to G6PD in Xq28. Genomics. 1993;17:496–498. doi: 10.1006/geno.1993.1354. [DOI] [PubMed] [Google Scholar]
- Gorlin JB, Yamin R, Egan S, Stewart M, Stossel TP, Kwiatkowski DJ, Hartwig JH. Human endothelial actin-binding protein (ABP-280, nonmuscle filamin): a molecular leaf spring. J Cell Biol. 1990;111:1089–1105. doi: 10.1083/jcb.111.3.1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartwig JH, Shevlin P. The architecture of actin filaments and the ultrastructural location of actin-binding protein in the periphery of lung macrophages. J Cell Biol. 1986;103:1007–1020. doi: 10.1083/jcb.103.3.1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotulainen P, Lappalainen P. Stress fibers are generated by two distinct actin assembly mechanisms in motile cells. J Cell Biol. 2006;173:383–394. doi: 10.1083/jcb.200511093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanova O, Margolis L, Vasiliev J, Gelfand I. Effect of colcemid on the spreading of fibroblasts in culture. Exp Cell Res. 1976;101:207–219. doi: 10.1016/0014-4827(76)90431-6. [DOI] [PubMed] [Google Scholar]
- Jefferson JJ, Leung CL, Liem RKH. Plakins: goliaths that link cell junctions and the cytoskeleton. Nat Rev Mol Cell Biol. 2004;5:542–553. doi: 10.1038/nrm1425. [DOI] [PubMed] [Google Scholar]
- Kaverina I, Krylyshkina O, Small JV. Microtubule targeting of substrate contacts promotes their relaxation and dissociation. J Cell Biol. 1999;146:1033–1044. doi: 10.1083/jcb.146.5.1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, Nakamura F, Lee W, Hong C, Pérez-Sala D, McCulloch CA. Regulation of cell adhesion to collagen via beta1 integrins is dependent on interactions of filamin A with vimentin and protein kinase C epsilon. Exp Cell Res. 2010a;316:1829–1844. doi: 10.1016/j.yexcr.2010.02.007. [DOI] [PubMed] [Google Scholar]
- Kim H, Nakamura F, Lee W, Shifrin Y, Arora P, McCulloch CA. Filamin A is required for vimentin-mediated cell adhesion and spreading. Am J Physiol Cell Physiol. 2010b;298:C221–C236. doi: 10.1152/ajpcell.00323.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreis S, Schönfeld H-J, Melchior C, Steiner B, Kieffer N. The intermediate filament protein vimentin binds specifically to a recombinant integrin alpha2/beta1 cytoplasmic tail complex and co-localizes with native alpha2/beta1 in endothelial cell focal adhesions. Exp Cell Res. 2005;305:110–121. doi: 10.1016/j.yexcr.2004.12.023. [DOI] [PubMed] [Google Scholar]
- Krylyshkina O, Anderson KI, Kaverina I, Upmann I, Manstein DJ, Small JV, Toomre DK. Nanometer targeting of microtubules to focal adhesions. J Cell Biol. 2003;161:853–859. doi: 10.1083/jcb.200301102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krylyshkina O, Kaverina I, Kranewitter W, Steffen W, Alonso MC, Cross RA, Small JV. Modulation of substrate adhesion dynamics via microtubule targeting requires kinesin-1. J Cell Biol. 2002;156:349–359. doi: 10.1083/jcb.200105051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee NPY, Mruk DD, Conway AM, Cheng CY. Zyxin, axin, and Wiskott-Aldrich syndrome protein are adaptors that link the cadherin/catenin protein complex to the cytoskeleton at adherens junctions in the seminiferous epithelium of the rat testis. J Androl. 2004;25:200–215. doi: 10.1002/j.1939-4640.2004.tb02780.x. [DOI] [PubMed] [Google Scholar]
- Lynch CD, Gauthier NC, Biais N, Lazar AM, Roca-Cusachs P, Yu C-H, Sheetz MP. Filamin depletion blocks endoplasmic spreading and destabilizes force-bearing adhesions. Mol Biol Cell. 2011;22:1263–1273. doi: 10.1091/mbc.E10-08-0661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mammoto A, Huang S, Ingber DE. Filamin links cell shape and cytoskeletal structure to Rho regulation by controlling accumulation of p190RhoGAP in lipid rafts. J Cell Sci. 2007;120:456–467. doi: 10.1242/jcs.03353. [DOI] [PubMed] [Google Scholar]
- Oakes PW, Beckham Y, Stricker J, Gardel ML. Tension is required but not sufficient for focal adhesion maturation without a stress fiber template. J Cell Biol. 2012;196:363–374. doi: 10.1083/jcb.201107042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straight AF, Cheung A, Limouze J, Chen I, Westwood NJ, Sellers JR, Mitchison TJ. Dissecting temporal and spatial control of cytokinesis with a myosin II inhibitor. Science. 2003;299:1743–1747. doi: 10.1126/science.1081412. [DOI] [PubMed] [Google Scholar]
- Thaiparambil JT, et al. Withaferin A inhibits breast cancer invasion and metastasis at sub-cytotoxic doses by inducing vimentin disassembly and serine 56 phosphorylation. Int J Cancer. 2011;129:2744–2755. doi: 10.1002/ijc.25938. [DOI] [PubMed] [Google Scholar]
- Tsuruta D. The vimentin cytoskeleton regulates focal contact size and adhesion of endothelial cells subjected to shear stress. J Cell Sci. 2003;116:4977–4984. doi: 10.1242/jcs.00823. [DOI] [PubMed] [Google Scholar]
- Xu Y, et al. Filamin A regulates focal adhesion disassembly and suppresses breast cancer cell migration and invasion. J Exp Med. 2010;207:2421–2437. doi: 10.1084/jem.20100433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaidel-Bar R, Ballestrem C, Kam Z, Geiger B. Early molecular events in the assembly of matrix adhesions at the leading edge of migrating cells. J Cell Sci. 2003;116:4605–4613. doi: 10.1242/jcs.00792. [DOI] [PubMed] [Google Scholar]
- Zaidel-Bar R, Cohen M, Addadi L, Geiger B. Hierarchical assembly of cell-matrix adhesion complexes. Biochem Soc Trans. 2004;32:416–420. doi: 10.1042/BST0320416. [DOI] [PubMed] [Google Scholar]
- Zaidel-Bar R, Itzkovitz S, Ma'ayan A, Iyengar R, Geiger B. Functional atlas of the integrin adhesome. Nat Cell Biol. 2007;9:858–867. doi: 10.1038/ncb0807-858. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.